Metabolic Markers
![]()
In this tutorial, we will walk you through how to identify metabolic task markers from single-cell data. We will use different approaches to detect important metabolic tasks that characterize distinct cell types in your dataset.
Here, we will use the results we previously generated for the Human Endometrial Cell Atlas (HECA) dataset (Mareckova & Garcia-Alonso et al 2023) by running our Quick Start Tutorial.
This tutorial includes following steps:
Loading libraries
[1]:
import sccellfie
import scanpy as sc
import pandas as pd
import numpy as np
import matplotlib.pyplot as plt
import seaborn as sns
import glasbey
import textwrap
## To avoid warnings
import warnings
warnings.filterwarnings("ignore")
In addition, we set up a folder to save our figures. This folder is stored in the settings of Scanpy:
[2]:
sc.settings.figdir = './results/marker-figures/'
Loading endometrium results
We start opening the results previously generated by running the scCellFie pipeline on the HECA dataset. If you haven’t run the pipeline yet, please follow this tutorial.
In this case, we will load the objects that were present in results['adata'] in that tutorial. This object contains:
results['adata']: contains gene expression in.X.results['adata'].layers['gene_scores']: contains gene scores as in the original CellFie paper.results['adata'].uns['Rxn-Max-Genes']: contains determinant genes for each reaction per cell.results['adata'].reactions: contains reaction scores in.Xso every scanpy function can be used on this object to visualize or compare values.results['adata'].metabolic_tasks: contains metabolic task scores in.Xso every scanpy function can be used on this object to visualize or compare values.
Here, we will name this object directly as adata. Each of the previous elements should be under adata., as for example adata.metabolic_tasks.
[3]:
adata = sccellfie.io.load_adata(folder='./results/',
filename='Human_HECA_scCellFie'
)
./results//Human_HECA_scCellFie.h5ad was correctly loaded
./results//Human_HECA_scCellFie_reactions.h5ad was correctly loaded
./results//Human_HECA_scCellFie_metabolic_tasks.h5ad was correctly loaded
In this case, we are interested in the metabolic task scores, which can be found in the adata.metabolic_tasks AnnData object.
[4]:
adata.metabolic_tasks
[4]:
AnnData object with n_obs × n_vars = 90001 × 215
obs: 'n_genes', 'sample', 'percent_mito', 'n_counts', 'Endometriosis_stage', 'Endometriosis', 'Hormonal treatment', 'Binary Stage', 'Stage', 'phase', 'dataset', 'Age', 'lineage', 'celltype', 'label_long'
uns: 'Binary Stage_colors', 'Biopsy_type_colors', 'Endometrial_pathology_colors', 'Endometriosis_stage_colors', 'GarciaAlonso_celltype_colors', 'Group_colors', 'Hormonal treatment_colors', 'Library_genotype_colors', 'Mareckova_celltype_colors', 'Mareckova_epi_celltype_colors', 'Mareckova_lineage_colors', 'Processing_colors', 'Symbol_colors', 'Tan_cellsubtypes_colors', 'Tan_celltype_colors', 'Treatment_colors', 'celltype_colors', 'dataset_colors', 'genotype_colors', 'hvg', 'label_long_colors', 'leiden', 'leiden_R_colors', 'leiden_colors', 'lineage_colors', 'neighbors', 'normalization', 'phase_colors', 'umap'
obsm: 'X_scVI', 'X_umap'
obsp: 'connectivities', 'distances'
Defining cell types
For metabolic marker identification, we need to specify which column in the AnnData object contains the cell type information:
[5]:
# Our column indicating the cell types in the adata.obs dataframe
cell_group = 'celltype'
Which can be visualized as
[17]:
sc.pl.umap(adata, color=cell_group, frameon=False)

Identification of metabolic markers
To identify markers, we can use different approaches. Here we show an approach based on TF-IDF, which comes from the Natural Language Processing field, and the logistic regression implemented in Scanpy.
Detection using TF-IDF
scCellFie implements a TF-IDF (Term Frequency-Inverse Document Frequency) approach, adapted from the SoupX tool in R.
We define express_cut=5*np.log(2) to determine which cells are expressing each metabolic task. This value (5*np.log(2)) indicates that all reactions in the task are over the threshold value of the determinant gene (or “active”). This condition is often rare in single-cell data due to data sparsity (rarely all reactions in a task are over their determinant gene’s threshold), but it is a good approach for identifying markers.
[7]:
mrks = sccellfie.external.quick_markers(adata.metabolic_tasks,
cluster_key=cell_group,
n_markers=20,
express_cut=5*np.log(2))
[8]:
mrks.head()
[8]:
| gene | cluster | tf | idf | tf_idf | gene_frequency_outside_cluster | gene_frequency_global | second_best_tf | second_best_cluster | pval | qval | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Conversion of carnosine to beta-alanine | SOX9_basalis | 0.558824 | 1.588612 | 0.887754 | 0.203806 | 0.204209 | 0.874235 | Luminal | 3.835604e-15 | 0.000000e+00 |
| 1 | Fructose degradation (to glucose-3-phosphate) | SOX9_basalis | 0.764706 | 0.705568 | 0.539552 | 0.493521 | 0.493828 | 0.913798 | preGlandular | 1.904306e-08 | 0.000000e+00 |
| 2 | 3'-Phospho-5'-adenylyl sulfate synthesis | SOX9_basalis | 0.127451 | 2.932873 | 0.373798 | 0.053160 | 0.053244 | 0.293322 | preGlandular | 3.009349e-03 | 0.000000e+00 |
| 3 | Biosynthesis of Tn_antigen (Glycoprotein N-ace... | SOX9_basalis | 0.088235 | 4.154814 | 0.366601 | 0.015606 | 0.015689 | 0.088235 | SOX9_basalis | 3.486177e-05 | 0.000000e+00 |
| 4 | Conversion of 1-phosphatidyl-1D-myo-inositol 4... | SOX9_basalis | 0.068627 | 2.717943 | 0.186526 | 0.066007 | 0.066010 | 0.192308 | Lymphatic | 5.137296e-01 | 6.318684e-181 |
By examining the distributions of TF-IDF scores, we can filter the results to identify more specific tasks:
[9]:
# Visualize TF-IDF score distribution
mrks['tf_idf'].hist()
[9]:
<Axes: >

[10]:
# Scatter plot of TF vs IDF with TF-IDF as color
scatter = plt.scatter(mrks['tf'], mrks['idf'], c=mrks['tf_idf'], cmap='YlGnBu')
plt.colorbar(scatter)
plt.xlabel('TF')
plt.ylabel('IDF')
[10]:
Text(0, 0.5, 'IDF')

scCellFie includes a filtering function that fits a hyperbolic curve, while the user can define some thresholds based on the tf-idf scores, and the ratio between the top and second best cell clusters (tf_ratio).
[11]:
x_col = 'tf'
y_col = 'idf'
df = mrks
tfidf_threshold = 0.3
tf_ratio = 1.2
# Visualization
plt.figure(figsize=(6, 6))
# Plot all points
plt.scatter(df[x_col], df[y_col], alpha=0.5, c='lightgray', label='All points')
# Plot selected points
filtered_mrks, curve = sccellfie.external.filter_tfidf_markers(df, tf_col=x_col, idf_col=y_col,
tfidf_threshold=tfidf_threshold,
tf_ratio=tf_ratio)
plt.scatter(filtered_mrks[x_col], filtered_mrks[y_col], c='red', label='Selected points')
plt.plot(*curve, label='Fit curve')
plt.xlabel('TF')
plt.ylabel('IDF')
plt.legend()
[11]:
<matplotlib.legend.Legend at 0x7fd0f89e8880>

A detailed list of the selected markers:
[26]:
filtered_mrks
[26]:
| gene | cluster | tf | idf | tf_idf | gene_frequency_outside_cluster | gene_frequency_global | second_best_tf | second_best_cluster | pval | qval | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 3 | Biosynthesis of Tn_antigen (Glycoprotein N-ace... | SOX9_basalis | 0.088235 | 4.154814 | 0.366601 | 0.015606 | 0.015689 | 0.088235 | SOX9_basalis | 3.486177e-05 | 0.000000e+00 |
| 20 | Presence of the thioredoxin system through the... | SOX9_functionalis_I | 0.320175 | 4.499821 | 1.440732 | 0.009537 | 0.011111 | 0.130323 | SOX9_luminal | 2.436788e-169 | 2.436788e-169 |
| 41 | Biosynthesis of Tn_antigen (Glycoprotein N-ace... | SOX9_functionalis_II | 0.167774 | 4.154814 | 0.697070 | 0.009330 | 0.015689 | 0.088235 | SOX9_basalis | 0.000000e+00 | 0.000000e+00 |
| 63 | Presence of the thioredoxin system through the... | SOX9_luminal | 0.130323 | 4.499821 | 0.586428 | 0.010076 | 0.011111 | 0.130323 | SOX9_luminal | 2.169252e-74 | 2.436788e-169 |
| 67 | Conversion of estrone (E1) to estrone sulfate ... | SOX9_luminal | 0.073548 | 4.105754 | 0.301972 | 0.015982 | 0.016478 | 0.073548 | SOX9_luminal | 1.432926e-20 | 1.030451e-259 |
| 101 | Glucose to lactate conversion | preGlandular | 0.544278 | 2.296725 | 1.250056 | 0.083174 | 0.100588 | 0.544278 | preGlandular | 0.000000e+00 | 0.000000e+00 |
| 102 | ATP generation from glucose (hypoxic condition... | preGlandular | 0.605766 | 1.987948 | 1.204232 | 0.118577 | 0.136976 | 0.605766 | preGlandular | 0.000000e+00 | 0.000000e+00 |
| 103 | AMP salvage from adenine | preGlandular | 0.601942 | 1.932106 | 1.163015 | 0.126902 | 0.144843 | 0.338397 | Cycling | 0.000000e+00 | 0.000000e+00 |
| 104 | 3'-Phospho-5'-adenylyl sulfate synthesis | preGlandular | 0.293322 | 2.932873 | 0.860275 | 0.043821 | 0.053244 | 0.293322 | preGlandular | 0.000000e+00 | 0.000000e+00 |
| 107 | GMP salvage from guanine | preGlandular | 0.103854 | 5.456934 | 0.566725 | 0.000358 | 0.004267 | 0.017722 | Cycling | 0.000000e+00 | 0.000000e+00 |
| 108 | IMP salvage from hypoxanthine | preGlandular | 0.103854 | 5.456934 | 0.566725 | 0.000358 | 0.004267 | 0.017722 | Cycling | 0.000000e+00 | 0.000000e+00 |
| 120 | 3'-Phospho-5'-adenylyl sulfate synthesis | Glandular | 0.647357 | 2.932873 | 1.898616 | 0.027442 | 0.053244 | 0.293322 | preGlandular | 0.000000e+00 | 0.000000e+00 |
| 140 | Synthesis of anthranilate from tryptophan | Glandular_secretory | 0.729369 | 2.805307 | 2.046104 | 0.041598 | 0.060488 | 0.729369 | Glandular_secretory | 0.000000e+00 | 0.000000e+00 |
| 147 | Synthesis of L-kynurenine from tryptophan | Glandular_secretory | 0.090210 | 4.927531 | 0.444514 | 0.004901 | 0.007244 | 0.090210 | Glandular_secretory | 1.518326e-177 | 1.518326e-177 |
| 148 | Phenylalanine to phenylacetate (via phenylacet... | Glandular_secretory | 0.087783 | 4.792850 | 0.420732 | 0.006044 | 0.008289 | 0.051948 | Glandular_secretory_FGF7 | 1.280152e-155 | 1.280152e-155 |
| 149 | Synthesis of N-formylanthranilate from tryptophan | Glandular_secretory | 0.078883 | 5.300553 | 0.418126 | 0.002902 | 0.004989 | 0.078883 | Glandular_secretory | 2.985949e-179 | 2.985949e-179 |
| 164 | Synthesis of alanine from glutamine | Glandular_secretory_FGF7 | 0.307359 | 3.305898 | 1.016099 | 0.035970 | 0.036666 | 0.081967 | preCiliated | 7.765291e-45 | 4.231413e-58 |
| 165 | Synthesis of L-kynurenine from tryptophan | Glandular_secretory_FGF7 | 0.194805 | 4.927531 | 0.959909 | 0.006762 | 0.007244 | 0.090210 | Glandular_secretory | 6.100311e-50 | 1.518326e-177 |
| 166 | Synthesis of N-formylanthranilate from tryptophan | Glandular_secretory_FGF7 | 0.142857 | 5.300553 | 0.757222 | 0.004634 | 0.004989 | 0.078883 | Glandular_secretory | 1.392133e-37 | 2.985949e-179 |
| 180 | Conversion of estrone (E1) to estrone sulfate ... | preLuminal | 0.328662 | 4.105754 | 1.349407 | 0.013731 | 0.016478 | 0.073548 | SOX9_luminal | 1.030451e-259 | 1.030451e-259 |
| 182 | Mevalonate synthesis | preLuminal | 0.368153 | 2.801823 | 1.031499 | 0.057994 | 0.060699 | 0.368153 | preLuminal | 1.489213e-145 | 0.000000e+00 |
| 184 | Synthesis of testosterone from androstenedione | preLuminal | 0.161783 | 4.681343 | 0.757364 | 0.007925 | 0.009267 | 0.131543 | Luminal | 4.782356e-116 | 0.000000e+00 |
| 200 | Mevalonate synthesis | Luminal | 0.690347 | 2.801823 | 1.934229 | 0.039422 | 0.060699 | 0.368153 | preLuminal | 0.000000e+00 | 0.000000e+00 |
| 205 | Synthesis of testosterone from androstenedione | Luminal | 0.131543 | 4.681343 | 0.615799 | 0.005134 | 0.009267 | 0.131543 | Luminal | 0.000000e+00 | 0.000000e+00 |
| 260 | Synthesis of thromboxane from arachidonate | MUC5B | 0.215539 | 4.655306 | 1.003399 | 0.008594 | 0.009511 | 0.128403 | Immune_Myeloid | 1.175419e-88 | 0.000000e+00 |
| 341 | Glucuronate synthesis (via inositol) | mPV | 0.416452 | 2.666280 | 1.110379 | 0.068004 | 0.069510 | 0.416452 | mPV | 1.042929e-82 | 1.737183e-230 |
| 342 | NAD synthesis from nicotinamide | mPV | 0.138817 | 6.707096 | 0.931062 | 0.000625 | 0.001222 | 0.065491 | ePV_1a | 3.589895e-98 | 3.589895e-98 |
| 343 | Synthesis of phosphatidylinositol from inositol | mPV | 0.141388 | 3.966842 | 0.560865 | 0.018402 | 0.018933 | 0.141388 | mPV | 6.861606e-31 | 2.793213e-56 |
| 360 | Glucuronate synthesis (via inositol) | ePV_1a | 0.518892 | 2.666280 | 1.383510 | 0.067519 | 0.069510 | 0.416452 | mPV | 2.032582e-128 | 1.737183e-230 |
| 362 | Ethanol to acetaldehyde | ePV_1a | 0.141058 | 6.662644 | 0.939819 | 0.000658 | 0.001278 | 0.141058 | ePV_1a | 5.120176e-101 | 5.120176e-101 |
| 367 | NAD synthesis from nicotinamide | ePV_1a | 0.065491 | 6.707096 | 0.439256 | 0.000937 | 0.001222 | 0.065491 | ePV_1a | 2.118539e-37 | 3.589895e-98 |
| 600 | Conversion of glutamate to glutamine | dHormones | 0.714286 | 2.658002 | 1.898573 | 0.069737 | 0.070088 | 0.590965 | Immune_Myeloid | 9.165935e-30 | 0.000000e+00 |
| 601 | Phenylalanine to phenylacetaldehyde | dHormones | 0.673469 | 2.347129 | 1.580719 | 0.095329 | 0.095643 | 0.673469 | dHormones | 1.542389e-22 | 0.000000e+00 |
| 640 | Conversion of glutamate to glutamine | Immune_Myeloid | 0.590965 | 2.658002 | 1.570787 | 0.050686 | 0.070088 | 0.590965 | Immune_Myeloid | 0.000000e+00 | 0.000000e+00 |
| 643 | Synthesis of bilirubin | Immune_Myeloid | 0.141089 | 4.639083 | 0.654524 | 0.004771 | 0.009667 | 0.025974 | Glandular_secretory_FGF7 | 0.000000e+00 | 0.000000e+00 |
| 644 | Synthesis of thromboxane from arachidonate | Immune_Myeloid | 0.128403 | 4.655306 | 0.597757 | 0.005082 | 0.009511 | 0.128403 | Immune_Myeloid | 0.000000e+00 | 0.000000e+00 |
| 645 | Pyridoxal-phosphate synthesis | Immune_Myeloid | 0.170792 | 3.473063 | 0.593172 | 0.025816 | 0.031022 | 0.170792 | Immune_Myeloid | 5.799610e-248 | 5.799610e-248 |
| 647 | Hydroxymethylglutaryl-CoA synthesis | Immune_Myeloid | 0.094988 | 4.220675 | 0.400912 | 0.011698 | 0.014689 | 0.094988 | Immune_Myeloid | 1.201930e-155 | 3.545350e-172 |
| 648 | Gluconeogenesis from Lactate | Immune_Myeloid | 0.059097 | 5.713844 | 0.337668 | 0.001222 | 0.003300 | 0.012987 | Glandular_secretory_FGF7 | 6.341532e-198 | 6.341532e-198 |
| 649 | Glutamine synthesis | Immune_Myeloid | 0.055384 | 5.918638 | 0.327796 | 0.000726 | 0.002689 | 0.017316 | Glandular_secretory_FGF7 | 2.344026e-203 | 2.344026e-203 |
| 681 | Conversion of 1-phosphatidyl-1D-myo-inositol 4... | Arterial | 0.260780 | 2.717943 | 0.708786 | 0.064951 | 0.066010 | 0.192308 | Lymphatic | 1.398377e-41 | 6.318684e-181 |
| 700 | Conversion of 1-phosphatidyl-1D-myo-inositol 4... | Lymphatic | 0.192308 | 2.717943 | 0.522681 | 0.065828 | 0.066010 | 0.192308 | Lymphatic | 1.244686e-06 | 6.318684e-181 |
[12]:
# Get unique list of filtered markers
tf_idf_mrks = filtered_mrks['gene'].unique().tolist()
len(tf_idf_mrks)
[12]:
29
Detection using Logistic Regression in Scanpy
We can also use logistic regression, as implemented in Scanpy, to identify metabolic task markers:
[20]:
method = 'logreg'
sc.tl.rank_genes_groups(adata.metabolic_tasks, cell_group, method=method,
use_raw=False, key_added=method)
sc.pl.rank_genes_groups(adata.metabolic_tasks, n_genes=20, sharey=True, key=method, fontsize=4)

[21]:
# Get the results as a dataframe
scanpy_df = sc.get.rank_genes_groups_df(adata.metabolic_tasks,
key=method,
group=None)
[22]:
scanpy_df.head()
[22]:
| group | names | scores | |
|---|---|---|---|
| 0 | SOX9_basalis | 3'-Phospho-5'-adenylyl sulfate synthesis | 1.646670 |
| 1 | SOX9_basalis | Biosynthesis of Tn_antigen (Glycoprotein N-ace... | 1.050843 |
| 2 | SOX9_basalis | Conversion of carnosine to beta-alanine | 0.983181 |
| 3 | SOX9_basalis | GDP-mannose synthesis | 0.850716 |
| 4 | SOX9_basalis | Synthesis of fructose-6-phosphate from erythro... | 0.804910 |
We can visualize these markers using dot plots:
[27]:
sc.pl.rank_genes_groups_dotplot(adata.metabolic_tasks, n_genes=5, groupby=cell_group,
key=method, use_raw=False, standard_scale='var',
)
WARNING: dendrogram data not found (using key=dendrogram_celltype). Running `sc.tl.dendrogram` with default parameters. For fine tuning it is recommended to run `sc.tl.dendrogram` independently.

We can filter the logistic regression markers based on their scores:
[28]:
# Visualize score distribution
scanpy_df['scores'].hist()
[28]:
<Axes: >

[31]:
# Filter based on score threshold
scores_threshold = 1.0
filtered_scanpy_mrks = scanpy_df.loc[scanpy_df['scores'] > scores_threshold]
[30]:
filtered_scanpy_mrks
[30]:
| group | names | scores | |
|---|---|---|---|
| 0 | SOX9_basalis | 3'-Phospho-5'-adenylyl sulfate synthesis | 1.646670 |
| 1 | SOX9_basalis | Biosynthesis of Tn_antigen (Glycoprotein N-ace... | 1.050843 |
| 215 | SOX9_functionalis_I | 3'-Phospho-5'-adenylyl sulfate synthesis | 1.249713 |
| 430 | SOX9_functionalis_II | Synthesis of quinolinate from tryptophan | 1.505565 |
| 431 | SOX9_functionalis_II | 3'-Phospho-5'-adenylyl sulfate synthesis | 1.135055 |
| ... | ... | ... | ... |
| 7101 | Venous | Conversion of lysine to L-Saccharopine | 1.190897 |
| 7102 | Venous | Synthesis of galactosyl glucosyl ceramide (lin... | 1.069577 |
| 7103 | Venous | Degradation of uracil | 1.018579 |
| 7104 | Venous | Degradation of cytosine | 1.018579 |
| 7310 | Arterial | Synthesis of nitric oxide from arginine | 2.254074 |
104 rows × 3 columns
[32]:
# Convert them to a list
scanpy_mrks = filtered_scanpy_mrks['names'].unique().tolist()
len(scanpy_mrks)
[32]:
61
Visualizations
We can combine the metabolic tasks from different approaches:
[33]:
both_markers = sorted(set(tf_idf_mrks + scanpy_mrks))
len(both_markers)
[33]:
81
Select only highly active markers
We can filter the markers based on their activity level by aggregating the metabolic activity to the cell-type level:
[34]:
# Aggregate the metabolic activity of markers into a cell-type level using trimean
agg = sccellfie.expression.aggregation.agg_expression_cells(adata.metabolic_tasks[:, both_markers],
groupby=cell_group,
agg_func='trimean')
Filter markers with an aggregated score of at least 1 in at least one cell type (you can adjust this threshold depending on how strict you want to be).
[35]:
both_markers = agg.T.loc[(agg.T > 1.).any(axis=1)].index.tolist()
len(both_markers)
[35]:
46
UMAP
Now we can visualize these metabolic task markers using UMAP:
[39]:
plt.rcParams['figure.figsize'] = (3,3)
plt.rcParams['font.size'] = 10
sc.pl.embedding(adata.metabolic_tasks,
color=both_markers,
ncols=4,
palette=palette,
frameon=False,
basis='X_umap',
wspace=0.7,
title=["\n".join(textwrap.wrap(t, width=45)) for t in both_markers],
cmap='OrRd'
)

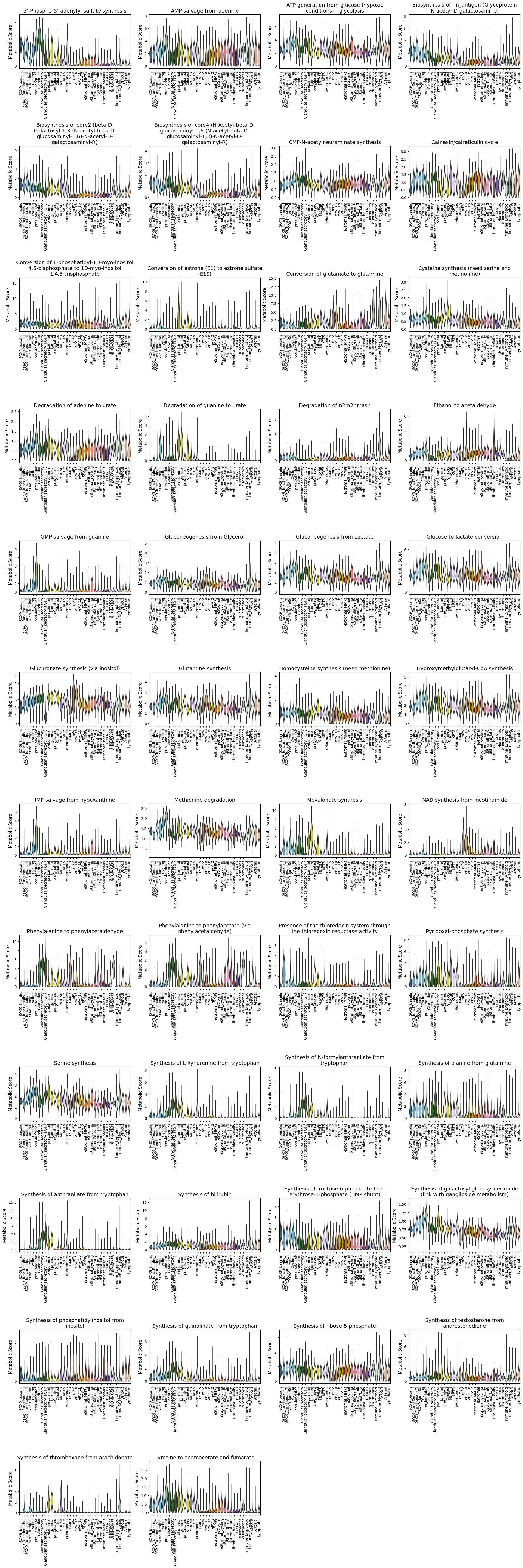
Violin plot
We can further inspect the metabolic score distribution across single cells per cell type for these markers:
[40]:
fig, axes = sccellfie.plotting.create_multi_violin_plots(adata.metabolic_tasks,
features=both_markers,
groupby=cell_group,
stripplot=False,
n_cols=4,
ylabel='Metabolic Score'
)
Heatmap
As a summary visualization, we can create a heatmap of the identified metabolic task markers across cell types based on the aggregated scores into a cell-type level (found in agg calculated before):
[42]:
filtered_agg = agg[both_markers]
[43]:
filtered_agg.head()
[43]:
| Task | 3'-Phospho-5'-adenylyl sulfate synthesis | AMP salvage from adenine | ATP generation from glucose (hypoxic conditions) - glycolysis | Biosynthesis of Tn_antigen (Glycoprotein N-acetyl-D-galactosamine) | Biosynthesis of core2 (beta-D-Galactosyl-1,3-(N-acetyl-beta-D-glucosaminyl-1,6)-N-acetyl-D-galactosaminyl-R) | Biosynthesis of core4 (N-Acetyl-beta-D-glucosaminyl-1,6-(N-acetyl-beta-D-glucosaminyl-1,3)-N-acetyl-D-galactosaminyl-R) | CMP-N-acetylneuraminate synthesis | Calnexin/calreticulin cycle | Conversion of 1-phosphatidyl-1D-myo-inositol 4,5-bisphosphate to 1D-myo-inositol 1,4,5-trisphosphate | Conversion of estrone (E1) to estrone sulfate (E1S) | ... | Synthesis of anthranilate from tryptophan | Synthesis of bilirubin | Synthesis of fructose-6-phosphate from erythrose-4-phosphate (HMP shunt) | Synthesis of galactosyl glucosyl ceramide (link with ganglioside metabolism) | Synthesis of phosphatidylinositol from inositol | Synthesis of quinolinate from tryptophan | Synthesis of ribose-5-phosphate | Synthesis of testosterone from androstenedione | Synthesis of thromboxane from arachidonate | Tyrosine to acetoacetate and fumarate |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Arterial | 0.256560 | 1.675679 | 2.481569 | 0.683852 | 0.442055 | 0.344145 | 0.754417 | 0.773945 | 2.522644 | 0.104100 | ... | 0.004131 | 0.796489 | 0.466867 | 0.699921 | 1.124336 | 0.031946 | 0.551582 | 1.033685 | 0.045549 | 0.030364 |
| Ciliated | 1.078283 | 1.451005 | 1.929947 | 1.445988 | 0.892795 | 0.846883 | 0.723723 | 0.565869 | 1.477182 | 1.089631 | ... | 0.704579 | 1.092265 | 0.682542 | 0.758055 | 0.308139 | 0.291273 | 0.635575 | 0.865189 | 0.193303 | 0.553913 |
| Cycling | 2.098761 | 3.000086 | 3.718694 | 1.807871 | 1.074159 | 0.971235 | 1.017814 | 1.237260 | 1.803018 | 0.326987 | ... | 0.273492 | 1.119333 | 1.211169 | 1.053797 | 0.542966 | 0.374733 | 0.957516 | 0.769935 | 0.106314 | 0.666761 |
| Fibroblast_basalis | 0.240646 | 1.469132 | 2.305450 | 0.710848 | 0.304962 | 0.355424 | 0.744974 | 0.695407 | 1.322372 | 0.140297 | ... | 0.000000 | 0.951126 | 0.491220 | 0.580029 | 1.478609 | 0.081673 | 0.513473 | 1.045122 | 0.000000 | 0.026769 |
| Glandular | 3.829955 | 1.562031 | 2.227763 | 1.779426 | 1.111685 | 1.377226 | 0.908321 | 0.626390 | 1.595914 | 0.025143 | ... | 2.093063 | 1.597565 | 0.794605 | 0.812703 | 0.527581 | 0.565066 | 0.695606 | 0.675938 | 0.201555 | 0.747315 |
5 rows × 46 columns
We then apply min-max normalization for better visualization:
[46]:
normalized_markers = sccellfie.preprocessing.min_max_normalization(filtered_agg, axis=0)
Followed by the heatmap
[50]:
# Create heatmap
plt.figure(figsize=(16, 9))
sns.heatmap(normalized_markers, cmap='YlOrBr', annot=False, linewidths=0.5,
cbar_kws={'label': 'Min-max scaled metabolic score'})
plt.title('Metabolic Markers across Cell Types')
plt.ylabel('Cell Type')
plt.xlabel('Metabolic Task')
plt.tight_layout()
