{
"cells": [
{
"cell_type": "markdown",
"id": "laden-daisy",
"metadata": {},
"source": [
"# Temporal Patterns"
]
},
{
"cell_type": "markdown",
"id": "brief-browser",
"metadata": {},
"source": [
"[](https://colab.research.google.com/github/earmingol/scCellFie/blob/main/docs/source/notebooks/temporal_patterns.ipynb)"
]
},
{
"cell_type": "markdown",
"id": "structural-empire",
"metadata": {},
"source": [
"In this tutorial, we will walk you through how to use General Additive Models (GAMs) to identify metabolic tasks following a trend across a trajectory defined by time points, pseudo-time, or ordered labels of cells.\n",
"\n",
"Here, we will use the results we previously generated for the Human Endometrial Cell Atlas (HECA) dataset [(Mareckova & Garcia-Alonso et al 2023)](https://doi.org/10.1038/s41588-024-01873-w) by running our [Quick Start Tutorial](https://sccellfie.readthedocs.io/en/latest/notebooks/quick_start_human.html)."
]
},
{
"cell_type": "markdown",
"id": "exterior-haiti",
"metadata": {},
"source": [
"## This tutorial includes following steps:\n",
"* [Loading libraries](#loading-libraries)\n",
"* [Loading endometrium results](#loading-endometrium-results)\n",
"* [Defining cell trajectory](#defining-cell-trajectory)\n",
"* [Running GAM](#running-gam)\n",
"* [Filtering GAM results](#filtering-gam-results)\n",
"* [Visualization of results](#visualization-of-results)"
]
},
{
"cell_type": "markdown",
"id": "postal-luther",
"metadata": {},
"source": [
"## Loading libraries "
]
},
{
"cell_type": "code",
"execution_count": 1,
"id": "extreme-harrison",
"metadata": {},
"outputs": [],
"source": [
"import sccellfie\n",
"import scanpy as sc\n",
"\n",
"import pandas as pd\n",
"import numpy as np\n",
"\n",
"## To avoid warnings\n",
"import warnings\n",
"warnings.filterwarnings(\"ignore\")"
]
},
{
"cell_type": "markdown",
"id": "guilty-patrol",
"metadata": {},
"source": [
"In addition, we set up a folder to save our figures. This folder is stored in the settings of Scanpy:"
]
},
{
"cell_type": "code",
"execution_count": 2,
"id": "indonesian-opportunity",
"metadata": {},
"outputs": [],
"source": [
"sc.settings.figdir = './results/GAM-Figures/'"
]
},
{
"cell_type": "markdown",
"id": "primary-metallic",
"metadata": {},
"source": [
"## Loading endometrium results "
]
},
{
"cell_type": "markdown",
"id": "wired-audience",
"metadata": {},
"source": [
"We start opening the results previously generated by running the scCellFie pipeline on the HECA dataset. If you haven't run the pipeline yet, please follow [this tutorial](https://sccellfie.readthedocs.io/en/latest/notebooks/quick_start_human.html).\n",
"\n",
"In this case, we will load the objects that were present in ``results['adata']`` in that tutorial. This object contains:\n",
"- ``results['adata']``: contains gene expression in ``.X``.\n",
"- ``results['adata'].layers['gene_scores']``: contains gene scores as in the original CellFie paper.\n",
"- ``results['adata'].uns['Rxn-Max-Genes']``: contains determinant genes for each reaction per cell.\n",
"- ``results['adata'].reactions``: contains reaction scores in ``.X`` so every scanpy function can be used on this object to visualize or compare values.\n",
"- ``results['adata'].metabolic_tasks``: contains metabolic task scores in ``.X`` so every scanpy function can be used on this object to visualize or compare values.\n",
"\n",
"Here, we will name this object directly as ``adata``. Each of the previous elements should be under ``adata.``, as for example ``adata.metabolic_tasks``."
]
},
{
"cell_type": "code",
"execution_count": 3,
"id": "loving-prediction",
"metadata": {},
"outputs": [
{
"name": "stdout",
"output_type": "stream",
"text": [
"./results//Human_HECA_scCellFie.h5ad was correctly loaded\n",
"./results//Human_HECA_scCellFie_reactions.h5ad was correctly loaded\n",
"./results//Human_HECA_scCellFie_metabolic_tasks.h5ad was correctly loaded\n"
]
}
],
"source": [
"adata = sccellfie.io.load_adata(folder='./results/',\n",
" filename='Human_HECA_scCellFie'\n",
" )"
]
},
{
"cell_type": "markdown",
"id": "supreme-portuguese",
"metadata": {},
"source": [
"In this case, we are interested in the metabolic task scores, which can be found in the ``adata.metabolic_tasks`` AnnData object."
]
},
{
"cell_type": "code",
"execution_count": 4,
"id": "crucial-channel",
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"AnnData object with n_obs × n_vars = 90001 × 215\n",
" obs: 'n_genes', 'sample', 'percent_mito', 'n_counts', 'Endometriosis_stage', 'Endometriosis', 'Hormonal treatment', 'Binary Stage', 'Stage', 'phase', 'dataset', 'Age', 'lineage', 'celltype', 'label_long'\n",
" uns: 'Binary Stage_colors', 'Biopsy_type_colors', 'Endometrial_pathology_colors', 'Endometriosis_stage_colors', 'GarciaAlonso_celltype_colors', 'Group_colors', 'Hormonal treatment_colors', 'Library_genotype_colors', 'Mareckova_celltype_colors', 'Mareckova_epi_celltype_colors', 'Mareckova_lineage_colors', 'Processing_colors', 'Symbol_colors', 'Tan_cellsubtypes_colors', 'Tan_celltype_colors', 'Treatment_colors', 'celltype_colors', 'dataset_colors', 'genotype_colors', 'hvg', 'label_long_colors', 'leiden', 'leiden_R_colors', 'leiden_colors', 'lineage_colors', 'neighbors', 'normalization', 'phase_colors', 'umap'\n",
" obsm: 'X_scVI', 'X_umap'\n",
" obsp: 'connectivities', 'distances'"
]
},
"execution_count": 4,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"adata.metabolic_tasks"
]
},
{
"cell_type": "markdown",
"id": "focused-orbit",
"metadata": {},
"source": [
"## Defining cell trajectory \n",
"\n",
"We can model the behavior of metabolic tasks across a trajectory of cells using GAMs. This trajectory can be defined by time points, pseudo-time, or pre-defined by ordered labels.\n",
"\n",
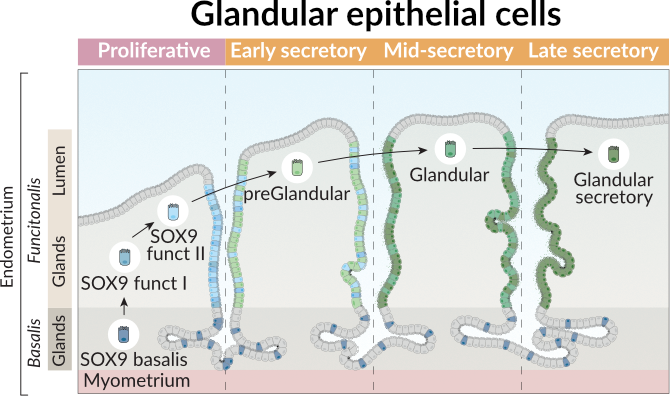
"To illustrate its application, we will focus on glandular epithelial cells in the endometrium, that differentiate across distinct phases of the menstrual cycle as shown in the figure below. In this figure, we observed the layers of the endometrium, the main glandular cell types and the trajectory they follow across the menstrual phases.\n",
"\n",
"{ width=50% }"
]
},
{
"cell_type": "markdown",
"id": "silver-banking",
"metadata": {},
"source": [
"Thus, we can define our trajectory based on this prior knowledge from the endometrial biology."
]
},
{
"cell_type": "code",
"execution_count": 5,
"id": "complicated-wagner",
"metadata": {},
"outputs": [],
"source": [
"trajectory = ['SOX9_basalis', 'SOX9_functionalis_I', 'SOX9_functionalis_II', 'preGlandular', 'Glandular', 'Glandular_secretory']"
]
},
{
"cell_type": "markdown",
"id": "modular-overall",
"metadata": {},
"source": [
"Next, we need to make sure that we use cells labeled with these cell types, but also that they are present in the proper menstrual phases. Additionally, we only consider cells from Control samples."
]
},
{
"cell_type": "code",
"execution_count": 6,
"id": "fundamental-metro",
"metadata": {},
"outputs": [],
"source": [
"def filter_donors(adata, only_control = True):\n",
" df = adata.obs\n",
" # First, include only cells in the menstrual phase where they are expected to be\n",
" cond_filter = (df.Stage.isin(['Proliferative', 'Proliferative Disordered', 'Proliferative Late']) & \\\n",
" df.celltype.isin(['SOX9_functionalis_I', 'SOX9_functionalis_II']) \n",
" ) | \\\n",
" (df.Stage.isin(['Secretory Early', 'Secretory Early-Mid', 'Secretory Mid', 'Secretory Late',]) & \\\n",
" df.celltype.isin(['preGlandular', 'Glandular', 'Glandular_secretory',]) \n",
" ) | \\\n",
" (df.Stage.isin(['Proliferative', 'Proliferative Disordered', 'Proliferative Late', \n",
" 'Secretory Early', 'Secretory Early-Mid', 'Secretory Mid', 'Secretory Late',]) & \\\n",
" df.celltype.isin(['SOX9_basalis', ]) \n",
" )\n",
" # Then, include cells that are only in Control Samples\n",
" if only_control:\n",
" cond_filter = cond_filter & (df.Endometriosis == 'Control')\n",
" return adata[cond_filter]"
]
},
{
"cell_type": "markdown",
"id": "hollywood-double",
"metadata": {},
"source": [
"We create the ``mt_adata`` variable to represent the ``adata.metabolic_tasks`` object after filtering the cells"
]
},
{
"cell_type": "code",
"execution_count": 7,
"id": "genuine-nelson",
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"(7974, 215)"
]
},
"execution_count": 7,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"mt_adata = filter_donors(adata.metabolic_tasks)\n",
"mt_adata.shape"
]
},
{
"cell_type": "markdown",
"id": "crucial-tribune",
"metadata": {},
"source": [
"## Running GAM \n",
"\n",
"GAMs extend linear models by allowing non-linear relationships between predictors and the response variable, while maintaining interpretability.\n",
"\n",
"A GAM models the expected value of a response variable $y$ as the sum of smooth functions of predictors:\n",
"\n",
"$$\n",
"y = \\beta_0 + f_1(x_1) + f_2(x_2) + \\cdots + f_p(x_p)\n",
"$$\n",
"\n",
"Where:\n",
"- $y$ is the response variable (in our case, **the inferred activity of a metabolic task**),\n",
"- $\\beta_0$ is the intercept,\n",
"- $f_j(x_j)$ are smooth functions (often splines) applied to the predictors.\n",
"\n",
"In this analysis, we fit a GAM of the form:\n",
"\n",
"$$\n",
"y_i = \\beta_0 + f(x_i) + \\varepsilon_i\n",
"$$\n",
"\n",
"- $y_i$: inferred metabolic task activity in cell $i$,\n",
"- $x_i$: an ordered index representing pseudotime, spatial progression, or a predefined trajectory of cell types,\n",
"- $f(x)$: a smooth function capturing non-linear trends in metabolic activity along the trajectory,\n",
"- $\\varepsilon_i$: residual noise.\n",
"\n",
"We use the **[pyGAM](https://pygam.readthedocs.io/)** library to fit this model with **penalized splines**, which help control overfitting by penalizing excessive wiggliness in the function $f(x)$.\n",
"\n",
"By fitting GAMs in this way, we can visualize and statistically assess whether a metabolic task shows structured variation across cell states, such as upregulation in specific cell types or gradual trends across a biological process (e.g. menstrual cycle)."
]
},
{
"cell_type": "code",
"execution_count": 8,
"id": "induced-disabled",
"metadata": {},
"outputs": [
{
"name": "stderr",
"output_type": "stream",
"text": [
"Fitting GAMs for each var in adata: 100%|██████████| 215/215 [00:25<00:00, 8.50it/s]\n"
]
}
],
"source": [
"gam_results = sccellfie.stats.fit_gam_model(\n",
" mt_adata,\n",
" cell_type_key='celltype',\n",
" cell_type_order=trajectory,\n",
")"
]
},
{
"cell_type": "markdown",
"id": "secret-aaron",
"metadata": {},
"source": [
"Typically scCellFie runs GAMs on single cells. Additionally, it allows running\n",
"GAMs on pseudo-bulks, by passing the following parameters:\n",
"\n",
"- ``use_pseudobulk=True`` : enables the use of pseudo-bulks instead of single cells.\n",
"- ``pseudobulk_agg='trimean'`` : specifies the aggregation method to summarize single cells into pseudo-bulks.\n",
"- ``n_pseudobulks=10`` : number of pseudo-bulks.\n",
"- ``cells_per_bulk=100`` : number of single cells included in each pseudo-bulk.\n",
"\n",
"Pseudo-bulks will be built on the ``cell_type_key``.\n",
"\n",
"\n",
"
\n",
"Note!\n",
"\n",
"If you are planning to use continuous values as cell labels (e.g. pseudo time), leave ``cell_type_order=None`` and specify the column where pseudo time values are located by using the parameter ``continuous_key``; for example, ``continuous_key=='pseudotime'``.\n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"id": "motivated-windows",
"metadata": {},
"source": [
"## Filtering GAM results \n",
"\n",
"After fitting our GAMs, we can simplify their outputs into a dataframe containing metabolic tasks (rows) and specific information from the models, contained in the columns."
]
},
{
"cell_type": "code",
"execution_count": 9,
"id": "useful-kenya",
"metadata": {},
"outputs": [],
"source": [
"results_df = sccellfie.stats.analyze_gam_results(gam_results, fdr_level=0.01)"
]
},
{
"cell_type": "code",
"execution_count": 10,
"id": "advisory-integration",
"metadata": {},
"outputs": [
{
"data": {
"text/html": [
"
\n",
"\n",
"
\n",
" \n",
"
\n",
"
\n",
"
n_samples
\n",
"
edof
\n",
"
scale
\n",
"
AIC
\n",
"
loglikelihood
\n",
"
deviance
\n",
"
p_value
\n",
"
explained_deviance
\n",
"
mcfadden_r2
\n",
"
mcfadden_r2_adj
\n",
"
gene
\n",
"
significant
\n",
"
adj_p_value
\n",
"
significant_fdr
\n",
"
\n",
" \n",
" \n",
"
\n",
"
Glycine degradation
\n",
"
7974.0
\n",
"
5.845167
\n",
"
0.029838
\n",
"
225703.731784
\n",
"
-112845.020725
\n",
"
7968.154833
\n",
"
1.110223e-16
\n",
"
0.708444
\n",
"
0.258058
\n",
"
0.741929
\n",
"
Glycine degradation
\n",
"
True
\n",
"
1.147586e-16
\n",
"
True
\n",
"
\n",
"
\n",
"
Serine degradation
\n",
"
7974.0
\n",
"
5.845167
\n",
"
0.024380
\n",
"
282270.796524
\n",
"
-141128.553095
\n",
"
7968.154833
\n",
"
1.110223e-16
\n",
"
0.703896
\n",
"
0.266481
\n",
"
0.733508
\n",
"
Serine degradation
\n",
"
True
\n",
"
1.147586e-16
\n",
"
True
\n",
"
\n",
"
\n",
"
IMP salvage from hypoxanthine
\n",
"
7974.0
\n",
"
5.845167
\n",
"
0.328057
\n",
"
21182.718189
\n",
"
-10584.513927
\n",
"
7968.154833
\n",
"
1.110223e-16
\n",
"
0.687279
\n",
"
0.283959
\n",
"
0.715884
\n",
"
IMP salvage from hypoxanthine
\n",
"
True
\n",
"
1.147586e-16
\n",
"
True
\n",
"
\n",
"
\n",
"
GMP salvage from guanine
\n",
"
7974.0
\n",
"
5.845167
\n",
"
0.328057
\n",
"
21182.718189
\n",
"
-10584.513927
\n",
"
7968.154833
\n",
"
1.110223e-16
\n",
"
0.687279
\n",
"
0.283959
\n",
"
0.715884
\n",
"
GMP salvage from guanine
\n",
"
True
\n",
"
1.147586e-16
\n",
"
True
\n",
"
\n",
"
\n",
"
Methionine degradation
\n",
"
7974.0
\n",
"
5.845167
\n",
"
0.045556
\n",
"
140316.366871
\n",
"
-70151.338268
\n",
"
7968.154833
\n",
"
1.110223e-16
\n",
"
0.674027
\n",
"
0.279506
\n",
"
0.720470
\n",
"
Methionine degradation
\n",
"
True
\n",
"
1.147586e-16
\n",
"
True
\n",
"
\n",
" \n",
"
\n",
"
"
],
"text/plain": [
" n_samples edof scale AIC \\\n",
"Glycine degradation 7974.0 5.845167 0.029838 225703.731784 \n",
"Serine degradation 7974.0 5.845167 0.024380 282270.796524 \n",
"IMP salvage from hypoxanthine 7974.0 5.845167 0.328057 21182.718189 \n",
"GMP salvage from guanine 7974.0 5.845167 0.328057 21182.718189 \n",
"Methionine degradation 7974.0 5.845167 0.045556 140316.366871 \n",
"\n",
" loglikelihood deviance p_value \\\n",
"Glycine degradation -112845.020725 7968.154833 1.110223e-16 \n",
"Serine degradation -141128.553095 7968.154833 1.110223e-16 \n",
"IMP salvage from hypoxanthine -10584.513927 7968.154833 1.110223e-16 \n",
"GMP salvage from guanine -10584.513927 7968.154833 1.110223e-16 \n",
"Methionine degradation -70151.338268 7968.154833 1.110223e-16 \n",
"\n",
" explained_deviance mcfadden_r2 \\\n",
"Glycine degradation 0.708444 0.258058 \n",
"Serine degradation 0.703896 0.266481 \n",
"IMP salvage from hypoxanthine 0.687279 0.283959 \n",
"GMP salvage from guanine 0.687279 0.283959 \n",
"Methionine degradation 0.674027 0.279506 \n",
"\n",
" mcfadden_r2_adj gene \\\n",
"Glycine degradation 0.741929 Glycine degradation \n",
"Serine degradation 0.733508 Serine degradation \n",
"IMP salvage from hypoxanthine 0.715884 IMP salvage from hypoxanthine \n",
"GMP salvage from guanine 0.715884 GMP salvage from guanine \n",
"Methionine degradation 0.720470 Methionine degradation \n",
"\n",
" significant adj_p_value significant_fdr \n",
"Glycine degradation True 1.147586e-16 True \n",
"Serine degradation True 1.147586e-16 True \n",
"IMP salvage from hypoxanthine True 1.147586e-16 True \n",
"GMP salvage from guanine True 1.147586e-16 True \n",
"Methionine degradation True 1.147586e-16 True "
]
},
"execution_count": 10,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"results_df.head(5)"
]
},
{
"cell_type": "markdown",
"id": "light-beverage",
"metadata": {},
"source": [
"With this dataframe, we can start selecting significant results:"
]
},
{
"cell_type": "code",
"execution_count": 11,
"id": "characteristic-works",
"metadata": {},
"outputs": [],
"source": [
"results_df = results_df.loc[results_df.significant_fdr]"
]
},
{
"cell_type": "markdown",
"id": "labeled-anchor",
"metadata": {},
"source": [
"### Adjusted McFadden pseudo R2\n",
"\n",
"In our dataframe, we can find the ``'mcfadden_r2_adj'`` column, which corresponds to the **adjusted McFadden pseudo R²**. This is a measure of model fit based on the log-likelihood of the fitted model relative to a null model (a model with no predictors). It is particularly useful for models with non-normal errors, such as logistic regression or generalized additive models (GAMs).\n",
"\n",
"- **Formula**:\n",
"\n",
"$$\n",
"R^2_{\\text{McFadden}} = 1 - \\frac{\\text{log-likelihood of the fitted model}}{\\text{log-likelihood of the null model}}\n",
"$$\n",
"\n",
"- **Interpretation**:\n",
" - A value close to **0** means the model performs no better than the null model.\n",
" - A value close to **1** indicates a strong model fit.\n",
" - **Adjusted McFadden R²** accounts for model complexity, preventing overfitting:\n",
"\n",
"$$\n",
"R^2_{\\text{McFadden, adj}} = 1 - \\frac{\\text{log-likelihood of the fitted model}}{\\text{log-likelihood of the null model}} \\times \\frac{n - 1}{n - p - 1}\n",
"$$\n",
"\n",
"Where:\n",
" - \\(n\\) is the number of samples.\n",
" - \\(p\\) is the number of predictors."
]
},
{
"cell_type": "markdown",
"id": "appreciated-content",
"metadata": {},
"source": [
"We can inspect the distribution of this coefficient, across the GAMs used to model each metabolic task:"
]
},
{
"cell_type": "code",
"execution_count": 12,
"id": "million-cargo",
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
""
]
},
"execution_count": 12,
"metadata": {},
"output_type": "execute_result"
},
{
"data": {
"image/png": "iVBORw0KGgoAAAANSUhEUgAAAh8AAAGdCAYAAACyzRGfAAAAOXRFWHRTb2Z0d2FyZQBNYXRwbG90bGliIHZlcnNpb24zLjkuMSwgaHR0cHM6Ly9tYXRwbG90bGliLm9yZy/TGe4hAAAACXBIWXMAAA9hAAAPYQGoP6dpAAAniUlEQVR4nO3df3BV9Z3/8ddNuLkxkht+FZIsAVGqWC3KQoPXWgqYkAKDsGamWnZbZFi73YnOSsatZRdKwo9CqSt0u5EqRXDXpnRx/LHaCoawhKFAlQjDD11WKA4oJC50yIVkuBxzP98/OtwvaSLkJOd+kpM8HzN3mPO5537O+5177rkvzv0VMMYYAQAAWJLS1QUAAIDehfABAACsInwAAACrCB8AAMAqwgcAALCK8AEAAKwifAAAAKsIHwAAwKo+XV3An4vH4zp9+rQyMzMVCAS6uhwAANAOxhhduHBBubm5Skm59rmNbhc+Tp8+rby8vK4uAwAAdMCpU6c0dOjQa67T7cJHZmampD8VHw6HPZnTcRy9/fbbmjJlioLBoCdzdnf0TM89FT3Tc0/l956j0ajy8vISz+PX0u3Cx5WXWsLhsKfhIyMjQ+Fw2Jd3aEfQMz33VPRMzz1VT+m5PW+Z4A2nAADAKsIHAACwivABAACsInwAAACrCB8AAMAqwgcAALCK8AEAAKwifAAAAKsIHwAAwCrCBwAAsKpT4WPlypUKBAJ64oknEmOXLl1SSUmJBg4cqL59+6q4uFj19fWdrRMAAPQQHQ4f7777rp577jmNHj26xfj8+fP1xhtvaPPmzaqpqdHp06f14IMPdrpQAADQM3QofFy8eFF//dd/rXXr1ql///6J8YaGBq1fv17PPPOMJk+erLFjx2rDhg3avXu39u7d61nRAADAvzr0q7YlJSWaPn26CgoKtGzZssR4bW2tHMdRQUFBYmzUqFEaNmyY9uzZo3vuuafVXLFYTLFYLLEcjUYl/enX/RzH6Uh5rVyZx6v5/ICeewd67h3ouXfwe89u6nYdPjZt2qT33ntP7777bqvr6urqlJaWpn79+rUYHzJkiOrq6tqcb8WKFSovL281/vbbbysjI8NteddUVVXl6Xx+QM+9Az33DvTcO/i156ampnav6yp8nDp1Sv/wD/+gqqoqpaenuy6sLQsWLFBpaWliORqNKi8vT1OmTFE4HPZkG47jqKqqSoWFhRqzfLsnc9p0uKzI9W2u7jkYDCahqu6Hnum5p6JnevaDK69ctIer8FFbW6tPP/1Uf/mXf5kYa25u1s6dO/Vv//Zv2rp1qy5fvqzz58+3OPtRX1+v7OzsNucMhUIKhUKtxoPBoOd//GAwqFhzwNM5bejM3yEZf8fujp57B3ruHejZP9zU7Cp83H///Tp06FCLsblz52rUqFF66qmnlJeXp2AwqOrqahUXF0uSjh49qpMnTyoSibjZFAAA6KFchY/MzEzdeeedLcZuvPFGDRw4MDE+b948lZaWasCAAQqHw3r88ccViUTafLMpAADofTr0aZdrWb16tVJSUlRcXKxYLKaioiI9++yzXm8GAAD4VKfDx44dO1osp6enq6KiQhUVFZ2dGgAA9ED8tgsAALCK8AEAAKwifAAAAKsIHwAAwCrCBwAAsIrwAQAArCJ8AAAAqwgfAADAKsIHAACwivABAACsInwAAACrCB8AAMAqwgcAALCK8AEAAKwifAAAAKsIHwAAwCrCBwAAsIrwAQAArCJ8AAAAqwgfAADAKsIHAACwivABAACsInwAAACrCB8AAMAqwgcAALCK8AEAAKwifAAAAKsIHwAAwCrCBwAAsIrwAQAArCJ8AAAAqwgfAADAKsIHAACwylX4WLt2rUaPHq1wOKxwOKxIJKK33norcf3EiRMVCARaXL73ve95XjQAAPCvPm5WHjp0qFauXKkvfvGLMsboxRdf1MyZM7V//37dcccdkqRHH31US5YsSdwmIyPD24oBAICvuQofM2bMaLG8fPlyrV27Vnv37k2Ej4yMDGVnZ3tXIQAA6FFchY+rNTc3a/PmzWpsbFQkEkmM//KXv9RLL72k7OxszZgxQ4sWLbrm2Y9YLKZYLJZYjkajkiTHceQ4TkfLa+HKPI7jKJRqPJnTpo78Ha7uubeg596BnnsHevYfN3UHjDGuno0PHTqkSCSiS5cuqW/fvqqsrNS0adMkSc8//7yGDx+u3NxcHTx4UE899ZTy8/P1yiuvfO58ZWVlKi8vbzVeWVnJSzYAAPhEU1OTZs+erYaGBoXD4Wuu6zp8XL58WSdPnlRDQ4Nefvll/eIXv1BNTY2+9KUvtVp3+/btuv/++3Xs2DHdcsstbc7X1pmPvLw8nT179rrFt5fjOKqqqlJhYaHGLN/uyZzdXSjFaOm4uBbtS1EsHrC+/cNlRda3efX9HAwGrW+/K9AzPfdU9Oy/nqPRqAYNGtSu8OH6ZZe0tDSNHDlSkjR27Fi9++67+ulPf6rnnnuu1brjx4+XpGuGj1AopFAo1Go8GAx6/scPBoOKNdt/Iu5KsXigS3ruygdOMvad7o6eewd67h382rObmjv9PR/xeLzFmYurHThwQJKUk5PT2c0AAIAewtWZjwULFmjq1KkaNmyYLly4oMrKSu3YsUNbt27V8ePHE+//GDhwoA4ePKj58+drwoQJGj16dLLqBwAAPuMqfHz66af6zne+ozNnzigrK0ujR4/W1q1bVVhYqFOnTmnbtm1as2aNGhsblZeXp+LiYi1cuDBZtQMAAB9yFT7Wr1//udfl5eWppqam0wUBAICejd92AQAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFa5Ch9r167V6NGjFQ6HFQ6HFYlE9NZbbyWuv3TpkkpKSjRw4ED17dtXxcXFqq+v97xoAADgX67Cx9ChQ7Vy5UrV1tZq3759mjx5smbOnKkjR45IkubPn6833nhDmzdvVk1NjU6fPq0HH3wwKYUDAAB/6uNm5RkzZrRYXr58udauXau9e/dq6NChWr9+vSorKzV58mRJ0oYNG3T77bdr7969uueee7yrGgAA+Jar8HG15uZmbd68WY2NjYpEIqqtrZXjOCooKEisM2rUKA0bNkx79uz53PARi8UUi8USy9FoVJLkOI4cx+loeS1cmcdxHIVSjSdzdnehFNPiX9u8uu86ss2u2HZXoefegZ57B7/37KbugDHG1bPToUOHFIlEdOnSJfXt21eVlZWaNm2aKisrNXfu3BZBQpLy8/M1adIk/fjHP25zvrKyMpWXl7car6ysVEZGhpvSAABAF2lqatLs2bPV0NCgcDh8zXVdn/m47bbbdODAATU0NOjll1/WnDlzVFNT0+FiFyxYoNLS0sRyNBpVXl6epkyZct3i28txHFVVVamwsFBjlm/3ZM7uLpRitHRcXIv2pSgWD1jf/uGyIuvbvPp+DgaD1rffFeiZnnsqevZfz1deuWgP1+EjLS1NI0eOlCSNHTtW7777rn7605/qoYce0uXLl3X+/Hn169cvsX59fb2ys7M/d75QKKRQKNRqPBgMev7HDwaDijXbfyLuSrF4oEt67soHTjL2ne6OnnsHeu4d/Nqzm5o7/T0f8XhcsVhMY8eOVTAYVHV1deK6o0eP6uTJk4pEIp3dDAAA6CFcnflYsGCBpk6dqmHDhunChQuqrKzUjh07tHXrVmVlZWnevHkqLS3VgAEDFA6H9fjjjysSifBJFwAAkOAqfHz66af6zne+ozNnzigrK0ujR4/W1q1bVVhYKElavXq1UlJSVFxcrFgspqKiIj377LNJKRwAAPiTq/Cxfv36a16fnp6uiooKVVRUdKooAADQc/HbLgAAwCrCBwAAsKrD33AKXMtNP/iN9W2GUo1W5Ut3lm3t0MeLP1o5PQlVAXDjyrGjs49nmzh2uMeZDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVfMkYAPRQXfFlf0B7cOYDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWOUqfKxYsUJf+cpXlJmZqcGDB2vWrFk6evRoi3UmTpyoQCDQ4vK9733P06IBAIB/uQofNTU1Kikp0d69e1VVVSXHcTRlyhQ1Nja2WO/RRx/VmTNnEpdVq1Z5WjQAAPCvPm5W3rJlS4vljRs3avDgwaqtrdWECRMS4xkZGcrOzvamQgAA0KO4Ch9/rqGhQZI0YMCAFuO//OUv9dJLLyk7O1szZszQokWLlJGR0eYcsVhMsVgssRyNRiVJjuPIcZzOlJdwZR7HcRRKNZ7M2d2FUkyLf3uDzvbs1f5m09X7dm9Bz+3n5+Odn45hyXiu8iM3dQeMMR26Z+PxuB544AGdP39eu3btSow///zzGj58uHJzc3Xw4EE99dRTys/P1yuvvNLmPGVlZSovL281XllZ+bmBBQAAdC9NTU2aPXu2GhoaFA6Hr7luh8PH3//93+utt97Srl27NHTo0M9db/v27br//vt17Ngx3XLLLa2ub+vMR15ens6ePXvd4tvLcRxVVVWpsLBQY5Zv92TO7i6UYrR0XFyL9qUoFg90dTlWdLbnw2VFSagqua7et4PBYFeXYwU9t7/nO8u2JrGq5PLTMcyrY4ff9+1oNKpBgwa1K3x06GWXxx57TG+++aZ27tx5zeAhSePHj5ekzw0foVBIoVCo1XgwGPT8jx8MBhVr7t47sddi8QA9t5MfH+xXJOPx0t3R8/X1hMe+H45hyXiu8uO+7aZmV+HDGKPHH39cr776qnbs2KERI0Zc9zYHDhyQJOXk5LjZFAAA6KFchY+SkhJVVlbq9ddfV2Zmpurq6iRJWVlZuuGGG3T8+HFVVlZq2rRpGjhwoA4ePKj58+drwoQJGj16dFIaAAAA/uIqfKxdu1bSn75I7GobNmzQI488orS0NG3btk1r1qxRY2Oj8vLyVFxcrIULF3pWMAAA8DfXL7tcS15enmpqajpVEAAA6Nn4bRcAAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABglavwsWLFCn3lK19RZmamBg8erFmzZuno0aMt1rl06ZJKSko0cOBA9e3bV8XFxaqvr/e0aAAA4F+uwkdNTY1KSkq0d+9eVVVVyXEcTZkyRY2NjYl15s+frzfeeEObN29WTU2NTp8+rQcffNDzwgEAgD/1cbPyli1bWixv3LhRgwcPVm1trSZMmKCGhgatX79elZWVmjx5siRpw4YNuv3227V3717dc8893lUOAAB8yVX4+HMNDQ2SpAEDBkiSamtr5TiOCgoKEuuMGjVKw4YN0549e9oMH7FYTLFYLLEcjUYlSY7jyHGczpSXcGUex3EUSjWezNndhVJMi397g8727NX+ZtPV+3ZvQc/t5+fjnZ+OYcl4rvIjN3UHjDEdumfj8bgeeOABnT9/Xrt27ZIkVVZWau7cuS3ChCTl5+dr0qRJ+vGPf9xqnrKyMpWXl7car6ysVEZGRkdKAwAAljU1NWn27NlqaGhQOBy+5rodPvNRUlKiw4cPJ4JHRy1YsEClpaWJ5Wg0qry8PE2ZMuW6xbeX4ziqqqpSYWGhxizf7smc3V0oxWjpuLgW7UtRLB7o6nKs6GzPh8uKklBVcl29bweDwa4uxwp6bn/Pd5ZtTWJVyeWnY5hXxw6/79tXXrlojw6Fj8cee0xvvvmmdu7cqaFDhybGs7OzdfnyZZ0/f179+vVLjNfX1ys7O7vNuUKhkEKhUKvxYDDo+R8/GAwq1ty9d2KvxeIBem4nPz7Yr0jG46W7o+fr6wmPfT8cw5LxXOXHfdtNza4+7WKM0WOPPaZXX31V27dv14gRI1pcP3bsWAWDQVVXVyfGjh49qpMnTyoSibjZFAAA6KFcnfkoKSlRZWWlXn/9dWVmZqqurk6SlJWVpRtuuEFZWVmaN2+eSktLNWDAAIXDYT3++OOKRCJ80gUAAEhyGT7Wrl0rSZo4cWKL8Q0bNuiRRx6RJK1evVopKSkqLi5WLBZTUVGRnn32WU+KBQAA/ucqfLTngzHp6emqqKhQRUVFh4sCAAA9F7/tAgAArCJ8AAAAqwgfAADAKsIHAACwivABAACsInwAAACrCB8AAMAqwgcAALCK8AEAAKwifAAAAKsIHwAAwCrCBwAAsIrwAQAArCJ8AAAAqwgfAADAKsIHAACwivABAACsInwAAACrCB8AAMAqwgcAALCK8AEAAKwifAAAAKsIHwAAwCrCBwAAsIrwAQAArCJ8AAAAqwgfAADAKsIHAACwivABAACsInwAAACrCB8AAMAqwgcAALCK8AEAAKxyHT527typGTNmKDc3V4FAQK+99lqL6x955BEFAoEWl2984xte1QsAAHzOdfhobGzUXXfdpYqKis9d5xvf+IbOnDmTuPzqV7/qVJEAAKDn6OP2BlOnTtXUqVOvuU4oFFJ2dnaHiwIAAD2X6/DRHjt27NDgwYPVv39/TZ48WcuWLdPAgQPbXDcWiykWiyWWo9GoJMlxHDmO40k9V+ZxHEehVOPJnN1dKMW0+Lc36GzPXu1vNl29b/cW9Nx+fj7e+ekYloznKj9yU3fAGNPhezYQCOjVV1/VrFmzEmObNm1SRkaGRowYoePHj+uf/umf1LdvX+3Zs0epqamt5igrK1N5eXmr8crKSmVkZHS0NAAAYFFTU5Nmz56thoYGhcPha67refj4c3/4wx90yy23aNu2bbr//vtbXd/WmY+8vDydPXv2usW3l+M4qqqqUmFhocYs3+7JnN1dKMVo6bi4Fu1LUSwe6OpyrKBnf/R8uKyoU7e/+vEcDAY9qqp762jPd5ZtTWJVyeWnfbuz+/QVft+3o9GoBg0a1K7wkZSXXa528803a9CgQTp27Fib4SMUCikUCrUaDwaDnv/xg8GgYs3deyf2WiweoOdewE89e/W4TsYxortz27Nf9olr8cO+nYznKj/u225qTvr3fHz88cc6d+6ccnJykr0pAADgA67PfFy8eFHHjh1LLJ84cUIHDhzQgAEDNGDAAJWXl6u4uFjZ2dk6fvy4vv/972vkyJEqKvLmtBQAAPA31+Fj3759mjRpUmK5tLRUkjRnzhytXbtWBw8e1Isvvqjz588rNzdXU6ZM0dKlS9t8aQUAAPQ+rsPHxIkTda33qG7d6t83OAEAgOTjt10AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVa7Dx86dOzVjxgzl5uYqEAjotddea3G9MUY//OEPlZOToxtuuEEFBQX68MMPvaoXAAD4nOvw0djYqLvuuksVFRVtXr9q1Sr967/+q37+85/r97//vW688UYVFRXp0qVLnS4WAAD4Xx+3N5g6daqmTp3a5nXGGK1Zs0YLFy7UzJkzJUn//u//riFDhui1117Tww8/3LlqAQCA77kOH9dy4sQJ1dXVqaCgIDGWlZWl8ePHa8+ePW2Gj1gsplgslliORqOSJMdx5DiOJ3VdmcdxHIVSjSdzdnehFNPi396Anv2hs4/rqx/PvUVHe/bz8c5P+3Yynqv8yE3dAWNMh+/ZQCCgV199VbNmzZIk7d69W1/96ld1+vRp5eTkJNb75je/qUAgoF//+tet5igrK1N5eXmr8crKSmVkZHS0NAAAYFFTU5Nmz56thoYGhcPha67r6ZmPjliwYIFKS0sTy9FoVHl5eZoyZcp1i28vx3FUVVWlwsJCjVm+3ZM5u7tQitHScXEt2peiWDzQ1eVYQc/+6PlwWVGnbn/14zkYDHpUVffW0Z7vLNuaxKqSy0/7dmf36Sv8vm9feeWiPTwNH9nZ2ZKk+vr6Fmc+6uvrdffdd7d5m1AopFAo1Go8GAx6/scPBoOKNXfvndhrsXiAnnsBP/Xs1eM6GceI7s5tz37ZJ67FD/t2Mp6r/Lhvu6nZ0+/5GDFihLKzs1VdXZ0Yi0aj+v3vf69IJOLlpgAAgE+5PvNx8eJFHTt2LLF84sQJHThwQAMGDNCwYcP0xBNPaNmyZfriF7+oESNGaNGiRcrNzU28LwQAAPRursPHvn37NGnSpMTylfdrzJkzRxs3btT3v/99NTY26rvf/a7Onz+v++67T1u2bFF6erp3VQMAAN9yHT4mTpyoa31AJhAIaMmSJVqyZEmnCgMAAD0Tv+0CAACsInwAAACruvx7PgD0Ljf94Dedun0o1WhV/p++w8LWRzA/WjndynaA3oIzHwAAwCrCBwAAsIrwAQAArCJ8AAAAqwgfAADAKsIHAACwivABAACsInwAAACr+JIxAAA6obNfnHeFzS/Q6+ovzuPMBwAAsIrwAQAArCJ8AAAAqwgfAADAKsIHAACwivABAACsInwAAACrCB8AAMAqwgcAALCK8AEAAKwifAAAAKsIHwAAwCrCBwAAsIrwAQAArCJ8AAAAqwgfAADAKsIHAACwivABAACsInwAAACrCB8AAMAqwgcAALDK8/BRVlamQCDQ4jJq1CivNwMAAHyqTzImveOOO7Rt27b/v5E+SdkMAADwoaSkgj59+ig7OzsZUwMAAJ9LSvj48MMPlZubq/T0dEUiEa1YsULDhg1rc91YLKZYLJZYjkajkiTHceQ4jif1XJnHcRyFUo0nc3Z3oRTT4t/egJ57h67o2atjUWe377YOPx/v2LeTKxn7tJs5A8YYT7t86623dPHiRd122206c+aMysvL9cknn+jw4cPKzMxstX5ZWZnKy8tbjVdWViojI8PL0gAAQJI0NTVp9uzZamhoUDgcvua6noePP3f+/HkNHz5czzzzjObNm9fq+rbOfOTl5ens2bPXLb69HMdRVVWVCgsLNWb5dk/m7O5CKUZLx8W1aF+KYvFAV5djBT3Tc7IcLiuysp3Pc/UxLBgMtvt2d5ZtTWJVycW+ndyek7FPR6NRDRo0qF3hI+nvBO3Xr59uvfVWHTt2rM3rQ6GQQqFQq/FgMOjqQdYewWBQsebesRNfEYsH6LkXoOfk8vpY1FFuj4s9YZ9g306OZOzTbuZM+vd8XLx4UcePH1dOTk6yNwUAAHzA8/Dx5JNPqqamRh999JF2796tv/qrv1Jqaqq+9a1veb0pAADgQ56/7PLxxx/rW9/6ls6dO6cvfOELuu+++7R371594Qtf8HpTAADAhzwPH5s2bfJ6SgAA0IPw2y4AAMAqwgcAALCKH10BgOu46Qe/6dLth1KNVuX/6Xs7etvHTtEzceYDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVhE+AACAVYQPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYlLXxUVFTopptuUnp6usaPH6933nknWZsCAAA+kpTw8etf/1qlpaVavHix3nvvPd11110qKirSp59+mozNAQAAH0lK+HjmmWf06KOPau7cufrSl76kn//858rIyNALL7yQjM0BAAAf6eP1hJcvX1Ztba0WLFiQGEtJSVFBQYH27NnTav1YLKZYLJZYbmhokCT98Y9/lOM4ntTkOI6ampp07tw59fms0ZM5u7s+caOmprj6OClqjge6uhwr6Jmeeyp6pmevnTt3zvM5L1y4IEkyxlx/ZeOxTz75xEgyu3fvbjH+j//4jyY/P7/V+osXLzaSuHDhwoULFy494HLq1KnrZgXPz3y4tWDBApWWliaW4/G4/vjHP2rgwIEKBLxJftFoVHl5eTp16pTC4bAnc3Z39EzPPRU903NP5feejTG6cOGCcnNzr7uu5+Fj0KBBSk1NVX19fYvx+vp6ZWdnt1o/FAopFAq1GOvXr5/XZUmSwuGwL+/QzqDn3oGeewd67h383HNWVla71vP8DadpaWkaO3asqqurE2PxeFzV1dWKRCJebw4AAPhMUl52KS0t1Zw5czRu3Djl5+drzZo1amxs1Ny5c5OxOQAA4CNJCR8PPfSQ/u///k8//OEPVVdXp7vvvltbtmzRkCFDkrG56wqFQlq8eHGrl3d6MnruHei5d6Dn3qE39Rwwpj2fiQEAAPAGv+0CAACsInwAAACrCB8AAMAqwgcAALCqx4SPiooK3XTTTUpPT9f48eP1zjvvXHP9zZs3a9SoUUpPT9eXv/xl/fa3v7VUqXfc9HzkyBEVFxfrpptuUiAQ0Jo1a+wV6iE3Pa9bt05f+9rX1L9/f/Xv318FBQXX3S+6Izc9v/LKKxo3bpz69eunG2+8UXfffbf+4z/+w2K13nD7eL5i06ZNCgQCmjVrVnILTAI3PW/cuFGBQKDFJT093WK13nB7P58/f14lJSXKyclRKBTSrbfe6rtjt5ueJ06c2Op+DgQCmj59usWKk8SbX3TpWps2bTJpaWnmhRdeMEeOHDGPPvqo6devn6mvr29z/d/97ncmNTXVrFq1yrz//vtm4cKFJhgMmkOHDlmuvOPc9vzOO++YJ5980vzqV78y2dnZZvXq1XYL9oDbnmfPnm0qKirM/v37zQcffGAeeeQRk5WVZT7++GPLlXec257/+7//27zyyivm/fffN8eOHTNr1qwxqampZsuWLZYr7zi3PV9x4sQJ8xd/8Rfma1/7mpk5c6adYj3itucNGzaYcDhszpw5k7jU1dVZrrpz3PYci8XMuHHjzLRp08yuXbvMiRMnzI4dO8yBAwcsV95xbns+d+5ci/v48OHDJjU11WzYsMFu4UnQI8JHfn6+KSkpSSw3Nzeb3Nxcs2LFijbX/+Y3v2mmT5/eYmz8+PHm7/7u75Jap5fc9ny14cOH+zJ8dKZnY4z57LPPTGZmpnnxxReTVaLnOtuzMcaMGTPGLFy4MBnlJUVHev7ss8/Mvffea37xi1+YOXPm+C58uO15w4YNJisry1J1yeG257Vr15qbb77ZXL582VaJnuvs43n16tUmMzPTXLx4MVklWuP7l10uX76s2tpaFRQUJMZSUlJUUFCgPXv2tHmbPXv2tFhfkoqKij53/e6mIz37nRc9NzU1yXEcDRgwIFlleqqzPRtjVF1draNHj2rChAnJLNUzHe15yZIlGjx4sObNm2ejTE91tOeLFy9q+PDhysvL08yZM3XkyBEb5XqiIz3/13/9lyKRiEpKSjRkyBDdeeed+tGPfqTm5mZbZXeKF8ew9evX6+GHH9aNN96YrDKt8X34OHv2rJqbm1t9e+qQIUNUV1fX5m3q6upcrd/ddKRnv/Oi56eeekq5ubmtgmd31dGeGxoa1LdvX6WlpWn69On62c9+psLCwmSX64mO9Lxr1y6tX79e69ats1Gi5zrS82233aYXXnhBr7/+ul566SXF43Hde++9+vjjj22U3Gkd6fkPf/iDXn75ZTU3N+u3v/2tFi1apH/5l3/RsmXLbJTcaZ09hr3zzjs6fPiw/vZv/zZZJVqVlK9XB7qblStXatOmTdqxY4cv35jnRmZmpg4cOKCLFy+qurpapaWluvnmmzVx4sSuLs1zFy5c0Le//W2tW7dOgwYN6upyrIlEIi1+qPPee+/V7bffrueee05Lly7twsqSJx6Pa/DgwXr++eeVmpqqsWPH6pNPPtFPfvITLV68uKvLS7r169fry1/+svLz87u6FE/4PnwMGjRIqampqq+vbzFeX1+v7OzsNm+TnZ3tav3upiM9+11nen766ae1cuVKbdu2TaNHj05mmZ7qaM8pKSkaOXKkJOnuu+/WBx98oBUrVvgifLjt+fjx4/roo480Y8aMxFg8Hpck9enTR0ePHtUtt9yS3KI7yYvHczAY1JgxY3Ts2LFklOi5jvSck5OjYDCo1NTUxNjtt9+uuro6Xb58WWlpaUmtubM6cz83NjZq06ZNWrJkSTJLtMr3L7ukpaVp7Nixqq6uTozF43FVV1e3+J/B1SKRSIv1Jamqqupz1+9uOtKz33W051WrVmnp0qXasmWLxo0bZ6NUz3h1P8fjccVisWSU6Dm3PY8aNUqHDh3SgQMHEpcHHnhAkyZN0oEDB5SXl2ez/A7x4n5ubm7WoUOHlJOTk6wyPdWRnr/61a/q2LFjiXApSf/7v/+rnJycbh88pM7dz5s3b1YsFtPf/M3fJLtMe7r6Ha9e2LRpkwmFQmbjxo3m/fffN9/97ndNv379Eh89+/a3v21+8IMfJNb/3e9+Z/r06WOefvpp88EHH5jFixf78qO2bnqOxWJm//79Zv/+/SYnJ8c8+eSTZv/+/ebDDz/sqhZcc9vzypUrTVpamnn55ZdbfFztwoULXdWCa257/tGPfmTefvttc/z4cfP++++bp59+2vTp08esW7euq1pwzW3Pf86Pn3Zx23N5ebnZunWrOX78uKmtrTUPP/ywSU9PN0eOHOmqFlxz2/PJkydNZmameeyxx8zRo0fNm2++aQYPHmyWLVvWVS241tF9+7777jMPPfSQ7XKTqkeED2OM+dnPfmaGDRtm0tLSTH5+vtm7d2/iuq9//etmzpw5Ldb/z//8T3PrrbeatLQ0c8cdd5jf/OY3livuPDc9nzhxwkhqdfn6179uv/BOcNPz8OHD2+x58eLF9gvvBDc9//M//7MZOXKkSU9PN/379zeRSMRs2rSpC6ruHLeP56v5MXwY467nJ554IrHukCFDzLRp08x7773XBVV3jtv7effu3Wb8+PEmFAqZm2++2Sxfvtx89tlnlqvuHLc9/8///I+RZN5++23LlSZXwBhjuuikCwAA6IV8/54PAADgL4QPAABgFeEDAABYRfgAAABWET4AAIBVhA8AAGAV4QMAAFhF+AAAAFYRPgAAgFWEDwAAYBXhAwAAWEX4AAAAVv0/gOD5Pi5BgOEAAAAASUVORK5CYII=",
"text/plain": [
""
]
},
"metadata": {},
"output_type": "display_data"
}
],
"source": [
"results_df['mcfadden_r2_adj'].hist()"
]
},
{
"cell_type": "markdown",
"id": "piano-society",
"metadata": {},
"source": [
"### Scale measure\n",
"\n",
"In our dataframe, we can find the ``'scale'`` column. The **scale** refers to the variance or the spread of the underlying distribution that is used for modeling. It indicates how much the observed data fluctuates around the fitted curve.\n",
"\n",
"- **Interpretation**:\n",
" - **Higher scale** values suggest that the data has more variability, implying a more spread-out distribution or greater fluctuations in the response variable.\n",
" - **Lower scale** values indicate less variability and suggest a more concentrated or stable response around the fitted model.\n",
"\n",
"- **Why use it?**: In GAMs, the scale can be informative about the nature of the data's distribution and helps in selecting which tasks or predictors might lead to clearer or more robust trends in the analysis. A higher scale may reflect data with more distinct or varied trends, while a lower scale may indicate more stable or predictable patterns.\n",
"\n",
"\n",
"We can inspect the distribution of this coefficient, across the GAMs used to model each metabolic task:"
]
},
{
"cell_type": "code",
"execution_count": 13,
"id": "maritime-cartridge",
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
""
]
},
"execution_count": 13,
"metadata": {},
"output_type": "execute_result"
},
{
"data": {
"image/png": "iVBORw0KGgoAAAANSUhEUgAAAikAAAGdCAYAAADXIOPgAAAAOXRFWHRTb2Z0d2FyZQBNYXRwbG90bGliIHZlcnNpb24zLjkuMSwgaHR0cHM6Ly9tYXRwbG90bGliLm9yZy/TGe4hAAAACXBIWXMAAA9hAAAPYQGoP6dpAAAlEUlEQVR4nO3de3BU9f3/8deGbDZEE2KIIckYLmIFxksQNDTqaCiEEByUmlYxaNFSUAeckky90BFJsDPgpehI01JnBNpKijoKVFQ0XOMlUAEziHUYQ/GCJKFAyZKkrkv2fP/wl/25JpBs2D372eT5mDkznHM++9n3ee/J5sXZ3azDsixLAAAAhomJdAEAAACdIaQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIwUG+kCesLn8+nIkSNKTEyUw+GIdDkAAKAbLMvSqVOnlJmZqZiYrq+TRGVIOXLkiLKysiJdBgAA6IGvvvpKF110UZfjojKkJCYmSvruIJOSkkI6t9fr1TvvvKNJkybJ6XSGdG50RL/tR8/tR8/tRb/t192eu91uZWVl+X+PdyUqQ0r7SzxJSUlhCSkJCQlKSkri5LYB/bYfPbcfPbcX/bZfsD3v7ls1eOMsAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJFiI12AqS4ve1uetu59lbQJPl96U6RLAAAgpLiSAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwUtAhpbq6WlOnTlVmZqYcDofWr18fsN/hcHS6PPXUU/4xQ4cO7bB/6dKl53wwAACg9wg6pLS0tCg7O1sVFRWd7q+vrw9YVq5cKYfDoaKiooBxixcvDhj3wAMP9OwIAABArxT0X5wtLCxUYWHhGfenp6cHrG/YsEHjx4/XxRdfHLA9MTGxw1gAAIB2Yf2z+I2NjXrjjTf0l7/8pcO+pUuX6vHHH9fgwYNVXFyskpISxcZ2Xo7H45HH4/Gvu91uSZLX65XX6w1pze3zuWKskM4bbqHug13a647W+qMRPbcfPbcX/bZfd3se7GPisCyrx7+NHQ6H1q1bp2nTpnW6/8knn9TSpUt15MgRxcfH+7cvW7ZMY8aMUUpKij744AMtWLBA99xzj5YtW9bpPGVlZSovL++wvbKyUgkJCT0tHwAA2Ki1tVXFxcVqampSUlJSl+PDGlJGjhyp/Px8LV++/KzzrFy5Uvfee6+am5vlcrk67O/sSkpWVpaOHTvWrYMMhtfrVVVVlRbujpHHFz1fMLi/rCDSJfRIe7/z8/PldDojXU6fQM/tR8/tRb/t192eu91upaamdjukhO3lnnfffVcHDhzQSy+91OXYcePG6fTp0/r88881YsSIDvtdLlen4cXpdIbtBPT4HFH1LcjR/oMYzscSnaPn9qPn9qLf9uuq58E+HmH7OykvvPCCxo4dq+zs7C7H1tbWKiYmRmlpaeEqBwAARJmgr6Q0Nzerrq7Ov37o0CHV1tYqJSVFgwcPlvTd5ZxXXnlFv//97zvcvqamRrt27dL48eOVmJiompoalZSU6M4779QFF1xwDocCAAB6k6BDyu7duzV+/Hj/emlpqSRp5syZWr16tSRp7dq1sixLd9xxR4fbu1wurV27VmVlZfJ4PBo2bJhKSkr88wAAAEg9CCl5eXnq6r22c+bM0Zw5czrdN2bMGO3cuTPYuwUAAH0M390DAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEhBh5Tq6mpNnTpVmZmZcjgcWr9+fcD+u+++Ww6HI2CZPHlywJgTJ05oxowZSkpKUnJysmbNmqXm5uZzOhAAANC7BB1SWlpalJ2drYqKijOOmTx5surr6/3L3//+94D9M2bM0CeffKKqqipt3LhR1dXVmjNnTvDVAwCAXis22BsUFhaqsLDwrGNcLpfS09M73ffpp59q06ZN+vDDD3X11VdLkpYvX64pU6bo6aefVmZmZrAlAQCAXigs70nZvn270tLSNGLECN1///06fvy4f19NTY2Sk5P9AUWSJk6cqJiYGO3atSsc5QAAgCgU9JWUrkyePFm33nqrhg0bpoMHD+q3v/2tCgsLVVNTo379+qmhoUFpaWmBRcTGKiUlRQ0NDZ3O6fF45PF4/Otut1uS5PV65fV6Q1p/+3yuGCuk84ZbqPtgl/a6o7X+aETP7UfP7UW/7dfdngf7mIQ8pEyfPt3/7yuuuEJXXnmlhg8fru3bt2vChAk9mnPJkiUqLy/vsP2dd95RQkJCj2s9m8ev9oVl3nB58803I13COamqqop0CX0OPbcfPbcX/bZfVz1vbW0Nar6Qh5Qfuvjii5Wamqq6ujpNmDBB6enpOnr0aMCY06dP68SJE2d8H8uCBQtUWlrqX3e73crKytKkSZOUlJQU0nq9Xq+qqqq0cHeMPD5HSOcOp/1lBZEuoUfa+52fny+n0xnpcvoEem4/em4v+m2/7va8/ZWQ7gp7SDl8+LCOHz+ujIwMSVJubq5OnjypPXv2aOzYsZKkrVu3yufzady4cZ3O4XK55HK5Omx3Op1hOwE9Poc8bdETUqL9BzGcjyU6R8/tR8/tRb/t11XPg308gg4pzc3Nqqur868fOnRItbW1SklJUUpKisrLy1VUVKT09HQdPHhQDz30kC655BIVFHz3P/1Ro0Zp8uTJmj17tlasWCGv16t58+Zp+vTpfLIHAAD4Bf3pnt27d+uqq67SVVddJUkqLS3VVVddpccee0z9+vXTvn37dPPNN+vSSy/VrFmzNHbsWL377rsBV0LWrFmjkSNHasKECZoyZYquv/56Pf/886E7KgAAEPWCvpKSl5cnyzrzJ1/efvvtLudISUlRZWVlsHcNAAD6EL67BwAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIQYeU6upqTZ06VZmZmXI4HFq/fr1/n9fr1cMPP6wrrrhC5513njIzM/WLX/xCR44cCZhj6NChcjgcAcvSpUvP+WAAAEDvEXRIaWlpUXZ2tioqKjrsa21t1d69e7Vw4ULt3btXr732mg4cOKCbb765w9jFixervr7evzzwwAM9OwIAANArxQZ7g8LCQhUWFna6b8CAAaqqqgrY9oc//EE5OTn68ssvNXjwYP/2xMREpaenB3v3AACgjwg6pASrqalJDodDycnJAduXLl2qxx9/XIMHD1ZxcbFKSkoUG9t5OR6PRx6Px7/udrslfffyktfrDWm97fO5YqyQzhtuoe6DXdrrjtb6oxE9tx89txf9tl93ex7sY+KwLKvHv40dDofWrVunadOmdbr/m2++0XXXXaeRI0dqzZo1/u3Lli3TmDFjlJKSog8++EALFizQPffco2XLlnU6T1lZmcrLyztsr6ysVEJCQk/LBwAANmptbVVxcbGampqUlJTU5fiwhRSv16uioiIdPnxY27dvP2sxK1eu1L333qvm5ma5XK4O+zu7kpKVlaVjx4516yCD4fV6VVVVpYW7Y+TxOUI6dzjtLyuIdAk90t7v/Px8OZ3OSJfTJ9Bz+9Fze9Fv+3W35263W6mpqd0OKWF5ucfr9eq2227TF198oa1bt3ZZyLhx43T69Gl9/vnnGjFiRIf9Lper0/DidDrDdgJ6fA552qInpET7D2I4H0t0jp7bj57bi37br6ueB/t4hDyktAeUzz77TNu2bdPAgQO7vE1tba1iYmKUlpYW6nIAAECUCjqkNDc3q66uzr9+6NAh1dbWKiUlRRkZGfrZz36mvXv3auPGjWpra1NDQ4MkKSUlRXFxcaqpqdGuXbs0fvx4JSYmqqamRiUlJbrzzjt1wQUXhO7IAABAVAs6pOzevVvjx4/3r5eWlkqSZs6cqbKyMv3jH/+QJI0ePTrgdtu2bVNeXp5cLpfWrl2rsrIyeTweDRs2TCUlJf55AAAApB6ElLy8PJ3tvbZdvQ93zJgx2rlzZ7B3CwAA+hi+uwcAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASEGHlOrqak2dOlWZmZlyOBxav359wH7LsvTYY48pIyND/fv318SJE/XZZ58FjDlx4oRmzJihpKQkJScna9asWWpubj6nAwEAAL1L0CGlpaVF2dnZqqio6HT/k08+qeeee04rVqzQrl27dN5556mgoEDffPONf8yMGTP0ySefqKqqShs3blR1dbXmzJnT86MAAAC9TmywNygsLFRhYWGn+yzL0rPPPqtHH31Ut9xyiyTpr3/9qwYNGqT169dr+vTp+vTTT7Vp0yZ9+OGHuvrqqyVJy5cv15QpU/T0008rMzPzHA4HAAD0FkGHlLM5dOiQGhoaNHHiRP+2AQMGaNy4caqpqdH06dNVU1Oj5ORkf0CRpIkTJyomJka7du3ST3/60w7zejweeTwe/7rb7ZYkeb1eeb3eUB6Cfz5XjBXSecMt1H2wS3vd0Vp/NKLn9qPn9qLf9utuz4N9TEIaUhoaGiRJgwYNCtg+aNAg/76GhgalpaUFFhEbq5SUFP+YH1qyZInKy8s7bH/nnXeUkJAQitI7ePxqX1jmDZc333wz0iWck6qqqkiX0OfQc/vRc3vRb/t11fPW1tag5gtpSAmXBQsWqLS01L/udruVlZWlSZMmKSkpKaT35fV6VVVVpYW7Y+TxOUI6dzjtLyuIdAk90t7v/Px8OZ3OSJfTJ9Bz+9Fze9Fv+3W35+2vhHRXSENKenq6JKmxsVEZGRn+7Y2NjRo9erR/zNGjRwNud/r0aZ04ccJ/+x9yuVxyuVwdtjudzrCdgB6fQ5626Akp0f6DGM7HEp2j5/aj5/ai3/brqufBPh4h/Tspw4YNU3p6urZs2eLf5na7tWvXLuXm5kqScnNzdfLkSe3Zs8c/ZuvWrfL5fBo3blwoywEAAFEs6Cspzc3Nqqur868fOnRItbW1SklJ0eDBgzV//nz97ne/049+9CMNGzZMCxcuVGZmpqZNmyZJGjVqlCZPnqzZs2drxYoV8nq9mjdvnqZPn84newAAgF/QIWX37t0aP368f739vSIzZ87U6tWr9dBDD6mlpUVz5szRyZMndf3112vTpk2Kj4/332bNmjWaN2+eJkyYoJiYGBUVFem5554LweEAAIDeIuiQkpeXJ8s688dzHQ6HFi9erMWLF59xTEpKiiorK4O9awAA0Ifw3T0AAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjBTykDJ06FA5HI4Oy9y5cyVJeXl5Hfbdd999oS4DAABEudhQT/jhhx+qra3Nv75//37l5+fr5z//uX/b7NmztXjxYv96QkJCqMsAAABRLuQh5cILLwxYX7p0qYYPH64bb7zRvy0hIUHp6emhvmsAANCLhDykfN+3336rF198UaWlpXI4HP7ta9as0Ysvvqj09HRNnTpVCxcuPOvVFI/HI4/H4193u92SJK/XK6/XG9Ka2+dzxVghnTfcQt0Hu7TXHa31RyN6bj96bi/6bb/u9jzYx8RhWVbYfhu//PLLKi4u1pdffqnMzExJ0vPPP68hQ4YoMzNT+/bt08MPP6ycnBy99tprZ5ynrKxM5eXlHbZXVlbyUhEAAFGitbVVxcXFampqUlJSUpfjwxpSCgoKFBcXp9dff/2MY7Zu3aoJEyaorq5Ow4cP73RMZ1dSsrKydOzYsW4dZDC8Xq+qqqq0cHeMPD5H1zcwxP6ygkiX0CPt/c7Pz5fT6Yx0OX0CPbcfPbcX/bZfd3vudruVmpra7ZAStpd7vvjiC23evPmsV0gkady4cZJ01pDicrnkcrk6bHc6nWE7AT0+hzxt0RNSov0HMZyPJTpHz+1Hz+1Fv+3XVc+DfTzC9ndSVq1apbS0NN10001nHVdbWytJysjICFcpAAAgCoXlSorP59OqVas0c+ZMxcb+/7s4ePCgKisrNWXKFA0cOFD79u1TSUmJbrjhBl155ZXhKAUAAESpsISUzZs368svv9Qvf/nLgO1xcXHavHmznn32WbW0tCgrK0tFRUV69NFHw1EGAACIYmEJKZMmTVJn78fNysrSjh07wnGXAACgl+G7ewAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMFPKQUlZWJofDEbCMHDnSv/+bb77R3LlzNXDgQJ1//vkqKipSY2NjqMsAAABRLixXUi677DLV19f7l/fee8+/r6SkRK+//rpeeeUV7dixQ0eOHNGtt94ajjIAAEAUiw3LpLGxSk9P77C9qalJL7zwgiorK/WTn/xEkrRq1SqNGjVKO3fu1I9//ONwlAMAAKJQWELKZ599pszMTMXHxys3N1dLlizR4MGDtWfPHnm9Xk2cONE/duTIkRo8eLBqamrOGFI8Ho88Ho9/3e12S5K8Xq+8Xm9Ia2+fzxVjhXTecAt1H+zSXne01h+N6Ln96Lm96Lf9utvzYB8Th2VZIf1t/NZbb6m5uVkjRoxQfX29ysvL9fXXX2v//v16/fXXdc899wQEDknKycnR+PHj9cQTT3Q6Z1lZmcrLyztsr6ysVEJCQijLBwAAYdLa2qri4mI1NTUpKSmpy/EhDyk/dPLkSQ0ZMkTLli1T//79exRSOruSkpWVpWPHjnXrIIPh9XpVVVWlhbtj5PE5Qjp3OO0vK4h0CT3S3u/8/Hw5nc5Il9Mn0HP70XN70W/7dbfnbrdbqamp3Q4pYXm55/uSk5N16aWXqq6uTvn5+fr222918uRJJScn+8c0NjZ2+h6Wdi6XSy6Xq8N2p9MZthPQ43PI0xY9ISXafxDD+Viic/TcfvTcXvTbfl31PNjHI+x/J6W5uVkHDx5URkaGxo4dK6fTqS1btvj3HzhwQF9++aVyc3PDXQoAAIgiIb+S8pvf/EZTp07VkCFDdOTIES1atEj9+vXTHXfcoQEDBmjWrFkqLS1VSkqKkpKS9MADDyg3N5dP9gAAgAAhDymHDx/WHXfcoePHj+vCCy/U9ddfr507d+rCCy+UJD3zzDOKiYlRUVGRPB6PCgoK9Mc//jHUZQAAgCgX8pCydu3as+6Pj49XRUWFKioqQn3XAACgF+G7ewAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYKeQhZcmSJbrmmmuUmJiotLQ0TZs2TQcOHAgYk5eXJ4fDEbDcd999oS4FAABEsZCHlB07dmju3LnauXOnqqqq5PV6NWnSJLW0tASMmz17turr6/3Lk08+GepSAABAFIsN9YSbNm0KWF+9erXS0tK0Z88e3XDDDf7tCQkJSk9PD/XdAwCAXiLs70lpamqSJKWkpARsX7NmjVJTU3X55ZdrwYIFam1tDXcpAAAgioT8Ssr3+Xw+zZ8/X9ddd50uv/xy//bi4mINGTJEmZmZ2rdvnx5++GEdOHBAr732WqfzeDweeTwe/7rb7ZYkeb1eeb3ekNbcPp8rxgrpvOEW6j7Ypb3uaK0/GtFz+9Fze9Fv+3W358E+Jg7LssL22/j+++/XW2+9pffee08XXXTRGcdt3bpVEyZMUF1dnYYPH95hf1lZmcrLyztsr6ysVEJCQkhrBgAA4dHa2qri4mI1NTUpKSmpy/FhCynz5s3Thg0bVF1drWHDhp11bEtLi84//3xt2rRJBQUFHfZ3diUlKytLx44d69ZBBsPr9aqqqkoLd8fI43OEdO5w2l/WsW/RoL3f+fn5cjqdkS6nT6Dn9qPn9qLf9utuz91ut1JTU7sdUkL+co9lWXrggQe0bt06bd++vcuAIkm1tbWSpIyMjE73u1wuuVyuDtudTmfYTkCPzyFPW/SElGj/QQznY4nO0XP70XN70W/7ddXzYB+PkIeUuXPnqrKyUhs2bFBiYqIaGhokSQMGDFD//v118OBBVVZWasqUKRo4cKD27dunkpIS3XDDDbryyitDXQ4AAIhSIQ8pf/rTnyR99wfbvm/VqlW6++67FRcXp82bN+vZZ59VS0uLsrKyVFRUpEcffTTUpQAAgCgWlpd7ziYrK0s7duwI9d0CAIBehu/uAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGCkkH8LMiJj6CNvRLqEoH2+9KZIlwAAMBhXUgAAgJEIKQAAwEiEFAAAYCRCCgAAMBIhBQAAGImQAgAAjERIAQAARiKkAAAAIxFSAACAkQgpAADASIQUAABgJEIKAAAwEiEFAAAYiZACAACMREgBAABGIqQAAAAjEVIAAICRCCkAAMBIhBQAAGCk2EgXAESToY+8EekSgvb50psiXQIA9AghBREz9JE35Opn6ckc6fKyt+Vpc0S6JACAQXi5BwAAGIkrKQAQAmd7KdDUK4a8FAjTcSUFAAAYKaJXUioqKvTUU0+poaFB2dnZWr58uXJyciJZEtDr/PB/+Kb+r/77+B8+ACmCV1JeeukllZaWatGiRdq7d6+ys7NVUFCgo0ePRqokAABgkIhdSVm2bJlmz56te+65R5K0YsUKvfHGG1q5cqUeeeSRSJUFAH0GH6m3RzT2WTKj1xEJKd9++6327NmjBQsW+LfFxMRo4sSJqqmp6TDe4/HI4/H415uamiRJJ06ckNfrDWltXq9Xra2tivXGqM1n5qXw3iTWZ6m11Ue/bRQNPT9+/HikSwha7OmWM++Lgp5Hi+6cG+3P48ePH5fT6bShqrM727lhsmB+Drvb81OnTkmSLMvq3sRWBHz99deWJOuDDz4I2P7ggw9aOTk5HcYvWrTIksTCwsLCwsLSC5avvvqqW3khKj6CvGDBApWWlvrXfT6fTpw4oYEDB8rhCO3/Stxut7KysvTVV18pKSkppHOjI/ptP3puP3puL/ptv+723LIsnTp1SpmZmd2aNyIhJTU1Vf369VNjY2PA9sbGRqWnp3cY73K55HK5ArYlJyeHs0QlJSVxctuIftuPntuPntuLftuvOz0fMGBAt+eLyKd74uLiNHbsWG3ZssW/zefzacuWLcrNzY1ESQAAwDARe7mntLRUM2fO1NVXX62cnBw9++yzamlp8X/aBwAA9G0RCym33367/vOf/+ixxx5TQ0ODRo8erU2bNmnQoEGRKknSdy8tLVq0qMPLSwgP+m0/em4/em4v+m2/cPXcYVnd/RwQAACAffjuHgAAYCRCCgAAMBIhBQAAGImQAgAAjNTnQkpFRYWGDh2q+Ph4jRs3Tv/85z/POv6VV17RyJEjFR8fryuuuEJvvvmmTZX2HsH0fPXq1XI4HAFLfHy8jdVGt+rqak2dOlWZmZlyOBxav359l7fZvn27xowZI5fLpUsuuUSrV68Oe529SbA93759e4dz3OFwqKGhwZ6Co9ySJUt0zTXXKDExUWlpaZo2bZoOHDjQ5e14Lu+5nvQ8VM/lfSqkvPTSSyotLdWiRYu0d+9eZWdnq6CgQEePHu10/AcffKA77rhDs2bN0kcffaRp06Zp2rRp2r9/v82VR69gey599xcL6+vr/csXX3xhY8XRraWlRdnZ2aqoqOjW+EOHDummm27S+PHjVVtbq/nz5+tXv/qV3n777TBX2nsE2/N2Bw4cCDjP09LSwlRh77Jjxw7NnTtXO3fuVFVVlbxeryZNmqSWljN/iR/P5eemJz2XQvRcfu5fFxg9cnJyrLlz5/rX29rarMzMTGvJkiWdjr/tttusm266KWDbuHHjrHvvvTesdfYmwfZ81apV1oABA2yqrneTZK1bt+6sYx566CHrsssuC9h2++23WwUFBWGsrPfqTs+3bdtmSbL++9//2lJTb3f06FFLkrVjx44zjuG5PLS60/NQPZf3mSsp3377rfbs2aOJEyf6t8XExGjixImqqanp9DY1NTUB4yWpoKDgjOMRqCc9l6Tm5mYNGTJEWVlZuuWWW/TJJ5/YUW6fxDkeOaNHj1ZGRoby8/P1/vvvR7qcqNXU1CRJSklJOeMYzvPQ6k7PpdA8l/eZkHLs2DG1tbV1+Iu2gwYNOuNrwQ0NDUGNR6Ce9HzEiBFauXKlNmzYoBdffFE+n0/XXnutDh8+bEfJfc6ZznG3263//e9/Eaqqd8vIyNCKFSv06quv6tVXX1VWVpby8vK0d+/eSJcWdXw+n+bPn6/rrrtOl19++RnH8VweOt3teaieyyP2Z/GBzuTm5gZ8yeS1116rUaNG6c9//rMef/zxCFYGhMaIESM0YsQI//q1116rgwcP6plnntHf/va3CFYWfebOnav9+/frvffei3QpfUZ3ex6q5/I+cyUlNTVV/fr1U2NjY8D2xsZGpaend3qb9PT0oMYjUE96/kNOp1NXXXWV6urqwlFin3emczwpKUn9+/ePUFV9T05ODud4kObNm6eNGzdq27Ztuuiii846lufy0Aim5z/U0+fyPhNS4uLiNHbsWG3ZssW/zefzacuWLQFp7/tyc3MDxktSVVXVGccjUE96/kNtbW36+OOPlZGREa4y+zTOcTPU1tZyjneTZVmaN2+e1q1bp61bt2rYsGFd3obz/Nz0pOc/1OPn8nN+620UWbt2reVyuazVq1db//rXv6w5c+ZYycnJVkNDg2VZlnXXXXdZjzzyiH/8+++/b8XGxlpPP/209emnn1qLFi2ynE6n9fHHH0fqEKJOsD0vLy+33n77bevgwYPWnj17rOnTp1vx8fHWJ598EqlDiCqnTp2yPvroI+ujjz6yJFnLli2zPvroI+uLL76wLMuyHnnkEeuuu+7yj//3v/9tJSQkWA8++KD16aefWhUVFVa/fv2sTZs2ReoQok6wPX/mmWes9evXW5999pn18ccfW7/+9a+tmJgYa/PmzZE6hKhy//33WwMGDLC2b99u1dfX+5fW1lb/GJ7LQ6snPQ/Vc3mfCimWZVnLly+3Bg8ebMXFxVk5OTnWzp07/ftuvPFGa+bMmQHjX375ZevSSy+14uLirMsuu8x64403bK44+gXT8/nz5/vHDho0yJoyZYq1d+/eCFQdndo/3vrDpb3HM2fOtG688cYOtxk9erQVFxdnXXzxxdaqVatsrzuaBdvzJ554who+fLgVHx9vpaSkWHl5edbWrVsjU3wU6qzXkgLOW57LQ6snPQ/Vc7nj/xUAAABglD7znhQAABBdCCkAAMBIhBQAAGAkQgoAADASIQUAABiJkAIAAIxESAEAAEYipAAAACMRUgAAgJEIKQAAwEiEFAAAYCRCCgAAMNL/AR/WmJBpA9mEAAAAAElFTkSuQmCC",
"text/plain": [
""
]
},
"metadata": {},
"output_type": "display_data"
}
],
"source": [
"results_df['scale'].hist()"
]
},
{
"cell_type": "markdown",
"id": "solar-affairs",
"metadata": {},
"source": [
"To identify metabolic tasks with meaningful dynamic behavior across the trajectory, we filtered for tasks that showed both good model fit and substantial variability. Specifically, we retained tasks with an adjusted McFadden pseudo R² > 0.2 and a scale parameter > 0.2.\n",
"\n",
"- A value above 0.2 for the adjusted McFadden pseudo R² indicates a relatively strong fit, suggesting the model captures a genuine signal rather than noise (McFadden wrote [on page 35 of this book chapter](https://elischolar.library.yale.edu/cowles-discussion-paper-series/707/) that **a value between 0.2 - 0.4 represents an excellent fit**).\n",
"\n",
"- A higher scale value indicates more overall variability in the task's activity across the trajectory, which can help prioritize more dynamic or biologically variable patterns.\n",
"\n",
"This dual threshold helps prioritize tasks with both high explanatory power and sufficient biological variability, reducing the chance of highlighting either overly flat or noisy trends.\n",
"\n",
"\n",
"\n",
"
\n",
"Note!\n",
"\n",
"For trajectories using continuous values (e.g. pseudo-time), instead of using the ``'mcfadden_r2_adj'``, we recommend using the ``'explained_deviance'`` to filter important metabolic tasks.\n",
"\n",
"
"
]
},
{
"cell_type": "markdown",
"id": "least-algorithm",
"metadata": {},
"source": [
"After filtering the metabolic tasks, we ended up with 15 important tasks."
]
},
{
"cell_type": "code",
"execution_count": 14,
"id": "special-mileage",
"metadata": {},
"outputs": [
{
"data": {
"text/plain": [
"15"
]
},
"execution_count": 14,
"metadata": {},
"output_type": "execute_result"
}
],
"source": [
"gam_tasks = results_df.loc[(results_df['mcfadden_r2_adj'] > 0.2) & (results_df['scale'] > 0.2)].sort_values(by='scale', ascending=False)['gene'].values.tolist()\n",
"len(gam_tasks)"
]
},
{
"cell_type": "markdown",
"id": "australian-hardwood",
"metadata": {},
"source": [
"## Visualization of results \n",
"\n",
"We can visualize the trends of these tasks through different approaches."
]
},
{
"cell_type": "markdown",
"id": "funny-evans",
"metadata": {},
"source": [
"### Violin plots\n",
"\n",
"For example, we can use violin plots to see how their distributions across single cells are changing across the trajectory. Violin plots in this case are useful for categorical labels (ordered cell types in this case)."
]
},
{
"cell_type": "code",
"execution_count": 15,
"id": "current-floor",
"metadata": {},
"outputs": [
{
"data": {
"image/png": "iVBORw0KGgoAAAANSUhEUgAAB9YAAAfFCAYAAADEPScWAAAAOXRFWHRTb2Z0d2FyZQBNYXRwbG90bGliIHZlcnNpb24zLjkuMSwgaHR0cHM6Ly9tYXRwbG90bGliLm9yZy/TGe4hAAAACXBIWXMAAA9hAAAPYQGoP6dpAAEAAElEQVR4nOzddXgUx/8H8Pdd3F1JQnD34Fq8WCkWPFiBUqRFirRFWqSFFr5tsUJxSimFUlwKBPfgbsGChwjEw83vj/x2ucvdRY4kdyTv1/PkSbJ2n93b3Zmd2ZlRCCEEiIiIiIiIiIiIiIiIiIiISCelsQMgIiIiIiIiIiIiIiIiIiIyZaxYJyIiIiIiIiIiIiIiIiIiygAr1omIiIiIiIiIiIiIiIiIiDLAinUiIiIiIiIiIiIiIiIiIqIMsGKdiIiIiIiIiIiIiIiIiIgoA6xYJyIiIiIiIiIiIiIiIiIiygAr1omIiIiIiIiIiIiIiIiIiDLAinUiIiIiIiIiIiIiIiIiIqIMsGKdiIiIiIiIiIiIiIiIiIgoA6xYJyIiIiIiIiIiIiIqIAIDA6FQKLR+7O3tUalSJYwfPx6RkZE61+3Tpw8UCgWWL1+et0GboP3790OhUKBRo0bGDkWnRo0aQaFQYP/+/cYORYsQArNmzUL58uVhY2Mjn4P6SMc6uz+TJ0/Ou50yQaZ8DuQUlUqFoKAgeHt7Iy4uTu9y+/fvxyeffIKyZcvCxcUFFhYWcHNzQ40aNTB06FDs2bMHQgit9XjPM22TJ0/O82vdPM8+iYiIiIiIiIiIiIiITELdunVRvHhxAGmVU48ePcLRo0fx/fffY+XKlTh06BCKFi1q5CiNJzAwEPfu3UN4eDgCAwONHU6+smDBAnz55ZdwcnLChx9+CEdHxwyX9/b2RkhIiNb0c+fO4fz58/Dy8kLLli215leuXDmnQjY5kydPxpQpUzBp0qQC/QLBkiVLEBYWhrlz58LOzk5r/osXL9CjRw/s3r0bAFCoUCHUrVsXTk5OiImJwaVLlzBv3jzMmzcPVapUwZkzZ/J6FwqE/HQ/ZcU6EREREREREREREVEBM2DAAPTp00dj2pMnT9CwYUPcuHEDX375JdavX68xf8aMGRg3bhx8fHzyMFIyxMqVKxEfH4+AgABjh6Jl3bp1AIC///4bzZo1y3T50qVL62wxPHnyZJw/f17v/ILOlM+BnJCQkICvvvoKvr6+GDhwoNb86Oho1KtXD9evX0fp0qUxf/58fPDBB1rLXbp0CXPmzMHatWvzImzKQUOHDkXXrl3h7u6eZ5/JinUiIiIiIiIiIiIiIoK3tzfGjBmDTz75BHv37tWa7+Pjw0r194QpV6bev38fAFCiRAkjR5K/mfI5kBNWr16N58+fY9y4cbCwsNCaP2zYMFy/fh1FixbF0aNH4eLionM75cuXx5IlSzBo0KDcDplymLu7e55WqgMcY52IiIiIiIiIiIiIiP6ft7c3ACA1NVVrXmbjDa9duxZNmjSBq6srrKysULhwYfTr1w83btzQufzjx48xYsQIlCxZEtbW1rC1tYW/vz+aNGmCH3/8UV5u2bJlUCgUaNGihd64Hz16BAsLC9jY2MhjxN+9excKhQKBgYEQQmDRokWoVq0a7Ozs4OTkhObNm+PYsWMa21m+fDkUCgXu3bsHAChSpIjGuN26xqtOSUnBDz/8gHLlysHGxgZubm7o0KEDrl69qjfeqKgoTJo0CZUrV4aDgwNsbW1RoUIFTJ06FfHx8VrLq1QqLFq0CHXr1oWzszMsLCzg6emJSpUqYdiwYbh7967G8vrG105KSsKsWbNQrVo1ODg4wNLSEt7e3qhevTq+/PJLvHz5Um/Murx8+RITJkxAuXLlYGtrCwcHB1SrVg0zZ85EQkKCzpjCw8MBaB7b3OzOXH0c5ufPn+Ozzz6Dv78/LC0t4e/vj2HDhiE6OvqdPiMlJQWrV69Gjx49ULp0aTg6OsLGxgalSpXC8OHD8ejRI53rqX9P586dQ4cOHeDu7g4rKyuULVsWP/30k9bY3wqFAlOmTAEATJkyReP8VO+FIqMx1uPi4vDNN9+gRIkSsLKygq+vL/r164eIiIhMx60OCwtDjx49EBAQACsrK7i6uqJFixbYvn27zuUDAwOhUChw9+5dhIaGonnz5nBxcYGNjQ2qVq2KlStXZn6AdZg7dy4AaPW8AQC3b9/GmjVrAABz5szRW6murkaNGln63JCQECgUCsyYMUPvMuvWrYNCodC5zRs3bmDIkCEoVaoUbG1t4ejoiLJly2LIkCG4dOmS1vLXrl1D3759UbhwYfl4N2nSRO75IT317+/evXvo3bs3fHx8YG1tjZIlS2Ly5Mla1ybw9t7Xp08fvHz5Ep9//jmKFSsGKysrNGrUSF4uNTUVCxcuRJ06deDk5ARra2uUKFECw4cPR0REhM5tZvV++ujRI4wcORJlypSR7yfVq1fH3LlzdaZJ+s5V9X2Ji4vD+PHjUbx4cVhZWclDS6SPNavYYp2IiIiIiIiIiIiIiAAAJ0+eBACUK1cuy+sIIdCnTx+sXLkS5ubmaNCgATw9PXHmzBksW7YMf/31FzZs2KAxDvaTJ08QFBSER48eISAgAC1btoS1tTUePXqEc+fOISwsDKNHjwYAdO/eHWPHjsV///2HGzduoGTJklox/Pbbb0hNTUWvXr3g5uamNb9v375Ys2YN6tevjzZt2uDcuXP477//cPDgQRw4cAA1a9YEABQvXhwhISFYv3494uLi0LFjR9jb28vbkV48kKSkpKBVq1Y4evQoGjRogDJlyuDkyZPYuHEjQkNDcfbsWa0xha9cuYKWLVviwYMH8PHxQb169WBhYYGTJ0/im2++wYYNG7B//344OTnJ6wwYMADLli2DtbU16tWrBw8PD7x8+RJ37tzB3Llz0aRJk0zHLlapVGjdujX27t0LR0dH1K9fH87Oznj+/Dlu3ryJWbNmoXv37nB1dc1wO5I7d+6gcePGuHfvHjw8PNCqVSukpKQgNDQUY8eOxV9//YU9e/bIlZotW7ZEYGCgzmObF+OhP3jwAFWrVkVKSgrq1q2LxMREHDlyBHPnzsWJEydw5MgRnS2fs+Lp06fo1asXnJycUKZMGVSsWBFxcXE4d+4cfv31V6xduxZHjx5F8eLFda6/a9cuzJ49G8WKFUOzZs3w+PFjHD58GKNHj8aDBw/wv//9T142JCREHl++UqVKGseuXr16mcYaFxeHDz74AKdOnYK9vT2aN28OGxsb7Ny5E9u2bUOrVq30rvvzzz9j5MiRUKlUqFy5MmrWrIknT55g//792L17N6ZMmYKJEyfqXHfp0qWYOnUqqlatipYtW+Lu3bs4fvw4QkJC5IrcrAoPD8eFCxfg5+eHUqVKac3funUrVCoVXFxc0KZNmyxvNytGjBiBlStXYuHChfjyyy9hZmamtcy8efMApHVVrm7NmjXo168fkpKSEBAQgFatWkGlUuHOnTtYuHAhPD09Ub58eXn5bdu2oVOnTkhMTESpUqXQoUMHPHv2DAcOHMC+ffuwa9cuLFmyRGec4eHhqFatmnxPTkhIQGhoKKZMmYI9e/Zgz549sLa21lrvxYsXCAoKQnR0NOrXr49q1arB0tISQNqLOW3atJHX/eCDD+Do6IijR4/i119/xZ9//oldu3ahatWqALJ3Pz148CDat2+PqKgoBAYGolmzZkhKSsLJkycxbNgwbNmyBVu3bs3WNRoTE4M6derg/v37qF+/PsqXL49jx45h5cqVOHDgAM6fP69xn80SQUREREREREREREREBULhwoUFALFs2TJ52ps3b8TDhw/Fr7/+KqysrISZmZnYsmWL1rohISFa6wohxIIFCwQA4e7uLs6ePStPV6lUYtKkSQKAcHZ2Fs+ePZPnTZkyRQAQAwcOFCqVSmN7ycnJYs+ePRrTvvrqKwFADB8+XCuu5ORk4e3tLQCIsLAweXp4eLgAIACIwoULi+vXr8vzUlNTRb9+/QQA0bx5c73HKTw8XGueEEKEhobK265SpYp4/PixPC8hIUG0aNFC3j918fHxolixYgKA+Prrr0VSUpI8Ly4uTnTr1k0AEH379pWn37t3TwAQfn5+Gp8juXLlirh3757GtIYNGwoAIjQ0VJ524MABOd7Y2Fit7Zw6dUq8ePFC5/7qUrNmTQFAtGvXTrx+/Vqe/uzZM1G1alUBQHTv3l1rvcyObVZJ51bDhg2ztBwA0adPH5GYmCjPu3//vihUqJAAINasWWNwLLGxsWLTpk0a36cQaefm+PHjBQDRqlUrrfWk7wmAWLhwoca8vXv3CoVCIczMzMSDBw907tOkSZP0xqTrHBBCiC+++EIAEGXLlhWPHj2SpyckJIhOnTrJ8aTf9s6dO4VCoRDu7u7iwIEDGvMuXLgg/Pz8BACxf/9+jXnS921hYaF1X1m2bJkAIJycnER8fLzefUnv999/FwBE586ddc7v1auXACCaNGmS5W3qou+eV7duXQFA/PPPP1rrXLx4UQAQHh4eGufa6dOnhYWFhVAoFOKXX34Rb9680Vjv7t274vTp0/L/T548EU5OTgKAmDp1qsZ98tSpU8LFxUUAEIsWLdLYjvr5/tFHH2kc1wcPHoiSJUsKAGLcuHEa60nfhXTcYmJitPZt7NixAoAoVqyYxvWbnJws+vfvLwCIIkWKaF0HmV3zjx8/Fm5ubkKhUIj58+drHJsXL16Ixo0bCwBiypQpOvc1/bmqvi8tWrTQ2JeXL1+KypUrCwBi+vTpOuPJCLuCJyIiIiIiIiIiIiIqYPr27St3x2tmZgY/Pz8MGzYMFStWxIEDB7LVylPqtn3ixIkarWcVCgUmTZqEihUrIjo6GosXL5bnPX36FEBaK2aFQqGxPQsLCzRp0kRj2pAhQ2BhYYEVK1YgLi5OY96GDRvw5MkT1K5dW24pmd6vv/6q0dLdzMwM06ZNAwAcOHAAKSkpWd5fdQqFAsuWLdNoeWltbS131b1nzx6N5VesWIHbt2+jTZs2+O677+SWoABga2uLRYsWwdPTE6tWrUJUVBSAt8eqatWqWi3mAaBMmTJZGk9b2k79+vXh4OCgNT8oKEhna39dDh8+jBMnTsgx29nZyfM8PDywaNEiAGnDAzx8+DBL28xtfn5+mDdvHqysrORpUlfwgPZ3lR0ODg5o166dxvcJpJ3L06dPh6+vL3bu3IlXr17pXL9Dhw5aY3w3btwYLVq0wJs3bxAaGmpwbOoSEhLk63DOnDnw8fGR51lbW2P+/PmwtbXVue6kSZMghMDChQvRoEEDjXkVKlTA7NmzAaRda7oMGzZM677Sp08flC5dGjExMTh9+nSW9+Ps2bMA0s59XV68eAEg7VzU5fz58+jTp4/Wz+HDh7P0+SNGjADwtmW6OqmL+gEDBmica1OnTkVKSgqGDh2KYcOGQanUrKItXLgwqlWrJv+/ePFixMTEoFq1avjqq6807pNBQUH46quvAACzZs3SGaONjQ0WLlwIGxsbeZqfnx9++uknAMD8+fORmJiotZ6FhQUWLVoER0dHjemJiYny/s6ZM0ejhwwLCwv88ssv8PLyQnh4ONavX68zJn3+97//ITIyEp999hk+/fRTjWPj5uaGlStXwsLCAnPnztUaGiEjdnZ2WLZsmca+uLi4YNy4cQAMu+ZZsU5EREREREREREREVMDUrVsXISEh8k/r1q3h7++PU6dO4YsvvsDNmzeztJ2HDx/i9u3bANK6qE5PoVCgb9++AKBROSiNPTxu3Dj8888/eP36dYaf4+vri06dOiEmJgarVq3SmKev22WJubm5Rjf0Em9vb7i4uCApKUkelz27AgICUKlSJa3pUoVf+nF8t23bBgAIDg7WuT17e3sEBQUhNTUVp06dAgCULl0aDg4O2L59O6ZNmyaPUZ5dVatWhZmZGZYuXYp58+bh8ePHBm0HgDw2csuWLeHl5aU1v1q1aqhUqRJUKhUOHDhg8OfkpCZNmuisNNb3XRni/PnzmD17NoYNG4Z+/frJFbapqalQqVS4deuWzvXatm2rc3pOxgakjY/++vVruLu7o3nz5lrzPTw80KxZM63pL168wMmTJ2FjY6M3Vmkc7qNHj+qcn5P7KL0kktUXQdJ78OABVqxYofWj7/tJ7+OPP4a/vz/27t2La9euydNjYmKwevVqmJmZ4dNPP5Wnv3nzBv/99x8AYODAgVn6DOka03VfBYD+/fsDAG7evIlHjx5pzW/evLnOF3HatGkDNzc3xMbG4syZM1rzq1SpgqJFi2pNP336NF6/fg1XV1ed36WtrS26du0KANl+ESSz+2KhQoVQokQJediKrAoKCtJ4eUTyLtcVK9aJiIiIiIiIiIiIiAqYAQMGYPny5fLP1q1bcefOHYwfPx6nTp1Cw4YN9bauVSdVTLi5uWm1cJQUK1ZMY1kA6NWrF3r06IEbN26gY8eOcHZ2RsWKFTFkyBDs27dP53aGDx8OQLOV6IULF3D48GF4eXmhU6dOOtfz8fHROy6vFLOulptZoa+luLTdpKQkjel37twBkLb/Uo8B6X+2b98OAHj+/DmAtNbQy5Ytg42NDb7++msULVoUvr6+6NChAxYtWpTpSwmSYsWKYc6cOXKrWV9fXwQGBqJbt274448/kJycnOX9lr7LIkWKZPh56ssaW2bflaHnAJA2bnmHDh1QuXJljBo1CnPnzsWyZcvkCttnz54BAGJjY/M8NnVS7wHqrY3T0zUvPDwcQggkJCTAyspK53nr6ekJ4O15m15O7mNMTIzGuum5u7tnGEubNm0ghJB/0veQkRlzc3MMGTIEwNsW6gDkHjXatWsHf39/eXpkZKTc04auMeF1yewac3Z2hqurKwDo7BUio2tT+o51rafv3MjNa166L9avX1/vffHKlSsA9H+nuuTGdWWe7TWIiIiIiIiIiIiIiCjfMTc3x9SpU7F48WI8fvwYK1euxGeffZYrn6VUKrF69WpMmDAB27Ztw5EjR3DkyBEsWLAACxYsQNu2bbFx40aYmZnJ69SqVQs1atTAyZMnceDAATRs2FCuZB84cKBWN9zqn5VbsrttlUoFQH9Lb3WFCxeW/+7YsSOaNm2KzZs349ChQzhy5Ag2btyIjRs3YuLEifjvv/9QoUKFTD9/2LBh6NKlCzZv3ozDhw/j8OHDWLt2LdauXYtJkybh0KFDOlt45ge5eR6MHz8eGzduROnSpfH999+jevXqcHd3l8/JOnXq4NixY3q7sc7N2HRJP/xCZvOk89be3h4dO3Y06DNzch+dnZ0B6H9RoWrVqli1ahXOnDkDlUqVK8f3k08+wbfffouVK1dixowZsLe3x/z58wHo7z3D1Og6H9W7js8r0vnVqVMnjWEldMlOLwW58b2zYp2IiIiIiIiIiIiIiACkVUQEBgbixYsXuHr1aqbLFypUCEBai8zY2FidLUil1ojSsurKli2LsmXLYsyYMRBCYN++fejevTu2bNmClStXyt3IS4YPH46ePXti7ty5qFSpEv744w+Ym5tj8ODBhuxunvP398e1a9fQv39/vS3s9XFyckKvXr3Qq1cvAGndWQ8bNgybNm3C0KFDs9zlupeXFz755BN88sknAIBr166hX79+OHbsGMaNG4cVK1Zkug3pu5S+W10y+t7zm3Xr1gEA/vrrL1SsWFFrfna6r85N0ndx9+5dvcvomie1vlYoFFi6dGmevwiQntQ6Xt8QDm3atMGoUaMQFRWF7du3a43tnhPc3NzQo0cP/P7771i5ciVKliyJ69evo2zZsmjcuLHWsra2toiPj8f169dRvnz5TLdfqFAhXLt2Te81FhMTg5cvX8rLppfRkBHSd+zn55dpHOrxZLZdQ695f39/3Lx5E2PHjkVQUFC21s1r7AqeiIiIiIiIiIiIiIgApLUclCpd7O3tM13ez89P7v53+fLlWvOFEPL0Dz74IMNtKRQKNGnSBN27dwcAnDt3TmuZLl26wMfHB//++y+mTZuGuLg4fPzxx/D19c001uyQWhqnpqbm6HY//PBDAG8rYt+Fv78/pkyZAkD3scqq0qVLY+zYsdnajjSe9s6dO+XxrtWdPXsW586dg1KpRIMGDQyO7X0hVXCq9zIg2bVrF168eJGjn2fo+VmtWjXY2tri+fPn2LNnj9b8Fy9eyGOBq/P19UXFihXx6tUr7Ny507Cgc1DVqlUBQO4ePL3ixYvL43WPHDlS7jo+p6kPTyF1Ca+rlw8zMzN57PrFixdnadvSNabvRZelS5cCAEqUKKGzInv37t3yEATqtm/fjsjISDg4OKBatWpZigVIG6/c3t4eL1++xObNm7XmJyQkYO3atQC07/WZna85eV/MbaxYJyIiIiIiIiIiIiIipKam4uuvv5YrAdu1a5el9UaPHg0A+O6773D+/Hl5uhACU6dOxblz5+Ds7Cy3kAaAlStXIiwsTGtbr169wv79+wHorqS0sLDAp59+itTUVPz4448AcqfbZakl5+XLl3N0uwMHDkThwoXx999/Y+zYsTrHsX/y5IlG5dvZs2fx119/ISEhQWvZLVu2ANB9rNLbt28ftm/fjpSUFI3pQghs3bo1y9sBgHr16qFmzZpISEjAoEGDEB8fL8978eIFBg0aBADo2rWrxljT+VWZMmUAAL/++qvG9OvXr+dKbwqGnp+2trYYMGAAAOCLL77QeCkiKSkJQ4cOlccCT2/q1KkAgL59+8rnnTohBE6cOIHdu3dnKyZDSBW3x44d07vMvHnzULx4cdy8eRN16tTR26PD3bt3dY41nhUVKlRA48aNcfXqVWzevBmOjo7o3bu3zmW/+uormJubY+7cuZg/f75WN+z37t3TuCd+8skncHR0xJkzZzB9+nSN5c+ePSt/H2PGjNH5eQkJCfj000817huPHj3CqFGjAACDBw+GtbV1lvfV2tpafmlg1KhRuHfvnjwvJSUFI0aMwJMnT1CkSBGt3jgyO1/HjBkDZ2dnzJ49Gz/99BOSk5O1lgkPD8fq1auzHG9uYVfwREREREREREREREQFzO+//y5XYANpXSqfP38eDx48AJBWCVSnTp0sbWvQoEE4evQoVq1ahaCgIDRs2BCenp44c+YMrl+/DhsbG6xZswYeHh7yOv/88w9CQkLg6+uLypUrw8XFBVFRUThy5AhiYmJQvnx5jYr49J83bdo0JCUloWLFirnSIrpjx44IDQ1Fz5490bx5c7i4uABIqwAqVaqUwdu1s7PDtm3b0KZNG8ycOROLFi1CxYoV4efnh/j4eNy4cQNXr16Fp6envP/37t1D165dYWNjg6pVq8Lf3x+pqam4ePEirl+/DktLS8ycOTPTz75w4QK++OILODo6omrVqvD19UVCQgLOnDmDe/fuwcnJCd9++22W92XNmjVo3LgxNm3ahCJFiqBBgwZISUlBaGgoYmNjUbVqVbkVb343adIkdOrUCd988w3WrVuHcuXK4dmzZzh06BDq168PX19fHD16NMc+r0WLFrCzs8O///6LevXqoUSJEjAzM0PdunW1hk9Ib9q0aThy5AjCwsJQvHhxNG7cGNbW1jh8+DCSk5MREhKCFStWyK2MJW3btsXPP/+MUaNGoV27dihevDhKlSoFJycnPH/+HOfPn8ezZ88wduxYNG/ePMf2VZciRYqgYsWKuHDhAq5evSq/2KDOxcUFR44cQffu3bF37140atQIfn5+qFy5MpydnZGQkICbN2/i4sWLEEKgQoUKBnVDPnz4cOzbtw8AEBISorenj+rVq2PJkiUYMGAAPvvsM8ycORPVq1eHSqXCnTt3cP78eUycOFFuRe7l5YU//vgDnTt3xldffYVVq1ahSpUqePbsGQ4cOIDU1FT07dtX732yd+/e2Lp1K4oWLYr69esjMTER+/btQ1xcHGrXri33dpEdU6ZMwenTp7F3716UKVMGH3zwARwcHHDs2DHcv38fbm5u+Pvvv7XOnczup35+fti0aRM6duyI0aNHY+bMmShfvjx8fHwQExODq1ev4vbt26hZsyZ69uyZ7bhzEivWiYiIiIiIiIiIiIgKmCNHjuDIkSPy/5aWlvDx8UFwcDAGDx4sd0OcFQqFAitXrsSHH36IRYsWISwsDHFxcfD29kafPn0wbtw4rcroUaNGoUiRIjh69CjOnDmDly9fwtXVFWXLlkX37t3Rt29f2NnZ6fw8T09PVK5cGSdOnNDZ7XJO+PTTT/Hq1SusXr0a27dvR2JiIgCgZ8+e71SxDgDlypXDhQsXsHDhQmzcuBEXLlzAsWPH4O7uDj8/P4wePRoff/yxvHytWrXw/fff4+DBg7h69SrOnj0Lc3Nz+Pn54bPPPsOwYcOyFFPbtm0RExODQ4cO4ebNmzh+/DhsbGzg7++PcePG4bPPPsvWmMtFixbFmTNn8OOPP+Lff//F1q1boVQqUapUKQQHB2P48OGwsbEx6Bi9bzp06IADBw5gypQpOH/+PG7fvo2iRYti8uTJGD16dI5XNHt5eWHHjh349ttvERYWhmPHjkGlUsmVrRmxt7fH/v37MWPGDKxduxY7d+6Eq6srmjVrhqlTp8oVru7u7lrrDh8+HI0bN8avv/6K0NBQ7N27F0qlEt7e3qhSpQpat26Njh075ui+6jN06FAMHDgQy5cvxw8//KBzGU9PT+zZswd79+7FmjVrcOTIERw8eBDx8fFwcHBAkSJFMHDgQHTq1AmNGzc2aOz4Jk2awMzMDCqVKtP7Ue/evREUFITZs2dj37592LJlC6ytrVGoUCF89tln6NKli8bybdq0wZkzZ/DDDz9g7969WL9+Pezs7FC/fn0MGjRI7u5elyJFiuD06dP46quvsG/fPkRFRSEgIADdu3fH2LFjDbo2rayssHPnTixevBgrV67EoUOHkJSUBH9/fwwbNgxjx47V2S19Vu6nDRo0wOXLlzF37lxs27YNp06dQlJSEjw9PREQEICePXvm2bmVEYVI39cAERERERERERERERGRibpx4wZKly4NJycnREREwNbW1tghEeULKSkpKF++PG7cuIGwsDB5LHNTFB8fj8DAQJibm+Pu3btaraTzyu+//45PPvkEzZs3x65du4wSg7rJkydjypQpmDRpEiZPnmzscPIdjrFORERERERERERERETvjYkTJ0IIgU8//ZSV6kQGCAsLg0ql0pj2+vVrDB06FDdu3EDFihVNulIdSBsvftq0aXj8+DEWLVpklBji4uIwY8YMAJDHLqf8jV3BExERERERERERERGRSdu8eTM2bdqEy5cv48SJE/D29saXX35p7LCI3ksdO3ZEfHw8KlSoAE9PTzx79gznzp2Th2RYvny5sUPMkv79++O3337D1KlTMxw+IqfNmjULly5dwuHDh3Hnzh20bNky18eVJ9PAinUiIiIiIiIiIiIiIjJpZ86cwdKlS+Hg4ICmTZti9uzZcHZ2NnZYlA/9/vvvOHz4cJaWdXd3x48//pjLEeW8kSNHYuPGjbhy5QqOHDkCpVKJwoULo2fPnhg9ejT8/f2NHWKWKJVKnD59Os8/d9u2bThw4ADc3d3Rp08fzJ49O89jIOPgGOtEREREREREREREREREAPr06YMVK1ZkadnChQvj7t27uRsQEZkMVqwTERERERERERERERERERFlQGnsAIiIiIiIiIiIiIiIiIiIiEwZK9aJiIiIiIiIiIiIiIiIiIgyUOAq1gMDA6FQKDR+rKysEBAQgODgYBw6dEhrnT59+kChUGD58uV5H3Aumzx5MhQKBSZPnpwj2zOVY2UqceQn0vWSH0n3BY6Fk6ZRo0ZQKBTYv39/ttbL6fuJKX0vOb1vBcHNmzcxdOhQlC1bFnZ2drC2toafnx+qV6+OoUOHYsOGDcYOMVOmdA6mt3z5cigUCvTp08fYoeh09OhRNG/eHK6urlAqle9tmmzqxzk77t69C4VCgcDAQK15pnyuZ0dERAR69eoFX19fmJub55vvjnJHfjnv6f1XqVIluVwiMjIyw2Wl51yFQoHKlStnuOypU6c0yj0OHz6sMV/K36r/mJmZwdXVFfXr18evv/6KlJSUd929HGfos0p+tmXLFtSvXx+Ojo7yd8njkzX79++HQqFAo0aNjBpHfspzEhUU6nUM69ev17tc06ZN39vn4bxmaP5cV57G2toanp6eqFSpEvr06YM//vgDiYmJuRN4LmA5pOnJqEwlLx0+fBgKhQJffvmlUeMo6Apcxbqkbt26CAkJQUhICD788EOoVCqsW7cODRs2xOzZs40dHhUATCDpXZhKYk6kyz///IMKFSpg3rx5ePbsGerWrYuOHTuiYsWKiIiIwLx58zBo0CCjxsh7cO559OgRWrdujT179qB8+fLo2bMnQkJCULx4cWOHpiU/vzRmTMY4rkIIdOjQAatXr4aLiwuCg4MREhKCevXq5WkceeF9vH+ZSsUFkak5deoULly4AABITk7G6tWrs7zu+fPnERYWpnf+kiVLsrQdLy8vuWyka9euKFq0KA4fPozhw4ejUaNGiIuLy3JMlPfOnTuHjh074tixY6hVqxZ69+6NkJAQeHt7Gzs0IqIC46uvvkJqaqqxw8gzpvqSm3qepnPnzqhVqxYSEhKwYsUK9OzZE/7+/li7dq2xw3wv8IUv01WvXj20bt0aP//8M27evGnscAosc2MHYCwDBgzQuDEkJiZi0KBBWLlyJb788ku0adMGJUuWNF6A9E5mzJiBcePGwcfHx9ihEBEVKE+fPkVISAiSkpIwatQoTJ06FdbW1hrLhIWFZfhGN2Xu448/Rq1ateDk5GTsULTs3r0b0dHR6N69O/744w9jh0P/r1ChQrh69SosLCyMHUquuHfvHk6ePImAgACcP38e5uYF9jGHiN4jUuV3oUKFEBERgSVLlmDEiBGZrhcUFITTp09j6dKlqFatmtb8hIQErF27Fj4+PjAzM8PDhw/1bqt06dJarei2bNmCjz/+GEePHsUPP/yAb7/9Nns7Rnnm33//RUpKCiZMmIBp06YZOxwykCnn7YkoY7a2trhx4wZ+//13DB482NjhFGi68jQAcPv2bUyePBmrV69Gt27d8PLlSwwZMiTvA6T3mimVqUyZMgXbtm3D2LFj8c8//xg7nAKpwLZYT8/a2hrz5s2DnZ0d3rx5wxPyPefj44PSpUvzoYSIKI9t3boVr1+/hq+vL3788UetSnUAqFatGmbMmGGE6PIPJycnlC5d2iRfILt//z4AoESJEkaOhNRZWFigdOnSKFasmLFDyRXSeVekSBFWqhPReyE+Ph5//vknAGDVqlWwt7fHxYsXcerUqUzXbd26Nby8vPDnn3/q7NZ0/fr1iImJQe/evWFmZpbt2Nq2bYuePXsCANatW5ft9SnvMN+VP5hy3p6IMia9EPftt98iPj7eyNGQLsWKFcOqVaswZswYAGnf2Z07d4wcFb1vTKlMpVq1aqhUqRI2bdrEoc2MhBXrauzt7VGqVCkA0HtChoeHo1evXvD29oaVlRWKFSuGr7/+GklJSXq3GxYWhh49eiAgIABWVlZwdXVFixYtsH37dp3Lq48nEhoaiubNm8PFxQU2NjaoWrUqVq5cqbG8SqVC0aJFoVAocOzYMb1xDBkyJMvjL6SkpGD16tXo0aMHSpcuDUdHR9jY2KBUqVIYPnw4Hj16lOk21L169QqLFy9Ghw4dUKJECdjZ2cHOzg4VKlTAV199hejo6Bw5FhJ9Y6yrd535/PlzfPbZZ/D394elpSX8/f0xbNgwvbEAwI0bNzBo0CAUK1YM1tbWcHJyQoMGDbLVZR+Q1kXqlClTAKS9YaQ+Bkz6LlZevnyJCRMmoFy5crC1tYWDgwOqVauGmTNnIiEhIVufq959eGpqKmbOnIly5crBxsYG7u7u6NKlC65du5bpdjZs2IB69erB0dERdnZ2qFu3rt7zGQBSU1Px+++/o1GjRnB1dYWVlRWKFCmCTz/9FA8ePNBaXr270JSUFPzwww9ynG5ubujQoQOuXr2qsU5oaCgUCgVKly4NIYTOOBITE+Hm5gaFQoErV65kup/37t3DDz/8gMaNG8vXr7OzM+rVq4fffvsNKpVK77pRUVH49ttvERQUBCcnJ9jY2KBo0aLo0qULduzY8U7HqE+fPihSpIgcY/pxhCSGXncZSUhIwOTJk1GiRAlYWVnBx8cHISEhcoFORrJ7L1SXF/dCY+1bfvP06VMAgIeHR5bXiY2NhaOjI8zNzXXeEyStWrWCQqHA/Pnz5WnqXZCdO3cOHTp0gLu7O6ysrFC2bFn89NNPWveE7NyDJdlJgyTr169Hy5Yt4eHhAUtLSxQqVAg9e/bUe/8JCwtDcHAw/Pz8YGlpCUdHRxQtWhQdO3bEpk2bNJbNqFuuPXv2oG3btvDy8oKFhQVcXFxQokQJ9OzZEwcPHswwZl3Wrl2LJk2ayPemwoULo1+/frhx44bOmCZNmgRA89hmddiKK1euYNKkSahbty4KFSoES0tLuLm5oWnTpnoL9g1JM6T8gCT9fVRXPjAuLg7jx49H8eLFYWVlBW9vb4SEhCAiIiLDmOLj4zFx4kSUKVMGtra2Gsfi5MmT+PLLL1GjRg14e3vD0tISXl5eaNu2Lfbs2aNzf9W/++zEZMgQItlNB7N7XHMiXyXtV8OGDQEABw4c0PmZ6veKQ4cOoW3btvDw8IBSqdTILz58+BDDhg1DiRIl5Jjq1q2L3377DW/evNH6fPXvIyYmBiNHjkRgYCCsra1RokQJ/PDDD/JxioiIwKBBg+Dv7w8rKyuUKlUKv/76a5b3VTqmWbl/qeehN23ahMaNG8PV1VU+Bg0bNoRCoZAr9XSZOXMmFAoFunTpIk9Tz0vfu3cPvXv3ho+PD6ytrVGyZElMnjxZK3/aqFEjfPDBBzq/n/TnY2pqKhYuXIg6derAyclJPo7Dhw/XeV5Lx0Q67xYvXoxq1arBzs4Ozs7OaNWqFY4fP57pcc3OPd6Q/KH69SeEwKJFi+Q4nZyc0Lx58wzzLpQ//f3334iNjUX58uXxwQcfIDg4GEDWunA3NzdHr169EBUVhY0bN2rNX7p0KQCgX79+BscntYTPTmFddvIyOfmsUrt2bSgUigy7d507dy4UCgU+/vjjHInh3r176NOnD7y9veV71aRJk5CYmJhpF7nZzSPqIt2Ply1bBgDo27evfD+Uht1Qv/e8efMGs2fPRpUqVWBvb681ZMuuXbvQpk0beHp6wtLSEr6+vggODsbp06d1fr76Ph4/fhytW7eGm5sbHBwc0LBhQxw6dEhedufOnWjSpAlcXFxgb2+PZs2a4cyZM1neV4kh+SbJypUrUb16ddja2sLV1RUtW7bUiFGfR48eYeTIkXI+zsHBAdWrV8fcuXN1dvusXhaVnfJDfXl7Q/K56qKiojBp0iRUrlwZDg4OsLW1RYUKFTB16lRWABLlkFatWqFhw4Z4/Pgx5syZkyPbfJdyeSEE/vnnH7Rp00a+V3p7e6NevXr44YcfdJYlZ7VMS7onHThwAADwwQcfaOTtly9fnivlszll2rRp8PX1RWpqqkHflRACS5cuRVBQEGxtbeHm5oYPP/wQR48ezXDoq3/++QcDBgxA+fLl4eLiAmtraxQpUgT9+vXD9evXsxVDXpwbgYGB6Nu3LwBgxYoVGt9x+v2Lj4/H999/j6pVq8rpTLly5fD1118jKipKZxx5kV9Ufx7esWMHGjVqBCcnJ7i4uKBNmza4ePGivOyaNWtQu3ZtODg4wNnZGR06dMDt27e1tplRmYr6M2lu15tI+vTpA5VKhQULFuhdhnKRKGAKFy4sAIhly5bpnF+8eHEBQAwfPlyeFhISIgCIESNGCEdHR1G4cGHRpUsX0bRpU2FjYyMAiPbt2+vc3v/+9z+hVCoFAFG5cmXRqVMnUa9ePWFpaSkAiClTpuiN8ZtvvhEKhUJUq1ZNdO3aVdSqVUsAEADEnDlzNNb56aefBADRvXt3nXHExMQIe3t7oVQqRXh4uDx90qRJAoCYNGmSxvIPHjwQAISTk5OoVauW6Ny5s2jVqpXw9fUVAISHh4e4efOm1udIxyr98T106JC8Xr169URwcLBo3ry5cHNzEwBE8eLFxYsXL3LkWGQUh7S//fr1E35+fsLLy0t06NBBtGrVSjg5OQkAonr16iI5OVlrm+vWrRPW1tYCgChdurT4+OOPRePGjYWdnZ0AIPr27avz2OsSEhIiKlWqJACISpUqiZCQEPln8eLF8nK3b9+Wj4GHh4fo2LGjaNeunXBwcBAARNWqVcXLly+z/Lnh4eECgChcuLDo0KGDsLCwEE2bNhVdu3YVRYsWFQCEvb29OHr0qNa60vGeOHGiUCgUom7duiI4OFjeD4VCIf755x+t9WJjY0WjRo3kbTds2FB06tRJlCpVSgAQbm5u4syZMxrrhIaGCgCiTp06omnTpsLW1la0bNlSdOzYUfj7+wsAwtnZWeNcFkKIChUqCABi9+7dOvd/6dKlAoD44IMPNKZLxzj99r777jsBQBQpUkQ0adJEdO3aVTRs2FC+fjt06CBUKpXW55w7d04UKlRIvoZatWolgoODRe3atYWNjY1o2LDhOx2jxYsXi44dOwoAws7OTuP8CQkJkZcz9Lpr2LChACBCQ0M1psfFxcnXnp2dnWjTpo3o3Lmz8PLyEm5ubqJ379467ydCmP69MK/3LT9btWqVACDMzMzEnj17srzesGHDBAAxYcIEnfNv3bolFAqFcHR0FK9evZKnS+fruHHjhKWlpShTpox8rZqZmcnpt7qs3oMNTYNSUlJEly5dBABhZWUl6tSpIzp37ix/po2NjdixY4fGOnv27BEWFhZyTJ06dRIff/yxqFGjhrCyshIfffSRxvLLli0TADSueSGEWL58uVAoFEKhUIiaNWuK4OBg0a5dO1G1alVhZmamdSwyolKp5HPf3NxcNG7cWHTt2lWULFlSABC2trYa+3Ho0CG9x3bUqFFZ+sz+/fvL6WyLFi3ke6d0jX3xxRda6xiSZmzcuFHOK0jHUf3n+fPnGse5ffv2omLFisLZ2Vm0bdtWfPTRR8LT01NOU6Ojo3XGVLNmTVG9enVhZ2cnPvzwQxEcHCyaNm0qL9ekSROhVCpFhQoVRKtWrUTnzp1F1apV5bj+97//ae2voTGp5wHSy6l0MKvHVYicy1c9f/5chISEiBYtWggAwsvLS+dnSveKIUOGCKVSKcqWLSu6du0qmjdvLtasWSOEEOLkyZPC1dVVABABAQEiODhYtGzZUo6zRYsWIikpSef38dFHH4kyZcoIT09P0bFjR9G8eXP5OWHo0KHi1q1bwtvbW/j7+4suXbqIDz74QL5Hff/991naVyGyf/8aOnSoACCCgoJEt27dRMOGDcXBgwfFhg0b5OtGlzdv3ojAwEABQBw4cECeLuWle/fuLdzc3ISXl5fo3LmzaNOmjfzd1a1bVyQkJMjrzJgxQ+/3o35vSExMFE2bNhUAhLW1tXzNSNexu7u7CAsL04pVOt+++OILoVAoRL169US3bt1E+fLl5fuXrjyqofd4Q/KH6tdfSEiIsLCwEI0bNxZdunSR76lWVlbi+PHj+r98ynfq168vAIjZs2cLIYQ4cuSI/PwQHx+vcx3pHvvdd9+JK1euCAAa6YoQb/NMdevWFUK8PdcPHTqksZx0Pad/NpFMnTpVABCOjo5Z2p/s5mVy8lnlt99+k+/T+kjp6+bNm985hsuXLwt3d3cBQPj6+oouXbqI1q1bCzs7O1GvXj1Rp04dnc9ThuQR9ZHS3GLFisn3XuneOmPGDCHE23tPQECAaNeunbC0tBRNmjQR3bp1ExUrVpS39fXXX8vP9HXr1hXdunUTlStXlvP0S5Ys0fs9jB49Wpibm4sqVaqI4OBgeT0rKytx5MgRMXfuXKFUKkWdOnU07nn29vY6y5QyYki+SQghhg8fLgAIpVIpGjRoILp27SrKli0rlEqlGDFihN7r4MCBA8LFxUUAEIGBgaJdu3aiRYsW8rTmzZtrlR0ZWn6oL29vaNmIEGnnqbSMj4+PaNmypWjbtq3w8vKSn13T5xeJKOvU09fjx4/LaWb6dKNJkyYC0F8XoYuh5fLJycmiQ4cO8j2vVq1aolu3bqJZs2ZyOWX6+0V2yrSuXr0qQkJC5PtIixYtNPL2Ul4jp8tnM5NZnkbdF198IQCIUqVKZeszhBDi008/lY9tw4YNRdeuXUW5cuWEmZmZGDVqlN4YzMzMhK2trQgKChIdOnQQ7dq1k8vh7ezsxJEjR/TuU07V2WTn3Bg1apSoW7euACCKFSum8R1LeQwhhIiMjJTTfUdHR9GuXTvRsWNHOY9UpEgRre8yr/KL0rk0btw4OX+jng9xdnYWt27dEmPGjJHLuzp16iSnm76+vlp1LhmVqUh5kbyoN5FcunRJABAlS5bUOZ9yFyvW1Zw/f15OSJYuXSpPVy8g/Oqrr0Rqaqo87+LFi3IhUvrKyJ07dwqFQiHc3d01CqSEEOLChQvCz89PABD79+/XGaOFhYXYsmWLxjwps53+YTs6OlrY2dkJS0tL8eTJE619+/XXXwUA0bZtW43p+m7SsbGxYtOmTVqFh8nJyWL8+PECgGjVqpXW5+ir0H7w4IHYs2ePePPmjcb0uLg4udB+yJAhWtsz5FhkFIe0vwBEnz59RGJiojzv/v37ckIiFbBKLly4IKysrIS1tbXYsGGDxry7d+/KGYYVK1Zo7YM++o69upo1awoAol27duL169fy9GfPnskPkPoqEHWREgCpcPL8+fPyvNTUVLliq3DhwhrHRoi3CYSzs7NWoZ+0L7pu5N27dxcARJs2bcTTp0815s2ZM0cAECVKlNC4rqSHRwCiSpUq4vHjx/K8hIQEuXB24MCBGttbvHixfLx0qVatmgCg9R3qy7idPHlSXLx4UWs7ERERcsK4bt06jXmvX7+WE+HevXtrVAAKkXat/vfff+98jDJKzCWGXnf6KtZHjx4tgLQKkIiICI3tffTRR/J3lv6cfh/uhXm9b/nZq1ev5HupQqEQjRo1Et99953Ytm2bePbsmd71bty4IRQKhfD09NS6/wgh5IeUYcOGaUyXzlcAYuHChRrz9u7dKxQKhTAzMxMPHjzQmJeVe7ChadCECRMEkFapeufOHY15f//9tzAzMxMuLi4iKipKnv7BBx8IAGL16tVacURHR4tjx47p/Pz0hW9FihSRH+7Te/r0qd4MuS4LFiyQ04uzZ8/K01UqlXz8nJ2dtb7XrBxbffbv3y9u376tNf3atWvy9XTixAmNeYamGUK8Tdv0kY6zVGgQExMjz3v58qX8EDl9+nS9MVWsWFEjJnXbt28Xjx490pp+9OhR4ejoKCwsLMTDhw9zJCZDKtYNSQeFyPy45ka+Sjrm+gpT1O8V8+bN05qfmJgoH4fBgwdrFJTfvn1brmRO//KP+vfRtm1bERcXJ88LCwsT5ubmckX+4MGDRUpKijz/33//lQsh1NfLTHbuX2ZmZmLTpk1a81NTU+VldN0XtmzZIp+/uj4bSHuZQP3+9+DBA7mgYty4cRrrZfb9CCHE2LFj5cIb9XMxOTlZfummSJEiWs8nUjw2NjZi7969GvNmzpwp36vT57EMvccbcl2o58ELFy4srl+/Ls9LTU0V/fr1E0BaJQ0VDNevX5fPP/V0tHTp0gKAWLlypc711CvWhRDyy2f37t2Tl/nqq680yjQMqVhXqVSiRo0aAoBo0KBBlvYpu3mZnHxWiYmJEba2tkKpVGqlm0KklfUAaS/3qN+HDY1Behbv2rWrRr714cOHckGorucpQ/KImdFX9iGE5r3Hz89P494j2bFjhwDSXmhKXwHy+++/y+fppUuXNOZJ34NCoRCrVq3SmDdy5EgBpFVa2Nvba7xsm5qaKr8kPmDAgCzvpxCG5Zu2bt0qgLSKi4MHD2rMmz59unx80l8Hjx8/Fm5ubkKhUIj58+drnCMvXrwQjRs3FoD2S9SGlh9mVrGe3XxufHy8/NLF119/rZF2xsXFiW7dugkgew1EiEhT+vRVqrRM/zK4IRXrhpbLS/ffwMBAce7cOY15KpVK7NmzR+OFGkPLtPSVG0pyunw2M9mpWF+9erV8X1XPE2Rm06ZNAkir/ExfES418tEXw9q1azXK9YVI+z7mzZsnAIhy5cppvZib03U22T039KVL6oKDg+V8jXrl9qtXr8SHH34oAO0XufMqvyidS1ZWVlr5kM6dOwsAonz58sLNzU3jeMTFxckvSE6dOlVjm1mpWM+LehOJSqUSzs7OAoBWmSflPlasi7SLdtu2bXKm09fXV+NmJ2WMq1WrprN16uDBgwUA8e2332pMlypF169frzOWdevWCQCiY8eOOmMcOXKkzvWkh+30DwVDhgzReMjWtc6uXbs0phta+O3r6yuUSqWIjY3VmJ7RQ50+cXFxwtzcXHh4eGjNM/RYZFax7ufnp7MA8/vvvxdAWot2dVJC8eOPP+qM4+TJk/I5klWZHXvpjSxbW1udFYSnT58WQNpbZlm9eao/WOt6mzsxMVGuEPvjjz805knr/fLLLzrXk1r8379/X55+5coVoVAohK+vr9a5ImnVqpUAoFGgKT08KhQKrcReCCG/CVq0aFGN6fHx8cLNzU0olUpx9+5djXnHjh0TAIS/v79WYmRIxm3Xrl0CgOjcubPG9P/9738CSHvLU1eil56hxygrFesZyei605VBjo+Pl3tK0NWK4vHjx3KLvvTntKnfC42xb/ndtWvX5GOT/qdy5cpiwYIFOq8P6VxPXzgXHx8vXFxchEKhENeuXdOYJ52vHTp00BlLy5YtdRZQZ6diKjvnYGRkpLCxsRHW1tY6C3aFeHuO/vrrr/K0smXLCgBZ7oVE30OOra2tcHJyytI2MiPli3Td91UqlahYsaIAIKZNm6Yx710q1jMitUYbM2aMxnRD0wwhsl6xbmdnp7Mgd+3atQKAaNy4sc6YdN2jskp6KE5fCWxoTIZUrGdEXzooRObHNTfyVVmtWE9/XCRSbxu+vr46X+5Zv369ACAcHBw0WmNL34e9vb3Wg7AQQrRr104Aaa0F1deTSC8RpC/Iykh27l/p87TqpErn/v37a82TCup/++03nZ9tY2Oj84URqULe0dFRY38z+34SEhKEvb29ADRbk0ri4uLkljH68qiff/65zm0HBQXpvFcZms/IiL7rQj0Prmv/Hj9+LBf86Oo5i/If6UWS9HlE6brUd62kr1iXCq4nT54shEjrbcLPz0/Y29vLZRrZqVhPTk4Wly9fFl27dpXPWV2ta3TJbl4mI9l9VhFCiF69eglA+8UyIYT4/PPPBZDWuvpdYzh48KB834+MjNRaT6rITR+joXnEzGS1Yl3fyxpShY++e2GbNm0EAPHJJ59oTJe+B135gMjISPlz0+fbhEh78QxIe1kqp+jLN0k9oYwdO1bnetILiemvOekaHTp0qM71Hj58KCwsLISHh4dGOaGh5YeZVaxnN58rvSDbpk0bnfG/evVKeHp6CnNz8xy5ZokKovTp67Vr14S5ubmwsrLSKJM0pGI9M7rK5Z8+fSq3Mj99+nSWtmNomVZmFet5WT4rRPYq1nfu3CmnUbqe3/SRXqgaP368zvnVq1fPcgzqateuLQCIy5cva0zPyTobQ86NzCrW7927J5RKpVAoFBoN9yQPHz6Uy1LVX0TIq/yidC7pyoecOXNGPgd0vXQv9e6WvkeFrFSs50W9iTrp/NH1Mj3lLnMUUH379pXHilBXrFgxbNiwAXZ2dlrz2rRpozUOFQCUKVMGADTG/Xvx4gVOnjwJGxsbtG3bVmcM0pgUR48e1Tlf33plypTBtWvXtMYZHD58OBYsWIDffvsN48aNg7l52te7d+9eXLt2DaVKlUKzZs10blOf8+fPY+/evQgPD0dcXJw8ZmBqaipUKhVu3bqFKlWqZHl7R48exaFDh3D//n3Ex8fLY61YWlri+fPniIqKgouLi9Z62T0WmWnSpAlsbW11bg/Q/C5VKpU8JrY07l16QUFBsLe3x9mzZ5GYmAhra+tsxaOLNB5by5Yt4eXlpTW/WrVqqFSpEs6fP48DBw6gR48e2dp+SEiI1jQrKysEBwdj9uzZ2L9/P7p37661jK7vwsrKCkWLFsXZs2cREREBf39/AMD27dshhMCHH34IBwcHnXE0atQI27dvx9GjR9GmTRuNeQEBAahUqZLWOrq+JwCwsbHBwIEDMWPGDCxYsADff/+9PG/evHkAgMGDB8PMzExnLLokJSVh9+7dOHXqFJ49e4akpCQIIfDq1SsA0BoLZ+fOnQCA/v37Z+lz3vUYZYWh1526M2fO4NWrV3B3d0fLli215nt7e6N58+bYvHmzxvT34V5ozH3Lr0qVKoXjx4/j5MmT2LZtG06cOIEzZ87g+fPnOHfuHD799FNs2LAB27Ztg6WlpbzeiBEjsH37dsydOxc9e/aUp69ZswZRUVFo1qwZSpUqpfMzMzpPdu7cme10IqvbTn8OhoaGIiEhAU2aNEGhQoV0rteoUSPMnz8fR48exdChQwEANWrUwJUrV9CjRw9MmDABtWrVks/d7KhRowb279+P3r17Y8SIEahSpQqUSmW2t/Pw4UN5TCld6YVCoUDfvn3xxRdfIDQ0FBMmTMj2Z+jz+vVr7NixA2fPnsWLFy+QnJwMAHj8+DEA7fuuJLtpRnYEBQXBx8cn29v29PRE/fr1M9x2ZGQktm3bhkuXLiEqKgopKSkAgJs3bwLQv7+GxpRd2U0HM2OsfJWkU6dOOqdL+a6uXbvCyspKa36HDh3g4uKCqKgohIWFoW7duhrzq1WrBk9PT631SpQoASBt/EFd+1GiRAlcvHgxw7Hw3oW+/QWAAQMGYPLkyVizZg1mzZol5wVu3bqF3bt3w9nZWeNerK558+bw9vbWmt6mTRu4ubkhMjISZ86cQZ06dbIU5+nTp/H69Wu4urrqvOfa2tqia9eu+PnnnxEaGqozj6rrXgUAvXv3xunTp7F//36d9ypDnjMMvS7Mzc315jWk8ysyMlLnsaX8IzU1FStWrACgPQZ67969MWHCBBw8eBC3b99GsWLFMtxWcHAwPv/8cyxfvhwTJ07Erl278PDhQ/Tr109nmYYuBw4c0FnOYWlpiRkzZmiMSZ4RQ/MyOfGsAqSV8axatQorVqzA+PHj5ekpKSn4448/AOgfcz47MUjjyrZs2RKurq5a22rdujWcnZ21xvw0NI+YUzp27Kg1LTU1FUeOHAEArbG9Jf3798fWrVsRGhqqc36rVq20prm6usppga75UtpoSNqXnXxTamoqDh8+DAB607PevXvj3LlzWtO3bdsGQH9epVChQihRogSuXLmCmzdvomTJkhrzs1N+mBXZzedmFr+9vT2CgoKwfft2nDp1Cs2bN89WPESkrVSpUujXrx8WLVqEb775BitXrnznbWanXD40NBTJycmoVq0aqlWrlum2c7NMKzfKZ3OKdAwB6LxP65KamiofA33l7927d8epU6f0buPWrVvYuXMnbt26hVevXuHNmzcAgKdPnwJIS7/Kli2bpXiA3D03suLgwYNQqVSoWrUqKlasqDW/UKFCaNGiBTZt2oTQ0FD5uTCv84sZ5UMym29IPiUv600AwM3NDcDb84jyToGtWK9bty6KFy8OIO0C9PT0RK1atdCyZUu9F3NAQIDO6Y6OjgCAxMREeVp4eDiEEEhISNBZOKfu+fPn7/x5QFoC3rx5c+zatQv//vuvXJAmJVhDhgzJcoIRFxeHXr16YePGjRkuFxsbm6XtPXv2DB07dpQfajLanq6bYHaPRWays73IyEh5P6UbX0YiIyP1Pihnh/RgVKRIEb3LFCtWDOfPn8/2Q5mzszOcnZ11zpM+7+HDhzrnZ+fY3blzBwCwZMkSLFmyJMOYdF0HmX1WUlKS1rwhQ4Zg1qxZWLJkCSZPngxra2s8f/4cf//9N6ysrPDJJ59kGIe648ePIzg4GPfv39e7TPpr4N69ewCA0qVLZ+kz3vUYZeRdrzt10vkQGBiodxld5+r7cC805r7ldzVq1ECNGjUAAEIInD17FrNmzcLatWuxZ88e/PzzzxgzZoy8fLNmzVCmTBmcOHECYWFhcoZf+u4yKmDM6XTC0G1L1/TevXszTXPVz4sZM2bgwoUL2LFjB3bs2AEbGxtUrVoVjRo1Qo8ePeRCs8zMnz8fbdq0wapVq7Bq1So4ODigevXqaNy4MXr16qV3X9KT0hU3Nzd5P9OTCvtzqgIXALZs2YK+ffsiMjJS7zL68h6GpBlZZej5ldF9BQAWL16ML774AnFxcXqXMXR/3+WclxiSDmbGWPkqib7vJLN8l0KhQJEiRRAVFaXznNf3fdjb22c4X3qAzonvS5eMzkEXFxf06tULv/32G5YsWYLRo0cDSLuPCCHQt29fnS+iAhnnTwMDAxEZGak3L6lLVvO96stmNaaczNsC73Zd+Pj4wMLCQu/nRUVF5dq5QKZj27ZtePLkiVzgqM7LywutWrXC5s2bsXTpUkybNi3DbTk4OKBTp05YsWIF9u3bh6VLlwLQX4Gsi5eXl/zCh1KphKOjI8qWLYt27dpl6yWP7OZlcvJZBUgreCxatCiuX7+Oo0ePygW4W7duxfPnz1GzZs0ciSErzw6FCxfWqlg3NI+YEzw9PXXezyMjI+V7jr57aGb33ozSv8jISJ3zpbQvu3mz7OabsrJ/+qZL31dmL0gCad9X+or1vC7DSn8spfh79eqFXr16ZbjtgvqsSpQbJk+ejNWrV+OPP/7A6NGjdVY4Hj58GL///rvW9Pbt26N9+/YADCuXz255ZG6XaeVU+ezvv/+uM50eN25clvdV3YsXLwCkPd9JaXtm38mLFy/k+7a+9F/f9Ddv3mDo0KH47bff5MpgXbL6XJ0X50ZWGPr8ltf5RV3pp/SMrm/+uzyj52W9ifp2o6Kish0rvZsCW7E+YMAAvW/k6pOdFl/SW0L29vY63wzO6c+TjBgxArt27cK8efPQqVMnPHjwAJs3b4a9vX229nf8+PHYuHEjSpcuje+//x7Vq1eHu7u73LKwTp06OHbsWIYJgroBAwbg8OHDqF27NqZMmYJKlSrBxcVFLljy9fXF48eP9W7PkGOREUO+S0B/Sxh1mWVG3hc58V1Ix65y5co6365WV7NmzXf6LImfnx86dOiAdevW4a+//kJISAh+//13JCUloVevXvDw8MjSduLj49G+fXs8ffoUffv2xaefforixYvD0dERZmZmuHHjBkqVKpXla0Cfdz1GGXnX6y4nvO/3wozkxL4VJAqFAlWrVsWff/6J+Ph4bN68Gf/++69GxbpCocCwYcMwZMgQzJ07F8uWLcOxY8dw9uxZBAYGZthjQ06nE4ZuWzovihcvrtWiNT31hxpvb2+cPn0aBw4cwJ49e3DkyBGcOHECR44cwfTp0zFjxgyMHTs2088vU6YMrl+/jt27d2Pfvn3yW7379u3Dt99+iyVLluhtsWNsERERCA4ORkJCAr788kv06NEDgYGBsLe3h1KpxO7du9GiRYs8yyvkxLZtbGz0zgsLC8OgQYNgZmaGH374AW3btkVAQABsbW2hUCiwaNEiDBo0yCj7C+ReOmjsfFVG38m7yOz7yO3vS5/M9nf48OH47bffsGDBAowcORKJiYlYtmwZFAoFPvvss3f67NzMXxgiJ66ld70ujHUekGmRCs4SExPRsGFDrflS4ePy5cvx7bffZtqaq1+/flixYgVmzZqF0NBQlCpVKtM8iLrSpUtj+fLlWd8BPbKbl8npZxWFQoE+ffpg4sSJWL58uVyxvmzZMgDQ2WPhu8SQUeW4rnmG5hFzQm6lfUDepX/vmm/KLun76tSpU6a9P0itxdQZswwLeBu/vh4Q1RUuXNjguIhIk4+PD0aMGIEZM2Zg/Pjxcu8R6m7duiX3XKMuMDBQrljP6XJ5XXK7TCunymcPHz6s83j16dPHoPTyzJkzANLSWqlxZVa+k8zoyxf8/PPPWLhwIby9vTF79mzUqVMHXl5ecm9m3bt3x59//pnl7zIvzo3clNf5xbx+Ts/LehMAiImJAYAsvYRKOavAVqznNqkFjkKhwNKlS/OsEKVly5YoWbIk9u/fj8uXL2PNmjV48+YNevXqpbfFmS7r1q0DAPz11186366TutnKiri4OGzfvh1KpRLbt2/XaikdFxeHJ0+eZHl7ec3d3R02NjZISEjAjz/+CHd39zz5XKl1lvT2ki7SvOy25IqOjkZ0dLTOVut3794FkJYBelfSdVC3bl3MnTv3nbeXVcOHD8e6deswb9489OzZEwsXLgSQcWvX9A4ePIinT5+iatWqcssPdfqugYCAAFy9ehXXrl1D06ZNM/2c3DpGOX3dSeeYdH7oomve+3AvfN/2LT+QutaX3hRWJ3WBunbtWvz444/ydfHpp5++F8dYOi9KlSqV7UJqhUKBRo0ayd2tJSYmYvny5fjss88wYcIEdOrUKdMuYYG0roZbtWold2kVGxuL2bNnY8qUKRg0aBA+/vjjTAsIpetCal2sKw9haBqkz5YtW5CQkICPP/4YP/zwg9b87OQ93gd///03hBAYNmwYvvzyS635xt5fQ9PBzBgrX5WZrOS7wsPDNZbND8qWLYumTZtiz5492LFjBx49eoTo6Gh8+OGHGd5vpGOhiyF5SemYZrTdzO454eHhqFy5co7Eo09uXRdUcDx+/Bjbt28HkJbGSt1w6/Lo0SPs3LkTrVu3znCbDRo0QPHixbFr1y4AuiuQ80pW8zK5VUYQEhKCyZMn46+//sLPP/+M2NhYuTVU165dtT7DkBiy8uwgtQxT9y55xNzi5uYGKysrJCUl4c6dOzrLfnI6v2coQ/JN6vt39+5dlCtXTmsZfd+jv78/bt68ibFjxyIoKOid489r/v7+uHbtGvr375/hsDBElPPGjh2LRYsWYfv27Th48KDW/D59+mTa4MOQcnmppey1a9eyFGdelGnlRPns8uXLcyzdTElJkY+t+hAYmX0n6unJvXv3dHbZri89kT7vt99+Q7t27bTmZ/f5IS/Ojax4l3oLY+cXTUVO1AlIvT1m9hId5TzTL6F+T/n6+qJixYp49eqVPOZyXpBa+wHA7Nmz5W5Msjs+18uXLwHofnN1165dOitD9ImJicGbN2/g6OiosyJ39erVJvsWFQCYmZnJ4zFLiVdOkN4kS01N1TlfSlx27typc5yMs2fP4ty5c1AqlWjQoEG2P3/VqlVa05KTk/HXX39pfP67+PDDDwEAmzdvztNuLevWrYtq1arh1KlT+Prrr3H//n1Ur15d7pI6K6RrQF8XLqtXr9Y5XepGcenSpfJ4ORkx9Bhldv7k9HVXrVo12Nvb48WLF9i9e7fW/KdPn+qc/j7cC9+3fTN1WTmvpO5zdVVy2NnZoX///khMTMT06dOxfv16WFtbo3///jkaZ2bXkKGaNGkCS0tL7N+/H8+ePXunbVlbW2Pw4MGoWLEiVCoVLly4YNB2HB0dMXnyZDg7OyM+Ph43btzIdB0/Pz+5Uk3XQ6wQQp7+wQcfGBRXehnlPYQQWLNmTY58jjrpLeecPg+yIqP9TUxMxIYNG/I6JA2GpoNAxsc1t/JV70rK9/z111860+ONGzciKioKDg4OOTYunaFy+v41YsQIAMDcuXOzNPQGAOzevVvnPW779u2IjIzUOk6ZxRwUFAR7e3u8fPkSmzdv1pqfkJCAtWvXAtB/z9GVt1WfnhN523e5LoiAtDT1zZs3qFmzJoQQen+kisPMuoWUDB48GG5ubvD09ETv3r1zcxeyRV9eJrfKCAICAtCkSRPExsbin3/+werVq5GamooOHTrAyclJY1lDY5CevXfu3Kmz280dO3bonJ6TecScYm5ujnr16gHQnd8DIL9ElFP5PUMZkm8yNzeXewf4448/dG5XX9ohPaebUl4lO973+IneZ05OTpgwYQIA6HwRKCsMKZdv3LgxLC0tERYWJrfKzsi7lGll9XkkJ8pnc9JXX32FR48ewcLCAl988UWW17OwsEDt2rUBQG+5xJ9//qlzekbf5eXLl3Hu3Lksx5HZ9nLq3AAy/44bNGgApVKJc+fO4fz581rzHz9+LJ9XmeUh8jq/aCretd5EpVLh6tWrAGD0MoqCiBXruWjq1KkA0t4Y37Jli9Z8IQROnDihs8LmXfTp0wdOTk5YunQpnj17hg8++EDnm1QZkcaz+PXXXzWmX79+HYMHD87Wtry8vODi4oLo6Gith5bjx49j/Pjx2dqeMUyaNAmWlpYYM2YMVqxYodGNqeTSpUv4559/srxNqULp8uXLOufXq1cPNWvWREJCAgYNGoT4+Hh53osXLzBo0CAAQNeuXbM0Rml63333HS5duiT/r1KpMHbsWDx8+BD+/v450g1QlSpV0LFjRzx48AAdOnTQ+fZeXFwc/vjjD50vD7wLqZD4+++/B5D9l0uka2Dv3r24cuWKxrxFixbJLyCkN2DAAPj5+eHs2bP45JNPtMZ/i42NxZ49e+T/DT1GHh4esLS0xJMnT+RMlbqcvu5sbGwwcOBAAMAXX3yBx48fy/MSEhLw6aefIiEhQee6pn4vfB/3zZTNnz8fISEhOHr0qNY8IQT++ecf+U3M9C2HJEOHDoVSqcTs2bORnJyMbt266exi8V1kdg82lJeXF4YNG4a4uDi0bdsWFy9e1FomKSkJmzdv1nhb+Mcff9Q5Xu+1a9fkN44z66YxPj4es2fP1jn20qFDhxAdHQ0zM7Mst9qUxlv+7rvvNB6UhBCYOnUqzp07B2dn5yyPjZYZ6b67fv16jevwzZs3mDhxos5z6l3l1nmQFdL+rlixAq9evZKnJyYmYsiQIRm23M0LhqaDQObHNTfyVe+qc+fOCAgIwKNHjzBy5EiNAoTw8HCMGjUKADBs2DC52z5jyenztlWrVihevDh27tyJ8+fPo1ixYvJDvj660sdHjx7Jx2nw4MEax0mK+ebNm0hJSdHanrW1tdz1/KhRozRae6akpGDEiBF48uQJihQporfl3YIFC7B//36NaXPmzMHJkyfh4OCQIy9ovct1QQS8raTMbCgMqXJcGh88M6NGjcKLFy/w9OlT+Pj4vHugBshOXiY3ywik8eWXLVuWYTfwhsbQoEEDVKpUCa9evcKwYcOQnJwsz1O/D+r6PEPyiLlNinfBggXYu3evxrzly5dj8+bNsLCwkJ+vjcXQfNPnn38OIK1sK31ecubMmXorGMaMGQNnZ2fMnj0bP/30k8b3LAkPDzfZF6oGDhyIwoUL4++//8bYsWM1jpnkyZMnWLx4sRGiI8r/PvvsMwQEBODEiRM4duxYttc3pFze09MTn376KYC0Zxv1Ml8g7Tl+3759ctfRgOFlWtl5HnnX8tmccOfOHfTu3RuzZs0CkPZCcXaHwRg+fDgA4JdffsHx48c15v388884ceKEzvWk73LevHkaz72PHz9G7969s/2ydF6dG9J3nP6ZRxIQEIDOnTtDCIFBgwbJLaeBtHLsgQMHIjExEXXq1JGH5wFMJ79oCt613uTy5cuIiYlByZIljd6zUIEkCpjChQsLAGLZsmVZXickJCTDdZYtWyYAiJCQEK15P//8szA3NxcARPHixUXr1q1F9+7dRbNmzYSnp6cAIMaOHaszxvDwcIPiEUKIzz//XAAQAMSGDRv0Ljdp0iQBQEyaNElj+oYNG4RCoRAARIUKFUTXrl1F48aNhYWFhWjcuLGoU6eOACBCQ0OzFNucOXPkeGrWrCm6desm6tatKxQKhejVq5fefTb0WOibrm9/JaGhoQKAaNiwoda8devWCVtbWwFA+Pn5iebNm4sePXqIDz/8UPj5+QkAIjg4WOd2dXny5Imws7MTAETdunVFnz59RP/+/cXSpUvlZW7fvi0fA09PT9GpUyfx0UcfCUdHRwFAVK1aVbx8+TLLnxkeHi4AiICAAPHxxx8LCwsL0axZM9G1a1dRrFgxAUDY2dmJQ4cOaa0rfX/6NGzYUOc5ERsbK5o0aSIACEtLS1G9enXRpUsX0blzZ1G9enVhaWkpAIirV6/K62T0PWQ1nqSkJOHl5SUACA8PD5GYmKh3WX3n2UcffSTH3bx5c9G1a1dRunRpoVAoxFdffSUAiMKFC2tt78yZM8Lb21sAEM7OzqJ169YiODhY1KlTR9jY2GjtlyHHSAghOnXqJAAIf39/0a1bN9G/f3/Rv39/eb6h152+7/L169eiRo0aAoCwt7cXbdu2FZ07dxbe3t7Czc1N9O7dW+/1Zer3wrzet/xM/bzz8PAQzZs3F927dxetWrUSgYGB8ryePXuKN2/e6N1O+/bt5WXDwsL0LqfvfJXou+9n5R5s6DmYkpIiunfvLgAIpVIpqlSpIjp27CiCg4NF3bp15c/dsWOHvI6Tk5MAIEqXLi0+/vhj0b17d9GoUSP53Ordu7fGZ+jKe0RFRcmfWalSJdGpUyfRrVs3Ubt2bTlNnzhxot5jmZ5KpRK9evUSAIS5ublo0qSJ6NatmyhVqpQAIGxsbMT27du11sssrdUnJSVFVKtWTb4OW7duLbp06SIKFy4sLCwsxNixY3WmDe+SZowePVoAEO7u7qJLly7yffTFixdCiIzzeEK8TVfTpwVZiSkqKko+x9zc3ET79u1Fx44dhaenp3BwcBAjRozQ+dmGxqRvuhA5nw5mdlyFyPl8VWbHPLN7hRBCnDx5Uri6usr7FRwcLFq1aiWsra0FANGiRQuRlJSksU5m30dm10NW0rL0cuL+ld7//vc/+Tr56aef9C4n7U/v3r2Fq6ur8Pb2Fp07dxZt27aVY6pdu7aIj4/XWjcoKEgAEKVKlRI9evQQ/fv310gfExMT5fyQjY2NaNWqlQgODhYBAQHydXL69Gmt7Upxf/7550KhUIgGDRqIbt26iQoVKggAwszMTPz9999a6xl6jzfkusjo+stqPPT+279/vwAgrKyssvQMV7VqVQFA/Pjjj/I06bz87rvvsvy50rmV/hlPup4zSquyI7t5mZx+VpEkJCQIFxcXeduBgYFCpVLpXNbQGC5evCinF4UKFRJdunQRbdq0EXZ2dqJu3bqidu3aAoA4cuSIxnqG5BEzk1E6kpV7jxBCfP311wKAUCgUol69eqJ79+7y+WdmZiaWLFmitU5m30Nm97TMnufTMzTfJIQQn332mXzMGzVqJLp16ybKlSsnlEqlvJ6u6+DAgQPC3d1dAGnlMY0bNxY9evQQbdq0kctPatasqbGOoeWH+qa/Sz730qVL8vOXs7OzaNCggejevbto3769KFu2rFAoFMLLy0vvdokoY/rSV8ny5cvl6zO7+X1Dy+WTkpJEu3bt5Hte7dq1Rffu3UXz5s1FoUKFdN6XDSnT2rp1q5wfbtOmjejXr5/o37+/VronxfSu5bOZkfI0Xl5eIiQkRISEhIhevXqJdu3aiZIlS8rH0sPDQ/z111/Z2ra6gQMHymmjlJ6UL19emJmZiS+++EIAEM2aNdNY5/jx43K5bvHixUWXLl1Ey5YthY2NjShXrpz4+OOPdZ4fOV1nk91zIykpSfj6+goAokqVKqJ3796if//+YubMmfIyL168EJUqVRIAhJOTk2jfvr3o1KmT8PDwEABEkSJFtL7LvMovvks+xJAylczyNTlZbyKZPXu2ACC+/PJLvZ9LuYcV61nwLhXrQqQ9dA0cOFCUKFFCWFtbC1tbW1G0aFHRokUL8csvv4iIiAidMb5LZdKOHTsEkFbhlpqaqne5jAr7Dh48KJo0aSLc3d2Fra2tKF++vJg2bZpISkrSezPIKLZ///1X1KlTRzg7Owt7e3sRFBQk5s+fL1Qq1XtRsS5E2g30iy++EOXLlxd2dnbC2tpaFC5cWDRq1Eh8//334tatWzrX0+fgwYOiadOmwsXFRSiVSp3nUWRkpBg/frwoU6aMfP5UqVJFfP/99zoLLTOingCkpKSIadOmidKlSwsrKyvh6uoqOnbsKC5fvqxzXUMTCCGEePPmjVizZo1o1aqV8PLyEhYWFsLNzU2UL19e9O3bV2zcuFEkJyfLy+dExboQQgQHBwsAYvz48Rkup+88S05OFrNmzRIVKlQQtra2wtXVVTRv3lzs3r0700KK58+fi6+//lpUqFBB2NnZCRsbG1G0aFERHBwsdu7cqbV8do+REGnnxqBBg0RAQICwsLDQeUwMue4y+i7j4uLEN998I4oVKyYsLS2Fl5eX6NGjhwgPD8/0+jLle2Fe71t+FhsbK/79918xbNgwUaNGDeHn5ycsLCyEjY2NKFasmOjWrVuWCgsXLFgggLQKmowYWrEuROb34Hc9B7dv3y46dOggChUqJCwsLISzs7MoU6aM6Nq1q1izZo2Ii4uTl129erXo27evKF++vHB1dRVWVlaicOHC4sMPPxQbN27UKhDWlfdISUkRCxcuFN26dROlS5cWTk5O8nHv2LGj2Lt3r844M7NmzRrRqFEj4ezsLCwsLIS/v7/o06ePuHbtms7lDa1YF0KIV69eiQkTJohSpUoJa2tr4enpKdq3by9Onz6tN214lzQjISFBfPnll6J48eLyA4v6d56bFetCpKUVQ4YMEcWKFRNWVlbC19dX9OzZU9y8eTPbBa+ZxWRIxbqh6WBmx1U9ppzKV+VExboQQty/f1989tlnomjRosLS0lI4ODiI2rVriwULFoiUlBSt5Y1RsS7Eu9+/0rt69aoAIGxtbUVUVJTe5dT3586dO6Jbt27Cy8tLWFpaiuLFi4uJEydq3NvU3bt3T3Tv3l34+PjIBSfpz5+UlBQxf/58UatWLeHg4CAsLS1FsWLFxLBhw8TDhw91blf9+l6wYIGoXLmysLGxEY6OjqJly5Y6C/iEMPweb8h1wYp1EkLIL6t16tQpS8tLL7yUKVNGnmbKFevZzcsIkfPPKpIhQ4bI94bM8iOGxCBE2nXdq1cv4enpKd+rJkyYIOLj40XRokUFAHH9+nWdn5mdPGJmcqJiXYi056ZWrVoJNzc3YW5uLr84deLECZ3L53XFuhCG5ZskS5cuFdWqVRPW1tbCyclJNG3aVISGhmaaf3j69Kn45ptvRNWqVeV0yc/PT9SpU0dMmjRJXLhwQWN5U6pYFyLt2WzmzJmidu3acl7ex8dHVK9eXYwZM0YcPXpU73aJKGOZVay/efNGftHT0Px+dsvlhUh7QX7NmjWiefPmws3NTVhYWAhvb29Rv359MWvWLJGQkKC1jiFlWosXLxZVq1aVX5bOaB/ftXw2M1KeRv3H0tJSuLu7i4oVK4revXuLP/74Q+e+Z4dKpZL329raWjg7O4vmzZuLgwcPipUrVwoAolu3blrrXbhwQbRr1074+PgIa2trUaJECfHll1+K2NhYg+ow8urcuHjxomjXrp3w8PCQnznTp0dxcXFixowZonLlysLW1lZYW1uLMmXKiAkTJuh8kTSv8ovvS8W6EIbVCQghRKVKlYRSqeTzo5EohBAClO/07NkTf/zxB6ZPn/7ed4tBOefu3bsoUqQIChcurLN7kfwkOjoafn5+SExMRHh4uEHd5dP7j/fC91e9evVw5MgRrFmzBt26dTN2OERE+drXX3+NadOmYeDAgfjtt9/0Ljd58mRMmTIFkyZNwuTJk/MuwAwoFAoAeK/H1yOi/CM8PBzFixeHg4MDXr58CaWSIzASEVHBVVDKZ/v164dly5bhp59+wsiRI40dDuVzYWFhCAoKwscff5ynQ+jRW8zh50MXL17EX3/9BXt7e3kcbqKCZsaMGYiLi0OXLl3ybaaNMsZ74ftrx44dOHLkCAICAvSOpUtERDnj8ePHmDdvHpRKpTwWLRER6RcXF6dzXNl79+6hR48eUKlUCAkJYaU6EREVePmpfPby5cuIi4vTmKZSqbB48WIsX74c1tbWbBhCeWLixImwtLTEDz/8YOxQCixzYwdAOWfAgAGIi4vDjh07kJqaiq+//hqurq7GDosozxw9ehRLly5FeHg49u3bB1tbW0ydOtXYYVEe473w/RQZGYmxY8ciKioK27dvBwDMnDkTFhYWRo6MiCh/GjduHCIiIrBnzx5ER0dj8ODBKFOmjLHDIiIyec+fP0f58uVRrFgxlCxZEo6Ojrh//z7OnDmDpKQkVKpUCd99952xwyQiIjKK/Fo+O2vWLKxbtw5VqlRBoUKFEBcXhytXruDu3bswMzPD/Pnz4ePjY+wwKZ87fPgwtm/fjjFjxqBEiRLGDqfAYsV6PrJkyRIolUr4+/tj9OjR+PLLL40dElGeunHjBpYsWQIbGxvUqlULP/zwA4oWLWrssCiP8V74fnr16hWWLFkCc3NzFC1aFKNGjUJwcLCxwyIiyrfWrl2L+/fvw9vbG59//jm+//57Y4dERPRecHd3x+jRo7Fv3z6cOnUK0dHRsLW1RcWKFdGxY0cMGzYMtra2xg6TiIjIKPJr+WxwcDBiY2MRFhaGc+fOITU1FZ6enggODsbnn3+OWrVqGTtEKgDq1avHYdBMAMdYJyIiIiIiIiIiIiLKx86cOYPJkyfj8OHDSExMRNGiRTFw4EAMHz7c2KERERG9N9hinYiIiIiIiIiIiIgon9q9ezfatm2LKlWq4JtvvoG9vT1u376Nhw8fGjs0IiKi9wpbrBMRERERERERERER5UOxsbEoWbIk6tSpg/Xr10OpVBo7JCIiovdWvm2xrlKp8OjRIzg4OEChUBg7HCIiIgCAEAKvXr2Cr68vH2azgOk5ERGZKqbpWcf0nIiITFVBSM/XrFmDp0+fYtq0aVAqlYiLi4ONjU2295fpORERmaq8TM/zbcX6o0eP4O/vb+wwiIiIdHrw4AH8/PyMHYbJY3pORESmjml65pieExGRqcvP6fmePXvg6OiIiIgItG/fHjdu3ICdnR169eqFOXPmwNraWud6SUlJSEpKkv+PiIhA2bJl8ypsIiKibMuL9DzfVqw7ODgASDuIjo6ORo6GiIgoTWxsLPz9/eV0ijLG9JyIiEwV0/SsY3pORESmqiCk5zdv3kRqaio++ugj9O/fHzNmzMD+/fvx66+/Ijo6Gn/++afO9WbMmIEpU6ZoTWd6TkREpiYv0/N8W7EudUfj6OjIhJ6IiEwOu03LGqbnRERk6pimZ47pORERmbr8nJ6/fv0a8fHxGDx4MH755RcAQIcOHZCcnIzffvsN3377LUqUKKG13vjx4zFy5Ej5f6nSguk5ERGZqrxIz/PnwDFERERERERERERERAWcjY0NAKBbt24a07t37w4AOHbsmM71rKys5Ep0VqYTERGlYcU6EREREREREREREVE+5OvrCwDw8vLSmO7p6QkAiIqKyvOYiIiI3lesWCciIiIiIiIiIiIiyoeqVasGAIiIiNCY/ujRIwCAh4dHnsdERET0vmLFOhERERERERERERFRPtSlSxcAwJIlSzSm//777zA3N0ejRo2MEBUREdH7ydzYARARERERERERERERUc6rUqUK+vXrh6VLlyI1NRUNGzbE/v378ffff2P8+PFyV/FERESUOVasExERERERERERERHlUwsXLkRAQACWLVuGjRs3onDhwpgzZw4+//xzY4dGRET0XmFX8ERElO9dv34dgwcPxsmTJ40dChERERno1atXGDFiBLZs2WLsUIiI6P+dOnUKn376KcLDw40dChFlwMLCApMmTcLdu3eRnJyMmzdvslKdiCgLTp8+jREjRuDly5fGDoVMBCvWiYgo3/vrr79w5MgRzJkzx9ihEBERkYGuXLmCffv2YcKECcYOhYiI/t8PP/yAw4cPY/369cYOhYiIiCjHff3119i3bx+2bdtm7FDIRLBinYiI8r3k5GQAQGxsrJEjISIiIkOpVCpjh0BEROlERUUBePvMRURERJSfREREAGBeh95ixToREeV7Qghjh0BERETviOk5EZHp4j2aiIiI8jPmdUjCinUiIiowFAqFsUMgIiIiA7Egg4jIdPFZi4iIiPIz5nVIwop1IiIqMFggT0RE9P5iOk5EZLp4jyYiIqL8jHkdkrBinYiICgy+WUhERPT+YkEGEZHp4rMWERER5WfM65CEFetERJTvpaSkAGCBPBER0fuM6TgRkeniPZqIiIjyM+Z1SMKKdSIiyveSkpIA8M1CIiKi9xkLMoiITBeftYiIiCg/UypZnUppeCYQEVG+l5ycDIAF8kRERO8zpuNERKaL92giIjJ1MTEx6NS5MyZOnGjsUOg9xLwOSVixTkRE+R4r1omIiN5/b968MXYIRESkB1usExGRqbtz5w6uX7uGjRs3GjsUInqPsWKdiIjyPakreKmCnYiIiN4/KpXK2CEQEZEefImZiIhMXWpqqrFDMGlMyzPG40MSVqwTEVG+J1Wos2KdiIjo/cUW60REpoeFzERE9L7gi7r6Xb9+HfXr18fmzZuNHYrJYu88JGHFOhER5XtShXpiUqKRIyEiIiJDsSCMiMj0SIXMfPmJiIhMHdMq/Xbt2oWYmBjMmjXL2KGYLL5MSBJWrBMRUb6XmJhWoS51CU9ERETvHxaEERGZLj5rERGRqePzROaYnuvHFuskYcU6ERHle1JLddUbFVJSUowcjXG9fv0akyZNQsuWLeHq6gqFQoHly5frXPbq1ato2bIl7O3t4erqil69euH58+dZ/qzNmzejatWqsLa2RkBAACZNmsTxrIiIyGBssU5EZLqkl5mJiIhMlfrzBJ8tKLvYYp0krFgnIqJ8LzHhbSFPQX/z8sWLF/j2229x9epVVKpUSe9yDx8+RIMGDXDr1i1Mnz4do0ePxrZt29CsWbMsjVW/Y8cOtG/fHs7Ozvj111/Rvn17TJ06FcOGDcvJ3SEiogKELUyIiExXQkKCsUMgIiLKkPrzBCvWichQ5sYOgIiIKDcJIZCQ+LaQJyEhAfb29kaMyLh8fHzw+PFjeHt74/Tp06hevbrO5aZPn464uDiEhYUhICAAAFCjRg00a9YMy5cvx8CBAzP8nNGjR6NixYrYvXs3zM3TshuOjo6YPn06RowYgdKlS+fsjhERUb7HXk+IiEyP1CNYfHy8kSMhIiLKGFusE1FOYIt1IiLK15KSkqB68zazHBcXZ8RojM/Kygre3t6ZLrdhwwa0adNGrlQHgKZNm6JkyZJYt25dhuteuXIFV65cwcCBA+VKdQAYMmQIhBBYv3694TtAREQFFlusExGZntevX2v8JiIiMlXqzxN8tqDs4hjrJGGLdSIiytfSV6QX9Ir1rIiIiMCzZ88QFBSkNa9GjRrYvn17huufPXsWALTW9/X1hZ+fnzxfl6SkJI3u+mNjY7MTOhER5WNsVUJEZFqSkpLkFuuvXr0ycjREREQZY1fw9C44xjpJ2GKdiIjytfQVs6yozdzjx48BpHUbn56Pjw9evnyZ4Vj1ma3/6NEjvevOmDEDTk5O8o+/v392wycionyKXcETEZmWmJgYnX8TERGZIvXnCT5bEJGhWLFORET5mlTAozAXGv+TfgkJaWPSW1lZac2ztrbWWMaQ9TNad/z48YiJiZF/Hjx4kK3YiYgo/2KrEiIi0xIVFSX/HR8fj+TkZCNGQ0RElDGpl5X0fxMRZQe7gicionwtMjISAGBmJ5Aao5D/J/1sbGwAQGer9MTERI1lDFk/o3WtrKx0VsgTERFxHEQiItPy8uVLrf+9vb2NFA0REVHG1CvT+TIYZRfHWCcJW6wTEVG+9vz5cwCAmb3m/6Sf1IW71KW7usePH8PV1TXDyu/M1vf19c2hSImIqCBhd41ERKblxYsXAABLawsA4EvMRERk0tQbgEgNR4iyimOsk4QV60RElK9J43mbOQmN/0m/QoUKwcPDA6dPn9aad/LkSVSuXDnD9aX56dd/9OgRHj58mOn6REREuqhXrLNQg4jI+J49ewYA8Axw0/ifiIjIFMXHx8t/ZzRMIZEubLFOElasExFRvhYREQEAMHcUUCjf/k8Z69ixI7Zu3aoxxvnevXtx48YNdO7cWZ6WkpKCa9euabROL1euHEqXLo1FixZpdNu7YMECKBQKdOrUKW92goiI8hX1inW2XiciMr6nT58CALwD3QHo7rGKiIjIVKhXrKv/TZQVfLmbJO80xnpycjL27NmDa9euIS4uDt988w2AtG40YmNj4e7uDqUy+3X3N2/exDfffIPDhw/j5cuXCAgIQPfu3TF69GjY2tq+S8hERFTA3LlzBwpzQGEJKGwE7ty5AyHEe/GWYW6ls3PnzkV0dLTcen/Lli14+PAhAGDYsGFwcnLChAkT8Pfff+ODDz7AiBEj8Pr1a8yaNQsVKlRA37595W1FRESgTJkyCAkJwfLly+Xps2bNQrt27dC8eXN07doVly5dwty5czFgwACUKVPmHY4KEREVVOkr1i0sLIwYjenJrXwDEZE+0kvLvsU8AbBinSinME0nyh2vXr3S+TcRUXYYXLG+efNmDBw4EM+fP5crKKRE/sKFC6hduzZWrVqF7t27Z2u7Dx48QI0aNeDk5IShQ4fC1dUVx44dw6RJkxAWFoZNmzYZGjIRERUwKSkpuHv3LpS2AgoFYGYr8Pr5azx9+hTe3t7GDi9DuZXOAsCPP/6Ie/fuyf//888/+OeffwAAPXv2hJOTE/z9/XHgwAGMHDkS48aNg6WlJVq3bo2ffvopw/HVJW3atME///yDKVOmYNiwYfDw8MCECRMwceLEbMdLREQEpKXrkuTkZNjY2BgxGtOSm/kGIiJ9Hj58CGs7K7j5Osv/E9G7YZpOlHtiY2Plv2NiYowYCb2P1HvlpILNoFfbjhw5gk6dOsHKygo///yzVkJeo0YNFC9eHBs2bMj2tletWoXo6Ghs27YN48aNw8CBA7Fs2TL07t0bmzdvRlRUlCEhExEVCJGRkezKSM2NGzeQmpoKM/u0rnqk35cvXzZmWJnKzXQWAO7evQshhM6fwMBAebly5cph165diIuLQ1RUFFavXg0vLy+NbQUGBkIIodFaXdK+fXucPXsWiYmJePDgAb777ju2LiQiIoMlJyfLf6tXshd0uZ1vICLSJTU1Fffv34ezhwOs7axgZWuJ8PBwY4dF9F5jmk6Uu16+fCn/zXomyi6VSmXsEMhEGNRi/bvvvoOzszPCwsLg7u6OyMhIrWWCgoJw4sSJbG9bemsofcG9j48PlEolLC0tDQmZiCjfW7VqFWbOnAlzc3P8999/cHd3N3ZIRnfhwgUAgLnD/1esO7yd3qRJE2OFlancTGeJiIjeV+qV6axYf4v5BiIyhoiICKSmpsLZ0xEKhQLOno64f/8+UlJS+DItkYGYphPlLvVrStf1VZBJ44dzHHH9+AxKEoNarJ84cQIfffRRhpU2/v7+ePLkSba33ahRIwBA//79ce7cOTx48AB//fUXFixYgOHDh8POzk7neklJSYiNjdX4ISIqSG7evAkgreWANHZ2QXfq1CkAgJlTWqbQ3FEAirfTTVVuprNERETvK7ZY1435BiIyhhs3bgAA3Hyc5d8pKSm4f/++EaMier8xTSfKPUIIPH36FA6ungCA58+fGzki06RQKIwdgsniSwckMahiPSkpCY6OjhkuEx0dDaUy+5tv2bIlvvvuO/z333+oUqUKAgIC0LVrVwwbNgxz5szRu96MGTPg5OQk//j7+2f7s4mI3mcJCQny3+wOPm3cm1OnTkFpDZj9/xCsCjPAzEHg8uXLJv0CVm6ms0RERO+r9GOsUxrmG4jIGK5duwYAcCvknPb7/8dZl6YTUfYxTSfKPa9evUJCQgI8A0tAaWaOx48fGzskk8IK9cxxjHWSGJQKFy1aNNPWfseOHUPp0qUNCiowMBANGjTAokWLsGHDBvTr1w/Tp0/H3Llz9a4zfvx4xMTEyD8PHjww6LOJiN5X6pXp6pXsBdWlS5cQHR0NcxfN8W8sXFVQqVQ4evSokSLLXG6ns0REZNpiY2Px6tUrY4dhctQr1lNTU40YiWlhvoGIjOHy5csAAA8/V43fly5dMlpMRO87pulEuUfq3dPZ0xeO7l6IiGBvn+rYGpso6wyqWO/YsSOOHDmCZcuW6Zz/448/4tKlSwgODs72tteuXYuBAwfi999/xyeffIIOHTpgyZIlCAkJwdixY/WOfWFlZQVHR0eNHyKigkS9Mp0V68D+/fsBABZumhlD6f/Q0NC8DinLcjOdJSIi09e8RXO0atXK2GGYHI6xrhvzDUSU14QQuHTpEpw8HGBlYwkAcC/kAqVSiYsXLxo5OqL3F9N0otwjNcR08vCBs6cvnj9/xvJTNewRLHNssU4SgyrWx4wZgzJlymDAgAFo1qwZ9u7dCwD48ssvUb9+fYwdOxaVK1fG0KFDs73t+fPno0qVKvDz89OY3q5dO8THx+Ps2bOGhExElO8lJibKfxf0jKEQArt27YLCDDB30axYV9oBShuB/fv3IykpyUgRZiw301kiIjJtKpUKca/jEB0dbexQTI56K3W2WH+L+QYiymvh4eGIiYmBd+DbcaDNLczg4e+CK1eumOxzFpGpY5pOlHvu3bsHAHD18YeLd1rdE3s9fotpd+bYqp8kBlWs29vb49ChQ+jatSv279+Pw4cPQwiBH3/8EUePHkWXLl2wZ88eWFlZZXvbT58+1fnmh9QigQUoRES6qVemq1eyF0RXrlzBgwcPYO6mgiJdSqdQABbuAvHx8Th8+LBxAsxEbqazRERk2lQqVeYLFVBssa4b8w1ElNfCwsIAAD5FPDSmexfxQEpKClutExmIaTpR7gkPDwcAuPj4w8XbX2MasZFWVrBinSTmhq7o4uKCP/74A7/88gtOnTqFly9fwtHREdWrV4eXl5fBAZUsWRK7d+/GjRs3ULJkSXn6n3/+CaVSiYoVKxq8bSKi/Ey9Mr2gV6xv3boVAGDpqTvDY+mpQtIDJbZu3YomTZrkZWhZllvpLBERmTb1h3UhBBQKhRGjMS1ssa4f8w1ElJdOnjwJAChUXPP+Uqi4F87vv4aTJ08iKCjIGKERvfeYphPljvDwcJhZWMLJwxtuhQoDAO7cuWPkqEyH1GKdlcf68diQxKCK9caNG6Nu3br47rvv4ObmhpYtW+ZYQGPGjMGOHTtQv359DB06FG5ubti6dSt27NiBAQMGwNfXN8c+i4goP1GvTC/I3fekpKRg2/ZtUFpqdwMvMbMHzOwFDhw4gJiYGDg5OeVxlBnLzXSWiIhMm/rDukqlgpmZmRGjMS2sWNeN+QYiyksqlQonTpyAg4sdHN3tNeb5FvOEQqnA8ePHMWTIECNFSPT+YppOlDtUKhVu3boFt0KFoVSawd2/KADg1q1bRo7MdLDFeubYuxxJDOoK/sSJEzq7a88JDRo0wNGjR1GtWjXMnz8fn3/+OW7fvo1p06ZhwYIFufKZRET5gXplekHODB0+fBhRL6Ng4aHdDbw6Cy8VUlJSsH379rwLLotyM50lIiLTpv6wzjfiNbEreN3yIt9w5swZtGvXDq6urrC1tUX58uXxyy+/5OpnEpFpunbtGqKiouBXylurVxVLawt4F3bHhQsX8OrVKyNFSPT+YlkAUe6IiIhAQkICPPzSKtTtnd1gY++ImzdvGjky0yGVJbPyWD8eG5IY1GK9dOnSuHfvXk7HIqtRo4ZJVnQQEZmyxMREKC3NoEp+U6C7gv/3338BAJbeGWd2LD0FEu8AGzduRLdu3fIgsqzL7XSWiIjofaTeSp0V62/ldr5h9+7daNu2LapUqYJvvvkG9vb2uH37Nh4+fJhrn0lEpuvw4cMAgIDSPjrn+5f2wePw5zh+/DiaNWuWl6ERvfdYFkCUO27cuAEA8ChcHACgUCjgHlAM966dR2JiIqytrY0ZnkmQKtaTk5Px5s0b9pymAyvWSWJQi/Vhw4Zh06ZNuHLlSk7HQ0REBkhNTUVKSgrMrNPelyqoLdYjIyNx4MABmNkLmNlnvKzSErBwVeHq1au4fv163gSYRUxniYiItKn3zpOcnGzESExLbuYbYmNj0bt3b7Ru3RpHjx7FF198gU8++QTff/89Zs6cmeOfR0Smb//+/VAqlfAr6a1zfuEyaUM4HjhwIC/DIsoXWBZAlDukcj/PgGLyNM/CxeUu4kmzLLkgN9jKCIcjI4lBLdaLFi2KRo0aoVatWhg0aBCqV68OLy8vrS6ggLSu3YmIKHdJmR8zGwukxCYV2Ir1bdu24c2bN7DJpLW6xNJbICUyrZX72LFjczm6rGM6S0REpE29Yp2FPW/lZr5hzZo1ePr0KaZNmwalUom4uDjY2NhAqTToHX0ies+9ePECly5dQqHinrCysdS5jLufC+ycbHHw4EG2eCPKJpYFEOWOq1evAgA8C5eQp3n9/99Xr15F+fLljRKXKYmPj5f/TkhIgJ2dnRGjMU2sWCeJQRXrjRo1gkKhgBACP/30k87EXcJxYYiIcl9cXByAtIp1QDMzVJBs3rwZUAAWnlkbk9bcVUBpCWzbvg2jRo2CublByWKOYzpLRESkLSGRrSh0yc18w549e+Do6IiIiAi0b98eN27cgJ2dHXr16oU5c+bo7TYzKSlJ40WI2NjYbH0uEZmm0NBQCCEQWN5P7zIKhQJFyhfCpSM3cfbsWQQFBeVhhETvN5YFEOWOa9euwcHVE7aOzvI0z8AS8jwC4hPeliXHxcXB3d3diNGYJg5HRhKDahAmTpyYYcJORER5S65YtzaHQqnA69evjRxR3rt16xauX78OC3cVlBZZW0ehBMw9VIiKiMLRo0dN5o1vprNERAWXEELn3wWdEAJJiW8ragtq7zy65Ga+4ebNm0hNTcVHH32E/v37Y8aMGdi/fz9+/fVXREdH488//9S53owZMzBlypRciYmIjGfPnj0AgCIV9FesA0DRiv64dOQm9uzZw4p1omxgWQBRznv58iWePHmC4tXqaUx38w2EmYUlh174fwnxb5+v+KylG4cjI4lBFeuTJ0/O4TCIiOhdSK2AzCzNobQyx6tXr4wcUd7bvn07gKy3VpdYeqqQHKHErl27TKZineksEVHBxcp03RITE6FSqaCwEBApigLbO48uuZlveP36NeLj4zF48GD88ssvAIAOHTogOTkZv/32G7799luUKFFCa73x48dj5MiR8v+xsbHw9/fPtTiJKPdFRUXhxIkT8CrsBgeXjLuH9S3mCRs7K+zavQtjxoxhd/BEWcSyAKKcd+nSJQCAV5GSGtPNzM3hWbg4rl+/jpSUFFhYZLGVTj6UmpqqUWnMivW31J/P1XvkojSjRo3CpUuXsGnTJr29meVHHBiNiCgfiI6OBgAorcyhtDRDVFSUcQPKY0II7Ny5EwozwMI1exUSZg6A0hrYu3cv3zwkIiKTwkr2t6TeeZRWaf8XxN55jMHGxgYA0K1bN43p3bt3BwAcO3ZM53pWVlZwdHTU+CGi99t///2HN2/eoHiVwpkuqzRTokhFf7x4/gJnzpzJg+hMkxCCz5hEREZ2+fJlAIBPsbJa83yKlkZKSgquX7+e12GZlPQV6axYf0t9XHUOR6Zt9+7dePToEZ4/f27sUPLUOw0mGxcXh3///Rfnzp1DbGwsHB0dUblyZbRv3x52dhm/vUpERDknMjISAGBmYw4za3NEP4/GmzdvCkzLgFu3buHBgwew8FBBkc1dVigACw8V4h7E4cSJE6hfv37uBGkAprNERAWPSqWS/2bF+ltS7zxKa4E3rxUFsneezORGvsHX1xeXL1+Gl5eXxnRPT08AKHAvc1LB9eDBA4T06YPBgwahS5cuxg7HKDZt2gSFUoHilTOvWAeAktUCceXYLWzevBnVq1fP5ehM0zfffIO9e/di7969sLW1NXY49B5hWQBRzjl79iwAwFdHxbpvifI4s/sfnD9/HuXLl8/r0ExG+gpjVqy/pX4sWLGun3o5RkFgcMX6hg0bMHDgQERHR2sU+CgUCjg7O2Px4sXo0KFDjgRJREQZk94KM7OxgLmtJRJVr/Hy5Ut4eHgYObK8ERoaCgCwcDesAsLCXYWkB0rs3bvXZCrWmc4SERVMb9680fl3QSdVpCv/v3c5qaKd0uRWvqFatWr477//EBERgVKlSsnTHz16BAAFJq9JdOzYMTx/9gzfffddgaxYv3PnDi5cuIDCZX1h52STpXV8inrA0c0eu3btwvjx4wtkxfKmTZsAADExMQVy/8kwLAsgyjkpKSk4f/483AoFwtreQWt+oZIVAABhYWHo0aNHXodnMtJXGLMC+S31inW+cKBfQatYN6gr+KNHj6Jr166Ii4vDgAEDsGbNGoSGhuLPP//EJ598gvj4eHTt2lVvt3BERJSznj59CgAwt7OEma2FxrSC4ODBg4Ai+93AS8wcAKUlcOjQIZNoHch0loio4FJ/IC1oD6cZiYmJAQAoLAUUSrDFuprczDdIFYhLlizRmP7777/D3NwcjRo1yoldICIT9/fffwMAStcsluV1FAoFytQshoSEBGzdujW3QnsvmMIzJr0fWBZAlLMuX76M+Ph4FC5XVed8Jw9vOHn44OSpUwX62UuqSLe2Sxt3i2OJv8WK9awpaNePQS3Wp0+fDisrKxw5cgSVKlXSmBccHIwhQ4agTp06mD59OrZs2ZIjgRIRkX4REREAAHNbS1jYW8nTCkI3RtHR0bhw4QLMHVVQGNgPi0IBmLmo8OzpM9y8eRMlS5bM2SCzieksEVHBpd5KPSUlxYiRmBa5Yt0i7Sc6Otq4AZmQ3Mw3VKlSBf369cPSpUuRmpqKhg0bYv/+/fj7778xfvx4+Pr65uSuEJEJio+Px7///gs7JxsUKVcoW+uWqVUUp3ZdxJ9//onOnTtDoVDkUpRE+QPLAohy1qFDhwAAhcsH6V0msEJ1nN+3GZcuXULFihXzKjSTkpycDACwsrVEYlwSW6yriY+Pl/+Oi48zYiSmTX0s+oLAoBbrx44dQ3BwsFYCL6lYsSK6dOmCo0ePvlNwRESUNQ8ePIC5rQWU5kqYO1jJ0wqCY8eOQQgBcwNbq0uk1u5HjhzJibDeCdNZIqKCS70yvaA9nGbk5cuXAAClOaCwEPL/lPv5hoULF2Ly5Mk4ceIEPv/8c5w9exZz5szB9OnT3yVsInpPbNiwAa9fv0a5uiWgNMteMaKtgw2KVw7ArVu3TOI5i8jUsSyAcsLFixfRsmVLNG7cuMC/gBEaGgpzC8sMK9aLVa0DANi3b19ehWVy5BbrtpYA2GJdnXrFemICXzjQp6C1WDeoYj0+Ph5eXl4ZLuPl5aVx0hERUe5ITEzEo0ePYOGUNuiopWNaxXp4eLgxw8oz0gOlod3AS8xdTKdineksEVHBJbUWSP93QRcZGQng/7uCtxB4/fo1C3z+X27nGywsLDBp0iTcvXsXycnJuHnzJj7//HODtkVE75eUlBSsXLkSFlYWKF/XsF69qjQuC0B7SAki0sayAMoJJ06cQEREBJ4/f479+/cbOxyjuXnzJm7evIkilWvB0tpG73KB5YNgZWOHHTt2FLjKQYn0XGX1/xXrbLH+lvr9Njk5mb3K6VHQGgUYVLEeGBiI//77L8Nl9u7di8DAQEM2T0RE2XD37l0IIeSKdXN7KyjMlLh9+7aRI8t9KpUKhw4dgtISUNq927aUFoCZg0BYWBji4ozbtQ/TWSKigku9spgVx289e/YMAKC0SvtRn1bQMd9ARLll/fr1ePLkCcrVKS63YssuN19nBJYrhNOnT+P48eM5HCFR/sI0nXKC+pBJBXn4pM2bNwMAytdrkeFy5pZWKFmzER49eoSwsLC8CM3kSGOH29illS2zYv2t9OOqc5z1t4R428itoL1wYFDFepcuXRAWFoaQkBA8evRIY97jx4/Rp08fhIWFITg4OEeCJCIi/W7cuAEAsHKxBQAolApYOlvj1q1bGuO05kdXr15FZGQkzF1VyInh+ixcVUhNTTV6gQ/TWSKigost1nV78uQJoAAUloDCSm0aMd9ARLkiPj4eixYtgqW1Bao0LvNO26rxYdqYtb/88otGISwRaWKaTjkhKipK598FSWJiIjZu3Ag7J1cUrVI70+UrNmoDAPjzzz9zOzSTJLXKtra30vif1I6FIt3/pFHvUNAq1s0NWWns2LHYuXMnVq1ahb/++gvFixeHl5cXnj59ilu3biE5ORk1atTA2LFjczpeIiJK5+rVqwAAS9e33RpZutriVeQL3L17F8WKFTNWaLlu7969AABzt5wpnDF3E8A9YM+ePWjSpEmObNMQTGeJiAou9Tfg2VLgrYiIh1BaCigUgNJK/P+0CFSvXt3IkRkf8w1ElBuWLl2KFy9eoHrLCrCxt36nbbkXckHxKoVx8exFbN++Ha1bt86hKN8PfJmAsoppOuUEqTLdysEJUVHRxg3GSLZu3YqYmBjU/jgEZuYWmS7vW6IcvIqUwr59+/Do0SP4+vrmQZSmQ6ostnOy0fif3h4LMxsLvIlP4bFRo16ZXtAaBRjUYt3W1hYHDx7E5MmT4efnhytXriA0NBRXrlyBn58fpkyZggMHDsDGRv/YFURElDOuXLmS1kr9/1usA4CVW9rfly9fNlZYuU4IgV27dkFhBli45ExBhZk9oLQWCA0NNWr3u0xniYgKLvXKdD60p0lMTMSTJ0+htElL783+//f9+/eNGZbJYL6BiHJaREQEli1fBntnW1T+4N1aq0tqt60McwszzJ49m+kbkR5M0yknREVFwczCEtYOLoiKelngxg1PTU3F0qVLYW5hiarNO2RpHYVCgeqtu+LNmzdYvnx57gZogl69egUAcHC20/ifIA8Xam6T9oIG8zBvqVesc4z1LLKyssLEiRNx69YtxMTE4MGDB4iJicGtW7fwzTffwMrKKifjJCIiHVJSUnD58mVYONtAaf72lm7tnpYRunjxorFCy3Vnz57F/fv3Ye6mgsIsZ7apUAAWngJxcXFya3hjYTpLRFQwqT+oc/y2NHfu3AEAKP//HULp961bt4wUkelhvoGIcooQAlOnTkVyUjJqt6sCC0uDOrvU4uBihyqNy+LZs2eYN29ejmzzfcEW65QdTNPpXUVGRsLSxh6WtvZ48+YNYmNjjR1SntqxYwcePHiACo1aw87JNcvrla7ZCM5ehbBhwwY8e/YsFyM0PdI5Yu9iC4VCUeDOmYxIFetmtpYa/5NmZTor1g3g4OCAQoUKwcHBISc2R0REWXTr1i0kJSXB2sNOY7qlqw0UZkqcP3/eSJHlvvXr1wMArHxy9s1bS++07f399985ut13wXSWiKjgUK9Y59vwaa5fvw4AMLNPq5hQWqb9SNNJE/MNRDmvIFWM7tq1C4cPH0ZAaR8UrxyQo9uu0qQsnD0csHr16nzdu1p6Ben8oZyVm2n6tGnToFAoUL58+RzfNhmPEAKRkZGwsE2rWAfSKtoLitTUVCxYuBBm5hao2a5HttZVmpmjdvveSE5OxuLFi3MpQtMUHR0NALC2s4a1nRViYmKMG5AJef36NQDA3M5S43+CRm8Y6uOtFwQGVawfOXIEI0eOxJMnT3TOf/z4MUaOHInjx4+/U3BERJSxs2fPAgCsPe01piuUSli52+L6jev5slD+yZMn2LFjB8xsBcyccnbbZjaAuYsKp0+fNlphD9NZIqKCS/0NeL4Nn0ZKj6WKdQBQ2qvw6NEjeQzJgoz5BqLcp1AojB1Cnnjx4gWmTpsKC0tzNOhUPcf329zCDA271IBKpcI333xT4MbjJMpMXqXpDx8+xPTp02FnZ5f5wvReef36NZKTk2Fpaw9L27QXMgpSxfq///6LB/fvo1KTdnB088r2+uXqNYeLtx/Wr1+Phw8f5kKEpkl6prKxt4K1nSVevnxp5IhMh9QtvoVDWsU6W/MTYGDF+uzZs7FlyxZ4e3vrnO/j44OtW7dizpw57xQcERFl7Ny5cwCg1WIdSKtsV71R5cvu4JcvX47U1FRYBaiQG2Vc1gFpb9wZ6w1VprNERAWX+hvwfBs+zfnz56FQAmZq2R1zx7RK9gsXLhgpKtPBfAMR5QSpC/iY6BjUalsZjm72ma9kgELFvVChXkncvHkTCxcuzJXPMDUFbXxjMlxepemjR49GrVq1EBQU9E7bIdPz/PlzAICVnSOs7NIq1gtKt+YJCQmYP38BLKxsULt9b4O2oTQzR/0unyA1NbVADVvy8uVLmFuYwcLKHDYO1oiOji5wLZD1kVrzWzhYa/xPmgrKS6gSgyrWT506hXr16mW4TIMGDfhGPBFRLhJCICwsDGa2FjB30B5jS2rFfubMmbwOLVc9fPgQ69b9BaU1YOGRO13qmTkBZo4Ce/fuNUp3+qaQzvbp0wcKhULvT0REhN51J0+erHMda2vrXIuXiCi/UK9MZ4v1tBYBN27cgJmDCgq1p1czx7TfYWFhxgnMhJhCvoEovysI40b++++/2Lt3LwoV90L5OiVy9bNqtakMJ3d7LFmyJN89r+rCinXKqrxI0w8ePIj169fjf//7n8HbINMlVaxb2jnAyt5RY1p+98cff+D582cIatUlW2Orp1eqRiN4FSmFbdu24dq1azkYoel6/vw5bBysoVAoYOtgA5VKxZ7B/l9UVBSUlmYws7WQ/6c0ZmZmOv8uCMwNWenZs2coVKhQhst4e3sXmLehiIiMISIiAs+ePYNdoIvOt8Lya8X6L7/8gpSUVNgWf6NRwJ6TFArApugbvD5njh9//BErV67M0zfvTCGdHTRoEJo2baoxTYj/Y+++46Oq0sePf+6dPimTTg2E3qWISFHAih3bomv96brrfndtu+7q2nftipVVQUAEVkEUBFF6C7333gMhvfcymbm/P4YJCTVl7tw7k/N+veZlnMzc80xI5ty5zznPo/DnP/+ZhISES8YHMHbsWEJDz+x0aWonWYIgCA1RM7HuLTvXlG3duhW32405svZiOmO4giTDxo0bNYpMP/Rw3iAIwa6srEzrEFSVnJzM+++/j8Vm5toHBiLJ6n72MVmMXPfgYGb/dwkvvfQSM2fOVKWHtF6IHutCXak9p7tcLp5++mmeeOIJevXqdcnHV1RUUFFRUf3/ogSy/nl/N6yh4VhCmk5iPT8/n2++mYQ9PIIBtz3QqGNJsszwB/6PGe88x6effsrXX3/toyj1ye12k5WVRUx8JAAhDhsAGRkZxMTEaBmaLmRnZ2OwGjFYPanUptRa4VKMxjPpZZPJpGEk/tegxHpERAQnT5686GNOnDhR62K6IAiC4FveHVq25ue/AGGwGDFH2tixYwdOpzMoJriNGzd6equHKartVvcyOsAU62bHjh3MnTuXkSNHqjpeTXqYZwcNGsSgQYNq3bdmzRpKS0t58MEH63SMe++9V5yEC4Ig1JMoBV/b2rVrATBG1J73JQMYwt3s37+f3NxcoqIavisl0OnhvEEQgl1paanWIajG6XTywgsvUFpayg0PDyYs0j89l5snxND/hp5sXrSbt956iw8++CBoy4iKcrpCXak9p48bN44TJ06wdOnSOj3+vffe4z//+U+DxhK0kZGRAYAl1IEl1FHrvmA2fvx4iouLuP7R57DY7I0+Xtsel9Ou95WsW7eO9evXn3N9LJjk5eVRVVVFSLgnoV4zsd6jRw8tQ9Ocy+UiNy8Xc4wdo81zXb0pLFSpq5q5hppJ9qagQXv9Bg4cyOzZs0lOTj7v90+ePMmcOXMYPHhwo4ITBEEQLsybWLc2u/AHKlvzMMrLy9m3b5+/wlJNZWUlb7/9Fkhg6+xSpbf62Wwd3EgG+Oijj/zaQ0ev8+y0adOQJIkHHqjb6l9FUSgsLBQ7NARBEOpBlII/Q1EU1qxZg2Q8U/q9JmOUgqIo1cn3pkqv5w2CEEyCeaHTl19+yZ49e+g6oD2d+iX4dezLb+hB83axLFiwgF9++cWvY/tTU2glIPiGmnN6Tk4Or7/+Oq+99hqxsbF1es5LL71EQUFB9e1CcQn6kZ6eDoAlzIHJFoJsMFbfF6xOnTrFDz/8QGTz1vS+7g6fHXfY7/+MJEl88sknQd3Sw7vwIjTSsyAh1OH5r6h25XnfdLvcGEPMSAYZg9Uofi411EysB8OGvvpoUGL973//O6WlpQwZMoSpU6eSlpYGQFpaGlOmTGHIkCGUlZXx/PPP+zRYQRAE4YwtW7d4dqVH2C74GG/SfcuWLf4KSzVff/01SUknsLR0Y/TThivZApYEF/n5+YwePdo/g6LPedbpdPLjjz8yePBgEhIS6vSc9u3b43A4CAsL46GHHqrTKumKigoKCwtr3QRBEJqS4uJiTwleKbgTOXVx9OhRUlJSMEa6z7ugzhTlWbi1atUqP0emL3o8bxCEYBOsrTnWrl3LN998Q0RsGFfffbnfx5cNMjc8NBiLzcw777zDsWPH/B6DPzidTq1DEAKEmnP6q6++SlRUFE8//XSdn2OxWAgPD691E/TNm0S3hkYgSRKWMEf171Gw+uKLL3A6nVw96k8YjL5L7sW16Uj3q0Zw4MABFixY4LPj6k11Yv10Qj0kwl7r/qbM+zMw2s0AGOwm0tKD+++pPmq2/BQ71utg6NChfPLJJ6SmpvLYY4/RunVrjEYjrVu35vHHHyc9PZ3PP/+coUOH+jpeQRAEAc+qweSTyVjiQi9aLs/azFMm3ru7PVAdPHiQb775BtkK1nb+XSVqaaVgCFOYO3eu33bE6XGeXbRoETk5OXUqAx8ZGclTTz3F119/zcyZM3niiSeYMWMGV1999SUT5e+99x4Oh6P6Fh8f76uXIAiCEBBKSkqQzQZkk6HJ71hfsWIFAKaY81c+ke0g2xRWrVpFZWWlP0PTFT2eNwhCsCkoKKj+Olj6rWdlZfHSSy9hMBq48dGrMFm02WkUFhXCNfdfSXl5Of/85z8pLy/XJA41icS6UFdqzemHDx9m/PjxPPPMM6SmppKUlERSUhLl5eU4nU6SkpLIzc1V6VUJ/pSamorRYsNosQJgDYsgOzs7aN+HDhw4wLx582jevhtdrhzu8+Nfde8fMBhN1cn7YORdeBF6OqHubQkT7Asy6sL7MzCGmE//10JpSWnQLrhsDLFjvY6effZZtm3bxpNPPkm/fv1o3749l19+Of/3f//H9u3b+etf/+rLOAVBEIQavDvQbc0vvnXbaDNhcljZtm1bwPZ1q6qq4vXXX8flcnlKwBsu/RxfkiSwd/GUnv/3v//ttySH3ubZadOmYTKZGDVq1CUf++yzz/Lf//6XBx54gHvuuYfPPvuMKVOmcPjwYb766quLPleUmhMEoakrKS1FMsrIRpFYX7p0KUhndqafTZLAFK1QWlrKxo0b/RydvujtvEEQgk1eXl711zWT7IHK5XLx4osvkpeXx+CRfYlpFalpPO0vi6fX1Z05dOgQH374oaaxqKGiokLrEIQAosacnpKSgtvt5plnnqFdu3bVt40bN3Lo0CHatWvHm2++qcKrEfxJURRSUlOxhp95T7eGR6IoStAmST///HMAhv3+yYtuPGooR2xz+t5wF6dOnWLmzJk+P74eeKsceEvB28OtSLIU9C0E6qI6sR7qTax7/puamqpZTHrV1HasN+rVXnbZZZe8QC4IgiD43ubNmwFPD/VLsTULo/BQFgcOHKBHjx5qh+ZzU6dOZd++fZibuzFFatOr2xACljYu0k94Voe//PLLfhlXL/NscXExv/zyCyNGjCA6OrpBx3jggQd4/vnnWbp0Kf/6178u+DiLxYLFYmloqIIgCAGvrLQU2WhAcXsSxk1Vamoq+/btwxjlRrrIp1ZTrJuKUzJLly7l6quv9l+AOqSX8wZBCEY1d3Lm5OTQvHlzDaNpvK+//prNmzfToXc8PYd00jocAAbf0Zf041n89NNPXHHFFdx8881ah+QzIrEu1Jev5/SePXsye/bsc+5/9dVXKSoq4vPPP6dDhw4+G0/QRkFBAaUlJcQ2T6i+zxYeBXj6kLdp00ajyNSxbds21qxZQ0KvK2jbQ712JgNHPsyuxN8YP348d955JzbbhVtyBiJv8ti7U12WZUIddpE85kwC3ZtQN51OsKenp9OlSxfN4tIjWW7wHu6A1LRerSAIQpDYsmULstmA+fRqwouxnk6+B2Kf9eTkZL766itkM1jb+7cE/NksbRQMIQo//PADO3fu1DQWf5szZw6lpaV1KgN/MfHx8aK8nCAIwiWUl5cjGWUko9ykL8QvWbIEuHAZeC9DGMgWWL58OVVVVf4ITRCEJujsxHog27BhA+PGjSM8OpTh912pyg6/hvCWpDdbTfz73//mxIkTWofkM015Phf0ISYmhjvvvPOcW0xMDGFhYdx555306tVL6zCFRjp16hQANkdU9X3er1NSUjSJSS2KojBmzBgArh71R1XHsodH0P/m+8jOzmb69OmqjqWF1NRUjCYDtjBr9X1hUSFkZmY2+c9X1Yl17471ULFj/UL0cj7pL/VKrLvd509q5Ofn87e//Y3evXvTu3dvnnrqKTIzMxsV2LZt27jjjjuIiorCbrfTs2fP6jdLQRCEpiw7O5ukpCSscaFI8qUnLW+5eO8u90ChKArvvPMOFRUVWDu6kDVu1SLJYOvkQlEU/vOf/6jSW8mf82x9fP/994SGhnLHHXc0+BiKopCUlERsbKwPIxMEQQguiqJQUVGBZJCQjVKTvhC/ePFiTxn4SyTWJQmMMW7y8/MD7lynsfR63iAIwaa0tLRWX/VATqxnZ2fzr3/9C9kgceOjV2GxmbUOqRZHTBjDRw2gtLSU5//xfEDPgzWTEZWVlRpGIgQCMacLvnAmsX6m0qB3x3qwtdnbuHEjW7dupePlV9GiQzfVx7vilvuwhYYz6dtvg65d16lTpwiNsNdKjIZFheByuZp8Ofi0tDRkswGD2VNCzbtzPVhbKzSG2LF+AWPGjMFkMlXvHPAqLy9n6NChjBkzht27d7N7926++uorhgwZQlFRUYOCWrx4MYMGDSIzM5PXXnuNzz//nNtuu616chAEQWjKNm3aBNStDDyA0W7GFG5ly5YtAbXScPny5axduxZjlPuSF9X9xegAcws3hw8f5scff/Tpsf05z9ZHVlYWS5cu5a677sJuP7dCwsmTJzlw4MA5zznb2LFjycrK4qabblItVkEQhEBXVVWFoihIBhlkmcrKShRFH3OgP6Wnp7Nr1y6MEe46Lawzx3ouRi9evFjlyPRDr+cNghCMvIn08Jhmtf4/0Ljdbl566SVycnIYdHtf4uKjLv0kDXTs25YeQzpx8MBBPv74Y63DabCayfTy8nINIxH0Tss5PTExkT179vjkWIL2vMnzWjvWIzxJ9mDKqyiKUt0qYcg9j/tlTIs9hP633EdBfn5Q7VovKSkhNzeX8Jja15gdMZ5NWsG2IKO+UlNTq5PpAMZQT+vKYKsAIdRfnRPrK1euJC4ujhtuuKHW/RMmTGDPnj10796d5cuXs2nTJu69916OHTvWoB3mhYWFPPLII9x6662sW7eOv/3tb/zxj3/k/fff58MPP6z38QRBEIJNdWK9Rd0S697HlpSUnJMA1auKigpGjx4NEtg6utFTNRlrOzeSCb748gvy8vJ8dlx/zbP1NWPGDKqqqi5YBv6RRx6hW7faq4Pbtm3LY489xieffMJXX33FAw88wFNPPUWfPn148sknVY9ZEAQhUHmroUgGCcngmfwCaVGcr1SXgY+t26ICQzjIZli6dGmT+Xnp9bxBEIJRdnY2ALFtOgKBm1ifNGkSGzZsoF2v1vS6urPW4VzUkJH9iG4ZwfTp01m2bJnW4TRIzcS62LEuXIyY0wVfqU6sR5zZsW6yhWA0W4IqQbp582a2b99Op/5X0yyhk9/G7TfiXmyh4UyZOpXS0lK/jasmb9uViNizE+ue/09KSvJ3SLpRWFhIUVFRdTIdwGA1IhllkVg/j6a2IaDOifVdu3YxbNiwc+6fMWMGkiTx/fffM3z4cPr378/06dNp1aoVc+fOrXdA06ZNIyMjg3feeQdZlikpKblgORxBEISmaOPGjRgsRsxRl+6v7uVNwm/cuFGtsHzqhx9+ICUlBUsrNwab1tHUJpvA2tZFcVEx48eP99lx/TXP1tf3339PXFwc119/fZ2f8+CDD7Jp0yb+/e9/89xzz7F582ZeeOEFVq1add5d74IgCIJHdWJdlpFOl1JrihfjlyxZ4ikDH123D+c1y8Fv3bpV5ej0Qa/nDYIQjLyJ9bgATqzv3buXL774gtAIO9foqK/6hRhNBm58ZAgms5E33nj9vBWx9K7m/K1GGzEheIg5XfCVkydPIkky1rDI6vskScLmiOZkcnLQJL6+/vprAAbd+ahfx7XY7Fx+0+/Iz8tj5syZfh1bLUePHgUgspmj1v3e/z927JjfY9ILb5UHU9iZHeuSJGEMNZN8KngWqvhKsLy/1FWdE+tZWVl06NCh1n1Op5MtW7bQsWNHLrvssur7DQYDI0aM4NChQ/UOaOnSpYSHh5OSkkKXLl0IDQ0lPDyc//u//7to6aSKigoKCwtr3QRBEIJNSkoKp06dwto8tF4XQ7xl4zds2KBWaD5TVFTEhAkTkExgaavPhVXmFgqyDWbM+IHU1FSfHNNf82x9rV+/noyMDAwGw3m/n5iYeM7J04QJE9i7dy+FhYVUVlZy+PBh3n//fcLC6l5lQRAEoSmqtWNdbpo71jMyMti+fTtGhxu5Hq1/veXgzy6jGqz0et4gCMHIm1iPiW+HJMnV/x8oysrK+Ne//oXL7eK6BwdhDbFc+kk6ENnMweCR/SgoKOTVV18NuAu2NZPpTXGRnFB3Yk4XfOXEiZNYwyORz7p+Y4uIprSkJCAXhp1t586dbNq0ifa9B9K8fRe/j9/vxnsw2+xMnjwlKN7bDx8+DEBU89qJ9Yi4cCRZatLvNd7d/KZwa637zeFWCgsKyc/P1yAq/Qq087TGqnNivby8/JzE9u7du6msrGTgwIHnPL5Zs2YNKolx+PBhqqqqGDlyJCNGjGDWrFk8/vjjjBs3jscee+yCz3vvvfdwOBzVt/j4+HqPLQiCoHfr168HwNYivF7PM1hNmKPtbN22Tff93aZNm0ZBQQGW1i5ko9bRnJ8kgzXBhdNZ5bNd6/6aZwVBEAT9OrNj/Uwp+GC4YFMf3pK/dS0D72VwnCkH3xQqnonzBkHwH28iIjQyBluYI+ASE2PHjiUpKYnew7rSqmMzrcOpl+6DOtC2e0vWrVvHnDlztA6nXmom1l0ul4aRCHon5nTBF0pLS8nOzsJeowy8lz0yFgiOst4TJ04EYODIhzUZ3xoaRt8b7iIrK5NffvlFkxh8ae/evUiSREyriFr3G00Gopo72L9/f5Odw7y79c9OrHv///jx436PSc+awmfwmuqcWG/RogX79u2rdd+6deuQJIkBAwac8/jCwkKio899I7+U4uJiSktLeeSRRxgzZgx33303Y8aM4cknn+SHH36oXkVztpdeeomCgoLqWzD1DREEQfBat24dAPaW9UusA9hbhOOsrGTbtm2+DstnSkpKmDp1KrIJLK30vdLNFKtgsCv88ssvpKWlNfp4/ppnBUEQBP2qqKgAQDLISAa51n1NxdKlSwEwxdTvPECSwBjtJicnhx07dqgQmb6I8wZB8B9vIj3EEUmIIzKgdqzv27ePKVOm4IgN48qbL7v0E3RGkiSGjxqA2Wpi9OjRAfWzr5mIaGrVZ4T6EXO64AveJJ89Ku6c79kjYoDAT6wfPHiQxMREWnftTeuu2s1p/W++D6PZwjfffBPQrT6cTie7d+8msrkDk8V0zvebtY2hrKysye5aP3jwIACWqNo9Ss2RtlrfFzya2gKMOifWhw4dytKlS1m1ahXgKSU1YcIEAG666aZzHr9z505at25d74BsNs8v5u9///ta9z/wwAPAmd2aZ7NYLISHh9e6CYIgBJOqqio2bNiAKcxyzmq5urC38rwvrlmzxteh+cysWbMoLCzE3MqFdP7K47ohSWBp46aqqoqpU6c2+nj+mmcFQRAE/aqdWPfsWNd7pRlfysnJYevWrRgcSr3KwHt5k/GLFy/2cWT6I84bBMF/vIl1e3gkdkcUhYWFAXEhXVEU3nvvPdxuN8NHDcBo1mk5sEsIcdgZeFsfioqKGDNmjNbh1FnNC8xN7WKzUD9iThd8wbu71rs7vaaQ08l2bz/tQHWmt/ojmsYR4oik97V3kJKSwrx58zSNpTH27t1LWVkZrTqeuxgDoGUHz/2bN2/2Z1i6sW/fPox2EwZr7UUHlmg74Pn5CWc0tUWEdU6sv/DCCxgMBq6//nr69etH+/bt2b17N7fffvs5fWBycnJYv349V111Vb0DatmyJeApa1NTXJznDzkvL6/exxQEQQgGu3fvpqioCFurhi0cssaFIpsMrF271seR+UZVVRX/+9//kAxg1vludS9TrIJsPbMgoDH8Nc8KgiAI+uUt6ymbZGSjZ4VZWVmZliH51bJly3C73dX90uvLGKEgmTx91oO9FJ04bxAE/8nJycFgNGGxhxLiiAQgNzdX46gubfny5ezYsYMOvdsEXAn4s3Uf1IHolhHMmTMnYHbO1UymB/ucJDSOmNMFX/BW+Q2NaX7O90KimwHSBSsBB4KDBw+yZMkSWnbqSUKvK7QOhwG3P4DBZObrr78OiMV25+NdzNO687m/MwCtO3nOHVavXu23mPQiLS2NjIwMLLGh53zP5LAimw1s375dg8j0pWZf9aZ2rlPnxHqPHj349ddfadOmDTt27CAnJ4e7776bSZMmnfPYr7/+mqqqKkaMGFHvgC6//HIAUlJSat2fmpoKQGzsuauuBEEQmgLviYy9laNBz5cMMtbmYRw7duyc91g9WLp0Kenp6Ziau3XbW/1skgzmli7KysqYNWtWo47lr3lWEARB0K/i4mIAJKOMbPYk1ktKSrQMya8WLVoE1L8MvJckgynaTWZmZtCXgxfnDYLgP7m5udgdkUiShN0RBaD7PuuKovDVV18hyzIDb+2tdTiNJssyg27vg6IojB8/Xutw6qTmBWaxY124GDGnC77gaScgERrT4pzvGUxm7JExHDhwoFYiLJB88cUXAFx17+NIkqRxNBAWGUOf60Zy6tQpZs+erXU4DZKYmIjRZLhgYt0ebiOuTTRbtmxp9GaiQOPdpW9tdm5iXZIkrHGhnDhxgoyMDH+Hpis1z28CdYFJQ9U5sQ5www03cOTIETIyMiguLmbmzJlERUWd87jnnnuOvLw8brzxxnoHNGrUKAC++eabWvdPnDgRo9HI8OHD631MQQhEycnJzJw5M2BPeATfW7lyJZJRxtai4a0uQuI9SXnvqkQ9+f777wGwtAqsFW6WFgqSAaZPn97oCyb+mGcFQRAE/SoqKgLAYDZWJ9abykWMzMxMNm/e7CkDb2n4cUxxnnPn+fPn+ygy/RLnDYLgH7m5uYScTqgHyo71zZs3c+jQITr0iccRG6Z1OD4R36UFMa0iWbp0CWlpaVqHc0k1E+viuo5wKWJOFxrD5XKxe88eQqJiMZrPfyId3jyegoICTpw44efoGm/r1q0kJibSpns/2vbsr3U41QaOfBiTxcbYceOqK48FisOHD3P48GHadGuJ6SKtYjr0jqeqqoolS5b4MTrtXWpzm/d+vVaF9ZfKysrqr0VivQ5iY2Mxmy/c9M5ut+NwOBq0eqhv3748/vjjTJs2jfvuu4+vvvqKUaNGMX36dP75z39Wl4oXhGD3z3/+k//85z9s3bpV61AEHUhPT+fQoUPYmochGxv01g2cmfhXrlzpq9B8Yv/+/ezYsQNjtBuDTeto6kcygqmZm7S0NJ/9XNWcZwVBEAT9ys/PB0C2GpFPX+AoKCjQMCL/mT9/PoqiYI5r3AI7Y4SnP/vChQubzId7cd4gCOopKyujtLQU++mEuj3c81+971ifOXMmAJcN66pxJL4jSRKXDeuCy+Xml19+0TqcSxI91oWGEHO60BAHDx6ktKQER8uECz7G0aItQMBdZ3a73YwePRqAYb//s65+90MckQy4/fdkZ2Xx7bffah1OvcydOxeAzpcnXPRxnfolgERAzLu+UllZyZo1azCFWTCFn3+hir215/r6ihUr/Bma7lRUVJz366ag4dkZFY0bN45///vfbNy4keeee47t27fz6aef8u6772odmiD4zd69e4EzF1iFpi0xMREAe3xEo45jDDFjjrazadMmXa2mnDFjBgCWloG5kt/S0pME8L4OQRAEQWgI73mfwWLEYPUk1vPy8jSMyD8UReHnn3/2lHKPa9y5gCR5FrwVFBQ0+QsdgiA0njeBfmbHuue/2dnZmsV0KU6nk1WrVuGICSMu/twdr4Gsfa94DEYDy5Yt0zqUS6qZTK+qqtIwEkEQgt26desAiGrT8YKPiYrvWOuxgeKXX35h7969dL/qRlp06KZ1OOe44pb7CYuK5dtvv9Vl283zKS8vZ/bs2djDrLTtfvFNrKERdtp0bcn27ds5dOiQnyLU1tq1aykuLiakbeQFF3KYwiyYo+ysWbOmyVSYO5+ysrLqr/WUZ/AHXSbWTSYTb7zxBklJSVRWVnL48GGee+45rcMSBEHQjDexHtK6Yf3VawqJj8DpdOrmZLqoqIjffvsN2aZgjAzMxLohBIwRbtatW8fJkye1DkcQBEEIUN4EjsFmwmAz1rovmG3bto3jx49jjHEjX7gSYZ2Zm3sWvP3000+NP5ggCE2aN4EecnqnekiE/nus79ixg5KSEhJ6ttLVzj5fMFtNtOrUjAMHDuh6cQPUTqaLxLogCGpasWIFkiwT1abTBR9ji4jGHhHDmjVrAmZnaUFBAZ9++ilmq41hv/8/rcM5L7PVxvAH/kJFRUX1znq9mzdvHgUFBXQb2AGD0XDJx/cc4vm98rbwDHbelmKh7SIv+rjQdpFUVVWxdOlSf4SlSyUlJdVfi8S6IAi6EmwfhIX6KykpYdOmTVii7RhDLlwSrK5CTu9618surt9++42KigrMLdwE8q+7uYVnUcCsWbM0jkQQBEEIVN4kgcFqxGgz1bovmHkv0ngrwDSWwe5Z8LZhwwaOHj3qk2MKgtA0VSfWI6Jr/VfP780HDhwAoFnbGI0jUUfzBM+/gfd16lXNvqM1vxY85syZo5trEoIQyJKTk9m1axeR8R0xWe0XfJwkScR27ElpaSmrVq3yY4QNN2bMGPLy8hh892OERep3Tus66DradO/HsmXLdP+zdblcfPvttxiMBnpd1blOz2nbrSWRzcL59ddfyczMVDlCbRUUFLBs+TLMETbMURf+ewIIax/d5Mrkn62oqOi8XzcFIrEuCDqnKIG5g1fwnTVr1uB0OhtdBt7LHGXDGGImcWWiLlbOz5o1CyQwNwvs33VTjIJsgtlzZjeZnq6CIAiCb2VnZyMZZGSzAdliRJIlXSdvfCE5OZlly5ZhCFUwhPvuuJZWnvOKqVOn+u6ggiA0ORkZGQCERnqSufbwCCTZUH2/HnkXFEW3aHy1Mz2KbuHZQab3hVM1d4SWl5drGIn+FBYW8tprr/HMM89oHYogBLzZs2cD0KJbv0s+tvnpx/z888+qxuQLO3fu5KeffiI2vj2X3/Q7rcO5KEmSuOGxv2MwGnn7nXd0vXN30aJFnDhxgi5XtMMebqvTcyRZou+13XE6nUyaNEnlCLW1YMECnJVOwjpGX3KzozHEjK1FONu2bePEiRN+ilBfCgoKzvt1UyAS64IgCDq3fPlyAELaRPjkeJIkYY+PoLCgkO3bt/vkmA114MABDh48iCnajdz4zfia8vSFdZOXm8eaNWu0DkcQBEEIQFlZWRhsJiRJQpIkDFZj0CfWv/nmG9xuN5Y2vq1cY4xWMNgVfvnlF1JTU313YEEIYlVVVcydO7e6DZUA6enpAIRHNwNAlg2ERcVU369HeXl5AHW+YB5obGFWAPLz87UN5BKacnnUSxE7+AXBNyoqKpg5cyZmWwixHXte8vGh0c2IaNWONWvW6DoR6HQ6efPNN1EUhRv/8A8MRh/0ilJZdKu2DLj9QdJSUxk3bpzW4ZxXVVUVY8eORTbIXH59j3o9t9PlCThiQvnxxx91fQ7UWLNmzUKSJUI7RNfp8eGdPZUUAmGxihpqnot5zz+bCpFYFwRB0DGn08mqVaswhVkwR/ruwog3Se9N2mtl7ty5AJibB/ZudS9vT9dff/1V40gEQRCEQKMoCtnZ2RjtZy4cGWwmMjIygraCUXJyMr/88guyXcEU49vXKElgaePG5XIxfvx4nx5bEILVli1beOWVV3j66aeDflFPXaWkpAAQHtui+j5HbAsyMjJ0W6XKm9A1W0waR6IO8+lWKXovOVozvsLCQg0jEQQhWM2ePZu8vDxa9roSg7Fu7/nxfa8C4Ntvv1UztEb57rvvOHToEH2uG0mrzr20DqfOBo18mMjmrZk6dSoHDx7UOpxzzJ49m6SkJLoP7EBYVEi9nmswyPQf0Qun08mXX36pUoTa2r9/PwcOHMAe76huy3YpIfERGKxG5vwyR7fnhWqqmUzPzc3VMBL/a1BivaCggF27dl1wxWVJSQm7du0SJ46CIAiNtGnTJoqLi7G3ibhkCZr6sDUPxWAxsnTpUs0u1rvdbhYtWoRkAmNkcCQMDKEghyisXLmy1g6F+hLzrCAIQtOTn5+P0+nEYDtTwsVgN1NRUdGoOUXPxowZQ1VVFdYE3+5W9zLFKRhCFGbPnq37ksGNIc4bBF+p+TtSXFysYST6cfz4cSz2UOzhEdX3RTZvjcvlIjk5WbvALsJk8lwMdrvdGkeiDneV53WZzfoueVbzYnNT28UlNJyY04W6Ki8vZ+LEiRhM5upkeV3EduhOSFQcc+bM4eTJkypG2DDp6el89dVXhDiiGHr/k1qHUy9Gs4UbHnsel8vF22+/rat5uKSkhC+/+hKTxUT/EZeubnA+nfslEN0ygl9++UWXCwcay9tWIaxTbJ2fIxlkQttHk5uTy9q1a9UKTbdqJtNFYr0O3nzzTYYMGYLL5Trv910uF0OGDOGdd95pVHCCIAhN3ZIlSwAIbRvp0+NKsow93kF6ejr79u3z6bHrateuXWRmZmKKcSMFUf0Uc6ybyspKVq1a1eBjiHlWEASh6cnKygLAaD+zOt67Ut77vWCyY8cOFi5ciCHM97vVvSQJrO3cuN1uRo8eHbQ7/8V5g+ArNcszi1LNnp/B8eNJxLRuV2uRc0zrdgAcPnxYq9AuymbzVDqrLA/OnVPOCs/r8r5OvcrJyQFAthjIyckJ2jlI8C0xpwt19f3335ORkUF836sw2+q++1iSZNoPHoHL5WLMmDEqRtgwo0ePpry8nOEP/gVrSJjW4dRbQq/+dB10HTt27Kiu0qkHEydOJCc7h77XdcMe1rD5U5IlBt/RD0VR+OCDD4JqXnM6ncybPw+j3YS9ZXi9nhvWyVM2/pdfflEjNF3znusgKeTk5mgbjJ81KJWxcOFCbrjhBsLCzv/mFh4ezogRI5g/f36jghMEgaCapIT6qaqqYtmyZRjtJiyx9SvRUxchp5P1ixcv9vmx68KbeDZFB9fvuPf1rF69usHHEPOsIAhC0+Mtu2yoUXbOYA/OxHpVVRVvv/02ALaOLlV2q3sZoxSMkW7Wrl2reQsctYjzBsFXapawbIrlLM926NAhXK4qmiV0qnV/3On/37t3rxZhXVLz5s0BKMoLzr7eRXmeKi7e16lX3h60lugQKioqxA7jGnxZjS/YiDldqIusrCy+Hj8esz2UtpcPrffzYzv0wNGiLYsWLWLz5s0qRNgwmzdvZvHixbTuchndh9yodTgNds2Df8VksfL555/rovLYyZMnmTJlCmGRIfQZ1rVRx4rv0px2PVuzefPm6s1gwWD9+vUUFhQS0i4KSa7fHGWJtGOOtLFy1aomV/HJm1g3hEBOdtNaRNigxPrJkyfp1KnTRR/ToUMHXZYTEYRAIz5wNF1bt24lPz8fe5tIVX4P7C3DkU0GFi9erMnEt379eiQZjBHBNenKISBbYO26tQ3+uYp5VhAEoenJzMwEaifWvbvXvd8LFtOmTePgwYOYm7sx1m9DQL1JEtg6eqrjvPfee7q4uOVr4rxB8JWKiorzft1Ubd26FeCc/q4t2ndDNhirv683rVu3BqAgW989yBuqIMtz0dr7OvUqNTUVg9WIOcJa/f+Ch8Fg0DoE3RJzulAXH374IWWlpXQYPAKjpf67jyVJovPwO0CSePvtt3WxmE5RFD755BMArn34mYC+Hh4WFcuVdzxEdnY2U6dO1TocPvjgA5xOJ4NH9sVoNjb6eINH9sVgNDB69GjKysp8EKH2Fi1aBEBou6gGPT+0XRTOykoSExN9GJX+ZWdnI5lAtig4nU6KioLz3PN8GpRYlyTpkh+yKioqLli2RhAEQbi0hQsXAhCa4Nsy8F6SwVMO/tSpU34vB19eXs7+/fuRQxWkIPtMLUlgcLjJzcklJSWlgccQ86wgCEJT402eG0PO3bEeTIn15ORkxowZg2wGa3v/9B002MES7yIjI4NPP/3UL2P6k7/PG9555x0kSaJnz4b1ZxT0q+bvUXl5uYaR6IO3V2Z897617jdZrLTs1IM9e/aQn5+vQWQX17lzZwCyTwVnX+/sFM/r8r5OPXK73aSkpGAMtWAMtQBw6tQpjaPSj0BOmKlNXAsQLmX16tUsXLgQR8sEWvTo3+DjhDdrTevLBnLs2DG+/fZbH0bYMImJiezZs4eug66jefsuWofTaP1vHkWII4opU6dSUFCgWRyJiYmsWrWK1p2b0/6yeJ8c0xETRt9ru5Gens6ECRN8ckwtud1u1qxZg9FuxhJtb9AxQtpEALBmzRofRqZ/nsS6gmT2/H91afgmoEGJ9a5du7Jw4cIL7sRzu90sWLCALl0C/01QEARBC06nkyVLlmC0m7A2C1VtHO9KvAULFqg2xvkcPHgQl8uFMTy4dqt7GcM8r2vPnj0Ner6YZwVBEJoeb8lYo91cfZ8xxFzre4HO5XLx2muvUVFRgbWjC9l06ef4iqWNgiFEYcaMGWzcuNF/A/uBP88bTp06xbvvvktIiO/bFAnaq7nrqKkn1gsKCtiyZQvN23clNCL6nO936DsYt9vNypUrNYju4rp164YkSWSczNY6FJ9TFIWMkzk0b96c6Ohz/130IjMzk8rKSkzhFkxhnsS62GF8hkisX5i4FiBcTHFxMf95801kg5Gu192NJDUotVOtw+CbsIY6GDduHMeOHfNRlA3z7bffIkkSQ+55XNM4fMVstXHlHQ9SUlzMTz/9pEkM5eXlvPfee8gGmavv6e/T996+13UnLCqEyZMnk5SU5LPjauHQoUPk5uZiaxXe4J+RyWHFGGJm3bp1TaYcelVVFfn5+chmBdnsec3e9nZNQYPefX//+99z6NAhHn/88XNW3BQUFPD4449z5MgRHnroIZ8EKQiC0NSsX7+egoICT28XFT902luGY7AYWbBggV9XPHsvKsj24DzZkE8vcGzoxRMxzwqCIDQ93hKxxtBzE+tpaWmaxORr//vf/9i6dSumWDfmWP+eA0gy2Lq4QIJXXnklqHrd+vO84R//+AcDBw6kf/+G75AS9KtmYr20NDj7c9fVkiVLcDqddB147Xm/3+XK4QDMmzfPj1HVTUhICF26dCHzRA6uquDa1VqQXURpYRn9+vXTOpSLSk5OBsAUZsEU7ikFLxLrQl2IawHCxXz00UdkpKeTMOBaQqObNfp4RouVLtfdhdPp5JVXXqGqqsoHUdbfvn372L59Ox36DSG6ZRtNYlBDr+G3YbGFMG3adE2qTEyaNInU1FR6D+tKZJxv+2+ZzEauuvNynE4n77//fkAnk3ft2gXQqI1tkiRhbRZKXl5ek6lQk5eXh6J4dquLHet19NRTTzF48GCmTJlCu3btGDFiBI8//jgjRoygXbt2TJ06lauvvpqnnnrK1/EKgiA0Cb/++isAYe0b1tulriSDTEhCJJmZmWzevFnVsWrynmTIVr8N6VeyzXNC2dBS8GKeFQRBaHqSTyVjsJmQjWd6pBjMRgwWY/UF+kB28OBBPh/zObIZbJ38UwL+bMYwsLb1lIR/9913NYlBDf46b1i1ahUzZ87ks88+803ggu4UFxdXf11SUqJhJNqbOXMmsmyg2+Drz/v9iLiWtO7amw0bNujyPfqKK66gyukiPSm4dg6lHPa0RtH74p4TJ04AYAq3Ygo1g4Quf0+0EsgJGLWJawHChaxdu5ZZs2YRFtuStv2H++y4Me260aLb5ezZs4cpU6b47Lj14a2i2fvaOzQZXy0Wm51uQ24gKyuTbdu2+XXs5ORkvvnmG0Ij7PS/UZ32TQk9W9GmWwvWrl3L8uXLVRnDH/bu3QuANaZxFbksp5/f0OqlgSY3NxcA2QTS6Up0IrF+CSaTiaVLl/L3v/8dl8vFkiVLmDx5MkuWLMHtdvPPf/6TRYsWYTL5sbafIAhCkCgsLGTZ8mWYI2yYoxrW26U+wjp4SujNnTtX9bG8vLvEZFNwfqCWjJ7/FhUVNej5Yp4VBEFoWpxOJymnUqrLxdZkDLOQnJwc0L00y8vLefHFF6lyVmHr4t8S8GeztFEwhCvMmzdPlztNG8If5w0ul4unn36aJ554gl69el3y8RUVFRQWFta6CfpX899Jy36gWtuxYwd79+6l4+VXERYVe8HH9b3+ThRFYdq0aX6Mrm4GDRoEwKlDwdFKxCv5oKeCy8CBAzWO5OJq7liXDDLGEIvYsV6D263NArtAIK4FCOdTWFjIG2+8gSwb6D5iFLLBcOkn1UOnYbdjCXXw5ZdfcuTIEZ8euy6Wr1iBxR5KQi99L5pqiC5XXgPg98Tz6NGjqaysZPAdfTFZjKqMIUkSV911OQajzIcffhiwbYSSkpKQZKm6wkxDmSNs1cdrCrxJdMl0phR8Xl6eliH5VYMbcVitVj766CNyc3PZvXs3a9asYc+ePeTk5PDBBx9gsZx7UUgQBEG4tAULFuCsdBLWMdovvccssSGYwq0sWbKkwYng+qouM+nbzwK6IZ1+XY0poSnmWUEQhKbj1KlTuFwuTI5zP8ybHFYqKyurS8UHos8++4yjR49ibuXGFKXtojpJAntXF5IR3nrrrYD+udak9nnDuHHjOHHiBG+99VadHv/ee+/hcDiqb/Hx8Y0aX/AP784TaFoXxs42ceJEAK649b6LPq7zgOGExzRj5syZuvt59e/fH7PZzMn9wdFKBMDlcnPqcAZt2rTR/XuKt0Kb8fSCOVOYubrvukBALxb0B3EtQDjbRx99REZGBu0GXk9oTAufH99ktdHt+ns0KQlfUFDAyRMnaNWlFwZj8C0YadW5J7LB6NddzOvWrWPFihW06tiMDn3ULa0fERtO7+FdSU1N5dtvv1V1LLWcPHkSY4gZSW7cNXhTuOe9ualUqMnPzwc8u9W9G8y89zUFDU6sexkMBnr06MHgwYPp3r07Bh+vmBIEQWhqZs2ahSRLhJ7eSa42SZII6xRNeXl5dfkltRmNp2fc4NywXv26ql9nI4h5VhAEIfgdO3YMAHPEuYl18+lku/cxgWbt2rV8//33GEIUbO31sUPNYANbRxclJSW8/PLLQXWBX43zhpycHF5//XVee+01YmMvvHu3ppdeeomCgoLqW1O5wBToMjMzPatPvF83Qbt27WLlypXEd+9Lq84Xr85gMBoZcNsDlJeXM2nSJD9FWDc2m43+/fuTdSqX0sIyrcPxiYykbCrLKrn66qu1DuWSUlJSkIwyBqvn86Ax1IKiKKSlBc9Ch8ZwOp1ahxAQxLUAATzn0rNnzyYsrhVt+g9TbZzohC606NGfffv2MXnyZNXGOZv3M05sfHu/jelPRpOZ6FZtOXLkqF/G8/Y8l2SJq+6+3C8bti6/vichDjvffPNNwM1zVVVV5OTkYAw1N/pYRrtnYUhTOYf2VreSapSCb0oVrxqdWBcEQRB8Z8+ePezfvx97fARGm/9WaoZ1jEGSJX788Ue/9Duz2TzlcZTguY5di/d1eV+nIAiCIFzM4cOHgTPl42oyR9pqPSaQFBQU8OqrryLJp3eJ6+jTpylOwRTrZuvWrZr1kwwUr776KlFRUTz99NN1fo7FYiE8PLzWTdA3b9IvLK4VstEUcBdGfUFRFD777DMAho76Y52ec9k1txEe05xp06frrgKGNwF9Yr++4mqoE/s8ryMQEutpaWkY7ebqhIbp9AV7vf2OaKWioqL6a3/ujBWEQFNaWsp//vMfTwn4G3+HLKu7uKLT1bdhCQnnq7FjOXHihKpjeRUXFwNgC3X4ZTwtWEPCKCkp9sv11unTp3P8+HF6DulEdIsI1ccDMFmMDLq9DxUVFXz88cd+GdNXcnNzURQFgw+uwUuyZ0FdVlaWDyLTP+/frmQ8s2O9KbX/qtNWumuvvRZJkpgyZQqtW7fm2muvrdPBJUli2bJljQpQEAShKfnhhx8ACO9St91AvmK0mQhpE8HBgwfZuXMnffr0UXW8mJgYAJQKCcKCb9u6+3SFP+/rvBQxzwqCIDRt1Yn1yOBKrL/77rtkZ2djbe/CEKp1NLVJEtg6uXEVynzxxX8ZOnQoHTt21DqsOvHnecPhw4cZP348n332Wa2EUHl5OU6nk6SkJMLDw4mKiqrXcQX9ycrKoqKiAocjCpezkpMnm16VgWXLlrF582Y69b/6krvVvYwmM1eP+iPzvnqLTz/9lNGjR6scZd1dffXVfPDBB5zcn0q3KztoHU6jndifgtVqpX9/fffgraysJDc3F1vLMwuKDHZPYr2p7GK7lJqJ9crKSp9UegtU4lqAcDHjxo0jLS2NhAHXqlIC/mwmq43O14xk92//46233mLChAmq73j2Jpv9kXTWkiRJuN1uVStP5ObmMnbsWKx2C1eMqNt5jK906teWPWsOsWjRIh544AH69evn1/EbypsE90Vi3Xuc7OxsnxxL77ztZCWjgiSDJJ9JtjcFdTpzSUxMRJKk6l6xiYmJdTq4P0pNCIIgBIu8vDzmL5iPyWHF1iLM7+OHd4ujOCmPadOmqZ5Yb9HC84HAXa7qMJpxl3vmP+/rvBQxzwqCIDRthw4dwmAxnvcDvTHEjGw2cPDgQQ0ia7hly5Yxf/58DOEKltb6vFAmm8DWyUXJHs+u7O+//z4gyqz687whJSUFt9vNM888wzPPPHPO99u1a8ezzz5bvctXCFzeUqwhkbEoLhdZR/eSm5vbZBZNlJaW8sEHH2Awmhj+4F/r9dzuQ25g+5LZLFy4kHvvvZcrr7xSpSjrp23btrRp04bkg2m4qlwYjPp/f7uQorwSctMKGD58uO77S3uT596SsOCZywEyMjI0iUlvysvLa31tt9s1jEZb4lqAcCHHjh1j6tSp2BzRJAyo24ILX4jr2JOY9t3YuHEjS5Ys4cYbb1R3vLg4AIrzgjcZWZSbTXR0jOqfM7766iuKi4u5+p7+WEP8O1dKksSQuy5n1qeL+OCDD5g+fTqyrKNyZRfgTYL7qmqswWaiMLWQiooK3Z+vNFZJSQkAkvfX2njmvqagTr/dbrcbl8tF586dq/+/Lrdg6lUnCIJ+vPHGG/z1r/W72BEIfvrpJ5yVThxd4zT5kGSNC8UcaWPx4sWqf+Bv397TO8lVEpwfBt2nzyO8r/OSjxfzrCAIQpNVXl7OyZMnMUfZzjv/S5KEOcJGUlISlZWVGkRYf8XFxbz77jueEvBdXOj52q8pWsHczM3evXuZPn261uHUiT/PG3r27Mns2bPPufXo0YM2bdowe/Zs/vCHP/j6JQoaOHToEAAh0c0JiWle676mYOzYsaSnp3PlHQ8S2axVvZ4rSRI3PPZ3JFnmrbfeqrUbV2vDhg2jstxJ2rHALkuatDcFgKFDh2ocyaV5L9IbaibWbU2r7+qllJWVVX/tTSg3VeJagHAhn376qed3Y9jtGIz+axUJ0HnY7cgGI5988glOp1PVseLj4zEYDGQkBec5R3lJEQVZqbRrl6DqOMeOHWPmzJ+IjAunxyBtqnA1axNN5/4J7Nu3jwULFmgSQ30lJ3sqNBlDfZMEN50+TkpKik+Op2fe+dubWJdkpUnN6fpfNiIIgnCWn3/+mVWrVmkdhk85nU6mT5+OwWwkrGO0JjFIkoSjezNcLpfqF5YTEhKwWCy4inV8pb0Rqoo8r6tr164aRyIIgiDo3dGjR3G73eftr+5ljrThcrk4fvy4HyNruC+//JLMzCwsbVwYAmATmrWDG9kEY8aMEUmPs8TExHDnnXeec4uJiSEsLIw777yTXr38W2pSUMf+/fsBCItrRVhsSwD27dunZUh+s3fvXqZOnUpk89YMvOOhBh2jWUInLr/pd5w4cYKvv/7axxE23PDhwwE4vueUtoE0kjexPmzYMI0jubQzu9/M1fd5k+w5OTmaxKQ3NXe0NaXdbYJQVzt27CAxMZHI+A5Et/P/dSWbI5rWvQeRkpLCzJkzVR0rJCSEbt26kXZkH5XlwZeUS96/A8XtVr2NyX//+19cLjeDbu+DbNAu5XflLb0xmgyMGTMmIBaFJyUlAWAK91Fi3eE5TqB8bm+Mc3asG5rWnC4S64IgCDowb948srOzCescg2zSrkRfWPsoDDYTP/74o6qrzIxGI5dddhmuYgl3lWrDaEJRwFUo07p16zr3WBcEQRCaLu+O0PP1V/fyfi8Qdo8eP37cU/rPBpZ4fZaAP5tsAks7F2VlZYwZM0brcARBEzt37cJktWNzRBHePB6A3bt3axyV+iorK3n11Vdxu93c9McXMZobfmH1qnv/QERcSyZNmsTevXt9GGXD9e3bl/DwcJL2pARs/9qKskpSDmfQs2fP6pLBeuZNnhtsZ7pvymYDkiw1mb6rl1KzB2tTuggvCHU1btw4ADoMHqFZ2f+EK67BYLbwzTffqL5rfejQobiqnBzeslrVcbSwf/0yQN2FYXv37mXp0qW0aBdL2x71q7rja2GRIfS8qjOpqamqL8rwhZ07dyIZZcwOq0+OZ4myVx832FVXn/HuWDfUrkgT7OrUY70xO0MDoUyTIAiCltxuN5MnT0aSJRzdtL1QIBlkHN3iyN2Wws8//8xDDzVsx0ZdDBgwgM2bN1OVL2GOCcyLPOfjKgbFSb16K4p5VhCEpmjp0qUsW7aMN998E5PJv+UN9eTw4cNA3RLrR44c8UtMjfHZZ5/hcrkIae9CCqBl3ObmCpWpCnPnzuWhhx7SddUZPZw31LUHrBAYsrOzST55kph23ZAkCWuoA2tYBNu2bUNRlKDu5fvFF19w5MgR+o24h/hufRp1LLPVxk1P/osZbz/Lyy+/zA8//IDNduH3dn8wmUwMHz6cuXPnknkyh2ZtA2/hb9KeFNwuN9dff73WodRJbm4uAAbrmXMbSZIwWI3k5eVpFZau1EysFxUVaRiJ9vQwpwv6cvToUdauXUtkfAccLdpqFofJFkKrXldycusqFi1axG233abaWLfddhtfffUVuxLn0eOqEaqN429lRQUc2bKa9h060K1bN9XG+fLLLwG48tbeujhn63ttN/atP8KEiRO45557dNtrPD8/n4MHD2JtHorko13+lthQJFliw4YNPjmenpWVlSHJVLd9kwwKTqcTp9PZJK7v1CmxPnz48Ab/UYqeL4IgCBe3evVqjh49SliHaIwh5ks/QWXhXWLJ35XOlClTuO+++1SbDK+66iq+/PJLqnKCK7HuzPGcjA0ZMqTOzxHzrCAITdHf/vY3AB566CF69OihcTTaqU6sX6IUPOh/x/revXtZvnw5BoeCMTqw5nZJAmt7NyW7JMaOHcvnn3+udUgXJM4bBF/bsmULABGt2lXfF9GqHekHtnP8+HHat2+vVWiq2rBhA5MnTyaqRTzD7v+zT47ZpltfLr95FFvmz+Djjz/m1Vdf9clxG+Omm25i7ty5HN52IiAT64e2esqp3njjjRpHUjfVO9attS+5ylZjddK9qSssLDzv102RmNOFs82YMQOA+L5XaRwJtO49mORtq5kxY4aqifX4+HiGDBnC2rVryTx5hLg22vQI97Wdy+dS5axk1O9+p1rCe8+ePaxevZpWnZrRsoM+qrrYQq30vKoz25buZebMmTz44INah3ReCxcuRFEU7K0dPjumbJSxtQxn//79nDhxgrZttVsco7bS0tLq3epwpiR8WVmZSKx7vf7667pY7SIIghCMvvnmGwAiejXXOBIPg8VIWOdo0vels3DhQm6//XZVxunRowdxcXFk5WSiuN0BtavtYpzZEmazuV6JdT3Os4mJiVxzzTXn/d769esZOHDgRZ+fkpLC3/72NxYvXozb7eaaa67h008/DdoLs4IgNFyglqb1lUOHDmEKs1y0FYzBbMQYatZ9Yt3bV9ia4EZn01qdGCMUDA6F5cuXc/DgQbp06aJ1SOelx/MGIbBt3LgRgMj4DtX3RcZ3IP3AdjZu3BiU52/Z2dm89PLLyAYDtz31BiaLb0qAAgy970+c3LuVGTNmMHDgQM13Wg8aNIioqCgObzvBoDv6YtCw92p9lRSUcupQBn379iU+Pl7rcOrEuyv97MS6wWqiKLewyezkuhiRWD9DzOlCTU6nk3nz52MJDScmQfvqSbbwSKLadGLHjh2cPHmSNm3aqDbWQw89xNq1a9n023Ru+8trqo3jL1WVFWxdNJPQ0DDuvPNO1cbxfv66YkQv1cZoiD7Du7J79SG+/fZbfve732E2a7+RrCZFUZg9ezZIEqHto3167LAO0ZSeKmDOnDk8++yzPj22npSWliIZalzLMZy5Pzw8XJug/KhOifV///vfKochCILQNG3fvp3t27djj4+46E41f4vo0ZzCA1lMnDiRW2+9FVn2/cUXSZK48cYb+e6776jKkzAF2M6283EVg7tEYtgNw7Db7XV+np7n2WeeeYYrrrii1n0dO1589XBxcTHXXHMNBQUFvPzyy5hMJj799FOGDRvGjh07iI727UmrIAiBrSkn1rOyssjNzSWkbeQlH2uOtJOZnEleXh6RkZd+vL+dOHGCxMREDOEKpojA/DeVJLC2cVOy28DUqVN55513tA7pvPR83iAEHkVRWLduHSarnbDYltX3R7XpBMC6dev4/e9/r1V4qnC5XPzrX/8iOyuLax9+mubtfLuIxmgyc/vT/2HqK0/w2muv06VLF02Twkajkdtvv50pU6ZwfPcpOvZRLzHia/s2HEVRFEaOHKl1KHWWm5sLkoRsOTuxbqz+frNmzbQITTcKCgrO+3VT1JTn9P/97398/fXX6OGjgM1u48svvtB8UeXmzZspLCigTb+rkVS4DtcQzbr2JefEIZYsWcIf/vAH1cYZMmQIXbp04cC6ZVx1zx+IaNby0k/Ssd2rFlCSn8sf//hHQkJCVBnj4MGDJCYm0rJDnG52q3tZQyz0HNKR7cv3M2fOHEaNGqV1SLWsX7+effv2EdI2EqPNt4vd7PERGG0mfvjhBx577LGgTTIXFxefd8d6aWmpNgH5mT7eoQVBEJqoiRMnAhCpk93qXsYQM6Htozh27JiqPTS9F0gq04NjhXZlumdaveOOOzSOxHeuvvpqHnrooVq3mJiLl5D86quvOHz4ML/99hsvvPBC9c71tLQ0Pv74Yz9FLghCoHC73VqHoJl9+/YBYIm69OI672P279+vakwNNX36dBRFwdI6sP89jZEKBrvCggXzyc7O1jocQVDd8ePHSU1NJapNp1oX8a1hEYRExbFx40YqKys1jND3vvjiCzZu3EinK4Zy+U2/U2WM6JZtGPHEPykuLuK5557T/CLj737neZ171ui78klNLpebfeuPEhoays0336x1OHWWnZ2NwWo8Zxey4fSFezG31E6m5+fnaxeIoBmn08mEiRMpKCggv0rS9FZQWkZGejrff/+91j8WVq5cCUBM++4aR3JGTLtuSJJcHZtaJEniiSeewO12sfG3aaqOpTZXVRWbfp2GxWpVtQz6V199BcDlN/RUbYzG6D28G0aTgQkTJ+B0OrUOp5qiKIwdOxaAyN4tfH582Sjj6NmM4uJivvvuO58fXw8URaG4uLg6mQ5nEutFRUXaBOVnddqxfjFr165lx44dFBYWEh4eTp8+fepV/lYQBKGpOnjwIKtWrcLaPAxrXKjW4ZwjolcLio7kMGHCBK655hpVypN17dqVbt26sf/AftwVbmSLz4fwG8UFlRkyMTExPp0H9TDPFhUVYbPZMBrrdtowc+ZMrrjiilo73bt27cp1113Hjz/+yLvvvqtWqIIgBKCmvGN99+7dAFhiL72LwRITUv2cwYMHqxpXfVVUVDD317nIFjDFBPa/pySBuZWbssNVzJs3j0cffVTrkOpFD+cNQmBZvXo1ANHtzi05G53QhZPbVrNlyxbdve801OLFi5k4cSJRLeK55cmXVC3B3H3IDaQe2cu2RbN44403+PDDDzUr+dy2bVuuuuoq1qxZQ8bJHJq10X8FqSPbTlBSUMrDDz9cr2pgWsvMyqxOotfk3RGXlZXl75B0p2YyvanvWL+YYJ7T161bR15uLq7mnXG17adtMIqCaedvLFq0iJdffhmr1XetQeprw4YNGM0WHC3005fZZLUR3qINO3ftoqSkRLXd1wA33HADbdu2Zc/K+Qy++/8RFnnxTR16dWD9Ugqy0njooYdUq9i4d+9eli9fTov2sbTurM8qKPYwT6/1HSv2M2vWLO6//36tQwJgwYIF7Nixg5CESCxR6pxfhHeOpWBvJpMmTeKuu+6iRQvfJ/C1VFpaisvlwmg689lfOv11U5nXG7xjfd26dXTp0oWhQ4fyzDPP8Morr/DMM88wdOhQunbtyvr1630ZpyA0WaLXUvDy9lbX2251L7PDSkjbSPbs2cPmzZtVG+eBBx4ABSpSA7uISmWGhFIFo0aN8knPPL3Ms96yRVarlWuuuYYtW7Zc9PFut5tdu3bRv3//c743YMAAjh49etHVixUVFRQWFta6CYIQ3JryjvWdO3cCYIm+9AUq6+nk+/bt21WNqSFWrFhBUWERpmaB2Vv9bKZYBUmGOXPmaB1KnenlvEEIPJ4daBLRbTuf872Ydt0AWLVqlZ+jUseBAwd45ZVXMNvs3PX3d7HY1V/cfM2DT9G6a28WLlxYXa1MK48//jgA25ft0zSOulDcCtuX78NgMPDII49oHU6dFRYWUlpSijHk3F6y3vvS09P9HZbuFBQUIJnOfC3U1hTm9Hnz5gHgjmmncSSAJOGOSaC0tFTVio2Xkpuby7Fjx3C0TEA2GC79BD+KbN0et8tV/dlFLQaDgT/84Q+4qpxsWzhT1bHUoigKG3+bjsFgVG2BrqIo1dUgr7ylt65zB32v7YbZamLs2LGUlJRoHQ4lJSV8/PHHSAaZ6P6tVRtHNhmI6t+KiooKRo8erdo4WsnLywNArnH52zuve78X7BqUxdi7dy833ngjhw8f5vrrr+edd97h22+/5d133+WGG27g0KFDjBgxorq0oSAIDdeUd3EFs+TkZBYtWoQ52o6tpX57rURe5kn6q3kR6OabbyYqKorKVBmlSrVhVKUoUJFswGw2V5dZbAw9zLNms5l77rmHzz//nF9++YW3336b3bt3c/XVV180qZObm0tFRcV5V2N670tNTb3g89977z0cDkf1Tct+lIIg+IfL5dI6BE04nU62b9+OOdKGwXLpiiAGqwlThJXtO7ZTVaWvCXPx4sUAmOOCY5GEbAJjlJsjR45w7NgxrcO5JD2cNwiBqaCggK3btuFo2RbzeZLMjlYJmKw2li9fEfCfS7Ozs3nqqaeoqKjgtr++TnSrBL+MazAaufO5t3DEtmDMmDEsW7bML+OeT//+/enTpw/HdiWTnaLvi55HdyWTm17A7bffTvPm+lyIfj4pKSkAmELPk1g/fd+pU6f8GpMe5eblIpkVJEPTuQBfV01hTi8tLWX58uUo1nAUe4TW4QDgivbsEJ8/f75mMezatQsAR8sEzWK4EO8OerUT6wC33norMbGx7Fg6h4qywOvVnLRrE9nJx7j11ltUm78SExPZvHkz7Xq21l1v9bPZQq30va47ubm5TJgwQetwGDNmDJmZmURc1hxTqLplU0PbRWFtFsqSJUtUb6Xgb962NlKNxLp8+tSnqVTmaVBi/c0336SyspL58+ezaNEi/vWvf/Hoo4/y4osvsnDhQubPn095eTlvvvlmowN85513kCSJnj312StCEAShISZPnozb7SayV3Ndryy0RIdgaxnO+vXrVfvgZrFYePTRR1GqoCJVvz+Li3FmSrjL4Z577rlk//G68Oc8eyGDBw9m5syZPP7449xxxx3861//YsOGDUiSxEsvvXTB55WVlQGef9ezeUuqeR9zPi+99BIFBQXVt+Tk5Ea+EkEQ9K6pJtZ37NhBRUUFthZhdX6OrXkYpSWl7NmzR8XI6qe8vJzVq1cj2xUM6lWG9DtTrCeJuHTpUo0juTQ9nDcIgWnVqlW4XS5iL9DLVZYNRCd0JS0tlUOHAqc399kqKip49tlnycjIYNjv/4+O/fxbStkeHsndz7+H2WrjX//6FwcOHPDr+F6SJPHXv/4VgM0Ld2sSQ1243W42L9yNwWDgySef1DqceklKSgLAFH5uKWnvfd7HNFVVVVUUFRYhmxQkk0JuXq7WIelKU5jTExMTqaiowBXdBt2UOrKF47ZHsGbNGs36A1cn1pvrb3OBNyZvGys1mc1mHvj976koK2HfmkWqj+drWxfPAuDhhx9W5fjl5eW8//77yAaZQbf3UWUMX+s9tAthUSFMmTJF0zlw165dTJ8+HXOEjcie6i/akySJ2EFtkQwSb771li527PtKWloaALL1zMJb2eL5uqlU5mlQYj0xMZF7772Xm2666bzfv+mmm7j33ntZsWJFo4I7deoU7777rqq9OwRBEPwtOzub2XNmYwq3EtImUutwLinidKn6SZMmqTbGfffdh8PhoOKUAbe+NuFdkuKG8hMGTCYjjz32mE+O6a95tr46duzIyJEjWbFixQUTYTabDfBcwDxbeXl5rcecj8ViITw8vNZNEITg5nQ6tQ5BE2vWrAHA3spR5+d4H+vtiawHO3bsoLy8HFN0YO9mPZspSgHJU45V7/R63iDo3/LlywGI7XD+xDpAzOnveR8baBRF4Y033mDXrl30HHozV9yqTX/P2DYduO2vb1BRUcHTTz9dvdPH36688kr69+/P8T2nSE/SJoZLObQlibyMAu6++25at1avTKsaDh8+DIA54tzEusFixGg3B/QiFV/w7lCXzJ6dbrk5uQFfEcOXmsKcvnDhQgDc0W00jqQ2d3RbnE6nZvPd3r17AQiL09/7nskWgs0Rze7du/3y93rPPfdgMpnYvnROQL0/FGSlcXzHBvr27UvXrl1VGeObb74hNTWV3sO7EhEXGNfLjGYjV915OVVVVbz33nua/Js6nU7eeOMNFEUhdnBbJIN/2pGaI2xE9GpBZkYGY8aM8cuY/uCt0CPX2FMlW2t/L9g16DeooKCAdu0u3gOlXbt2je6T849//IOBAweet0+rIAhCoJo+fTrOSicRPZohyTpZnXsRtuZhWKLtLFmyRLWydSEhITzxxBMoTqhIDqxe65XpEu4yGDXqvvOWP28If82zDREfH09lZeUFV1pGRUVhsViqVy/W5L2vZcuWqsYoCEJgqays1DoETaxYsQLZKGNtVr8d65JB1tXF1E2bNgFgjAici151IRnBEKqwc+dOSkv1XYZSz+cNgn6Vl5ezZs0aQqLisEfGXvBx0W27IBuMmpYwb4zJkyczb948Wne5jBv/8A9Nq4V1vHwIQ+9/kvT0dP7+979rsrBMkiSeffZZADb8tkN3CYsqp4vNC3djtpj585//rHU49eat8maOsp/3++YoG2lpaeTn5/sxKn3JzMwEPCVjZbNCZWUlhYWFGkelH8E+pxcVFbFm7Vrc9giw6Ssp6I7y7Mr2Jv79Orbbza5du7BHxmKyXngjgpbCm8eTn5/vl3YWUVFRXH/99WQnHyP9mDZVXhpi98r5KIrikxaR53Py5EkmTZpEaISd/jcEVnXnhJ6taNu9JevWratuI+ZPU6ZM4ciRI4R3jcUad277IzVF9mqOOcLK9OnTqytTBLqTJ08CINvPnEdKBs/cfuLECa3C8qsGZS9atmzJhg0bLvqYjRs3NurC+apVq5g5cyafffZZg48hCIKgN+Xl5cyYMQOD1URoh2itw6kTSZJw9GiO2+1m2rRpqo1z//33E9csjsoUGfe5m511SXFBxQkDVpuVJ554wmfH9cc821DHjh3DarUSGnr+E1FZlunVqxdbtmw553sbN26kffv2hIXVPYkkCELwO1+Fi2B39OhRjh8/jq2VA9lY949kssmArWU4hw8f1k0pWe/FAaNDX8kZXzA6FKqqqjh48KDWoVyUns8bBP1av3495eXlxHbocdHHGc0Wotp04uDBgwG3A2XdunV89tlnhEXFMfK5tzCazu177W8DbnuA7kNuYPv27bz//vuaxNCnTx+uueYaUo9mknzw3MWwWtq77jBFeSU8+MCDxMXpu2/s2byJMVO4FYPFeN7HWGI9FTmD5cJ6Q3hLxEoWBcla+z4h+Of0xMREnJWVuKP0tVsdAGso7pAo1q9f7/eFC8eOHaO4uBhHCx3+XE7zxrZjxw6/jHfnnXcCsHe1/xc6NISiKOxdvYiQkBCuv/56VcYYPXo0lZWVXHXX5ZguMM/olSRJXHVXfwxGAx999FF1RUt/SEtLY9y4cRjtZqL7+b8ihGSQiR2cgKIovPPOO0HRCu/YsWMgn9ml7iXb3KSkpDSJzRMNSqzfcccdJCYm8tprr53zR1BeXs4bb7zBihUrGDlyZIOCcrlcPP300zzxxBP06tWrTs+pqKigsLCw1k0QBEFv5s+fT0FBAeFdYup1IV1roQkRGO1mZs2apVpPGKvVyrPPPIvigrLjgfGzKT8p466EJ/7whE96q3upPc/WRVZW1jn37dy5k7lz53LjjTciy55/o5MnT57TJ/Lee+9l8+bNtZLrBw8eZPny5aqt3BUEIXA1hQ9dZ5s/fz4Aoe3q3xLG+5wFCxb4NKaGUBSF/fv3IdsVJIPW0fieIcyzWGD//v0aR3JxejhvEAKPt/LFpRLrnsd0r/WcQJCens4LL76IbDBy59/fIcQRpXVIgOfC8og/vkhcQid+/PFHfv31V03iePrpp5EkiY3zdupm17qzwsm2pfsIDQ3lD3/4g9bh1Nvhw4cpKirC2uzCO+Fsp3fJnW8RclPh3e0qW8/0Y/XHDthAEexzuvf81R2tvz7i4ClP73K5WLp0qV/H3bp1KwARrS5erUBLES09sXljVduAAQOIiori4MZE3G79JyLTj+6nICuN66677qLtDxtqw4YNJCYm0qpTM9r10l+7gLpwxITSe3hX0tPTmTp1qt/G/eijj6ioqCCqfytkszYfWq1xoYR1jGbfvn3MmTNHkxh8RVEUjh49imxTOLsQlBziWWh4/PhxbYLzowZlLl577TXatWvHu+++S5s2bbjtttv4wx/+wG233Ubbtm156623aNeuHa+99lqDgho3bhwnTpzgrbfeqvNz3nvvPRwOR/UtPl6fE7QgCE3bjBkzQJII73Lhcot6JMky4V1jKS0tZd68eaqNc9ttt9G1a1ecGTJVRaoN4xPucqhMkYmLi+WRRx7x6bHVnmfr4r777uPWW2/lnXfeYcKECfztb39j8ODB2O32WrtrHnnkEbp161bruX/5y1/o0KEDt956K6NHj+azzz7jhhtuoFmzZjz//POqxSwIQmCqqqrSOgS/crvdzJs3D9lkwN46ot7PD4mPQDbK/Pbbb5onQ3JycigsLMIQoo+kjK8ZTpe20/uFAT2cNwiBxeVysXLlSiyh4YQ1a3XJx8e07w5IAZNYr6qq4oUXXqAgP5/rHn2WFu3V6XPaUCazhTufextrSChvvvmmZ9ePn3Xq1Ilbb72VrFN5JO3RRyWC3WsOU1ZczmOPPYbD4dA6nHrz7jK2tbhwdS5LbCiSQWb9+vX+Ckt3vHOqwaZgOF0xXy9VePQgmOf0/Px81q1bhzskCqz6rGLnLQfv7wWs3veEyNYd/DpufYTGNsdks7Nu3Xq/fAYxGo3ccMMNlBTkknpor+rjNdahzasAGDFihCrH/+qrrwAYMrKfpm1tGqvfdd2xhVmZPHmyahu3atqzZw+LFy/GGhdCaDttF1lGXd4a2WTgyy+/9OuOfV9LT0+nuLj4vNcAvPcdOnTI32H5XYMS69HR0WzYsIFHH32U4uJi5s+fz7fffsv8+fMpKiriscceY8OGDURF1f+XNScnh9dff53XXnuN2Ni6J55eeuklCgoKqm/Jycn1HlsQBEFNe/fuZd++fYS08ez+DjRhnWKQZIkff/pRtTFkWeaFF14AoOyoAZ1snjivsiQZxQXPPfc3n69GVXOeras777yT7OxsPvnkE/7yl78wY8YM7r77brZs2XJOIv1sYWFhJCYmMnToUN5++21ee+01evfuzcqVK+s1twuC0DRonRz2t61bt5KSkkJIQmSDqtfIJgP2tpGcPHnSb6UYLyQ1NdUTk/USDwxQ8unp3fs69UoP5w1CYNmzZw+5ublEJ3RFki79PmS2h+JoEc/WrVspKtL56ldg/PjxbN++nW6Db+Cya27XOpzziohryc1/eony8nJeePFFTaq3PPnkk8iyzOZFuzWfi50VTnas2I/DEc6DDz6oaSwNtWqVJ6lib3nhvtGyUcbaPJQDBw6QkZHhr9B05fDhw0iyZ471XoDXe8sVfwrmOX3x4sW4XC7c0W21DuXCLCG4w2LZtGkTmZmZfhmyoqKC9evXY4+MxaaT6irnI0kyUW06k5aWypEjR/wy5rXXXgvA4a2r/TJeYxzZtgabzcaVV17p82Nv376d7du3065na2Ja1b/imZ6YrSZ6D+1CUVERP/30k+rjffHFF4Anqa31ggSjzYSjWxxZWVmejXcBylu19GKJ9aYwrze41m5MTAyTJk2ioKCAnTt3snr1anbu3ElBQQHffPNNg0vivvrqq0RFRfH000/X63kWi4Xw8PBaN0EQBD355ZdfAAjv5LuS4f5ktJmwxzs4eOCgqhPkFVdcwXXXXYerQKIqR5+rMKuKwJkh06NHD2699VZVxlBrnq2rZ555ho0bN5KTk4PT6SQ1NZX//e9/dOzYsdbjEhMTz3shrnXr1vz0008UFBRQVFTEr7/+es5zBUEQgOrWEk3F7NmzAQjrGN3gY4Sffq73WFrxtg2RzcG5OEIygGQ8f3sUvdH6vEEILKtXey5Qx7S/+GLJmqLbdcPlcrFu3Tq1wvKJ3bt3M378eBxxLbjx8ec1v4h6MZ2uGEqf60Zy8MABxo4d6/fxExISuOmmm8hOySPlsLZJ3gObjlFeUsFDDz1MSEiIprE0RF5eHlu2bsEaF4LBarroY0PiI4DAaq3gK5WVlezfvx85REGSQbKAZPL83QpnBOuc/ttvvwES7mj99hEHcMe0RVEUv+1aX79+PaWlpXVqzaI1b2uYJUuW+GW8/v37Y7PZOL5zo1/Ga6iCrDRyUk4wcOBALBaLz4//888/A9B7uL4q8DRUjyGdMJoMzJ49W9WFfQcOHGDt2rXYWoRha6aPKhmOHs2QzQamTJmC0+nUOpwG2bdvHwCG83S+8d63d6/+q0w0VqOvYplMJnr16sWQIUPo1asXJtPFTyAv5vDhw4wfP55nnnmG1NRUkpKSSEpKory8HKfTSVJSErm5uY0NWRACip4vBAh1V1VVxYIFCzDYTNgusoJd78I6eD7AqVkOHuC5555DlmXKj8u63LVefswzfT7//POqJ4R8Oc8KgiDokcEQhM25L6CgoIBFixZhclixxl24B+ulWJuHYQq3sGDBAoqLi30YYf0UFhYCnoviwUoyKhQUFGgdRp2J8wahLlavXo1sMBAVX/dFjzEJXQBYs2aNWmE1WkVFBa+88gput5tb/vwKFrv+E7TDH/wrkc1bM2nSJE2Si48++igAOxL3+31sL7fbza5VB7FYLNx///2axdEYy5Ytw+1yE9L20jsJQ9pEALBw4UKVo9KfXbt24XQ6MYZ7PuRLEhjC3Zw6dYr09HSNo9OfYJrTk5OT2b59O25HMzD7vv+0L7mj2oAk88svv/ilmodnwQE069xb9bEaK6ZdNwwmM7/Nm+eXn43ZbObKK68k+9RxinL8U0GgIY7v2gTAVVdd5fNjO51OlixZQlhUCC3aB0cFSIvNTELPVhw7dkzVkuFTpkwBIKJXc9XGqC+DxUhYpxiysrIC9jxg586dABjCzn0PkAwghyjs2bMn6Fv+NSobUFFRwezZs3n99dd59tlnef3115k9ezYVFRUNOl5KSgput5tnnnmGdu3aVd82btzIoUOHaNeuHW+++WZjQhZ0yB/9NAKZ1iXZBN/YunUr+fn5hCZEIsmBu1jC3ioc2WxgyZIlqv5uJiQk8Lvf/Q5XqYQzQ18/L2eeRFW+zNChQ7niiitUHcvX86wgCIIeGY1GrUPwm3nz5lFZWUl455hGLZ6UJImwTjGUl5czf/58H0ZYP9W94YK56IAM5eVlWkdRJ+K8QaiLnJwc9u3bR0Sr9hhMdW9PFRrbArM9lDVr1ur2M+qECRM4fvw4/W66l/iu+k9QAJitNm7+00u43W7eeOMNv+9e6t69O5dffjknD6RRlKvNtZmUwxkUZBdz++23ExERoUkMjTV37lyQqFP/VqPdjK1FGFu3buXUqVN+iE4/vBUvjJFn3kNMp79uyn3nzyfY5nRvlSV3TDuNI6kDoxlXZCsOHz5cvTNTLbm5uSxbtozQmOaExrZQdSxfMJjMxHXsRfLJk2zZssUvYw4aNAiApD3+Ga8hTuz2xDZ48GCfH3vv3r2UlJSQ0KNVUG2+S+jRGoBNmzapcvzc3FwWLlyIOcKGrYW+Nrg5usWBBNOnT9c6lHpzOp3s2LkD2a4gX2Ctl9GhUF5ezv792i3a9IcGX8WaO3cuf/rTn8jKyqr1oUqSJOLi4hg/fjy3316/XlY9e/Y8bznDV199laKiIj7//HM6dOjQ0JAFHZo/fz4vvvgiL7/8Mr///e+1DkcQVJOYmAicWZ0eqCSDjL2Vg1PHT3Hs2DFV35P/+Mc/8vPsnyk/Aaa4KurQ/lF1igLlSZ5AnnrqKVXHUmOeFQRB0KNA3n1TH4qiMHPmTCRZIqxDw8vAe4V1jCFveyo//fQTo0aN8kGE9Vc9PwXPNZ7z0mkOsRZx3iDU1dq1awGIPr0Dva4kSSa6bRfS9m/l4MGDdO2qr3Kkx44d45tvviE8pjlX/+4JrcOpl9ZdL6PP9XeyY+kcvvvuOx577DG/jn/PPfewdetWDmw6xhU39fLr2AD7Nx6rjiMQHTlyhO3bt2NrFY7RXrfFKmEdYyhLK+Lnn3/mmWeeUTlC/Vi1ahWSDMaIM/OUMcrzdWJiInfddZdWoelKsM3pVVVVntaIRjPuqNZah1Mn7tj2GHKT+fnnn+nRQ70S7T///DNVVVW07HVlwCRNW/W6krT9W5k+fbrqm00ABg4cCEDS7i30GnaL6uPVl9vt4uS+bbRu3ZrWrX3/++3dHdyyfZzPj60l7+777du38/DDD/v8+L/88gtVVVXEdGmpu78tU6gFe+sIdu/ezf79++nWre6tmbS2a9cuykrLMLe68AdkY4RCZapnwVyvXv4/r/SXBqUpli1bxj333EN+fj6PP/44kydPZsGCBUyePJnHHnuMvLw87r77bpYvX16v48bExHDnnXeec4uJiSEsLIw777wzqP8xmqKlS5cCZ3qFCOcK9rIZTcW6deuQTYZGlX3VC3trB3DmopxamjVrxn2j7sNdDs4sfZwEuQrAVShx7bXXqnrio9Y8KwiCoEdNZcf6rl27OHz4MCFtIy/Zf7UujDYT9vgIDhw4oFkPs+p/O7cmw/uHov92BeK8QaiPVatWARCdUP/EeHQ7z3NWrlzp05gaS1EU3n//faqqqrj+0ecwW+1ah1RvQ+/7EyGOSMaOHUtGhn/7nV9//fXYbDaO7Djh13EBnJVVJO05Rfv27VVNXqnJu+PM0bXuSQ/PuYCRmTNnnqn+EuROnjzJwYMHMUS6kWpMqwabp2zs2rVrKS0t1S5AnQjGOX316tVkZmbiim4Lsr7PqbwURzMUSwi//fabar+XlZWVTJs2DaPFSotul6syhhrCW7QhrFlrli1b5peqG+3atSMuLo4Te7aguPX3oSPj+CHKigurd9b72tGjRwGIbhWhyvG1Ehphx2I3c+zYMZ8f2+128+NPPyEbZUI7XLqSjBYcXTwLC3766SeNI6mfFStWAGCKunBi3RSpgHTmscGqQYn1N954A5vNxpYtW5gwYQKPPPIII0aM4JFHHmHixIls2rQJq9XKG2+84et4hSCjtxVDeiRK5Qe+nJwcjh07hrVZKJJBB9uuG8nWIgzwlLdX26OPPorBYKAiWR+91suTPf9+Tzyh7i4YMc8KgtCU6D1p6SveD83hnWN8dkzvsbT6QB4a6lkwqLg0Gd4/XBJhYWFaR3FR4rxBqKvKykpWr1mDPSIGe2T934ui23ZGkg26u1C2evVq1q9fT/s+A+l4+RCtw2kQa0gYQ+//M2VlZfz3v//169g2m42rrrqKvIxC8jIL/Tr2qUPpVDldXHfddQF5fSg7O5s5c+ZgCrNgb+Wo8/Nko0x451jy8vI8O3mbgAULFgBgjj33g705xk1FRYXu3lu0EIxz+o8//giAOy6AqtBKMq7Y9pSWlqrWdunXX38lKyuLVj2vxGi2qDKGGiRJom2/objdbiZPnuyX8YYMGUJpYR4ZSYdVH6++ju/cCKhTBh7gxIkTyLJMeFTgb9SqSZIkImLDOHnyJG4fL5jYsGEDp5KTCWkXhcGsz0X8tlbhmEIt/DbvN4qLi7UOp07cbjeLFi1CMtauPHM2z/fd7NmzJ6hb3jQow7N9+3buu+8+evbsed7vX3bZZYwaNYpt27Y1KjivxMRE9uzZ45NjCfriLWmk1x5tehAob67ChXnL9gTDbnXw9IQzhprZsWOH6n+7zZs35+abb8ZVIlGVr+2FFlcpVOXKXH755apXT/H3PCsIgqClQLyQXl8FBQUsWLAAk8OKtbnvkrS2luGYwizMnz+foqIinx23rry9cJVKvw/tF4oCbie67/krzhuEulqzZg2lJSXEduzZoPdeo8VKVJuO7N27l+TkZBUirD+3282nn36KLBu45sG/ah1Oo/S8+ibiEjoxd+5cDh/2b/Jg2LBhACQfSPPruN7xhg8f7tdxfWXy5MlUVlYS0bM5kly/vylH9zhko8zEiROprAzSifQ0RVH47bffkAxgij73GoKpmee+X3/91d+h6U6wzenJycmsXbsWd1gsij1C63DqxR3bHiSZH36Y4fNrX06nkwkTJiAbTcT3u9qnx/aHuE69sEfE8PPPP/ulysrQoUMBOLp9nepj1deRbeswGo2q7VjPyMjA7rAiB8FGrbOFRoTgdDrJy8vz6XG///574MyucD2SJImwzjGUlZYxZ84crcOpky1btpCeno4p1n3Jdq3mJjCvN+gv0m63Ext78V/MuLg47PbAK78lCHojEuuB78CBAwBYYoLnPdESE0Jubi5ZWVmqj/XAAw8AUJmqbeKlMs0zZXrjUZOYZwVBaEqawgLLOXPmUFlZSXjnWJ8uJJAkibAusZSVlTF37lyfHbeuvH0E3eXBuThCqQAUVOmX6EvivEGoK+/7RLMufRp8DO9z9XKhbMmSJRw5coQeQ28iulWC1uE0iiTLDL3vSRRFYezYsX4d29snN/Vopl/HTT2aSUiIne7du/t1XF9IT09n2vRpmEIthHWMrvfzDVYT4V3jSE9Pr97RG6x27NhBUlISxmg30nk2DxpsYAhXWL9+Penp6f4PUEeCbU7/8ccfURQFd7OOWodSf2YbrsjWHDx4gB07dvj00PPmzSMlJYVWPQdgCdF3ZaTzkWSZhAHX4nQ6mTRpkurjDRo0CLPZzKHN+mpFU5idQfqx/Vx55ZWEhIT4/PiKopCZmUlIuM3nx9aD0AjP68rM9N25x9GjR1m1ahXWZqFYYnz/b+JL4V1ikY0Gpk6dGhCtgGfOnAmAudmlKwyYYhQkI8yaNQuXKzjL2zUosX799ddX98a+kKVLl3LDDTc0KCih6WkKO5Xqo+YF5sJC/5ZiE3zPu9vAHBkYH3zqwhLpOfk5dOiQ6mP17NmTbt264cyRcWu0kF9xgzNDJjo6mmuuuUb18cQ8KwhCUxLsifWqqiq+//57ZKPcoAvvlxLeMQbJIPP999/7/UNrixYtMJlMuEqC81ze+7ratm2rcSQXJ84bhLpIT09nRWIiYXGtCItt0eDjxHXsidFiZebMmTidTh9GWH+KojB58mQkWWbQyEc0jcVX2l02gBYdurF06VK/VgVo2bIlzZs3J+NEtt/GrCx3kpteQO/efTAa9Vmq9WL++9//4qx0Etm3ZYNbvkX0ao7BbGTcuHEUFBT4OEL98C4csLS48MV4cws3brebWbNm+SssXVJrTt+8eTNPPfUUPXr0ICQkhDZt2jBq1ChVr+mUlZUx6+efwWTFHanvRYoX4l0QMH36dJ8ds6qqivHjxyMbjLTtP9xnx/W3Zl37YHNE89PMmarvWg8JCeHqq68m6+RRclKSVB2rPg5s9LSvuPHGG1U5fmFhIU6nkxBH8FxPrsl+esGALzdtff311wBE9Gzus2OqxWAxEtYpmrS0NN0sWL2QrKwslixZghyiYAi/9OMlA5iaucnIyCAxMVH1+LTQoDO/jz76iMzMTB555JFzTvSTk5N5+OGHyc7O5qOPPvJJkILQ1JSVlVV/rUVZT8G3kpKSMFiMGKyBd7HgQkwOK+Dp9aM2SZK46667QIHKTG0u3DtzJNxOGDlyJCaTSfXxxDwrCEJTEuyJ9cWLF5OWlkZYpxgMFt+fCxisRsI6RpOcnMyyZct8fvyLMZlMdOnSBVeJhOLb1ni6UFXkOe+4UDlWvRDnDUJdTJkyBbfLRXzfqxp1HIPJTMueA8jKyuK3337zUXQNs2fPHvbs2UOn/lcT0aylprH4iiRJ9L/lPhRF8fsu5m7dulFSUEZpUblfxstOyaseN9Ds3r2buXPnYom2E9o+qsHHMViMRPRuTkFBAePGjfNhhPqRkZHBwoULPRfjL9KG3hyrIJlgxowZlJf753dQj9Sa0z/44ANmzZrFddddx+eff86f/vQnVq1aRb9+/VRrv7pgwQKKCgtxxXUA2aDKGGpTTpewX7x4MdnZvll4NH/+fJKTk2nZawCW0DpkqHRKlg20u/I6nJWVfum1fssttwCwZ9VC1ceqC0VR2LNyPmazmeuuu06VMbwJZ3u4VZXja83XifWDBw+yYMECLDEh2FtfZMLRkYhezZGNMmPHjtV1W5jp06dTVVWFpZWbuu6PtbT0XCCYOnWqipFpp06J9WuvvbbW7eGHHyYyMpLvv/+eDh060LFjR4YMGULHjh3p0KED06ZNIyIigocffljt+AUhKNUs/15SUqJhJEJjKYpCcnIyxjBzUFVmMIX5L7EOnhNok8mEM0ObnkKVGZ5/u5EjR6pyfDHPCoIgBCeXy8XXX3+NJEs4ujdTbZyIHs1AgnHjxuF2+zfD3adPH3CDKwiLLFXlS8iyrLvEujhvEOrLW2ra5oiiWefejT5em75XIxtNjB07loqKCh9E2DDexH7va+/QLAY1dL5iKLbQcObNm+/XSiRdunQBIDc93y/j5aZ5xunUqZNfxvMVl8vFO+++A0D0gPhGf853dI3D5LAybdq06mp3wWTy5MlUVVVhbX3xi/GSAcwt3eTl5TWpXev+mtP//ve/c+LECcaMGcMTTzzBq6++yurVq6mqquL999/3+etSFIUffvgBJAlXXACWgfeSJFzNOuJyuXzye+l2uz291Q2GgN6t7tWsSx9sjih+/OkncnNzVR1r+PDhOCIi2LNqAa4qbSvmAKQd2Uf2qeNce+21OBzqJHG9JdJDwoNzx3qIw3eJdUVR+OSTTwCI6tcyYK7BG+1mwrvGkpaW5tPKGL5UWlrKDzN+QDad6Z1eFwY7GKPcbNu2jd27d6sYoTbqtGXiYtv1q6qqOHbsGMeOHat1/86dOwPmF1gQ9KbmxQktL1QIjZeTk0NFRQUhLSK1DsWnjGFmANLS0vwynsPhYOjQoSxbtgxXCRj82CbH7YSqXJkePXrQvn17VcYQ86wgCEJwmj9/PseOHSO8cwymMItq45jCrYR1iObw4cMsWrSIm2++WbWxznb11Vfz3Xff4cyRMUYEz7Z1txNchRJ9+/RW7WJZQ4nzBqG+Ro8eTWVlJT2uvRHZ0Phdg5bQcOL7DOHElkQmT57Mk08+6YMo60dRFFasWIEtzEHbHv38Pr6aDEYTna4Yxq4Vv7J//36/Le5JSEgAID+ziNad1C+hmp/lqc7Xrl071cfypVmzZrF3z15CO0Rja9b43siSQSZmQDxpSw7zzjvv8O233wbN+/WpU6eYMeMHZBuY4i59Md7Syk1liszX479m5MiRhIaG+iFKbflrTh88ePA593Xq1IkePXqwf//+eh2rLvbu3cv+/ftxR8WDObD7Q7ujE+DkTn6aOZMnnngCQyPm0ZUrV5KUlETLnldgDdXX+WVDyAYDbfoN5eCKOfzwww/85S9/UW0ss9nMXXfeyeTJkzm0eRXdBqmzS7yuti35GYBRo0apNkZOTg4Q/DvWfVENYvXq1axbtw57q3DsLQPrbyuiVwuKjuQwbtw4br/9dqKiGl4JRw0///wzRYVFWBNcSPXc72aNd1OcKzNp0iQ+/fRTdQLUSJ1+FG63u0G3YG1ML/hOsHxY8LWaveqqqqo0jERorPT0dACMIWaNI/Etg9mIbDb4LbEOcNtttwFQ6edd684sCRS49dZbVRtDzLOCIDRlwXo+WFpaymeffYZslInsrX6J4sjenh6vn376qV9LqPbv3x+73Y4zWyaYqvo7czzz/7Bhw7QO5RzivEGoj2XLlrF48WIcLRNo1qWPz46bMOBaLKEOvh4//pykjz+kpKSQlpZGmx6XIxuCp+WWV7vLrgBg06ZNfhvTm1gvyPZPOzrvOG3btvXLeL6Qm5vLZ59/hmw2EN3fdz2j7a0chLSNZOvWrbrvs1pXiqLw4Ycf4nRW1flivGwCS7yLvNw8vvrqK/WD1AEt53RFUcjIyCAmJuaCj6moqKCwsLDWrS5mzpwJ4CkDH+gMRlzRbclIT2fdunWNOtSMGTMAiO97tS8i04UWPfpjstr46aefal3PVsOoUaOQJIlti2aqOs6lFOfncHDDCjp06ED//v1VG8ebcLaFBmli/fTramy1A6fT6WmPIUlEXxHvi9D8ymAxEtmnJcXFxbqb+5xOp2fBnwHMLev/Yd/gAEO4wrJlyzT5vKAmbWrqCoJwUcF6gbkpCtbEOnjK1aSl+y+xfvXVVxMaGoozy78X7iszZGRZ9uvuP0EQBCHwgssOhgABAABJREFUTZw4kczMTBw9mvnlPMAUZsHRPY60tDQmTZqk+nheZrOZG2+8EXd5cJWDd2ZISJJU3c9REAJRZmYmb7zxb2SjiW7X3+vTz5lGs4Wu192Fs7KSF154we+V1o4cOQJAi/Zd/TquvzRv7+k77n2d/tCqVSsAinL9046uKLeEiIgIQkL8WI6skT7++GOKCouI6tsKo83k02PHDIhHNhr46KOPKCgo8OmxtbBo0SJWrFiBMcKNKbbuH+AtrRVkG3z//Xfs3LlTxQiF77//npSUFO67774LPua9997D4XBU3+LjL520Ki8vZ+HChSiWEJRw9Voh+ZP79AKBOXPmNPgYmZmZrFu3johW7QiNDo6fC3iqrDTv2o/s7Gw2bNig6ljx8fEMHz6clEN7SD2yV9WxLmb7ktm4qpw89NBDql7D97aKtdiD75oygPn0PFpU1LgFfT/++CPHjx8nvEss5ojArJAR3jkWc4SVn376iUOHDmkdTrVffvmFzMxMzC3cyA047ZEksLZxoygKEydO9H2AGhKJdUHQIaPReN6vhcDjTaybgjGxHmKisKCQsrIyv4xnsVi4/vrr/Xrh3jOWxIABAy66ilsQBEFoOH/3BPeHI0eOMGnSJEyhFiJ6qV9O1yvyshYYQ8xMnDjRryvC77jD09+4Ii04Pl66SqEqX2bAgAG0aNFC63AEoUGcTifPP/88BQX5dBp6GyFRsT4fI6ZdN1pdNoiDBw/yzjvvoPhx9WtKSgoAEc1a+W1MfwqPjkM2GDl16pTfxnQ4HNjtdopyi1UfS1EUinJLaNlS/YouvrJp0ybmzp2LJdpOeBff/z0ZQ8xE9m1BXl5ewJdLTUtL4z9v/gfJALbOF++tfjZJBnuXKtxuhX/961/ViSXBtw4cOMBf//pXBg0axKOPPnrBx7300ksUFBRU35KTky957JUrV1JSUoI7ui31+sfXMcUegWJzsCIxscG/k8uWLUNRFJ9Wj9GLZl37ArBkyRLVx3r44YcB2LLgJ9XHOh9nZQU7l87BERFRXVlTLd4qZCZzcOYGZFnGYJQbVW2ttLSUcePGYTAbieoTOOcUZ5Nkz257t9vNf//7X63DATxVlCdOnIgkgyW+4ddsjFEKhlCFefPmcfLkSR9GqK1G/VWeOnWKFStWkJqaet7VyZIk8dprrzVmCCHI+fODdyCxWM704LRag7PcS1PhLZVuCMrEuuc1paen+60v3q233sqcOXOozJQxOtRPxFRmStXjakHMs4IgNAXB1vbG5XLx+uuv43K5iB3YHtnY+H7GdSWbDMQMbEP6siP8+9//ZvLkyciy+snu/v370759e44nHcPd3o0c4Kc9Famen9nFdnDpkThvELwUReE///kPO3bsoHnXvrTqdaVqY3UaeitFGcnMnj2bTp06VV/wVpt3d5PFHji7netDkmUs9hBKSvyzexw87xEtWrQgJV39ZH55SQXOyqqASaxXVFTw5ptvgiQROzgBSVYnWejo1oyio7nMmjWL2267TdUSw2qprKzk+eefp7ioGFsXF4YGbB40OsDSxs2pk6d4/fXX+fjjj5tcZUc15/T09HRuvfVWHA4HM2fOvGjPcIvFUusaZV14k6vu6MBp83BJkoQrug3OU7tZtWpVgyoabdy4EYCYhC6+jk5z4c1aYbKFsGHDRtXH6t+/P926dePgxkQKf59BeIx/d//vW7OI0qICnnzySdWv2ZtMni3CrqrgbOekKAquKjdmc8M/PE6bNo38/Hyi+rXCYA3sBQi2luFYm4WSmJjInj176Nmzp6bxzJs3j5SUFMytGvf5XpI8c3rpPomJEyd6zqeCQIN/2/75z3/y+eef1+rpoihK9YmO92vxwV24mKZ2YlxXNSfm+p7ACvriTaybQgP8CvN5GEM9v5tpaWl+S6xfccUVxMbGkpOVhdLBXac+bQ2lKFCZKWM2m7n++uvVG+gCxDwrCEJT4e/ywWr73//+x+7duwntEE1Ia4ffxw+JjyC0XRTbt29n+vTpPPjgg6qPKUkSDz74IG+99RYVKTK2doFbhcDtBGe6TLNmzbjmmmu0DqfOxHmDUNPYsWP55ZdfCG8eT9fr71H1c7fBaKLX7Y+w5YcvGT16NHFxcYwYMUK18byqFw0F8WJ9xe32y+Komlq0aMHRo0dxVlRhsqh3gboor7R6vEDw9ddfc+LECRw9mmGJtqs2jiRLxA5uS8q8/fz73/9m1qxZAXVNSFEU3n33XXbv3o25mRtzs4b/fVoT3LgKPUnab775hieeeMKHkeqbmnN6QUEBN998M/n5+axevdrni1ucTidr1qxBsYai2P1/HqwmJbI1nNrNypUrG5RY379/P5aQcKzhkSpEpy1JknG0aEPasf0UFBTgcKj3by9JEg8//DAvv/wy25fMZtjv/6zaWGdTFIWti2ZhNBq5//77VR8vNDQUgPLSStXH0kJluROgwS1hnE4n//vufxgsRhzd4nwZmiYkSSKqbytSFx5k6tSpfPjhh5rF4nK5GD9+PJIM1kbsVvcyxSgY7Apz587lySefrG4/FMgadIY+YcIEPv74Y6655hpmzpyJoig8+uijTJ8+nT//+c8YjUZ+97vfsXz5cl/HKwQZsWP9/MSO9eCRlpaGZJCRVbwooRXvjnXv4gF/MBgM3HLLLbidUJWr7sIcVwm4SySuueaa6pNZfxHzrCAITYk/d+Op7dixY4z57xiMNhMxAy7dg1ItMVfGY7Ca+PTTT/1Wbu2OO+4gMiqSylQZJYCLEFSmSiguePTRRwOmJZM4bxBqmjZtGmPHjsUeEU3vO/4fBqNv+0CfjzXUQe+R/w+D2cJLL73EunXrVB+z+mJzSeP6cuqV2+2ioqzU7/3HvRc6C1UuB1+UU1xrPD3bv3+/p71LmIWovurvsLfGhODo3owTJ04wbtw41cfzpe+//55Zs2ZhCFOwdapfCfizSRLYu7mRrTBmzJgmM4epOaeXl5dz++23c+jQIX777Te6d+/u8/j379/vKQPvCIxFM/Wh2MJRzDY2btxU7+vZLpeLtLQ0bJHB22LQHuF5bf5oYTJixAiio6PZteJXnJX+W6R9ct92spOPcdNNN/mlXWTbtp6qD/kZfuqH6Wd5GQXAmddZXytXriQ3J5fQjtHIJv9ViVOTtVko5kgbS5cuJS8vT7M4Fi1axMmTJzE1dyP7YH2fJIGlrRuXy8U333zT+APqQIMS6+PHjychIYEFCxZw1113AZCQkMB9993Hl19+yeLFi5k9ezZZWVk+DVYQmgpvqRcQPdYvJhAWZqSmpmIMMQdldQbj6V343v6G/uLtYeQt064WZ4Zcazx/EvOsIAjBzul0Vn+dn5+vXSA+5HK5eO2113BWOokZ1BaDhovqDFYTMQPbUFFRwauvvuqXPvZWq5VHHn4EpQoqUgPzvEepgsoUAw6Hg3vuuUfrcOpMnDcIXrNnz+a9997DEhpOn7v+gNnuv8WhYbEtuez2R3Ep8Oyzz7J161ZVx/PusizISld1HK0U5WShuF1+L5UeH+9ZFFaYrW5iveB0Yt07nl45nU5efe01XC4XMYPb+q29S1TflpjCLEyaNIm9e/f6ZczGWrlyJaNHf4hsgZAeLiQf/KhkM4T0qAJZ4cUXXwiYn0VjqDWnu1wu7rvvPtavX89PP/3EoEGD1AifXbt2AaCExapyfE1JEu7QWHJysuu9waS0tBRFUTBZGtAbIUAYLZ6NYf5YNG02m7n33nspKy7k4IYVqo/ntWPZLwB+2a0O0KWLp21AelK2X8bzt4ykHAA6d+7coOcvWLAAgPBOwbNgRZIkwjrF4HQ6WbZsmSYxuN1uJkyYAJJvdqt7mWIVZJvCnDmzyczM9NlxtdKgxPqBAwe46aabapWkqtkbcdiwYdx666189NFHjY9QaBICIUGqlWBMyPpKzYvyelReXk5ubm51AjrY1Oyx7k9dunShU6dOVOXIuFXaEaco4MyUiYiIYMiQIeoMchFinhUEIdjl5ORUf52bm6thJL7z3XffsWvXLkLbRxHSJkLrcAhNiCQ0IbK6JLw//P73vyciIoKKZINqc7SaKk5JuJ3whz/8AbtdvVK/vibOGwTw9EF84403MNtC6HPXE9gc0X6PIbJ1ey677WEqK53831/+ws6dO1Uby9uKKvvUMdXG0FJW8lEAv7Xc8qreHZep7u447/ETEhJUHaexJkyYwKGDBwnvHIu9RbjfxpWNBmKHJOB2u3nllVeorNR3GeCDBw/yj3/+A0VSsPeo8snuNi9DKNi7uSivqODpp5/2+/UHf1NrTn/++eeZO3cuN998M7m5uXz33Xe1br5y+PBhANz2CJ8dU0+UkAgAjhw5Uq/neTdQuV0BeIJcR+7TrQsa0y+7Pu69915kWa5OdqutpCCPw5tX0rVrVy677DK/jBkfH098m3iSD6YFZZ/1pL0pSJLE4MGD6/1ct9vNpk2bMIaaMUcE14IV++l2cps2bdJk/FWrVnHkyBHMcZ6qMb7i7bXudFYxdepU3x1YIw3eRhEREVH9dUhISK2LY+BJfCxdurTBgQlNi0ge11bzpFnvyWMtVVRU+O2ErSG8q6+8CehgY7SbQfJ/Yl2SJG6//XY++eQTnJkSlpa+X5hTlSvhroRb7r2lVgUJfxLzrCAEh8OHD/Pee+/xxhtvNLjEWTDKzj6z6v7s97dAdPLkScaMOV0C/so2WodTLWZgG8rSi/nss88YNmwYrVu3VnW8kJAQnnjiCT766CMqTsrY2gdOr3V3JVSkGIiKivLbLhRfCvTzhtmzZ/Pxxx/j8kN1Ba1cNWQIo0ePVuXYS5Ys4eVXXsFosdLn7icIjW6myjh1EZ3QhZ63Psjued/x5z//mW+++UaVcsPx8fGEhoaRemS/z4+tB2lHPa+rR48efh3Xm8jPUzmxnpdRgMlk8vuO/Po4cOAA48ePxxhqJvoKdefP87E1D8PRLY6j+48yduxYnn32Wb/HUBc5OTk89dRTlJeVE9LDhTHM92OYohVs7V1kHc3i2WefZfLkydhswZVIqUmNOX3Hjh0A/Prrr/z666/nfP+hhx6qd5znU13R0OLfNhb+olg8v+D1rdxotVoJCQmlorhAjbB0oaLI89qioqL8Ml7z5s0ZMmQIq1evJicliehWCaqOt3f1QtwuF/fcc4/f8hiSJHHD9TcwadIkju1KplO/BL+M6w8FWUWkHM3g8n6XEx1d/8WgJ0+eJD8/n9AO/l9IqjZTmAWj3VT9vu1PiqIwceJEwJME9zVznEJFEvz444/88Y9/xOFw+HwMf2nQjvVWrVrV6pfRoUMHNm7cWOsxe/bs8XsvKCHwiIT6+ZWWlp73a6F2dYOKCv/10WkI74cfo02bxKzaJFnCYDVpkhC57bbbkGWZyowGTWOXVJnheW+64447VDn+pYh5VhCCx6effsrmzZsDrkem2mqWrwz0MmCKovDvf/+byspKoge20bQE/NkMVhMxV8ZTXl7OW2+95ZcqUffddx/NmzenMkXGXa76cD5TfsLTG/6vf/1rwF2sD4bzhoULF1JQUECRU6LQZQy6W3FREYsWLVLls92aNWt44YUXkI0m+tz1BGGx2icqYzv0oMdN91NSUsKTTz7JsWO+31UuyzKXX96P3NQTFOcFX4nUk3u3YTAY6d27t1/Hbd26NRaLhZzUfNXGUNwKuWkFdOjQQbet75xOJ6+8+goul4vYIQma9W6NurxVdUn4PXv2aBLDxVRVVfGPf/yD9PR0rO1cmGLUO88wt1Iwt3Czb98+3n777aCtfKnWnJ6YmIiiKBe8+UphYSEYzSAHR7/jc5g8G2ca0sqqU6eOlORm4XLquwJFQxVlpWK1WmnVqpXfxrz77rsB2JU4T9VxFEVh98p5WCwWbrnlFlXHOtuoUaOQZZmdKw8E1fvertUHQfFUPGuI5ORkAMwOH26p1glJkjA5rKSnp/t9w+XmzZvZuXMnphg3BhUKuEkymFu7KCsrY9q0ab4fwI8adAbrXQ3kNXLkSN5++22efPJJ7rjjDtasWcOCBQsCqi+dIOhJQUHBeb8WqFUCTe+J9by8PABkqz4vFviCbDFUv05/io2NZdCgQaxduxZXKT6d7N1VUJUj0759e1V219SFmGcFIXh45/HiYnV7lQaamon1QO/7PHfuXDZv3kxImwhC20ZqHc45QhIisR91sG7dOubPn8+tt96q6nhWq5Vnn32Wl156ibJjMiHd9b8D2VUMlWmeud97gS6QBMN5w4kTJ1DMdpy9/Xux0l8MSVsxZBzm1KlTDe4jeT67d+/mb3/7GwoSfe74f4Q38/+u2gtp1rk3LqeT/Ut+4k9PPsn3331Hs2a+3Uk/aNAgVq5cybGdG7lsuLrvbf5UVlxI2pF99O59md8XxBgMBjp37sy+/ftwudwYDL5fyFyQXYSzssqnfwu+NmnSJA4dPER4F/+WgD+bbDQQe1UCqQsO8vrrrzNjxgzNKqqdz1dffcWWLVswxbqxxKub8JEksHV04yqRmDt3Lv3796/uQR5MAn1Odzqdnn+sYCV53hNrVhqtq/79+7Njxw7yTh0jpl1XX0emqYqSQoqz0xgyZAgGg/8WVQwbNoyIyEj2rV3MsPufRDaoc/01/eh+clJOcPPNNxMe7t85oVWrVtx4440sXLiQ43tO0b5XvF/HV0NhTjH71h+hdevWXHvttQ06RlpaGuDZ3R2MjKEWFKWI9PR04uP982+uKApjx44F1Nmt7mVpoVCZDFP/N5WHHnqIsDAVSt34QYPOkB9++GE6dOjAiRMnAPjnP/9Jnz59mDBhAnfccQcffPABbdu2Va3MmRA8vCutgmnFlS/U7DWqRdJSz2qu1NJ7n7Hycs82Ldmozq5qPZCNhurX6W933nkngM93rTszJRS35/haVdUQ86wgCMGuZrWTnJycgD0XLCgo4KOPPkI2GXRVAr4mSZKIGdgG2SjzwYcfenYSqeyWW26hd+/eOLNkqvJVH65RFAVKjxhAgRdffFG3uycvJhjOG7Kzs1FMgVUpoD68r61mG4zGSktL46mnnqKiopKetz5ERCv/9uKui5Y9+tPx6lvISE/nr399irKyMp8ef/jw4QAc2pTo0+Nq7ei2tbjdrgZfbG6sHj164KpykavSrvXMZM85QM+ePVU5fmMlJSXx9ddfYwwxE3259otVbM3CCO8Sy+HDh5kyZYrW4VTbsWMHEydORLaBvbPbL7lUSYaQ7i5kE7z73rvVOxaDSaDP6TabDYK4j7j3tTWkupF3zso4uMOHAelDxsGdwJnX6C8mk4lbb7mFkvxcju/arNo4e1YvBM5ch/S3p59+GoPBwPpfd1DlDPxe6+t/3YGrys2zzz7b4MVi3s+0siU4q2PIZs/rKioq8tuYa9asYcuWLRij3aq0dfGSDJ5d68VFxUyaNEm9gVTWoGzE8OHDWbBgQXWfyNDQUDZs2MCPP/7Iu+++y7Rp09i9e7foIylckjdpJUrC13b2xWbhjJqJ9YasEPUndxD3iKxJq9d5zTXXEBYWijNDxpf5mMoMGVmWue2223x30HoS86wgCMHOu3AwPCqEiooKnydb/OXzzz8nPz+fyD4tMYaYtQ7ngkyhFiL6tCQvN5cvvvhC9fFkWebll19GkiTKDhtQdHxK5MyUcBVIXH/99QwePFjrcBok0M8b3G63pxKVSruMdOH0a/NVKXin08nf/vY3cnNz6XzNSF3vfGvTbyitLhvIwYMHePPNN3167FatWtGzZ0+Sdm+mpCB4FqTvXbMYgBtuuEGT8S+77DIA0k+oU2I/I0m/iXVFUXjvvfdwOp3EXBlffWFba9GXt8ZoNzFu3DhSU1O1DofKykpee/01FBTsXauQ/Pj2LVvA2tFFeVk5b775ZsAuzryQQJ/TY2JiwO2CKn1vhGkoqdLzmSU2Nrbez73ssstISEgg88geKkuDp5qZorhJ2b0Rs9nMzTff7PfxvS0cvclvX6tyVnJg/TLi4uK48sorVRnjUtq0acPDDz9MQVYRWxbrry1IfRzfc4qjO0/St29fbrzxxgYfp6SkBPBs+ApG3vMPf1U+dDqdfPTRRyCBrZ36H94trRRkC0ydOjVgF8n5bJufyWTi3nvv5cUXX+T+++/Xdf82QdC7mjvWc3Nzg+6DQmPUTKbrPbFut3vqk7urdHw1uZHcVa7q1+lvnt5Gt+KugKp83yzOcZWCq1DiqquuatAHJTWJeVYQApNYPHh+3hL54TGepdD+2EXta3v37mXmzJmYI204usVpHc4lRXSPwxxhY8aMGRw4cED18bp37859992Hq1Si4pQ+/w7cTig/ZsBqs/Liiy9qHY5P+eq8YfPmzTz11FP06NGDkJAQ2rRpw6hRozh06JDPYhWfdervv//9L3v37qVljytofdlArcO5KEmS6DzsDhwtE/jtt9/47bfffHr8O+64A7fLxZ6V8316XK3kpiVzcu9WrrjiCr/2qa2pT58+AKQdU6dVS+qxTCwWC1276m9ByMqVK1m3bh321g5C2uinvYtsNhDVvzUVFRV89tlnWofD1KlTSTqehLmlG6MGlfJNsQrGaDcbNmxg0aJF/g/AzwLpWkD79u0BkEqDZ7FTTVJZPnDmddbruZLEQw89hLvKSfKOtT6OTDtZR/dRmpfF7bffjsPh8Pv43bp1o1OnThzZuobyYt/v7j26bS1lxYXcdtttfi1zf7a//OUvtG7dmu3L95GeFJit1EqLyln502bMZjP/+c9/kOWGpya97WGlIK0SK59uxeOvNrjffPMNx44dw9zSjcEPU4wkg7WDi8rKyoBdJNeg37z27dszZsyYiz7myy+/bNAkIwjCmYvLRpMZl8vls50NwaBmMr3m7nU9iomJAaCqJDhX6iqKgqvUWf06teBdmVqZ7psL9pXpcq3jakXMs4IQPALxA4I/eFeYh0Z4FmcFWg96t9vNu+++i6IoxAxsgyTrM3FckyTLRF8ZXyt2tT3zzDPExMRQccKAS4dFCcqPybgr4emnnqZ58+Zah9Ngap43fPDBB8yaNYvrrruOzz//nD/96U+sWrWKfv36sWePb3bMGAwGLBZLkygd64sFqfv372fKlCnYI2LoPFzbc9a6kg0Getx0P0azhfff/8Cn7c5uv/12QkJC2Lb4Z1xV+v58WBdbF85EURTuv/9+zWJo3bo1sbGxpB3L9PlcUV5aSU5aPr1799ZVr3DwXGv4+OOPkWSJ6Cv018M2tF0UltgQFixYwM6dOzWLIz09na+//hrZDLYEbTYRePutSzKMHv1hUF0zC/RrAb179wZAKgzMxN+lSIWZmM1munTp0qDnjxw5kpiYGJJ3rA2KXeuK282x9UuQDQYee+wxTWKQJImRI0ficlayf/1Snx9/9+mFe1qVgfey2Wy88847SEgs+d86yksD61qz4lZYPm09pYVlPPvss7Rr17gWRt72sIHwObwhpNOJdX+0wd21axfjxo1Dtvh3XjfFnFkkN23aNL+N6ysNSqwnJSWRn59/0cfk5+dX94MRhEsRF51r8/astoVHAARseVQ11JxQ9N5j3Vuay1mgTQ9ytbnKnLgrXSQkJGgWQ69evUhISKAqR0Zp5LVYRQFnpkxYWKjf+0KdTcyzghB8xLlObaWlpZ4yY6FWIPDOdebNm8euXbsIbReFrZmKDch8zN4inJCESLZv387CheqUS6wpLCyMV155BcUNZYd827qlsZx5EpXpMt27d+eBBx7QOpxGUfO84e9//zsnTpxgzJgxPPHEE7z66qusXr2aqqoq3n///QZGfK7o6GgkZ2C9D9SH97VFR0c36jiKojB69Gjcbjddrrsbg0m/LSjOZguPpP2gERQU5DN+/HifHTc0NJRRo0ZRlJvJrhW+3Q3vb4U5Gexa8Svx8fGa9VcHT5JiwIABlBSUkZ/p24oyqUczQIEBAwb49Li+8Ouvv5KUlERY5xjMDqvW4ZxDkiRiTif8L5V4VdOHH35IeXk51nYuv5aAP5vBCuZ4N5mZWXz99dfaBeJjgX4tYMCAAZjMZuS8U1qH4nvlxcil+QwcOBCzuWHzr9Vq5c9//jOuygqOb1zm4wD9L23/Vkpy0rnrzjs1bU/g2U1uZFfiPJ8etyg3i+O7NtGnT59GJ4J9oV+/fvzlL3+hKLeEpd+tC6gWpFsW7+HkgTSGDRvGww8/3OjjeXMnwbpjXTJ4Fgx4X6da8vPz+ec//4nL7fJ7axdJAntnN7IZPvr4I3bv3u2/wX1Atd+8goICz6pzQbgIcZH5/LwTo/H0hRKXy6VlOLpSswSK3hPrDoeDNm3aUJ5ZjOIOvt/18gzP6lpvDz4teFemKi6ozG7cKsWqfAl3Bdx0080BMX+JeVYQAosoCV9bWVkZRpMRk8W3fYf9obS0lE8++QTZKBPdv7XW4dRbdP/WSAaZjz/++P+zd9/xUVVpA8d/d0omvfcCoST03otUAbGhgIKIYK/rir13bKti2deyNiwoK4q6qKB0REF67zWk996m3Pv+MUwkkkDKzNw7yfnuh8/CZObeJ2Myz73nOec5bpnQcOGFFzJ27FisRTqndZhpLsUGlUf06HQ6nn32WQyGFry392lNvW4YOnToWYPHSUlJdOvWjQMHDjgrPBISEpDMFSC3zFXrUpX9ujk+vnmfGdu2bWPLli2Et+tCaEIHZ4TmVnE9B+MTFMaiRYvIy3Pe/t3XX389vr6+/PHdfKrKnd8G1l1++/oDbFYLd9xxh+qfS459ZNMOZzn1uOmHs2sdXyssFgvvv/8+kl5HSM8YtcOpl3ekP77xQWzevJmtW7e6/fyrVq1ixYoV6IMUjFHqj3F4J8jofOCzzz5j3759aofjNloeC/Dz82PUyJHoKoqQSvNAkVvMH33OMYBm7yM+efJk2rVrR/ruPynLz3bG264Kq7ma4xt+xcfHhzvvvFPVWMLCwhg1aiTZJw6RfdJ52xXtWbcURZa58sornXbM5rr55psZMWIEpw5ksHnZbrXDaZDje1LZ8useEhIS7KvunTA24ig461pqYf309+XKwrrVauXBBx8kIyMD77Y2DMEuO1W9dF7g09mG1WrlnnvuITfXc7qdNPhK/bfffqv175MnT571GNgLgKmpqXz55ZckJyc3P0KhRXN8kIrB5roppwvs4v35y5nt37W+xzrA4MGDWbRoEVW5ZR61oq0hytPs++Oqvdpg4sSJvPXWW1hyJEzRTb+5t+TYf88uvfRSZ4XWKCLPCoLQmpSXl+PlbawprHtSK/jPPvuMvLw8QnrHYvDznNWiDkZ/E0HdosjencmCBQu45ZZbXH7Oxx57jE2bNlFxvAxjqBWdymPBVSd1yJVw0003aHKP34ZQ87pBURSys7Pp1q1bvc+prq6uNSHWsdVVfTp27MimTZuQKopR/Ju3qluLpMoioqNjmr0v7oIFCwBIHKjeaubm0On1tO0/koOrvuPrr7/mrrvucspxQ0NDuf3225k3bx6//fc/jL/pAacc151S9m5l/+/L6dGjB5dccona4TBkyBAAUg9l0eOCprU8rsupQ5n4+fvRvXt3px3TGZYuXUpGRgZBXSMx+Go7t4f0jqUirZgPPviA/v37u+28WVlZPPPMM0h68E22oYVhKnssVsp2wcMPP8x///tf/P391Q6r0VraWMC0adNYsWIFxv3Ob8uttpCQEMaNG9esYxiNRh588EHuvPNOjqxbQu8rb/bIcd+Tm1dTXV7K3XffTWRkpNrhMHXqVFatWsWuVUucch0gyzZ2r/kRPz8/LrroIidE6Bx6vZ6XX36Za665hu0r9xMSGUSnAeqvpq9PbloBqxZsxMfHh7feeougoCCnHNcxMV8yqLfvvSvpTn9frpwI/8orr/Dnn39iDJcxtVFvspwxRMG7vY3cY7n885//ZP78+Xh7a69z0N81uLA+atSoWkXQzz77jM8++6zO5yqKgiRJTm0NJ7RMYsV63Ryz0y1m+6wkre09pqYz29x4wkr+iy66iEWLFlF2LL9FFdZli42KlCLatGlDly5dVI0lLi6O3r17s3PnTmSzvYVMYykyWPJ0REVH0bt3b6fH2BAizwqC0JoUFxdj8jHi7WuvsJ6v6KYV+fn5zJ8/H4OvF8HdotQOp8lCekRTeiSPjz76iKlTpxISEuLS80VGRnL//ffz7LPPUnlMh19X9doWWkuhOl1HmzZtuP3221WLo7nUvG748ssvSU9P57nnnqv3OS+99BLPPvtsg4/pKLJJZfktr7BeXY5krqRHj+HNOkxeXh5r1q4lMCqeoJg2TgrO/aI79+Ho70v5/vvvuf3229HrnTMgOnPmTH7++Wd2rvofHfoOpUOfoU45rjtUlpWw9P0X0esNPPXUU+h06q++io6Opn379qQeOYXNakPvhIHrkvwyinNLGTt2rOor8s+kKArz589H0kkEd4tWO5zz8g73wyc2kI0bN3LgwAG33I9XVlZy7733UlRUhE+yDb2vy0/ZYIZgMCXIpKSk8PjjjzNv3jynfa64S0sbCxg4cCCzZ8/m6NGjaofidJMnT3ZKt4ALLriAkSNHsm7dOvKO7SeiY/2TFbWoojCX1O3riYuLY/bs2WqHA9i7LMXGxrL/j+WMnHEnJp/mfVCd2LWJkrxspk2bhq+vhj70sG+39c477zBjxgzWLtpEYJgfMe3Vn9zwd+XFFSz96DesFhvzXn+DpKQkpx3bUXB2tExvaRwr1l1VWP/qq6/473//i95fwbezrPpkOVOcglwus3fvXh5//HFeffVVTVwPn0uDr2SfeuopJElCURSee+45Ro4cWecetHq9ntDQUEaPHq16sUXwHKLAXpujkG45vcpDSzedQuP069eP2NhYso5nE9ovHr2pZfy3LD2Wj2y1cfnll2tiZu24cePYuXMnlnwJU0zjP0+sRRKKFcaPG69a4hZ5VhCE1sJms1FcXExUYhje/vaBqYKCApWjapiPPvqIyspKIoa0RWf0rEHbM+mMekJ6xpC36RTz58/nvvvuc/k5J0+ezE8//cS2bduw5CsYw9x//a8o9hbwKPD00097xEz4+qh13XDw4EHuuusuhgwZcs6B1EcffbTWz1VJSQkJCQn1Pt+x6lJXko0crd1VeE2hK7G3ee3Xr1+zjvPLL78g22zEdHXfClVX0Bu9iEruRfqeTWzbts1p3a+MRiMvv/wy06ZPZ+n7LzJr7ocERWi3pbeDLNv4+b25lBbkMmfOHE110Rg+fDiff/45mSdyiU9qfsH51IGMmuNqyR9//MGxY8cI6BjmMZ1ogrtHU5lRwueff85LL73k0nNZrVYeeeQR9u7di1e0jFczusS5inc7GVsZrF69mtdee42HHnpIE+MUDdXSxgIkSeKBBzyvc4i7PfTQQ2zYsIEj638iNDEZvcFzFlUd/u0nZNnGgw8+qJltCXQ6HVdffTVvvvkm+3//lT7jmte+fcfy7wG4+uqrnRGe07Vt25Y333yTW265hV/mr2fKnAkEhmmnY4fFbGXpx79RXlzBAw88UOdnWnOYzWYkg86jPusbwzFh4MwOYM7yxx9/8Morr6DzAr/uNiQNDGtIEvgkychVsHz5ctq3b++0zlau0uAKzzPPPFPz93Xr1nHDDTcwa9YsV8QktEIt9UOwqRzvh6MVvNZn6LjTmT8rnvBzo9PpuPbaa3n11VcpOZSr6f3aGkqRFYr3Z+Pl5cVVV12ldjgAjB07lldffbXJhXVLvlRzHLVoLc9u2bKFzz77jDVr1nDy5EnCwsIYPHgwc+fOPW/buU8//ZQbbrihzq9lZmYSHa39lSCCILhOcXExNpsNH39vfP3thc38/HyVozq//Px8Fi1ahNHfRECS56+oDUwOp2hvFgsXLuTGG28kODjYpefT6XQ89dRTTJkyhcqjYAi2uv0m3pwhYSuVmDRpkupb2TSXGtcNWVlZXHLJJQQFBfHtt9+ec0WgyWRq1EBrdHQ07dq148SpVJBtoNPACI+T6IoyARg2bFizjrNs2TIkSUdkck9nhKWqqM59SN+ziWXLljn1d7Fjx4488fjjPPXUU3z3+qPMePrdZq9Yc7X1X3/I8R0bueCCC+q9flbLBRdcwOeff07K/gynFNZP7tdmYX3hwoUABHX1nE40PjEBeAX78Ouvv/LAAw8QFuaa6xJFUXj++edZvXo1hhAZnyT1V7XVRZLAr6tM2U6JBQsWEBISwq233qp2WA2mtbEAwT3atGnDzJkzmT9/Pqk7fidxwGi1Q2qQ/JTD5J84yODBgxkzRltb01x55ZW8++67bPt1Mb0vvKLJ48YFmakc3/Unffv21fS2CwMGDODJJ5/kmWeeYelH65gyZzxGk/oTNBRFYc1/N5GbWsCUKVNc8nlmsViQdBpMSE7i+N7O3BLXGVJTU3nggftRkPHrpv4WbWeSdODbVaZsh47333+fzp07qzpOfz5NqtatWbNGJHjBKTyhMKoGR4tz/emV6p6wl7i7nDnJwFMmHEyZMoXAoECK92Ujm7Xfvv58So/nYympZvLkyYSGhqodDmBvB9+hQwdsRTqUJnSXtRZIBAQGqNYG/u+0kGdfeeUVFi9ezNixY3nrrbe49dZb+e233+jbty979+5t0DGee+45vvjii1p/XF24EQRB+xyr030CTPgE2AvrhYWFaobUIF9//TVms5mg7lFIHnINci6SXkdQtyiqqqr4+uuv3XLO9u3bc+ONNyJXQdUp976HshmqTuoJDAzk/vvvd+u5Xc0d1w3FxcVMnDiRoqIifvnlF2JjY51+jlGjRoHNilSc7fRjq0a2oivOJDExkcTExCYfJiMjg927dxPSpiNePs3bp10LgmPbYvIPYuXKlU4fMLzyyiuZMWMGuaeOseStJ7Fp+F56x4rv2fTjl7Rr186+ckhjuaVfv374+vqScrog3hwWs5WMo9l06tRJU5Nss7OzWb9+Pd6RfphCtT0J40ySJBHYOQKLxcJPP/3kknMoisJrr73Gd999hz5Awa+rjKStH9FaJAP49bCh84Z///vfNRMmPI0WxgIE97n11lsJCQkhZctazJXlaodzXooic3T9UnQ6HQ8++KDmxvVDQ0OZOHEiBRkpnNyzpcnH2bH8O8C+zYzWTZkyhZkzZ1KQVcyarzdpoivw7t8OcXRHCv369ePxxx93yc+Jl5cXik3979VVFNn+vXl5Oa+TTlVVFXPmzKGsrByfZBuGQKcd2ml0RvDrZp+A/9hjj5GSkqJ2SPVq1iXRjh07eOihh7j88su58MILax5PSUlh0aJFHtPSUVCPFj7stcjR5sPbL6DWvwVqrYzxlH2z/Pz8uPGGG7FVWynal6V2OM2i2GSKdmbi5eXFLbfconY4tQwbNgzFBtbixl2w2SpBrpIYMniI5n6m1Myz9913HykpKbz99tvcfPPNPPHEE6xfvx6r1drg/dwmTpzIzJkza/3x5La7giA4h2M/dW9fEyZfr1qPaZXNZmPx4sXoTQYCOoarHY7TBCaFo/PSs3jxYmTZPfue33zzzcTGxlKdqsNW4ZZTAlB5XIdihTlz5rh8T3m1uOq6oaqqissuu4zDhw/z008/0bVrV2eFXMv48eMB0BWccsnx1SAVZYLNyrhx45p1nF9//RWAqBawWh2wr7zv2J2ioiI2b97s9OM/9NBDjB49mhO7N7P0/RdrOsFpyaFNa1j52ZuEhYfz3nvvERAQoHZIZzEajQwbNoyinBKKcpuXp9MPZ2G12BgxYoSTonOOZcuWoSiKR+Z2/3ahSHodP/74o0uO//HHH/P555+j91Pw62FD8oBd7XQm8OtpRecFL774IsuWLVM7pCYTY+6tg7+/P7fffjtWcxUpW9eqHc55ZR/aTVleJpdffrlmV3I7iuFbly1q0uurK8rYs+5nYmJiGD3aM7oI3HffffTr14+jO06xb8NRVWPJTsljw5IdRERE8Nprr9Vsd+tsPj4+KDZZk9d4ziBb7N+Xj4+P04751ltvcfjwYbxiZbyitFuT0/uBT7KNiooKHn74YadPwnWWJhfWH3roIfr3789rr73GTz/9xJo1a2q+pigKM2bM4Isvvmj0cbds2cI//vEPunXrhp+fH23atOHqq6/m8OHDTQ1V8ACiwF5bWVkZAP6hEbX+LXjminWAa665hvDwcIr35WCt1GZCaIjiQ7lYyqqZMWMGkZGRaodTi6ONpK24ca9zFOK11hLWVXm2oYYOHXrWzMikpCS6devGgQMHGnyc0tLSmi4cgiAIAJWVlQAYvAwYjHqQoKLCjRXWJtiyZQs5OTn4tQtBZ/Cc64/z0Rn1+CWGkJmZybZt29xyTh8fHx566CFQ7MVud7CWgCVbR9euXZkyZYpbzulurrpusNlsTJs2jY0bN/LNN98wZMgQZ4ZdS7du3UhISEBfmAY27a4ybgx93kkALr744mYdZ9myZUg6PREdujkhKm2I6tQbgKVLlzr92Hq9nn/961/07duXAxtWsHz+PE2NORzbsZGf3nkOX19f3nv3XeLi4tQOqV4jR44E4OS+9GYdx/F6Z++x2lyrV68GScKvredNuNKbDPjEBHDo0CEyMzOdeuxffvmFt956C5336VXg6ncWbjC9z+niusG+2m3Hjh1qh9Roao8FCO41depUYmJiSN+1EXOFdsd/FVnmxKaVGAwG7rjjDrXDqVfnzp3p378/J3ZtIj/9ZKNfv2vNT5irKrnmmmswGDxgRhH2iXCvvfYawcHBbPjfDopy1Jm0bqm2svLLjaDAq6++Sni46yatxcTYt1q1lplddg41Wcvsiywd32dzbdq0iQULFqD3VfDpoP3JCF6RCl7RMvv27eOjjz5SO5w6NWkkY/78+bz22mtceuml7N69m0cffbTW1xMTExk4cCBLlixp9LGd0XpW8BxaaxmjFUVFRQCExiQA9taLgp2nFtZ9fX258847ka02Cnc1v5WeGmxmK0W7MgkICODmm29WO5yz9O7dG0mSsJY0csX66cJ63759XRFWk7gyzzaHoihkZ2c3+OJ49OjRBAYG4uvry+WXX86RI0fO+5rq6mpKSkpq/REEoWVxdOLRG3RIkoTBoMds1vYN8W+//QaAvwcOvJ+P43v6/fff3XbOMWPGMGjQIKz5OiyFrr0fUBSoPGbvSPPII4941PVjQ7nyuuH+++9nyZIl9raaBQUsWLCg1h9nkiSJyy+/HGxWdAWpTj22KixV6Ioy6dKlCx07dmzyYVJSUjhw4ABhickYvT2nVfX5BEYn4BMYyqpVq1ySA7y9vfm///s/unbtyq5V/2PNl+9ooriesm87/3vzCYwGA++9+y5dunRRO6RzuuCCC9DpdJzc2/TCuiIrnNyfQVhYGN27d3didM1TWVnJrt278I7wQ2/yjOLJ3/nGBwH2AXNnOX78OE8++cTp1ura2n+1ofR+4NPNhtVm5d577yU/P1/tkBpMq2MBgus4OkLarBZObV+vdjj1yjm2l4rCXK644gqXbAnkTI7tFLb+8m2jXifbrGz/dTE+Pj4eNxk3PDycZ555BqvFypr/qtMSfvMvuynOLeWGG26gX79+Lj1X27ZtATAXVbn0PGpxfF9t2rRp9rGqqqp45plnQAKfzjZNb+tyJp8OMjoTfPDBBxw7dkztcM7SpLfx3dMX/4sXL6Z79+519vrv3LlzgwbQ/84ZrWcFz6GFG1stcrR0CottW+vfQu2fGXe1LHWWK6+8knbt2lF6OA9Liecl/qK92diqrdx8880EBQWpHc5ZgoKCaNeuHbZSHY35aLGWSvj6+tKhQwfXBddIrsyzzfHll1+Snp7OtGnTzvk8X19frr/+et555x2+//57HnroIVatWsXQoUNJTT33QPlLL71EUFBQzZ+EhARnfguCIGiAo4BiMNqLnXoPKKxv3boVyaDDO9Jf7VCczjsqAEmvY+vWrW47pyRJPPTQQ0iSRNXxxuXtxrLkSdhKJCZMmECfPn1cdyIVufK6YefOnQD8+OOPXHfddWf9cbZJkyYhSRK63ONOP7a76fJOgiJz5ZVXNus4K1asACAquZcTotIOSZKITOpBeXk5f/75p0vOERAQwPvvv0+HDh3YuvRr/lg83yXnaaiMo/v47rVH0Enw9ttve8RnUmhoKL169SLzRC5V5U3boi4nNZ+KkkpGjhypqclNhw8fRrbJmML91A6lybxPx96YjmLnYrPZeOyxx6iqqsa3kw29B8/lMQYreLe3kZ+fzwsvvKB2OA2m1bEAwbUmTZpEWHg4GXs2YTVrbztQRVE4tfU3dDodN954o9rhnNfIkSNJaNOGfb8to7K04YvVjmz7nZK8LK688koCAzW4AfV5jB07lvHjx5N5IpfDW0+69dwFWcXs+e0wCW0SuOuuu1x+vl697NfFlZktczFOZWYJPr6+Ttly4YMPPiAtLQ1TvIxBezsP1UsygE+SDavVynPPPae5OmKTrmj379/PuHHjztkOIyoqipycnEYf21mtZwXPIlau15aXl4dvYDABYZE1/xbsrNa/2kJ6Wntpg8HAPffcgyIrFOz0rFXr1koLJftziIyMZMaMGWqHU69u3bqhWEFu4LwFxQZyuUSXLl00NcjjyjzbVAcPHuSuu+5iyJAhzJ49+5zPvfrqq5k/fz6zZs3iiiuu4Pnnn+fXX39t0KDGo48+SnFxcc2f8xXiBUHwPI4tbowm4+n/N1BeXq5mSOdks9k4dvwYXkHeSHrt5Apn0Rl0GINMHDl61K03q8nJyUyaNAlbmYQlxzX3AooMVSf0NddgLZUrrxvWrl2Loij1/nG2mJgYhg4diq40Fyo9uGuXoqDPOYaXl1ez28CvW7cOSdIRltjZScFph6O1/bp161x2jpCQED766CMS2rRhw3fz2fZr41avOUte2gm+feVBZKuF119/3aXbKjjbqFGjUGSFUwebdg/raAOvtb1qHe3TjYEeuCT7NGOgNwAZGc4ZX1iyZAn79u3DGCVjDNfWAHZTmOIU9EEKK1asYMuWLWqH0yBaHAsQXM/Ly4trZ8zAUl1J5n73bM/UGMWZKZRkpzJ27FiPWPyg0+m4dsYMrBYzu1Y3vLvDtmXfIEkS1157rQujc60HH3wQb29vNi3dhdXivnHzjT/uQJZlHnn4kTonBDlbr1698PXzpSKtWHMF1+aylFRhKa5i0MCBzd6j/sSJE8yfPx+dN3i39awFigDGMAVjuMz27ds116mlSSNDBoPhvKtKMjIy8Pd3zoqOhrSeFa1jPZMoqNctLy8Pv6BQ/IJCAcQF8xksFkudf/cUY8aMoUuXLpQdL8BcVKl2OA1WvC8L2Wrjtttuw9vbW+1w6tW5s33A0VbWsM8W2+k6jtZaMLo7z55PVlYWl1xyCUFBQXz77bfo9fpGH2P48OEMGjSIlStXnvN5JpOJwMDAWn8EQWhZHO04fQPs+cTH35u8vDzN3hAXFhZirjbXDF63RMYAbyorKmq2I3KXu+66C6PRSNVJPYoL7vPN2RJyJVx11VUeMQjYVFq7bmiuq666CgB9jvZa/jWUVJKDVFXKRRdd1KxOT1VVVezZs4fA6HiM3j5OjFAbAqMTMHiZ2LbNtUWE8PBwPvzgAyIiIln9+dsc3vKbS8/3d2WFeXz7ygNUlZcyd+7zmttn/Hwc8TZ1n/WT+9IxmbwYNGiQE6NqPse4oae2gQeQjDqQJKeMgdpsNj744D9IevBp53mD73WRJPDpYC8svf/++ypH0zAtLacLDTd16lS8vEyk7foDxRUXxs2QuvMPAGbOnKlyJA13xRVX4Ofnx86VPyDbrOd9fvbJI6Qd2s2IESOc0n5bLdHR0cycOZOyogr2bXBPZ4usk7mk7M9g4MCBjBgxwi3nNBqNjLtwHJbSaiozS91yTncpOZwLwEUXXdSs4yiKwksvvYTVasWngw2p8UO5muDTUUbSw+vzXtdUzbdJhfUePXqwevXqeleLVlRUsHLlSqftpdCQ1rOidazQUlRVVVFcXIx/aAT+oRGAKKyfybEv69//7ikkSeL2228HoGhftsrRNIzNbKXkUB6RkZHNbmXpao4WObbyhhbWpVqv0wp359lzKS4uZuLEiRQVFfHLL780ay+thIQEsbWFIAikpKQAEBBqHxAMDPOnsrKS3NxcNcOql+PmTefloXeiDaAz2b+30lL3DkpER0czbdo05CowZzl3wq0iQ3WKHm9vb2699VanHltrtHTd4AwjR44kIiICfd5JkM8/EKpFupyjgL2LT3OcOnUKm81GQGScM8LSHEmnwz8ilpMnT7q8G1lcXBzvvfcuPj4+/PTOc2SfdM9gs9Vi5vt5j1GSn8N9993HpZde6pbzOlO7du1ISEjg1MEsbLbGFXtKC8vJzyhi0KDB+Phoa3KIYxWYImtzYl+DKUqzV7SBfZ/2tLR0jJGyR+6rXh9DABhCZDZv3lxzDaplLS2nCw0XEhLCJZdcTEVhHgUp2mn1X11WQu7RvXTq3NkjtjBx8PPz4/LLL6ckP4dj2zec9/k7VnwHwDXXXOPq0Fzu+uuvx9/fnx2r9mM1u/5aesuvewG4++67XX6uMzn+WxXvy3LreV1JNtsoPZJPaGgo48aNa9axVq1axcaNGzGEenYXGp0JTG1tFBYU8t5776kdTo0mFdZvvPFGDh8+zO23335WYaukpITrr7+erKwsbrnllmYH2NDWs6J1rNBSONqRBYRGEhAagSRJNY8J1Jq5q/X9WOszatQoEhMTKTuWj61K+6vuS4/kIVtsXHfddU65YXeljh07AiA3sKOwfLqwnpSU5KqQmsSdefZcqqqquOyyyzh8+DA//fQTXbt2bdbxjh8/TkREhJOiEwTBUx08eBCjyUBgqH1f0LDY4JrHtahmJX0L7rSk5nd28803YzKZqE517qp1c5aEXG0fcDlX57OWQCvXDc5iMBiYOnUqWM3o8k+pHU7jWarQF6bTqVMnevbs2axDFRfb2+EbfTx3D+jzMfr4YbPZarYJcaVOnTrx6quvYrOYWfL2U1RXVrj8nGu/epfMYwe48soruf76611+PleQJIkRI0ZgrjSTfbJx29SdOmAfy3DXCrbGcHTG8oR78vrYquwFk+Z0xnBYvnw5AF5R2lop6wxeUfZruRUrVqgcyfm1tJwuNI6jUJi66/yFYHdJ270RRZa5dsYMj+s8O336dAB2rfnxnM+rrqzgwIZVxMfHe9RWLfUJCgpixowZVJRWsf9P13aAyj6VT+rBTIYMGULv3r1deq6/69atGwMHDqQivYTKrJaxar1obxa2aivXXXdds1rqm81mXnvtNZDsK749nSlOQecDCxcu5MSJE2qHAzSjsD59+nQ+/vhjIiIi+PjjjwEYOHAgcXFxfPvtt8yePdt+M9wMjWk9K1rHCi2FY1JIcFQsBqMX/iERYqLIGc5s/+6phXWdTsf06dNRZIXSY9pevasoCiWH8zB6GTW/Wh0gLCyMoKAgbBUNXLF+eiytffv2Loyq8dyVZ8/FZrMxbdo0Nm7cyDfffFPvzUVmZiYHDx6s9btZ16rTpUuXsm3btma3MhIEwbOVlZVx7NgxIhJCkXT2z+rINmEA7Nq1S83Q6uXYAkWxum+POneTrfabbZPJ/UvUwsLCuPrqq+2r1p2017qiQHWqHpPJdM7J2S2FFq4bnG3y5MnodDp0HtgOXpd7HBSZq6++utkD0L6+vgBYq6ucEZomWavt22O5a7upESNGcNNNN1GYlcaaBf/n0nOd3LOF7b8uJjk5mccee8zjChJnGj58OAApBxq3l7djX3bH67XE0erXUuy5v1+O2J3RtvjPP/9EZwR9CxxONYTaC+t//vmnypGcX0vM6ULDdenShb59+5J/4iDlBep3L7VZzGTs2URwcDAXX3yx2uE0Wvv27enduzcndm+mNL/+9/PQn6uxVFcyZcoUdLomlcs057rrrsPX15cdqw9gc+F97NZf9wCo1iHsvvvuAyB/S6rHd6CxlFVTvD+byKioZm+7sHjxYtLT0zHFyei11TCoSSQd+LS3YbPZePfdd9UOB2hiYR3gq6++4j//+Q/t2rUjPT0dRVHYunUrbdq04b333uOTTz5pVmDObD0raJdjBZBW99RUg2PWTWi0fTuDkJh4MjMzqary3Js9Z/L0PdYdLr30UoxGI2XH89UO5ZzMBZVYiqu4cOyFTpkF72qSJNGhQwfkSqlBq97kCh0xMTE1g5Za4uo8ez73338/S5YsYeLEiRQUFLBgwYJafxweffRRunTpQnr6X3suDh06lKuvvpp//etf/Oc//+G2225j0qRJJCQk8Nhjj7k0bkEdiqKwatUqsrJaTgsuwTW2b9+OLMvEto+seSw6MRxJJ7F161YVI6tfWJi98G+t8NzrjvOxVVqQJInQ0FBVzn/99dfb9xRN1eGM2wJLroRcBVOmTKn579fSqX3d4GzR0dGMGDECXVk+UkWR2uE0nKKgzz2Ot4+PUwag27dvj16vpyhdGysznM1mMVOanUb79u3dOrHnrrvuolOnTuxe8yMZR/a55BxWi5kV8+eh1+t56aWX3DZxwFX69++Pl5cXaYcafq0n22TSj2TTtm1b4uK0t51Bu3bt8Pb2piq3ge3ONKgqzx57t27dmnWc4uJi0tPT0QXILbJBj84IOl+FAwf2e8T4Y0vL6ULjOLqbpGxdp24gQMa+rZgry7nmmmtUmYDrDJMmTUKRZQ5sXFXvc/b9sRxJkjxyu5b6BAcHc80111BeXMEBF61az0ktIGV/BgMGDKB///4uOcf5dOvWjUmTJlGdX0HRfs/YcrUuiqKQuyEF2Spz3733Nuu60WKx8NFHHyHpwdTG81erOxjCFPQBCr/88gvHjx9XO5ymF9YBbrnlFnbt2kVZWRlpaWmUlJSwb98+brvttmYF5ezWs4J2efKMbVc5dsye7MLiEgEIj0tEURTNtLlQ25kTDDx1xTrY2/IMHDiQ6vwKrOXa/T7KTxUCMGHCBJUjabh27dqBAnLluZ+nWEGu1t5q9TO5Ks82xM6dOwH48ccfue666876cy7Tpk3jyJEjvPjii9x999388ssv3HLLLWzZsoWoqCiXxy64365du5gzZ07NbGFBqI9jpVBc0l+fBV7eRiITQtm9ezcVFa5vy9tY3t7ehIWFYS2tPv+TPZSltJqIiAjVtnyJjIzkkksuwVYhYS1o3v2BfbW6Dp1Ox6xZs5wUoWdQ87rBFaZMmQLgUavWpZIcpKoyLp44EX9//2Yfz8fHh1GjRlGak05hmvoDSM6WsX8rVnM148ePd+t5DQYDTzzxBADrv/nIJefY+9syCrPSmDFjBsnJyS45hzt5e3vTp08fctMLqCpvWD7MSS3AXGXRbFtdg8FAr169MBdWemw7+KrMEoBmt991bD+oawGr2uqj81EoKSnV5LVmXVpaThcabuTIkXTs2JGsA9upKGrc9hvOZLNaSNm6Bh9fX4/ed3zcuHEYjUYObFhZ59dLC/NIO7CT/v37Ex0d7eboXGv27Nn4+vqydcU+LC7Ya33TzzsBuOOOO5x+7MZ48MEHCQ8Pp3BHBtWF5xkM1qiSw3lUZpQwatSoZk/O/e2338jJycErWkan7R1dG0WSwJRgnyjw7bffqhxNMwvrDj4+PsTGxjrlxrGhrWeFlsETZoq626FDhzB4mQiJiQcgoq19z2it7jvqbuXl5XX+3RONHDkSgIrTN8NaVJlZgl6v96jP4nbt2gFgqzz3wLytsvbztcyZebah1q5di6Io9f5x+PTTT1EUhcTExJrH5s6dy44dOygqKsJsNpOSksK7774riuotWEmJ/XNsz549KkciaN369esxmgxEJ9be8zqhUwxWq5WNGzeqFNm5dezYEUtpNXILbAcvm21Yy8wkJSWpGoejCF6d0bzCuq0EbGUSY8eO1eQKSXdQ47rBFYYPH054eDj6/BSQPeN3T5drL347cwulm2++GUmSOLT6e6zmljPBp7K4gOMbfiUgIKBmD1R36t27N8OGDSNl71ayjh9y6rEVWWbzj19h8vbmpptucuqx1TRo0CBQION4w9oTpx/N/ut1GjV06FAAKjI8b29W2SpTmV1GcnIy4eHh53/BOTgWMEgtowNynRzfm6d1g2wpOV1oOJ1Oxz/+8Q8URebYH7+oFkfqjt+pLith5rXXEhISoloczRUUFMSgQYPIPnmYkryzVzQf3fY7iqIwbtw4FaJzrZCQEGbPnk1FSSW71jm3rpB2OIvUQ1kMGzaMAQMGOPXYjRUUFMSzzz6LYpPJWXsM2eIZ9w0O1fkV5G9OJTg4mCeffLLZC1FXrFgBgFd0y1mt7mAMU5CMsHzFctXripq7ZGpo61mhZXCsOJbllveL3hQWi4WjR48SkdAenU4PQGRb+yDnoUPOvdn3VIWFhXX+3RP17dsXgKrsMpUjqZtik6nOq6Br166abJVeH0ehXD7PRHT59D7snlBYFwRBaAmOHz/OyZMnadM5Br1BX+trid3sBdDVq1erEdp5de7cGbDf9LY01fn2iYqO71EtycnJ9O/fH2uBrmbyW1NUZ9hvcWfMmOGkyAS1GAwGLrvsMrCakYoat6+zKqwW9IVptGvXjl69ejntsN27d+f666+nvCCHvUu/QrY5f8WRu5kry9n1v0+xVlfx6KOPqrZlg2NCz97fljr1uKmHdlGUk8ElF1/coraj6NevHwAZRxtWWM88llPrdVo0bNgwACrSi1WOpPGqsktRrHLN99AcjqKZ4pkL9xtENktIkkRgYAvcRF5occaMGUOfPn3IObKHwlT3d+6pKismZcsaQkNDufHGG91+fmcbM2YMAMd2bDjra47HRo8e7daY3GX27NmEhYWxY+V+youdcy8r22T++GE7Op2Oe++91ynHbK4RI0Zwww03YC6uIndDiupF14ayVVvJXnsMZIWXX36ZyMjI87/oPLZt24bOC3R+TghQYyQdGIJlsrOyychQ9/7Q0NAnNqVVriRJNW2tG+rM1rM//vjjWV+fOXNmo+MQtCs1LQ2AgoIClSPRhpMnT2I2m2uK6QDh8e3Q6fRixfpp2dnZdf7dE3Xs2BEvLy/MGm1TYy6uQpEVj9uOo23btgDIlRJQ/4WUo1V8mzZt3BDV+bkrzwqCIKhl2bJlALTvdfbnbkRCKIFh/qxatYonn3xSc3vR9uzZE4CqnDJ8ogJUjsa5qnLsE/ycWQhsqmnTprF161bMmTp82jd+4q1sAWuejg4dOmi6kOMMreW6YdKkScyfPx993kmsoQlqh3NOusI0kG1cdtllTt/y7J///CfHjh3jt99+Y/ePX9DjkmvRG72ceg53qS4rYef3H1NekM0NN9xgnzyhkkGDBhEeHs6hzWsZO3uO0/67Hd60FqBF7dUK9kkeRqOR7JTztyZWZIWslHzatWun6ZWOycnJREREUJBRhKIoHrVdYUWGvWPU8OHDm32s+Ph4/P39qSgqRVFocfusKzaQyySSk5NV2/bmXFpLThcaTpIkHnnkEa6ZMYODq75j4Mw56A3u+dlVFIVDq3/Aaq7m3nufaBHdEhyfkyd2b6bPuL+6ClktZlL37yApKanFtYF38PPz45577uGpp55i4487uXDm0GYfc9/Go+RnFjF16lQ6derkhCid4+6772b37t1s27YNrxAfQnrGqB3SOSmyTPbaY1hKq7n99tudMlFOlmWys7PRB8otLpc7OLatyc7OVrVDXYML6ydPnkSv12MwNPglTbJ27VqXHl/QjqqqKk6ePAlAfn4+eXl5zW5f5ekOHz4MQOTp9u8AxtNt4Y8cOeJxN3qukHZ6MobeoCM1NVXlaJpHr9eTmJjI0ZPavBmyFNtbpGl5D/K6xMbGotPpsFWee3aircr+u6SVwrq78qwgCIIaZFlmyZIleHkba1ann0mSJJL6tmXbin2sXr262fuKOVtNl5msUuih7Rv0xqrMKkWSpJrvUU1jxowhKCiI0uxivBPlRrekNWdLKDJMnTq1xV8zt5brhg4dOtCpU2cOHT4MVjMYtFtM1uWnALjk88tgMDBv3jzuu+8+fvvtN7Z98z49L5uFd0Cw08/lSiU56exe8hnVZcVcd911qq9y0uv1DBs2jP/973/knjpW6z68OU7s3oyfvz99+vRxyvG0wsvLi65du7Jn7x6sFhsGo77e5xbmlGCuNGti0ta5SJLE4MGD+fHHH7EUV+EV7DmbjFdmlmAymZq9vzrYfxfGjBnDkiVLsBZJGEM8Y6VfQ5lzJBQbjB07Vu1Q6tRacrrQOF27dmX2rFnMnz+fo+uX0mn0JLecN3P/VvKO72fw4MFMmuSec7paTEwMbdu2Je3gThRZRtLZbzKyjh3EUl2l6S1LnGHSpEksWrSIvdv20mVwB+I6Nn2bxorSSjYv3U1gYCB33323E6NsPqPRyLx587jmmmvI2J6OMcgb/7banNynKAp5m1KpzCzlwgsvdNo+9RaLxb5aX3N9yp1H0tmvUdTe2qXRb/GoUaP4/PPPKSkpobKy8rx/BKE+Bw8eRLb9tefFvn37VIxGG44ft+/JFxaXWOvx8Lh2lJSUkJ+fr0JU2pKamopfkC+h0cGkpaV5TGuX+sTExCCbbZrc/8VaYd+qITY2VuVIGsdoNBIZFYlyni0o5SoJnV6nuX2/RZ4VBKEl2rx5MxkZGXTs3QajV92Dhp0H2idyfffdd+4MrUHCw8Pp0KEDVdllKLaWs4WRbJWpyimnU6dOBAUFqR0OXl5eTJo0CdkMloLGFcYVBSxZOoxGo6orYN2tNVw3TJx4ESgyusJ0tUOpn6UaXUk2PXv2dNnKCZPJxJtvvslVV11FaU46Wxb+m/yUwy45l7MpikL6nk1s+/pdLBWlPPDAAzz44IOamAAzcOBAANIO7XbK8cqLCyjMSqN/v34tskjWo0cPZJtMfsa5t2XLOZVf83ytc0yAqMorVzmShpOtMubCKrp3746Xl3MmHF177bUAVJ3Q4eHDLLUoNqg+pcdkMjF16lS1wzmn1pDThca56667SE5OJm3XBnKO7nX5+cryszm8dgkBgYE8//zzmsjTztKvXz+qysvITTte81jaYXvu79+/v1phuYVOp+OJJ55AkiTWL96KrRn3sxuX7KS60sw999xDaGioE6N0jtDQUP7973/j6+tLzm8narqzaU3xvmxKDuXSuXNnXnjhBXQ651TCTSYToaGhyFUt53f37xzfm9r1igb/F9u/fz/33HMPO3fuZPr06cTGxnLvvfeyZ88eV8YntGB799ovCGK69a/179bMsQI7JKZ2m0PHv0+dOuX2mLTEarWSlZVFYJgfgWH+VFVVefw2Ao4uDbZK7W1mZqu079/oifsCxsbEIldL5xwQUKohKjIKvb7+lRbuJPKsIAgtmaNY3mVwh3qfExQeQGzHSDZt2kR6uvYKaEOGDLEXonM9Z+D9fKpy7BMFhgwZonYoNa644goAzFmNGwywlYGtXKpZ9d7StabrhvHjxwOgK0hTOZL66QrTQVFqYnUVo9HIk08+yZNPPoliqWbnD59w9I9fak1Y1xpLVSX7li3k4KrvCPD3591332X27NmaGax3rKjOOOqcif6ZRw/UOm5L49gmLCf13Pfhuae/3q1bN5fH1FydO3cG0OwWbXWxFFeBoji1BW/Xrl2ZOHEitlKp0TlYy6pO6ZCrYNasWURERKgdTp1aU04XGsdkMvHqq6/i4+PDgRXfUFGY67JzWaur2PPTF9gsZp5/7rkW1xrdkZczjx2oeSzz6P5aX2vJunXrxrRp0yjIKmb3uqZtN5txLIdDW0/QvXt3pkyZ4uQInSc5OZk33ngDHRJZq49hLlF3ZfPflZ0oIH9rGlHRUbzzzjv4+vo69fjdu3dHrpSwVTj1sJqgKGAt1BEQGKBqG3hoRGG9c+fOvPbaa6SlpbF48WKGDBnCO++8Q+/evenfvz/vvfcexcXFroxVaGEOHLAnsrjuA2v9uzVLS0vDYPTCP7h2ITMo0t5yVIuDzO6Un5+PzWbDP9gX/xB70snKylI5quZx7FWkxRXrjpgCAjxvL9mIiAhQQKlnvoKigGyWiIyMdG9g5yDyrCAILVVJSQmrVq0iJCqIyDbnnqzVZZC98L5kyRJ3hNYojuJz5ek9TVsCx/eipcJ6UlIS3bt3x1qgQzY3/HXmLPutbUtpWXk+rem6ISEhgeTkZHQlWWCzqh1OnXSF9qK/O9oMS5LE1VdfzYIFC2iTkEDKljVsW/QeFUXn3/fa3YrST7D5q7fIPryLvn378u233zhl/0hnatOmDf7+AWSfcM7q/+yT9uN4QkG5KRyF9by0c69Yz00vRK/Xk5SU5I6wmiUmxj7eYitvRNJRmau6yz3wwAP4+flRdVyPfJ4OcJ7AWgbVqTpiY2O55ZZb1A6nXq0ppwuN1759e5577jms1VXsWvIplirnV8sUWWbvsoVUFOZyww03aHbbhOZw5OWs44dqHss6foioqKhWszXt3XffTWhoKFuX76O8uHGTyWSbzPrFW5EkiSeeeEIzi5TqM3ToUJ5++mlsVRayVhzBqpEFbZVZpeT8fhJ/f3/ee/c9l4xLX3755YA9/7U0lhwJuRouu/QyjEajqrE0+t3V6/VcccUVLFmyhNTUVF588UXKy8u56667iI2NZebMma1+Va3QMIcPH0Zv9CIwOgGTfxCHjxxROyTVpaenExgefdbs/eAI+42eY3/x1sqxOt030Adff28Aj2+P75iVJlu011bWUVj38fGcfeYcHBfFSj1jI4oNkLW5Gl/kWUEQWprly5djNpvpPLDdeVcotu+ZgNFkZMmSJZrb7qV///4YDAYqWlJhPbMEo5eX5vYBnjRpEij2PVEbQpHBkqsjIiKCoUOHujg6bWkt1w2jRo0C2YZUkq12KGeTrehKsunYsSPx8fFuO23Xrl355ptvuOKKKyjJTmXzl2+RsW+rJj47ZdnG8Y3L2f7tfzCXl3DXXXfxySefaHL1myRJdOnSmYLMU1iqm7+iyVFY79KlS7OPpUWJiYmYTCby0utfsa7ICvkZhXTs2NFpbcpdyTGRXLZq7568Po57dcdEfWeJjIzkwQcfRLFC5VHPHpBXFKg8pAcFnnnmGY8Y12gtOV1ovIsuuoibb76ZisI89vz8pdM71RxZ/zP5Jw9ywQUXcM899zj12FrRvn17jEYjuSlHAagsLaa0IKfF5uu6BAYGMmfOHCzVFv78eWejXrv/z2PkZxYxefJkj5k8eOWVV3LnnXdiKa0ma+UR1Re1mQsryV59DINOz9tvv+2yyYdjxowhuVMy5iwdlsKW04FGNkPVMT3ePt7Mnj1b7XCat419VFQUDz/8MAcOHGDFihWEhoaycOFCdu7c6aTwhJbKYrFw/Phx/EKjkCQd/uHRZGZktOoZmGVlZRQUFBAcfXYbi+Ao+2MpKSnuDktTioqKAPD2NeHtbwLw+J8Zx0CDFvdrVWT7oJzJZFI5ksYLDg4GQLbUfQHhWMkeEhLipoiaRuRZQWg5tFDoUMuSJUuQJImkvonnfa7Ry0CHXgmkpaWxfft21wfXCL6+vvTs2RNzQYXqN+XOYDNbqS6ooHevXnh7e6sdTi0TJ07EYDBgyW7Y7aqlQEKxwKWXXqr51ROu1JKvG0aMGAGArihT5UjOJpXkgmyridGdfH19ef7553n99dfx9TZxYMU37Pvlv1jN6i01rSotYvu3H3Bi0yri4uL4/LPPuP322zX9u9m5c2cUWSY39Vizj5V98jCRkZGav89oKr1eT3JyMgVZJcj13MOW5JdhqbY6tU25K8my9u7FG8oVsU+ePJkBAwZgydNhyfPcAXlzuoStTOLKK6/UVGeehmrJOV1omrvvvptx48ZRmHqUQ6u/d9r9ZequDaTu+J2kpCT+9a9/aTpfN4fRaKRDhw7kph4/nfPte617QmcVZ5o0aRJdunTh0NYT5GcUNeg1lmoLW3/dg6+fL3fffbdrA3Sy22+/nSlTplCdX0H2b8drxrrdzVphJnPVEWxmKy+++CIDBgxw2bmMRiPPPfscer2eygN6bC1gJzvFCuV79MgWuHfOvarvrw7NLKwDbNmyhTvuuIOpU6eSnp5ObGysW2dpC57pwIEDmM1mAqPtPyuBUfb/37Vrl5phqerwYfvM9vC4xLO+FhgWhZe3T81zWitHEd3k64XJ16vWY57K0bZErcR+LorNHpMnrDL4O8feqko93UIdhfXAwEA3RdR0Is8KguDJjh8/zo4dO0joFI1/cMP2Dus0oB0AP/zwgwsja5p+/fqhyEqL2Ge9OqccFPv3pDVBQUGMGDECW5nUoIEAS7Z94P+yyy5zcWTa11KvG7p3705gYBC64kz7MkQN0RXbi/3Dhw9XLYbx48ezePFi+vTpQ/ahnWxZ+G/K8t2/ur/g1BG2fPU2xRknufjii/nmm288Yt9Sxx7b2Seb10WvsrSY0vycmnbpLVVycjI2q42inLo7uOSl29vEe0ph3dEZT+9tUDmShtN728cRCgvP3ZK/KSRJ4sknn8RgMFB5TI/igfMOZDNUpegJCgri/vvvVzucJmupOV1oGp1OxwsvvED37t3J2LeFlC1rmn3MvBMHOLJ2CWFh4bzzzjtO74KhNR07dsRSXUlJfg756SeB1ldY1+l0zJkzBxTYtKxhdZjdvx2morSKG66/QZOdP8/F0bp+2LBhVKQWk7c51e0xyBYbWauOYi0zc99993HRRRe5/JzdunXjhRdeQLZA+W4D1lKXn9JlZDOU7dFjK5OYNm0a11xzjdohAU0srOfl5fHGG2/Qs2dPBg8ezCeffMLYsWP5+eefSUlJoW/fvs6OU2hh1q5dC0Bogj15hSR0rPV4a+RYlRXT8eybcEmnI7p9F44dO+bxheTmcNzw+vib8PHzrvWYp6oprNu0NUAIf62iV3vPkqZwtPOrt7BulWo9T2tEnhUEoaX45ptvAOgyuEODXxPbIZKgiACWLVumueueHj16AFCd5/mF9ap8+/fQs2dPlSOp26WXXgqAOefct6yKFawFOjp16tTqBsYcWsN1g16vZ8iQwUjV5VBdpnY4tUjF2fj4+NC7d29V44iJieGTTz7hhhtuoKIwl21fv0PeiYNuO3/qzg3s/P5jFJuFp59+mpdfftljBugdhfWcZhbWHYV5x/FaKkfBPD+zqM6vOx5PTk52U0TN4+gMaPD3nE5tRn/75HdXdTVs164dM2fORK6C6gzPW7VelaJDsdpX+Dom3XuK1pDThabz8fHh3//+N7GxsRzb8CvZh5q+QK00J529S7/C29ubd999h5iYGCdGqk3t27cHID8jpaaw3q5dOxUjUsfQoUPp378/J/emn3fVusVsZfe6gwQFBTFr1iz3BOhkBoOB119/neROnSg5mEPxwRy3nVtRFHJ+P0l1fgVXX301119/vdvOfckll/Dkk0+CVaJ8lwFzruflc1s5lO0wYCuRuPzyy3n00UfPu72guzS4sC7LMj/99BOTJ08mLi6uZsbf66+/Tnp6Ot9++y0TJ05Ep/PsPXgE17NYLHz//Q8YTT6EJtpvtIJjE/H2D2Lp0qWUl3v+QGVTrFmzBp1OT5uudV8kJ/YcgCzL/Pbbb26OTDuys+2rLvyCfPEL8qn1mKdy7POlyVbwNhmdXofB4Dkz9x1qBvHq6dSr2P72PA0QeVYQhJamtLSU77//Dr8gXxK7N3x1jSRJdB+WRHV1dU1hXisce/BVF1SoHEnzmU9/D1rdV/CCCy7A19cXS650zgXKljwJRba3j29NWuN1w6BBgwDQaWmfdXMluspi+vfvr4nJqAaDgfvuu4/XX38dvQS7l3xG5gHXbquhKApH//iFw2v/R1hYGJ9/9hlTp07VzKBXQ7Rr1w4vL69mr1h37K/eWgrreelFdX7dMUjvKYX13bt3A2AKa1hnHS0wBJjQeelrYneFm2++Gf8Af8yn9PVOWNciuQrMmTratm3L5MmT1Q6nQVpjTheaLjw8nPfeew9/f3/2L19EceapRh+juryEXUs+Q7FZ+de//tXiO604tG3bFoDCzFQKMu0rl9u0aaNmSKq5+eabAdixZv85n3dw83Eqy6u59tpr8fX1nDz5d35+fvzfv/9NSGgI+ZtTqcysu+uOsxXuzKA8pZCBAwfyyCOPuP36+Oqrr+bf//4/vL18qNivp+KIrmZMXMsUxT6xr2y7AbnKPlFu7ty5mtqqosEZOT4+nkmTJrF27VpuuukmNm/ezO7du5kzZw7h4eGujFFoYX766Sfy8nKJ6T4QvcE++CDpdMT1GkJ5eTmLFi1SOUL3O378OLt37yaxxwB8/OtuTd150BgA/ve//7kzNE1JTbVf9ASG+eMf7ItOp6t5zFM5Lkq0uFerbJHx8fbxqEExBz8/P+AcK9ZttZ+nBSLPCkLL5omfpc319ddfU15eQc8Ryej1jRsI7DKoAyYfLxYsWEBlZaWLImy8qKgo/Pz8sBRVqR1Ks5mLqggKCtJsOz9vb29Gjx6NXCkhn2Perfn0/q/jx493U2Ta0BqvGwYPHgyAVOy+VSbnoyuxx+Io+mvF+PHj+fTTTwkIDGD/8kVkHdrpsnMd37iclC1rSExMZOHChXTv3t1l53IVo9FIUlISeanHkG1NryDmpBwFWn5h3dEdJD+z7jbkeRmFhEeEExoa6s6wmuz3339H0kl4R2pn0vX5SJKEd1QAJ0+eJC0tzSXnCAoK4sYbbkS2QHW651zHVqXoQIF//OMfmpjw1BCtMacLzdO+fXvmzZsHKOz56XOqyxpeJJStVnb/+DnVZcXcd999jBo1ymVxao2jiF6UnU5RdjoREZEeXSxujqFDh9KhQweO7TxFZVnd97aKorD39yMYjUamTZvm5gidLyYmhrffehu9Tk/2byewVphder7ytCIKd2USFx/PvHnzVMtJI0aMYOHChSQlJ2HO0FG2Q6/p1vByNZTv01F5RE9gQBBvv/02t956q+bG1Bq8DDErKwuj0UivXr04efIkTz311HlfI0kSP//8c7MCFFoWq9XKhx9+iM5gpE3f2nvQxfcczKmt6/j000+ZPn16zUre1uCrr74CoNeY+veFDI6KpW23fmzatInDhw97zOxvZzpy5AjeviZ8/E1IkkRQRABHjx5FURTNfbg2lKMtmWzWYGHdbCU4OELtMJqkprBuk4A6lrlZaz9PC0SeFQShJSkuLmb+/Pl4+5noNrTx7bm9vI30GJHM1l/3snDhQm688UYXRNl4kiTRrl079h3Y79HXH4qsYC2tpl3Pzpr+HkaPHs3PP/+MJV9C7392PldsYCvU0bFjRxISElSIUD2t8bohPj6eqKgosgtysCkKaOBnVyq1F9b79++vciRn6969O/M/+YTrr7+eA78uwjsgmODYRKeeI2PfFk5uXk1iYiKffvqpZifqNESnTp3Yt28fhVlphMUlNukYuaeO4u8fQGxsrHOD05iAAPv3mJ9xdmG9utJMaUE5PYf1dn9gTZCRkcHu3bvxjvZHb/KsTm1+bYKpSC3i119/5aabbnLJOa699lq+WPAFRWmFeMVY0Xm55DROYysHc7aO5E7JHjXhrjXmdKH5hgwZwkMPPsjLL7/M3mVf0WfyLegasJrzyPqfKMlKZdKkSR7b2rupHPcLBVmpFOdl06d3L5UjUo8k2ferfvHFFzm05QS9R5/dxSzrRB6F2cVceumlHjNZ7nx69+7NQw89xIsvvkj22uPEXtQJSef8ewpLWTW560/i5eXFW2++qfq2JB06dGDhVwt58803WbBgAWU7DJjiZbzbykgaWQSuKGDOlqg+pke22idVv/DCC0RGRqodWp0addVosVhYt25dg5+v5UEaQR1r1qwhNTWV+F5DMfnVXpltMPkQ13soJzet4qeffuKqq65SKUr3ys3N5fvvvyckKp4O/Yad87kDLplOyr5tfPzxx7zyyituilAbKioqSElJIbZDZM1nS3hcMEe2p5Cenk58fMNbzGqJ48LEWmFROZLaFEXBVmklrL1nDo6dd491W+3naYXIs4IgtBTvvvsuJSUlDJvUFy/vps3M7jWyM/t+P8IHH3zAZZddRkSENiZ7xcfHs3fvXmyVFgy+Gh9hroe1wowiK5q/fho2bBh6vR5LgYJ327MnIVqL7W3gR44cqUJ06mtt1w2SJDFgwAB++uknqCoFn7o7fbmTriQHf39/za5QTk5O5u233+amm29m37KFDJo5B4PJORPYywuyObTmfwQFBfH+++97dFEd/mpbnpt6vEmFdZvVQkHmKXr36uXxv2sNkZyczNq1a6kqr8bb76+9yQsyi2u+7gm+//57FEUhoIPn/fz6tQ0mb5OO7777jhtuuMElbcJ9fX256867mDt3LlUndfgma28LOwdFgcqj9tXq9993v8e1TW9tOV1wjhkzZrB7926WLl3Kyc2raD/k3BNKco/uI23XRjp16swTTzzR6n6O/Pz8CAoKIv3wXhTZRlxcnNohqeriiy/m1Vdf5fC2k3UW1g9vOwHAFVdc4ebIXGv69Ons2LGDZcuWUbgrg9A+zv05UGSFnN9OYKu28vRzz9VsoaM2k8nEww8/zNixY3nqqadITU3FkqfDJ8mGMeQce6+5ga0SKg/rsBbp8PPz48EnHmTy5Mma/oxqcGH9xIkTroxDaCWWLFkCQHyvoXV+Pb7nEFI2r+bHH39sNYX1+fPnYzabGXP5teh0554i1K7XIKISk/nll1+4/fbbadeunZuiVN+BAwdQFIWIhL9myEUkhHJkewr79u3T/MBwfWJiYgCwllerHElttkorik0mOjpa7VCaJDDQPtBab2HdYk/Mas8YPJPIs4IgtBQ7duxg4cKFhEQF0n1441erO5h8vBh0aS/Wfr2Z559/nrfeeksTN1aOARhLabXnFtZL7dcdWr9+8vf3p0ePHuzctRPFCtLf7l6thfafB0eL8NaktV439O/fn59++gldSQ6y2oV1cyVSVSn9Ro7U1H5/f9e/f3/uvOMO/u///o8Tm1aRNOJSpxz38Nofka0Wnn/++RYxMO1ob56bepzOg8c0+vUFGaeQbTY6duzo7NA0qVOnTqxdu5a8jELik/66Z3SsYtfKIPK5VFZW8t///he9yYBfYoja4TSa3suAf7tQTh05xbp16xg9erRLzjNlyhS+/vprjhw5gle0jEH9OU11suRKWIt0jBw5kqFD6x5z1KrWmtOF5pMkiaeeeordu3dzcvNqwtt1ITC67i5O5ooyDq5ajMlk4tVX/4W3t7ebo9WGmJgYDh48COCxY57OEhQUxIgRI1i1ahWF2cWERP01RirbZI7tSiUyMpIBAwaoGKXznfl7k747HZ/YQHyinLfwqmhPJlU5ZUycOFGTkxL69+/P4sWLee+99/jss88o3w1eUTLeHWR0bu5Wr8hQnSZRnaJHkWHUqFE8/vjjHvG72eDCetu2bV0Zh9AK2Gw2Nm3ahF9YFH6hda84MvkFEBSbyK7du6moqGjx+5zk5eWxaNEigiJi6HbBRed9viRJDJ18Pd/Pe4wPP/yQF1980Q1RasOePXsAiGrz10zyqDbhNV+bMGGCKnE1V0BAAKGhoZQWn2PzUBVYiu372XrqZ7+/vz86nQ7ZUveMO/l0wV1LhXVPfa8FQRDOVFBQwEMPPQQSjJ4+CL2hecWmLoM6cHTHKdasWcNXX33Ftdde66RIm87RQtBaWg1OvAF3J4uHFNYBBgwYwM6dO7GWSmfNpLeWSBgMBnr37q1OcCpqrdcNjpbrUmkORKlbwNSV5gLabAP/dzfccAM//PADabv/pO2A0Xj5NG87pOKMFApOHeGCCy5wWTHP3Tp06ABAXlrTClyO17WWwrpjRXp+RlGtwnpeRhHw10QFLVu8eDFFRUWE9IpB18zrFbUEd4+m9Ege//ngP4waNcolExANBgNPPfUUs2bNovKQHv9+NiSNLQaXLVB1TI+3tzePPPKI2uE0WmvN6YJz+Pn5MXfuXK6//noOrvqOAdfcjVRHx4ajvy/FXFnOI4880qoWav1dZGSkKKyfYdy4caxatYrju9PoN+6vMdLM47lUlVczedKFHtcBpCH8/f15+eWXmT17Nrm/nyT+8q7ojM2/FqjOL6dwVybR0dGa7grh4+PDfffdx8SJE3nmmWfYv38/1kId3h1tGMMVt+y4ZS2FysN6bGUSYWFhPP7441x44YWafc/+ruX9VgialZ6eTmVlJYFR5x7AC4yKR7bZOHbsmJsiU8/nn39OdXU1gy6fid7QsHkuHfsNJ7JtEj///DOpqakujlA7du/eDUBUYnjNYxHxIeh0upqveaqkpCQsJdXIFu3ss15daC+se8KASF30ej3BwcEo9XTYV8z2/28pewQJgtqs1nraQwitSnV1Nffeey9ZWVkMurgX0YnNb90uSRJjZwzBL9CHf736LzZs2OCESJsnMTERAHNxlbqBNIMjdk8YVOvZsycAtpLajysyyGUSnTp1arUrblqjNm3aEBERgb4k1973V0VSiX1/dU9YxePl5cV1112HbLWQdXBHs4+XvnczYC/YtxRhYWGEhISIwnoDOe4T8zOLaj2en1mEwWDQfH6pqKjgww8/ROelJ6hrlNrhNJlXkDf+7ULZt3cfa9euddl5evfuzTXXXIOtQqIqRXtDyZVHdchmuPvuuz1i0qAgOFu/fv2YPHkypbkZZB7YdtbXS3PSydy/ja5duzJ9+nQVItSOM7cX8/RtbJxhxIgR6PV6Tu5Pr/X4yX1pAIwZ0/guPp6id+/e3HjjjVhKq8nfln7+F5yHYpPJ+f0kiqwwd+7cmm6qWtalSxe+/PJLHnjgAYySiYr9eir223OqqygyVJ7QUbbDgK1MYsqUKSxZsoRx48Z5TFEdRGFdcKOsrCwAvAPP3WLLO9Be6MrMzHR5TGoqLi5m0aJFBIRG0mPkxAa/TpIkBk+6DlmWmT9/vgsj1A5FUdixcwf+wb74B//VxcDgZSAsLph9+/ZhsWhrj/LG6NatGwDVedpZtV6da4+le/fuKkfSdBERESjmuhOybJYIDAzEy8szW/gKgtYUFhaqHYLmKSoXgFzNZrPx2GOPsX37dpL6JdJnzNl7tDWVX5APF914ATqdjnvvvZf9+/c77dhN4SgmVOdXqBpHc1TnV6DT6WpWaGqZ41rEVlY7p9sq7DflnnytIjSeY591LJX2fdZVpCvJxs/PT7P7q//dRRddhE6nI+9Y8z5DFUUm78QBoqKiPGK1fmMkJSVRlJ2OpbrxE6dyTh2rOUZr0KZNG0wmL/JPr1AH+7VOYVYx7du3x2h0cy/RRlqwYAEFBQUEdY1Cb2pwM09NCukdC5LE22+/jc3musn699xzD7GxsZhTddi0M3SBpUDCkqOjV69emuhsJAhq+cc//oHJZOLEnyuR//ZZcHzjCgAeeOABTW9f4w5nLrARhXV7J9U+ffqQk5JP1RnblJ46kImvry99+/ZVMTrXu+OOO+jYsSMlB3OozG7evUXh7kzMhZVMnz6dQYMGOSlC1zMYDMyePZvvv/+eAQMGYMnTUbbNgCXP+UVuWzmU7dBTfUpHXGwcH3/8Mc8884xHTEL4O1FYF9wmLy8PAJPfuX9RvPz8az2/pVq4cCHl5eUMuGQaekPjbjqTB44gNCaBH374gZycHBdFqB1ZWVnk5eYR1Tb8rK9Ftw3HbDbXtPHxRI72pZXZZeoGcpqiKFTllBEWFubRs72jo6NRrHXvs65USzX72wuC0HzFxcVqh6BZLb2g7vDaa6+xfPly4jpGMWb6IKfPNI5qG86464ZSWVnJnXfeQXp682eUN1VgYCAdOnSgOrccxSarFkdTyVYZc145ycnJ+Pk1rx20O4SHhxMaGoqtvPbPlHz63452xELr4Rio0pVkqxfE6f3V+/fv7zED1KGhoXTq1InirFNnDbg3RkVhLpbKcgYPHuxRq0oaIjk5GUWWyU1tfPe8nJQjREZGEhwc7PzANEiv19O+fQcKs0tQZPu1TllhBeYqi+YnFxQXF/PJJ5+g9zES3M1zV6s7eAV5E9AxjKNHj7J06VKXncfX15cnnngCRYHKY9oYTnbEotPpeOqppzzm81gQXCEiIoKrrrqKqtIico7sqXm8vCCHvBMH6Nevn0d02XG1M/N0a8nZ5zNs2DAURSH9mL3GUFZUQWFOCQMHDtT8RLnm8vLy4rnnnkOn05H7RwqytWn399WFlRTtySI6Opp7773XyVG6R0JCAh999BEPP/wwBrwo36en8qgOxUlDHtVZUs0q9alTp7J48WIGDhzonIOrQBtXQkKr4BgA9Q4IPufzfALsK9ozMjJcHZJqSktL+fzzz/ENCKLn6Msa/XqdTs+gy2disVj4+OOPXRChtuzatQuAqMSzZxI6WsN7cjv4/v37o9PpqMwsOf+T3cBSUo213MygQc4vjLiTo3Au/23BiaPYLgrrguA8rlwd4+kcn6Oe/Hl6Pl9//TULFiwgLCaYi268oNn7qtenXY94LpjSn/z8Au666y7Ky9VbLjV8+HBki42KTHVXzDZFZUYJslVm2LBhaofSYElJSciVEsoZHzWOQrvWCziC8zkK6/qMA0hFmar80afa708GDx6s5lvRaN27d0e2WqgobPrk7NKcjJpjtTSOTmJZxxo3abusKJ/S/Jya17cWSUlJWM1WSgrs+bggqwhA891QPv/8c8rLywnuHu2U/VS1ILR3LJJO4v3333fpFk0XXHABQ4cOxVqow6qB4QtLroRcYW8jKybaCQI1XRsy9m2ueSxj3xYAZs6cqUpMWnPmylhPXCXrCo4JFxlH7JNWM04X2D256NkYPXr04LrrrsNSUkXRnsZ3UFYUhdwN9hbwTz/9NL6+vud/kUbpdDpmzpzJokWLaN++PdXpOsp26pGrz//a+igyVBzSUXlIj79vAG+//TZPP/20R0zyPxfP7nckeJQDBw4A4Bd27hnBvqGRgFTz/Jbogw8+oLS0lFEz7sTL26dJx+g2fDx//u8LFi1axPTp0zW/h1lz7N27F4CoNnUU1tvaH9uzZ89ZX/MUAQEB9OjRg917dmOrtqreiq4izb7ydMiQIarG0VyO1fa2Kgm9/18rRm2Vtb8uCELz+fv7qx2CoJJ9+/bx8ssv4xPgzcW3jMTk49otNroPS6I4r5Rdaw/y3HPP8corr7j0fPWZMGECn332GaVH8vCLD1IlhqYqOWLvCjVhwgSVI2m4xMRENm3ahFwJ+tMfN7bTnfhb8jWwULe4uDjatm1LSkoKxkPrVI1l6NChqp6/sRwTUcrysvEPb9ok07L87FrHakl69eoFQNqh3fSdMKXBr8s4bL9f7dmzp0vi0irH529hdjFB4f4UZNsrrVourFdWVrLwv/9F72MksFPE+V/gIQx+XgQkh3Pq4CnWrVvH2LFjXXau66+/ng0bNmDO1mEIVLdzjznLPsnuhhtuUDUOQdCK+Ph4BgwYwJYtW6kuL8XL15/sQ7sICgpm5MiRaoenCWeOXYhxDLuuXbtiMpnIOpkLQNYJ+/+39DbwZ7rzzjtZvnw5WXuy8G8Xildww+s1JYdyqc4t5+KLL2b48OEujNJ9OnTowMKFC3nhhRdYsmQJZTsM+Ha3Ymjkr4xsgfJ9emzFEt26dWPevHnExsa6Jmg3E4V1wS0sFgubNm3CJzD0vCvWDV4mAqLi2LFjB+Xl5R4/e+Xvdu/ezRdffEFwVFyjbtb/Tqc3MPraf/Dd64/w9NNP88knn2AwtMxf6Z07d6LT6QiPDz3ra4Fh/nj7mdi5c6f7A3OikSNHsmvXLiozSvBvd/b36U4VaUVIksQFF1ygahzN1aZNGwDkytqPy5X2m29RWBcE5xEzvc+vJbaEt1gsPPbYY1itVi6+bgQBIe65ZhtyaW+yU/JZunQp48aN48ILL3TLec/UvXt3unfvzt59ezEXV+EV5O32GJrCXFRJRWoRffr0oUuXLmqH02CO4o2t8q/JcnKlRHBwsGjh2Eq9+uqrbNiwQdUY4uLiaN++vaoxNJaj4Fle0PQ2+uWnC+taLp42VXx8PFFRUaQe2ImiKA3uNnNq/3aAVtdi1/HzX5hdTGK3OIpOF9a1/HuxYsUKSktKCOkVg87Qspp4BnWJouRgLosWLXJpYX3QoEEEBARQXqjuknVFBluxjs6dO5OQkKBqLIKgJWPHjmXLli3knzxIQEQc1WXFTLj88hbf0ruhziyme3m5dlK4pzAajXTr1o0dO3dgMVvJTsnHZDK1qk4gju1O7rrrLnL/PEXshOQGXQdaKy0Ubs8gICCABx980A2Ruo+vry9z584lOTmZ119/nfJdBvy6WzE0cF2BXA3luw3YKmDixIk899xzeHt7xrhJQ7TMKpwLlJeXY7FYVI3Bz8/PY5Pgxo0bKSkpIaFPw2btRLTvyvHsNFavXs1llzW+VbpW5eXl8cCDDyIrChNvfQSDsXkJvEPfoXQZeiE7NqzkzTff5IEHHnBSpNpRVlbG3r17iWgTitHr7I8sSZKIaR/BiT1ppKenExcXp0KUzTdq1Cjefvttyk8VqVpYt1Vbqcwqo1fPnoSFnd0hwJO0bdsWcBTS/ypoOQrtYoWbIDhPUVGR2iEIKvjqq684fvw43YcnEZ8U7bbz6vQ6xl4zmP/+aymvvPIKw4cPd/sNmiRJ3Hrrrfzzn/8kf0sq0WM7ar7dv6Io5G06BcDNN9+scjSN8/fJcooCcpVE26S2KkYlqKlLly4eNTlEK/5asd74NpcOZXmZhIdHtMhJLZIkMWTIEH744QeyTx4mul2n875GURSO7/yTgIAAunbt6oYotSMxMRGA4lz7tihFOSXo9XpN35P/9ttvAPh38Ox73bp4BXljCvdl8+bNVFRUuKwVrU6no2vXrmzatAlFBkml+Qlylb243tq2YBCE83F0nyxMO4612r43oqd12HGlPn36MHPmTDEm+Dddu3Zl+/bt5KUXUpBVRLeu3T22DtVUI0aMYOzYsaxatYqyEwUEtD//tULBtjRsZiv3PHQP4eHhbojSvSRJYvbs2cTFxfHggw9Svgd7cT343K+Tq6FslwG50t5V5t5779X8eEljicL6eeTl5fHqq6+ydOlStUMhOCSE+++7j0mTJnncD+I333wDQEzXfg16fnTnPhz/cwXffPNNiyms5+fnc/vtt5OZkcGI6beR0KV3s48pSRLjb7yf3FPH+OyzzwgKCuLmm2/2uJ+Pc1m7di02m422XepvE9K2aywn9qSxcuVKZs+e7cbonKdjx44kJCSQnpaBYpOR9OrcnVakFoGiMGbMGFXO70wJCQnodDpsFbVXidoq7L8fjsK7IAjNt2bNmpq/Hzp0iE6dzj8Q3Vq0xJXqAAUFBbz//vt4+5kYOLGX288fFBFA79Gd2bZiH59//jm33nqr22MYNWoUQ4cOZcOGDQ2+8VZT2bF8KjNLGTlypMd1pfmrsG6fLKdUA/JfjwuC0DAhISFEx8RQkJXaqBXZDtXlpVSVFDJ41CjXBKgBI0eO5IcffuDott8bVFjPSztBUU4GEydObHUD0I77raLThfXivFLi4+M1/T7s3rMbg68RY4BJ7VBcwic6kKK8LA4dOkSfPn1cdp6ICHsbfcUCkkpvpWK2/39kZKQ6AQiCRrVr146goGCKM05irbLPSnXl54GnMZlMPPzww2qHoTmOMZxjO09hs8qtdkzn4Ycf5vfff6dgazp+CcHojPp6n1uVV07p0Xy6du3K1KlT3Ril+1144YW89dZb3DPnHir2gV9vK/p6GhYqVijfq0euhDvuuIM77rijRdWqHFpW3yMnUhSF77//nssvn8TSpUuRfYOxhSao9kcOiaeopIwnn3ySW265hVOnTqn9FjXY0aNHWbduHUGxiQRENGwPBZ+gUMLbdWbHjh3s2LHDxRG63rFjx7hu1iwOHTpEvwlTGXTZtU47tsnXn6kPv0pQRAxvv/02L730kurdFZxFURQWLlwIEiT1Taz3ee17tsFg1PP1119jtVrdF6ATSZLEhRdeiGyxUZFZqloc5aeKAFzaOs5djEYjcfFxKJW1k7dcKeHl5UVMTNP2lRQEobbDhw+zadOmmn9/8cUXKkajPY7Cuiyruwels73xxhuUlZUx4KIeePuq00Kvz5iu+Ab68OGHH5Kamur280uSxBNPPIGvry95G09hLq5yewwNZS6qJO/PU/j7+/PYY4953I1tbGwsOp0O+fRbbKsS27oIQlP179cPc0UZ5QU5jX5tUdpxoGXvuTls2DC8vb05+OeaBk2OO7TJPrlQjW1J1GY0GomOjqYkvwxLtYWK0irNt+QuyC9A7+vlcXmwofS+9kkNBQUFLj2Po5WyouLwi2Kz/zdsadtHCkJzSZJEly6dqSwuIO/EAYKCgsT4l3Bejm1cTuxNA1rmlj8NERMTw4033oi1wkzRvvq3TlIUhfzN9jGIhx9+GL2+/gJ8SzFixAhefOFFZCuU7zXUeQ2gKFBxSIetTGL69OkttqgOorBep61btzJz5kyeeuopSisqsSb2x9p9ArakYar9sSYPx9xzInJIHJs2bWLSFVcwb948zbdetdlsvPjiiyiKQruBjVsBmzjQXtjz5EKxLMssWrSIadOmk3rqFMOm3MiYWf90+gdKYFgUM555l4g2HVi4cCEzZ87k+PHjTj2HGlasWMHu3btp3yOBoHD/ep/n7etFl0EdSE1NZdGiRW6M0LkcgzHlKYWqnF+22KhIL6Fjx44tZjV3u8R2yOa/bvgVxV5Yb9OmDTqdSIGC4Ay//PILAD0vm41vcDjLly/32ElOrmCz2QCoqtJu0bWxfv75Z3744Qci24TSbUhH1eLw8jYy/Iq+VFVV8dBDD6nyHickJPDcc88hW2xkrz6KrVp7P/u2KgtZq48hW2Xmzp1LbGzDJrpqidFoJDIq8vSK9b9awmu53bAgaJWjHWz+iQONfm3e6de05JayPj4+jBw5koKMFHJSjp7zuYqisP+PFfj6+npcJxBnSUhIoKyogsIc++RwrX8um0wmZKtN7TBcRrHaJ3KaTK5dRh4QEHD6fC49zTk5zu2IRRCEv8yZM4eZM2cyc+ZMnn/++RZb2BKcxzEOXFpQXuvfrdH1119PREQExfuysVXVXZOqSCumKqeMcePGtegJp383ceJEbr/9duQqqDhy9ri6OUvCkqdj8ODBPPLIIy36s0dUFU6zWq2sWrWKWbNmccMNN7B7925sYW3sxeyojqCFHwIvX6xJw7EkDcOi82L+/PmMGz+eV155RZVVOudjtVqZO3cuW7ZsIaJjd0LbJjfq9UHRCcR2H8CBAwd47LHHMJvNLorUNfbv38+sWbN4/vnn0XmZmHz/ywybcoPLPlACQsK59pn36Dn6Mvbv38+UKVN44403qKiocMn5XC0zM5Pnnn8Og1HPkMt6n/f5/Sd0x+Trxbx58zh8+LDrA3SB7t27ExkZScWpIhTZ/a2DK9KLUWwy48aNc/u5XcXRItbm2JPVYr8Bd+wHKAhC82VlZQEQEBmLf0QMlZWVlJSUqByVdhQXFwNQWKjOpClnW7lyJU8++SQmHy8uvHYoOpW2LnHo2KctXQZ1YO/evdxzzz2qXPdMmDCBG2+8EXNxFVmrjyJbtdOdQLbayFp9FEtJFbfddptHd6SJjYlFNkv2SXLV9utpT5wkIAhqGzFiBAaDgexDuxr1OpvVQt7x/SQkJJCc3Lh7e09z6aWXArDv91/P+bzMo/spyk5n7Nix+Pj4uCM0zXF8DmedyAW0X1hPTEzEWlKNbGmZxfXqAvt1kKsLIo7Vr3KVemOlttOT7cRKXEE4W7du3Xj44Yd5+OGHGT16tNrhCB4gKCgIP/+/OoC05vssX19fbrnlFmSLrc5V64qiULgzA0mS+Mc//qFChOq67bbb6NWrF5YcHZbCv64DZAtUHdcTGBjICy+80OJX8bf6wnpRUREfffQRF02cyJw5c9ixYwdycByWbuOxdRwKXr5qh1ibJKGEJmDpeTHWtn2plCUWLFjAJZdcwp133snvv/+uib08jx49yk033cS3335LQGQsXcdNbVJBOXnUJILj2vHLL78wc+ZM9u3b54Jonevw4cPcd999TJs2jV27dtFl6IVc//KndOw3zOXn9vL24aJbHmLy/S/jFxrBJ598woSLLmL+/PkeVWDPzMzkpptvoriomOFX9iMo/PwzkH38vRlzzWCqq6u59bZbOXr03KsLtEin0zF27Fhs1Vaqst3fDr48pQhoGW3gHRwDCn9f4ab1FoWC4EmCg4MBMFeWY64sR6fTERgYqG5QGlJRaf/g8aQ8XBebzca7777L/fffj6SHiTeNIDhSG/+dR0ztT2L3ODZs2MB1112nSteee+65h4kTJ1KVXUb22mMoNvWL64pNJmv1Mapyyrnsssu466671A6pWWJiYkCx76mqVNsfi46OVjcoQfBAQUFBjBgxgtLcDEpzMxv8upwje7Caq7n44otb9OoTsLeDDw4J4cCGFci2+pfkOgrvl112mbtC0xxHUTPzeG6tf2vViBEjUGSlZgu0lkS22KhMK6Zdu3Yu3yqlW7duAFiLVCysFztaXndRLQZBEISWJDrqr3ur1n6fNXnyZCIiIig5lHvWZLyqrFKq8yu46KKLalrotyYGg4EnnngCSZKoOq7DUYqsTtGhWOEf//gHkZGR6gbpBq22sF5eXs68efMYN24cb731Ftm5+diikjD3vBhrpwtQ/EPVDvHcdHrk6GQsvS7F2nEoNv9w1q9fzx133MGUKVNYv36920NSFIXt27dz3333MXnyZLZv305kck/6Tr0dg6lps7f1BiO9r7ypZuX69OnTueuuu9i4caOm9ipVFIWtW7fyz3/+kylTprBixQpiO3Zj+pNvc9k/niYgJNyt8XTsN4wb//UFF1x9C1VmC/PmzWPCRRfxzjvvkJ+f79ZYGkNRFFatWsVVV00l9VQq/Sd0p2sjWsy26x7PiKkDyM/LZ8aMGXz//fea+jlpCMdq8TI3t4OXrTIVacW0bdu2Ra1AcayYcOzJ6phRLwrrguA8jo4y0un/AR67hYuzKYpCaYl9olSxB6/i379/P9deey3vvfce/iG+TLprLLEdtHOjpDfouej6C+hxQTKHDx/mqquv4qOPPnLrz6FOp+OFF15gxIgRVKQVk73uOIqK1yCKTSZr7XEqM0oYPXo0zz77rMcXwhw353K1/c+ZjwmC0DiTJ08GIH3Ppga/Jn3PJiRJ4sorr3RVWJphNBq5eOJEyosKSNm3vc7n2KwWDm5cRUREJAMHDnRzhNrh+BzOSc2v9W+tuvTSS9Hr9RTtzlSlS5wrFe3LRrbKTJo0yeU5v1OnTkRFRWLJ06GosPhfrgJrsY7evXvXTPIVBEEQmic83F6/MJlM+PpqbLGpm5lMJqZPn45stlF6vKDW14oP2icTXnfddWqEpgmdO3dmwoQJ2MokbCWg2MCcrSM6OpqpU6eqHZ5btMrCeklJCdOvuYb58+dTqeixtu2Luc/l2BL7gY82Vt40mKRDDmuDtetYLN0nYAtP5MjRo9x555188sknLj+9oigcPHiQd955h0suuYTZs2ezYsUKAiLj6X3FjfS4+FoMXs3b20lvMNLlwqn0nXobwXHt+e2337j11luZMGECb775Jnv27FGteGqxWFi2bBkzZszghhtuYM2aNcQld+eqh1/j2mffo02XPqrEBWD0MjHkilnc9tY3DJ96E9VWmffff5/x48fzzDPPcOzYMdVi+zvHxISbbrqJOXPmUF5Rzqhpgxh4Uc9GH6v7sCQm3HABMjaeeuopZsyYwfr16z2mwN63b1/CwsMoT3FvO/iK9GJki43x48d7/MD7mf4qrJ9esV5V+3FBEJpHURR+//13DCZv/MIiCYptiyzLbNiwQe3QNKGkpATb6ZVu5WVlHretTW5uLk8//TTTp09n3759dB7Ynqvuv4iIeO1NQNXpdVwwuT8TbxyB0aTnrbfeYtIVk1i1apXbujkZjUbmzZvH4MGDKT9VRPba46qsXLcX1Y9RkVrEsGHDeO211zAajW6Pw9kcAz2KWUI2SwQEBrh8D1lBaKmGDx9OdHQMWQe3Y62uOu/zS3MzKM44yfDhw1vNdfQll1wCwP4/VtT59RO7NlNZVsLFF09s8a0uzyUiIgL4a09Wx7+1Ki4ujqlTp2IurqL4wNntXT2VpaSK4r3ZhIWFcc0117j8fHq9nqlTr0KxQnWG+8cPqlJ1oMBVV13l9nMLgiC0VBMmTCAmJoZJkyapHYomTJ48Gb1eT+mRvJrHbNVWKlKL6NKlC927d1cxOvVNmzYNsBfULXkSihWmTJnSIsYeGsKgdgBqWLduHSdPnEDxDsQa3wN0OqSSHLXDcgo5NAHFJxBD6m7mz5/PjTfe6PRzVFZWsmXLFn7//XfWrVtHRkYGAHqjFzFd+xHTbQDBsYlOL86FxLen31W3UZyVSsbezeQc2c3HH3/Mxx9/TGRkJCNGjOCCCy5g8ODBLp9VVVFRwaJFi1iwYAHZ2dlIOh2dBo5iwCXTiU3q5tJzN5bJ15+hk69nwCXT2bv+F7YtW8TixYtZvHgxw4YN46abbmLAgAGqxJaVlcWKFSv49ttva9q2tusez+DLehPSjPayHXomEJkQyqafd7Fv2z7uvPNO4uLimDJlChMmTKjZd1uL9Ho9F024iC+//JKKjBL84oPcct6yY/YVBhMnTnTL+dzF0brIsbLNsSdra29pJAjOkpOTQ3p6OpFJPdDpDYS368zJzavZvn17i9pWoqkc+887ZGZmunzPS2eoqqris88+4+OPP6ayspKw2GCGX9mPuI5Raod2Xu16xBOXFMWWX3azZ/0R5syZQ//+/XnwwQfp2rWry89vMpn497//zT//+U82btxI1trjRI9qj+Smvehlq0z22mNUpBUzfPhw3nzzTby8vNxyblcLDbVP6JAtgEUiNFx7EzwEwVPo9XqmT5/Gm2++SeaBbST0Pve2ZWm77BPm3FGw04oePXoQHx/P0a3rsZqrz1owcPDPVQBcfPHFaoSnGWFhYef8txbdddddrFi5goLtGfjEBGIK9exVeYpNJnv9CWSrjUceecRtqwyvvfZavvjiC0pPleAVZUXnpssNWxmYM3W0a5fY4sYvBEEQ1HTVVVeJCUtnCA8PZ9CgQWzYsAFruRmDnxflp+wL4VrD1kjn07dvX0JCQiguKKxZUODoxNsatMrCet++ffEPCKCstATj0T/UDsdlRo0a5bRjpaSksH79en7//Xe2bN2KudpepTKYfIju3IeIjt0Ja5uM3uj6K+mg6ASCohNIHnU5BaeOkHtsH3nHD/Dtt9/y7bffYjAY6NevH8OHD2f48OF06NDBaR90FouFRYsW8eGHH5Kfn4+Xjy/9J15N3wlTCI6Mdco5XMVo8qbPhVfQe8zlHNu5ka1LF/HHH3/wxx9/MGTIEO65556afbJcxWq1sn//fv744w/WrFnDgQMHAHsL1+T+iXQflkx0onPa5geE+HHhzKH0HtOFvb8f4ci2k7z99tu8/fbbtO/QnrFjxjJs2DB69uypuZlUkyZN4ssvv6T0SK5bCuvWSgsVacV07dqVpKQkl5/PnXx9fQkMDKCs2t6CWRZ7sgqCU2Vn21f6eAeGnP7/0FqPt3YnTpwAICA0ktKCHE6cOKH5wvqOHTt44oknOHXqFL6BPoy6fBCdB7ZDp/OcRlde3kaGXdGPbsOS2PjjTrZu3co111zD7Nmzueuuu1y+ytnb25u3336bOXPm8Mcff5C1+ihRozuiM7j2PZStMlmrj1KZUcKIESOYN29ei1rR7SisKxZ7cT0kJETliATBs02ZMoV333uPtF0biO81BEmq+zPKUlVB9sGdtGnThmHDzl2Ab0kkSWL8+PF88sknpOzbToc+Q2q+ZrNaOLZjI3Fxca1+f+czC+ne3t4e0To2JCSEuc/P5c477yRr9VHiLu6MwdczJ6EpikLuxhSqc8u57LLLuOiii9x27oCAAO655x6ef/55Ko/q8Ovq+i49igIVh/WgwCOPPIrB0CqHtQVBEAQ3GTt2LBs2bKAirZjAThFUpBXXPN7a6XQ6BgwYwPLly5FzJcLCQlvVnvOavAKprq7mqaee4osvvqCwsJCePXsyd+5cp814iIuL49tvvmHdunUe0x66scLCwhg/fnyzjnH06FGWLl3KqlWralYTAwRExBKb2ImwxE4ExrRBp1On7ZneYCSifVci2ndFkWVKstPIP3mI/JOH2LRpM5s2beL1118nISGBsWPHMnHixGatVCopKeG+++5j06ZNePn4MmzKjfSfeBUmX38nfleuJ+l0dOw7jI59h5F5/CC/f/MRGzduZPPmzTzxxBNO3QdDURSOHTvGpk2b+PPPP9mydQvlZfYWcXqDjjZdYmnXPZ4OvRLw9nPNwG94bAijrh7I0Mv7cHx3Kif2pnHq0Ck+/PBDPvzwQ3x8fOjbty+DBw9m0KBBdOrUSfXiQZcuXejSpQsHDh7EUlaN0d+1g+Ilh3JRZIUpU6a49DxqiYyMojTFvsexYpbw9/f3iAEfLWhOPk5PT+fee++1X2DJMqNHj+aNN95oVRdZLZmiKOzbt49XXnkFgMCoBAC8fP3xDghm9Zo1LFy4kEmTJrXq37fVq1cD0GvMZfz+7cesXbvWqRMfnW3Dhg3ceeedyLJMnzFd6D++O0aTtiafNUZwRCATbxxB+pFs1n6zmfnz53P06FHeeustl0+q8/b25q233uL+++9n3bp1ZK0+SvQY1xXXZYvNXlTPLGXs2LG8+uqrmps42FyBgfZuRnK1BAoEBbmnq4/g+vtzQR3BwcFcPHEiP/zwA4WpxwhtU/cE28z9W7FZLVxzzTWq3ye52+jRo/nkk084uv2PWoX1tEO7qa4oY8yUK1v9aqUzP4s9acLTBRdcwL333ssbb7xB5oojxIxPxuDjWXlTURQKtqVRejSfnj178tRTT7k9hqlTp/Lzzz+zfft2zDkKXpGu3X6nOlXCVipx6aWXMnToUJeeqyUS+VwQBKFxBg4cCEBldikByeFUZZcRExNDQkKCypFpQ3JyMsuXLwdFolOnzq3quliThfXrr7+eb7/9ljlz5pCUlMSnn37KxRdfzJo1axg+fLhTzhEXF8eMGTOccqyWZvv27bz99tts27YNsLd4j0zqQXi7LoS2TcbkF6ByhGeTdDqCYtoQFNOG9kPGYa4spyDlMHknDpJ54iCffvopn376KT169OAf//hHky7AH374YTZt2kTygJFMuPlBfAI8fzAvpn1nrnr4NVL2been/3uGZ599tqatflMVFBSwdu1a/vzzTzZt2kRBQUHN14IjAujWK4n4pCgSOsXg5e2+G1cvbyOdB7an88D2WM1W0o5kk3Y4i7Qj2TUr98E+MDBgwAAGDRrE6NGjiYpSp+3trFmzePTRRynel034INe1rpctNkoO5hAUFMRll13msvOoKSIigqNHj6LI9sJ6eKxzuiK0Bk3Nx2VlZYwePZri4mIee+wxjEYjb7zxBiNHjmTnzp0e0SJS+IuiKOTl5XHixAmOHj3Kvn372Lp1a81WMNFd+hKZZN9bSpIkuo6/mt0/fc6LL77I66+/Tp8+fejduzdJSUl06NCBhISEFtOaui4VFRXs2LGDxYsXs2LFCqISkxl0+UwObFzJ4sWLqaio4IorrqBv3754e3urHW4tc+fOBQkuv3OMR7R9b6i4pCiufmAiv376O+vXr2f16tVMmDDB5ec1mUy88cYbPPjgg6xatYqsVUfsxXWjcyelyhYbmSuPUJVdxvjx43n55ZdbXFEdziisn94OOiBAe/ckLZU77s8FdUybNo0ffviB9D2b6iysK4pC+p5NmEwmLr/8chUiVFePHj0ICgri5O7NKIpSM1h4YtcmwF6cbe18fHwwmbyorjZ73ISnG264gZycHL788ksyfzlkL677ecY1qiIr5G0+RcnBXNq1b8fbb7+tynWlTqdj7ty5TJkyhaojlegDrOh9XHMuawlUndQTGRnBI4884pqTtHAinwuCIDRO27ZtCQ4Opjy3HGuZGVuVhd69e6sdlmbEx8fX+ffWQHOF9c2bN/Pf//6XV199lQceeACwF5i6d+/OQw89xIYNG1SOsGVbuXIlDzzwADabjbC2ycT1GExoYjJ6g2cNznn5+BHduQ/Rnfsg26wUnDpK+p5N7N27l9tvv51nn32WK6+8slHHPH78OCZffybc8hA+/k3f/1uL2nbry5ArZrPyszc5fvx4owvrsizz888/89NPP7Fp05/YTu+r4R/sS6cB7YhLiiI+KRr/YG2sWjR4GUjsFkditzgAKkorST+STfqRbNKOZrNy5UpWrlzJiy++SN++fbn44ouZPHmyW9uMTZgwgf/7v/8j43AmQd2iXLZqvXh/NrYqKzNvmomPj4vugFXmKOIqZpDN9j1yhPNrTj5+9913OXLkCJs3b2bAgAEATJw4ke7du/P666/z4osvuuV7EM7ParVSUFBAbm4ueXl55OTk1PzJzs4mIyOD9IyMmi1gHIzevkR16k1s9wGExNfeciUkoQNDZj9I+p4/yTmylz//tHctOVNERARxcXFER0cTGRlJZGQkUVFRhIeHExERQXh4OH5+fm55DxrLYrFQWFhIYWFhzR7zjj9paWkcOXIEq9UKQHynnlz6j6fQGwxMeeBf/PjOsyxbtoxly5bh5eVFx44diYuLIz4+nri4OOLi4oiIiCA0NJTg4GC3F0ftRQOoqjDXKiC0BFazFfn09YmiuHY11ZmMRiOvvvoqjzzyCMuXLydz1VFixjqvuC5bbGSuOEJVThkTJ07kxRdfbLFtUf397V2i5Cr7z6UorLuHuD9v2bp160anzp05fHg/5spyvHxq597ijJNUFOZx+eWX10xuaU30ej2DBg1i+fLlFOVkEBJlv388uXcrJpOJvn37qhyhNvj7B1Bdne9xn8uSJPHwww9jMpn45JNPSP/pAJEj2uETo+2fdVuVhex1x6nMLKVz58785z//qdkuRQ0JCQk8+eSTPPbYY1Ts1+Pf24bk5MaWshkq9hvQSTpefvkVj5vEoQUinwuCIDSeJEl06tSJTZs2UZ1n78LbqVMnlaPSjsjIyJq/t7bxds2Nunz77bfo9XpuvfXWmse8vb256aabeOyxx0hNTRWtFlxo+/bt2Gw2AqMS6HHpdW7ZM93VdHoD4e06E5rQkb2/LCT36F62bt3a6ML6wIED+eGHH/j0kRu44OpbSB44Ai9vbRSKmyM/PYUdK79nx/Lv0OsNTZp19e9//5uPPvoIgKjEcDr2bkNi1zgCw/09YlDeN8CHpL6JJPVNBKC0oJxTBzM4uuMU27ZvY9u2bezZs4fnnnvObd+P0Wjknnvu4aGHHiJ/axrRozo4/RzWcjNFe7MJDQtl1qxZTj++VjgK67Zy+387NQcdPElz8vG3337LgAEDaorqAJ07d2bs2LEsWrRIFNbdqKCggBMnTpCWlkZaWlpN0dxRRC8sLDxnkdHLxw/v4EiCA0PwC43ALzQK/4gYfEMizvl56OXrT7tBF9Ju0IVYqiopzUmjPD+HsvwsqkoKKCspZNfuPezcubPeY/j4+BAREUFERASRkZFEREQQGxtLfHw88fHxJCYmotc3b9ROURQqKytrCuUFBQUUFhZSVFRU8/czHy8sLKKsrLTe4xlNPkQkJtO2Wz8Sew4goXPvmvcpOCqWmc+8x6kDOzi5Zwspe7dz5Nhx9u/fX+/x/P0DCA0NISTE/ic0NLTm72c+FhwcTEhISLPb7j/yyCM88MAD/Dp/PaHRQXQblkSHngn4BnrmxCvZJpN5IpcDfx7j2K5UbFYbo0aNYvTo0W6Nw2g08sorr6DT6fjll1/IXHmEmAuTml1cP7OofskllzB37twWW1SHvwrp8ul5Po5Cu+Ba4v68ZZMkiSuvuIKXX36Z7EO7SOhdu7Nb5oHtAFxxxRUqRKcNAwcOZPny5aQe2ElIVBzVFWXkphxlwIABmEyu3bLLUziuxzzxc1mSJObMmUNcXBwvvfQSmcuPENwrhpAe0Uh67W19UJFRQu4fJ7GWmxk7dixz587VxPt+2WWXsWvXLr7++mvK9+vw6yYjOentU6xQvkePXA0PPHBfrXtMoeFEPhcEQWia9u3bs2nTJspTi2r+LdidOfG2tU3C1dzIy44dO0hOTj7rP4RjP4OdO3fWmeirq6upPmM1VUlJiWsDbaGuvPJKfv75ZwqyU1n/wfOEtkkiLLETBpO2WpU2hs1iJj/lMAUnD2E1VxMYGMhVV13V6OM888wztG3blnfeeYel77/Aivmv07HfBbTvPcjjVvQDlObnsH/DSrJPHAKgXbt2vPTSS3Tr1q1Rx6murmb+p/MB6NCrDR162X8/c9MKyE0rONdLNc3k40W3oR0Jjgxg34aj/PDDD9x99921ZmK52oQJE1i4cCE7duygcE+m01etlxzJQ7bYuHfOvS16D2RHYd2SK9X6t3BuTc3Hsiyze/dubrzxxrO+5hiYLC0trXdFS2Py+fXXX09WVlaDvh9PFh0dzaefftqo15w8eZLrr7+e/Pz88z5X0unx8vPH5BuAyT8IL79AvP0D8fILRF/HiumyvEzK8jIbFQ+Al58/oX4da/6tyAqWynKqy4qpLi+huqyE6vISzBVlWKsrqays5NSpU5w6dareY953333ccMMNjYqjuLi42asydHoDPgFB+AWHEhwRS1BkDEERMfgEBNUU0iuKCzm0aU2dr49KTCYqMdle2C8poig3k+LcTIpzMikvzqeytBjZZqOsrJSystJzvgd1GTVqFC+99FKjB1pHjhzJwoUL+fjjj/n1119Zv3gr6xdvJSI+lLZdYwmJDkL70+XAXG0h9VAWaYeyqK40A9C+Q3uunXEtU6dOVWWPYIPBwEsvvYQkSSxbtozMlUcI6ty8a4riAzlU5ZRx6aWXMnfu3GZPNNE6o9GIyWSqyRGt7cZdLeL+vOWbOHEi/3r1VTL2bsbL94wV6wrkHNlDdHQ0/fr1Uy9AlTlWpR/atAYvbx/y01NQFIU+ffqoHJl2OCZparXb0PlIksTVV19Np06deOCBB8jamUF5SiHB3aORdNq58qnIKKH0SB56vZ577rmHm266SVMLGR555BGysrJYt24d5ftx2n7r1Wk6bGUS06ZNa9ELAlxN5HNBEISmiYuzdyyqzCyt9W+BWvUELUz0cyfNFdYzMzOJiYk563HHY479PP/upZde4tlnn3VpbK1BUlISP//8MwsWLGDZsl84fmwfucf2qR2WUyS0acNFEyYwe/bsJrWN0uv13HzzzUycOJGff/6ZH3/8kQMbVnBgwwoXROseBoOB0aNHc8kllzB69Ogm7XlrMpl4+aWXee211zi26xTHdjVu8N8TBAcH889//tOtRXWw71f2zDPPMHXqVAq2pbvkHEOGDGHSpEkuObZWOPKHOdteSImOjlYzHI/R1HxcUFBAdXX1eV9bX+skkc+dIycnp0FFdQBFtlFdWkx1aTFkp7k4Mufatm1bowvr7777brNbHco2K+VF+ZQX5ZNz8kizjuUKa9eu5cMPP+Tee+9t9Gs7duzISy+9xIMPPsjSpUtZv349W7ZsITdtrwsida34+HhGjBjBuHHj6Nevn+qDzwaDgRdffBFFUfjll1+oyi5r9jEdK9VbelHdITw8nPR0+zWRmCjnHuL+vOULDQ1l6JAh/P777+xd+tVZX584cboqE5K0okOHDgQFBXFi16aavdUB0Qb+DPHx8eTm5np8Z7BevXrxww8/8MYbb/D111+Ts/6E2iGdpXPnzjz//PN07txZ7VDOYjAYeO2117jnnnvYsGED1obdijTI5MmTefTRR1W/lvNkIp8LgiA0TWJiIgC2Sgt6vV4U1s9wZo0tJCRExUjcT1LcuclgA3To0IFOnTqxdOnSWo8fP36cDh068MYbbzBnzpyzXlfXDLqEhASKi4vFaoZmOHXqFNu2bavZK9QT6fV6evXqRfv27Z16Ea4oCvv37+fAgQNu3avTWXx8fBg+fDjBwcFOOV51dTWrV6+mrKz5g8Ra4u3tzZgxY1SdfX/48GF27drl9ON6eXkxbty4Fr1aHez7IS9fvpyKigq8vLy48MILVf3vWVJSQlBQkObzU1PzcWpqKm3atOGVV17hoYceqvW1Tz75hJtuuokdO3bUu+2EyOfOc/ToUXbs2KF2GC4TFBTE6NGjG70PeV5eHv/5z38ICwtrkYW5vLw8SkpKuPXWW512Y1NRUcGWLVvIyclxyvFczWAw0KdPH9q2bavJAVir1crq1aspLi5u1nGCg4MZPXp0i27//ncHDx5kz549+Pj4MG7cONXbMHtKTm8OcX/eOmRnZ7N+/fqz7muNRiMXXnhhq1uB8nf79u2rtXVLYGAg48eP12SOUUN6ejqbN29m1KhRLWZQdc+ePRw8eFDtMGoJCAhg7Nixjb72dTez2cyqVaucNjYUGhrK6NGjXTrBR+Rzkc8FQRDqY7VaWblyJaWlpSQmJootSf7mzz//pKioiAsvvFD1sQl35nPNjcL4+PjUStgOVVVVNV+vi8lkUn1gpSVq06YNbdq0UTsMTZIkiW7dujW6dXpLZTKZmDhxotphtEjJyckkJyerHYbHMhqNXHLJJWqH4XGamo8djzfltSDyuTN17NiRjh07nv+JrUx4eDiPP/642mF4FF9fX0aOHKl2GC2GwWBg/PjxaofhkTp37qzJVXotmbg/bx2ioqKYOnWq2mFolrjvP7e4uDiuvPJKtcNwqh49etCjRw+1w/BIXl5eYmxIg0Q+FwRBaBqDwcBFF12kdhiaNXjwYLVDUIXm+nnFxMSQmXn2vp2Ox2JjY90dkiAIgiC0Ok3Nx6GhoZhMJpHLBUEQBKEFEPfngiAIguD5RD4XBEEQBOfRXGG9d+/eHD58mJKSklqPb9q0qebrgiAIgiC4VlPzsU6no0ePHmzduvWsr23atIn27dsTEBDg9HgFQRAEQXA+cX8uCIIgCJ5P5HNBEARBcB7NFdanTp2KzWbjgw8+qHmsurqa+fPnM2jQIBISElSMThAEQRBah4bm41OnTp21/+DUqVPZsmVLreL6oUOHWL16NVdddZV7vgFBEARBEJpN3J8LgiAIgucT+VwQBEEQnEdze6wPGjSIq666ikcffZScnBw6duzIZ599xsmTJ/n444/VDk8QBEEQWoWG5uNZs2axbt06FEWpeezOO+/kww8/5JJLLuGBBx7AaDQyb948oqKiuP/++9X4dgRBEARBaAJxfy4IgiAInk/kc0EQBEFwHs0V1gE+//xznnzySb744gsKCwvp2bMnP/30EyNGjFA7NEEQBEFoNZqajwMCAli7di333nsvc+fORZZlRo0axRtvvEFERISbohcEQRAEwRnE/bkgCIIgeD6RzwVBEATBOSTlzCVmLUhJSQlBQUEUFxcTGBiodjiCIAiCAIj81Fji/RIEQRC0SuSohhPvlSAIgqBVIkc1nHivBEEQBK1yZ47S5Ip1Z3DMFygpKVE5EkEQBEH4iyMvtdB5bU4n8rkgCIKgVSKnN5zI54IgCIJWiXzecCKfC4IgCFrlznzeYgvrpaWlACQkJKgciSAIgiCcrbS0lKCgILXD0DyRzwVBEAStEzn9/EQ+FwRBELRO5PPzE/lcEARB0Dp35PMW2wpelmUyMjIICAhAkiRVYykpKSEhIYHU1FTRJudvxHtTP/He1E+8N3UT70v9tPTeKIpCaWkpsbGx6HQ6VWPxBCKfewbx3tRPvDf1E+9N3cT7Uj+tvTcipzeclvI5aO9nSSvE+1I/8d7UT7w39RPvTd209r6IfN5wIp97DvHe1E28L/UT7039xHtTPy29N+7M5y12xbpOpyM+Pl7tMGoJDAxU/YdLq8R7Uz/x3tRPvDd1E+9L/bTy3ohZ8A0n8rlnEe9N/cR7Uz/x3tRNvC/109J7I3J6w2gxn4O2fpa0RLwv9RPvTf3Ee1M/8d7UTUvvi8jnDSPyuecR703dxPtSP/He1E+8N/XTynvjrnwupuEJgiAIgiAIgiAIgiAIgiAIgiAIgiAIwjmIwrogCIIgCIIgCIIgCIIgCIIgCIIgCIIgnIMorLuByWTi6aefxmQyqR2K5oj3pn7ivamfeG/qJt6X+on3RnAG8XNUP/He1E+8N/UT703dxPtSP/HeCM4ifpbqJt6X+on3pn7ivamfeG/qJt4XwVnEz1L9xHtTN/G+1E+8N/UT7039Wut7IymKoqgdhCAIgiAIgiAIgiAIgiAIgiAIgiAIgiBolVixLgiCIAiCIAiCIAiCIAiCIAiCIAiCIAjnIArrgiAIgiAIgiAIgiAIgiAIgiAIgiAIgnAOorAuCIIgCIIgCIIgCIIgCIIgCIIgCIIgCOcgCuuCIAiCIAiCIAiCIAiCIAiCIAiCIAiCcA6isC4IgiAIgiAIgiAIgiAIgiAIgiAIgiAI5yAK64IgCIIgCIIgCIIgCIIgCIIgCIIgCIJwDqKwLgiC4IEqKiqYPHkyX375pdqhaNLEiRP56quvqKysVDsUQRAEQaiXyOfnJvK5IAiC4ClETq+fyOeCIAiCpxD5/NxETrcThXU3UhSF1atXs2zZMkpLS9UORxA8QkVFBWFhYbz66qtqh6Ipvr6+rFy5koqKCrVD0aTjx48zc+ZMoqKimD17NitXrkRRFLXDEloIkc8FofFEPq+byOfnJvK54EoinwtC44l8Xj+R0+sn8rngSiKfC0LTiJxeN5HPz03kdDuD2gG0VI8//jgbNmxgzZo1gD3Jjx8/ntWrV6MoCm3atGHVqlV06NBB5Uhdb/v27Y1+Td++fV0QifYUFBQ0+jWhoaEuiES7fH19MRgM+Pn5qR2K5gwfPpyNGzdyyy23qB2K5hw6dIgtW7awYMECFi1axIIFC4iOjmbGjBlce+219O7dW+0QBQ8h8vlfRD6vn8jn5yfyef1EPq+fyOeCs4h8XpvI6XUT+fz8RD4/N5HT6ybyueAsIp/XJvJ5/UROPz+R0+sn8nn9RE63k5TWOJ3ADTp37sykSZN45ZVXAPjmm2+YNm0aL7zwAr169eK2225j1KhRfPHFFypH6no6nQ5Jkhr0XEVRkCQJm83m4qi0oTHvjUNreW/OdOedd3Lw4EFWrVrV6PerJTt+/DgTJkxg2rRp3H777cTHx6sdkibJssyKFStYsGAB//vf/ygvL6dLly7MmjWLGTNmiPdNOCeRz/8i8nn9RD5vGJHP6ybyecOIfC40h8jntYmcXjeRzxtG5PP6iZx+fiKfC80h8nltIp/XT+T0hhE5vW4inzdMa87porDuIgEBAcybN69mVss111zDnj172Lt3LwAvvvgi7733HqmpqWqG6RafffZZo18ze/ZsF0SiPc8880yjk9bTTz/tomi067fffuPOO+8kPDycW265hcTERHx8fM56XmuZdekQEBCA1WrFbDYDYDAYMJlMtZ4jSRLFxcVqhKdJRUVF3HbbbXzzzTeA/UJ71KhR3HvvvVxyySUqRydokcjnfxH5vH4inzeMyOd1E/m88UQ+FxpL5PPaRE6vm8jnDSPyef1ETm8ckc+FxhL5vDaRz+sncnrDiJxeN5HPG6+15XTRCt5FDAYD1dXVgH1G2KpVq5g1a1bN16OiosjLy1MrPLdqTsKWZZm0tDSio6Px8vJyYlTa8Mwzz6gdgkcYNWpUzd/Xr19/1tdb26xLhylTpojZhA30+++/s2DBAr799lsKCgro3r07s2bNwmg08sknn3D55Zfz+OOP89xzz6kdqqAxIp//ReTz+ol83jAin9dN5POGE/lcaCqRz2sTOb1uIp83jMjn9RM5vWFEPheaSuTz2kQ+r5/I6Q0jcnrdRD5vuNaa08WKdRe54IILsFgsLFu2jO+//55bbrmFlStXMnr0aACeeuop5s+f32pm0DVVdnY2sbGxrFixgjFjxqgdjqCShs7AbC2zLoWG2b9/PwsWLGDhwoWcOnWKyMhIZsyYwXXXXXfWfi+33norixcvJj8/X51gBc0S+dw5RD4XQORzoWlEPhecQeRz5xE5XRD5XGgKkc8FZxD53HlEPhdA5HShaUROFyvWXeapp57isssuIzw8HIBhw4bVJHmAn3/+mQEDBqgVnkdpyXM/tm/f3ujXtLbWKyCSt9B4vXv3Zs+ePZhMJiZNmsS7777LhAkT0Ol0dT5/9OjRfPTRR26OUvAEIp87j8jntYl8LgjnJ/K54CwinztXS83pIp83jMjnQmOJfC44i8jnztVS8zmInN5QIqcLjSVyup0orLvIuHHj2L59OytWrCA4OJhp06bVfK2wsJARI0YwadIkFSMUtKB///4NbivSWluvCOeXlpbGjh07KC4uRpbls75+Zlus1iI4OJgPPviAq666isDAwPM+f9KkSZw4ccINkQmeRuRzoSFEPhecQeTzs4l8LjiLyOdCQ4h8LjiLyOm1iXwuOIvI50JDiZwuOIPI52cTOd1OFNZdqGvXrnTt2vWsx0NCQnjjjTdUiEjQmvnz56sdgseoqqpi8eLFbN++vc5kJkkSH3/8sUrRqaOqqorZs2ezePFiZFlGkqSa2aZnXjy2tiRfVVXF5MmTSUpKalCCB/D19aVt27YujkzwVCKfC+cj8nnDiXx+NpHP6ybyueBsIp8L5yPyecOJfF43kdPPJvK54GwinwsNIXJ6w4mcfjaRz+smcvpfRGFdEFTUnHYrsiyTlpZGdHQ0Xl5eToxKe1JSUhg9ejQnT54kODiY4uJiQkNDKSoqwmazER4ejr+/v9phut1jjz3Gd999xwsvvMCQIUMYNWoUn332GTEx/8/efUc3Vf9/HH/dtHTRDaVAGS1FoKyyKVKk7DItW0Qse1SmDBH9WooioIjsKRsVQUWUoVJkCshShmzKkk2hBQqUjs/vj/4SGnJvmrZJbtq+HudwtLk37btpkudNbpJbAjNnzsSNGzewatUqtce0OicnJ4wfPx6zZs3Ca6+9pvY4RFQAsOemYc/lsefy2HMisjb23DTsuTI23RB7TkRqYNNNw6bLY8/lsekvyH/wPWWbRqOBvb09nj9/rvvazs7O6D97e76ugXLu7t27CAgIwN69e9UexeLGjh2LxMREHDhwAOfOnYMQAt999x0eP36MadOmwdnZGb/99pvaY1rd999/jz59+uC9995DlSpVAAB+fn5o3rw5Nm3aBE9PT8ybN0/lKdVRpUoVXL58We0xKA9iz8na2HP2nD1Xxp5TTrHnZG3sOXsOsOlK2HPKKfac1MCms+nsuTI2PQNLYyYfffQRJEnSxVv7NZElaT+CJL/7448/EBUVhXr16uH+/fsAMn53R0dHjB07FqdPn8bIkSOxefNmlSe1rjt37qBevXoAAGdnZwBAUlKSbnnnzp0xadIkLFiwQJX51DR58mS8+eabaNKkCZo3b672OJSHsOekBvacPWfP5bHnlFPsOamBPS/YPQfYdCXsOeUUe05qYdMLdtPZc2VsegbuWDeTiRMnGv2aiHLuyZMn8Pf3BwC4u7tDkiQkJibqljdo0ABjxoxRaTr1+Pr6Ij4+HkDG8Uq8vLxw9uxZtG/fHgDw8OFDPHv2TM0RVTN37lx4e3ujVatWCAgIQEBAgG5DSEuSJGzcuFGlCclWsedElsOey2PPlbHnlFPsOZHlsOfK2HR57DnlFHtOZFlsujz2XBmbnoE71smmubq6Ijo6GuXKlVN7FFJRmTJl8N9//wEA7O3t4efnhwMHDqBTp04AgFOnTsHJyUnNEVVRv3597N27F++99x4AoH379vj8889RokQJpKen48svv0RISIjKU6rj+PHjkCQJZcqUQVpaGi5cuGCwDl/lTGQ97DkB7LkS9lwZe05ke9h0Ys+Vseny2HMi28OeE8CmK2HPlbHpGbhj3UxWrVqVo/O9/fbbZp7Etl29ehVXr15FaGio7rRjx47hiy++QHJyMnr06IGIiAjdssKFCyM6OlqFScmWNG3aFBs3btRdF3r37o0pU6bgwYMHSE9Px+rVqwvcbQkAhg8fjvXr1yM5ORmOjo74+OOPsX//fvTq1QsAEBgYiNmzZ6s8pTp4rBfKKfbcNOw55QR7Lo89V8aeU06x56Zj0ym72HNlbLo89pxyij03HXtOOcGmy2PPlbHpGSRRUA4YYWEajSbb55EkCWlpaRaYxnZFRETg8ePHiI2NBQDcvn0bQUFBeP78Odzc3HDnzh2sX79e96ooUnb79m2UKFECsbGxaNq0qdrjWNTVq1dx6NAhtGvXDo6Ojnj27BmGDh2KH374AXZ2dmjXrh1mz54Nd3d3tUdVXXp6Ok6cOAE7OztUqlRJdxwqIjINe24a9tx82HP2XA57TpQ77Lnp2HTzYM/ZcyVsOlHOseemY8/Nh01n0+Ww55QZd6ybyZUrV3J0vrJly5p5EttWsmRJjBgxQvcxGp9//jk++ugjnDx5EgEBAQgPD8fjx4+xb98+lSe1fQUp8kQ5tWvXLmzevFl3H122bFm0bdsWjRs3VnkyslXsuWnYc/Nhz4myxp5TdrHnpmPTzYM9J8oae07ZxZ6bjj03HzadKGsFvel8WYWZFMRg58T9+/dRrFgx3debNm1C48aNERgYCADo1KkTJkyYoNZ4RDZt9+7dOTrfa6+9ZuZJbN/z58/Ro0cP/PTTTxBCwNPTEwCQkJCAL774Ah07dsS3336LQoUKqTso2Rz23DTsOVHOseemY88pp9hz07HpRDnHppuGPaecYs9Nx54T5Rx7bjo2PQN3rJNV+fj46F7FkpCQgAMHDmDq1Km65ampqUhNTVVrvDzF1dUV0dHRKFeunNqjmN2kSZOyfR5JkvC///3PAtPYjrCwMEiSZPL6QogC+xFYMTEx2LBhA8aMGYPRo0fD19cXAHDnzh188cUX+PzzzzFp0iR8/PHHKk9KlDex5+bDnutjzw2x5+w5kSWx6ebBnusrCD0H2HRTsedElseemw+brq8gNJ09Nx2bnoEfBW9Bt27dwtKlS3H06FEkJiYiPT1db7kkSdi+fbtK06mjT58+2LhxIz788EPs3LkTW7duxblz5xAQEAAAiIqKwq5du/Dvv/+qPKn1Xb16FVevXkVoaKjutGPHjuGLL75AcnIyevTogYiICPUGtCIeQ0nerl27cnS+gvIRLJkFBAQgLCwMy5cvl13eu3dv7Ny5E5cvX7buYJQnseeG2HNl7PkL7Lk89tx07DmZE3suj02Xx56/wJ4rY9NNw56TObHn8thzZWz6C2y6PPbcdGx6Br5j3UKOHz+OsLAwPH36FBUrVsSJEydQuXJlJCQk4Pr16wgMDETp0qXVHtPqpk6dinPnzmHMmDFwcHDA9OnTdYFPTk7GunXr8Oabb6o8pTqGDx+Ox48fIzY2FkDG8VyaNGmC58+fw83NDd9//z3Wr1+PTp06qTyp5b28UUwZCmKsc+rmzZuoX7++4vL69etj7dq1VpyI8ir2XB57row9f4E9l8eem449J3Nhz5Wx6fLY8xfYc2VsumnYczIX9lwZe66MTX+BTZfHnpuOTc+Q/ZeokEnGjx8PV1dXnD17FrGxsRBCYNasWbh27Rq+++47PHjwQO/jWAoKX19f/Pnnn3jw4AEePnyIESNG6Jalp6dj+/btmDhxonoDqujgwYNo0aKF7utVq1bh6dOnOHbsGK5fv45mzZph+vTpKk5IlHeUKlUKO3fuVFy+a9culCpVynoDUZ7Fnstjz5Wx50Tmw56TubDnyth0eew5kfmw52Qu7Lky9lwZm05kPmx6Br5j3UL+/PNPjBs3DmXKlMH9+/cBvHhFUNeuXbF3716MHTs2xx8zkdd5eHgYnObs7Izg4GAVprEN9+/fR7FixXRfb9q0CY0bN0ZgYCAAoFOnTpgwYYJa45EN6tu3b5brSJKEpUuXWmEa2xIZGYno6Gh4enpi1KhRKF++PCRJwvnz5zFz5kysX78eMTExao9JeQB7bhx7bog9p+xiz5Wx52Qu7HnW2HR97DnlBJsujz0nc2HPs8aeG2LTKbvYc2VsegbuWLeQ9PR0+Pr6AgA8PT1hZ2enCz4AVKtWrUDc8FatWgUA6NWrFyRJ0n2dlbffftuSY9kkHx8fXLlyBQCQkJCAAwcO6L3KMjU1FampqWqNp6qAgABIkmR0HUmScPHiRStNZBv++OMPg8slLS0NN2/eRFpaGnx8fFC4cGGVplPXhAkTcPHiRSxevBhLlizRHUMoPT0dQghERkZyo5lMwp5nYM9Nx54rY8/lsefK2HMyF/b8BTbdNOy5MvZcGZsujz0nc2HPX2DPTcemK2PT5bHnytj0DNyxbiEBAQG4dOkSAECj0SAgIACxsbHo1q0bAGDfvn3w9PRUcULr6N27NyRJwhtvvAEHBwf07t07y/NIklQgI9+8eXPMnj0b7u7u2LlzJ9LT0xEREaFbfurUqQJ7nKDGjRvLxuzKlSv4888/UbVqVdSsWVOl6dRz+fJl2dNTUlKwaNEizJw5E9u2bbPuUDbCzs4OK1aswLvvvostW7boNqDLli2LNm3aoHr16ipPSHkFe56BPTcde66MPZfHnitjz8lc2PMX2HTTsOfK2HNlbLo89pzMhT1/gT03HZuujE2Xx54rY9MzSEIIofYQ+dGYMWPw888/49y5cwCAL7/8EqNHj0bTpk0hhMDOnTsxevRofPbZZypPalmZb1iZv86Kdv2C5Pbt2+jUqRP2798PBwcHTJs2TXc8nOTkZPj5+eHNN9/E7NmzVZ7Uthw7dgytWrXCmjVr0Lx5c7XHsSlRUVG4cuUKNm/erPYoVnf16lX4+PjA2dlZdvnTp09x9+5dlClTxsqTUV7Dnmdgz03HnucMe66MPWfPKffY8xfYdNOw5znDnhtXUJvOnpO5sOcvsOemY9Nzhk1XVlB7DrDpWtyxbiEPHjxAXFwcqlevjkKFCkEIgcmTJ+OHH36AnZ0d2rVrhwkTJsDBwUHtUcnGJCYmwtnZWe+68fTpU5w7dw6lS5eGt7e3itPZpujoaGzatAlHjhxRexSbsmjRIowZMwaPHj1SexSrs7Ozw+rVq/Hmm2/KLv/uu+/w5ptvIi0tzcqTUV7DnlNOsefZx57LY8/Zc8o99pxyij3PPvZcWUFtOntO5sKeU26w6dnHpssrqD0H2HQtfhS8hXh5eaF27dq6ryVJwocffogPP/xQxakoL/Dw8DA4zdnZGcHBwSpMkzf4+vri1KlTao9hc7Zt2wYXFxe1x1BFVq8ZS0lJ0R0DhsgY9pxyij3PPvZcHnuujD0nU7HnlFPsefax58oKatPZczIX9pxyg03PPjZdXkHtOcCma3HHupXFxcUhOTkZQUFBao9iFU2bNs32eSRJwvbt2y0wjW1ZtWoVAKBXr16QJEn3dVYK4rFwjImPj8fSpUtRqlQptUexukmTJsmenpCQgN27d+Po0aMYP368ladSz8OHD5GQkKD7Oj4+HlevXjVYLyEhAWvXrkWJEiWsOB3lN+x51thz49hzfey5IfY8A3tOllTQeg6w6UrYc/MoyD0H2PTM2HOyJvbcNAWh5wCbbi4FuensuT423RA/Ct5CZs+ejX379mHt2rW60/r06aO7I69Zsya2bNmCYsWKqTWiVYSFhUGSpGyfb8eOHRaYxrZoNBpIkoSnT5/CwcHBpFfySJKU7z9GQ47SxmJCQgLOnDmD58+fY/Xq1ejRo4eVJ1OX0nXGy8sLgYGB6N+/PwYMGJCj22BeFBMTo7jh8zIhBD755BNMmDDBwlNRXseeZ2DPlbHnpmPP5bHn+thzsgT2/AU2XR57bjr2XBmb/gJ7TpbAnr/Anitj003Hpstjz/Wx6Yb4jnUL+eqrr9CkSRPd17/99htWrlyJQYMGoVq1avjwww8RExODefPmqTil5e3cuVPtEWzWpUuXAEB3XBft12QoPT3dIFSSJCEgIADNmzdH3759UalSJZWmU096erraI9iUli1bwtXVFUIIjBs3Dj169ECtWrX01pEkCYULF0bt2rVRp04dlSalvIQ9z8CeK2PPTceey2PP9bHnZAns+Qtsujz23HTsuTI2/QX2nCyBPX+BPVfGppuOTZfHnutj0w1xx7qFXLlyRe/jZ9atW4eAgAAsWLAAAHDr1i2sXr1arfHIBpQtW9bo1/QCNxbJFA0aNECDBg0AAElJSejcuTOqVq2q8lSU17HnlBX23HTsOZmCPSdLYM8pK+y56dhzMgV7TpbAnpMp2HTTselkCjbdEHesW8jLn7D/+++/4/XXX9d97e/vj1u3bll7LJvy6NEjJCYmyr4CqEyZMipMRGTb5I5dYoqCeHuKjo7W+zoxMRGurq6ws7NTaSLKq9jzrLHnRNnDnpuOPSdzYc9Nw6YTZQ+bbhr2nMyFPTcNe06UPey56dj0DNyxbiEVKlTAhg0bMHjwYPz222+4ceMGWrdurVv+33//wdPTU70BVbRgwQLMmDEDcXFxiusUhGOaKB3DxBhJkrB9+3YLTGNbtMdGyq63337bzJPYFn9//xwdu6Ug3J7kHD58GB9++CF2796N58+f4/fff0fTpk1x79499OvXD6NGjUJYWJjaY5KNY8+VsecZ2HNl7Lk89jx72HMyB/bcODadPTeGPVfGppuOPSdzYM+NY88zsOnK2HR57Hn2sOncsW4xY8aMwZtvvgkvLy8kJSUhKCgIrVq10i3/448/UKNGDfUGVMnChQvxzjvvoFWrVujbty8++OADjBo1Ck5OTlixYgV8fX0xfPhwtce0CrljmGTl5Vdm5le9e/c2OE17Wb18GWS+DPN75JctW6b3+6anp2PWrFm4cuUKevbsiYoVKwIAzpw5g2+++Qb+/v4F5vb0sn379qFp06bw8/PDW2+9ha+++kq3rGjRokhMTMSiRYvyfeQp99hzeez5C+y5MvZcHntuOvaczIU9V8amZ2DPlbHnyth007DnZC7suTL2/AU2XRmbLo89Nx2bnoE71i3kjTfeQJEiRbBlyxZ4enoiKioK9vYZF/f9+/fh7e2NXr16qTyl9c2ZMwetWrXC1q1bER8fjw8++ABt27ZF06ZNMW7cONSpUwfx8fFqj2kVPIaJskuXLul9nZCQgMjISHh4eGDYsGF6MZszZw4ePXqElStXqjGqVb288TN58mQ8e/YMFy5cQJEiRfSWTZw4EaGhoQX2I7AmTJiAoKAgHDhwAI8ePdKLPAA0adKkQFxnKPfYc3ns+QvsuTL2XB57bjr2nMyFPVfGpmdgz5Wx58rYdNOw52Qu7Lky9vwFNl0Zmy6PPTcdm/7/BJEVOTo6innz5gkhhEhMTBSSJImtW7fqlk+dOlWUK1dOrfHIRvXu3Vs0b95cpKenGyxLS0sTzZo1E71791ZhMnWVKlVKfPHFF4rLP//8c1G6dGkrTmQ7XFxcxKxZs4QQQty7d09IkiS2b9+uW75kyRLh7Oys1nhEeR57TjnBnstjz5Wx50SWx6ZTdrHnyth0eew5keWx55QTbLo89lwZm56B71gnq/Lw8EBqaioAwN3dHS4uLrh27ZpuuZubW4F9tU9mjx49QmJiItLT0w2WlSlTRoWJ1PXTTz9h8uTJsh/jo9Fo0KlTJ3z44YcqTKau+Ph4PHnyRHH5kydPCsyrUV9WqFAh2duP1vXr1+Hq6mrFiYjyF/bcNOy5PvZcHnuujD0nsjw2PWvsuT72XBmbLo89J7I89tw0bLo+Nl0ee66MTc/AHesWdPz4ccyZMwdHjx6VvcOWJAkXL15UaTp1VK1aFceOHdN9HRISggULFqBNmzZIT0/HokWLUKFCBRUnVNeCBQswY8YMxMXFKa6TlpZmxYlsgxACZ86cUVx+6tSpAnMsnMxCQkIwc+ZMtG7dGrVr19ZbdvjwYcyaNQv169dXaTp1hYSE4Pvvv8fIkSMNliUlJWH58uVo3Lix9QejPIk9N8SeG8eey2PP5bHnythzMif2XB6brow9l8eeK2PT5bHnZE7suTz23Dg2XR6bLo89V8am/z/13iyfv+3YsUM4OjqK4sWLi3bt2glJkkSzZs1Ew4YNhUajEdWqVSuQH6OxbNkyUa9ePfHs2TMhhBB79+4VTk5OQqPRCI1GIxwdHcWmTZtUnlIdCxYsEJIkifDwcPHpp58KSZLEu+++KyZMmCBKliwpatasKZYvX672mKqIjIwU9vb24osvvhBJSUm605OSksT06dOFvb29iIyMVG9Alfz777/Cx8dHaDQa8eqrr4rIyEgRGRkpXn31VaHRaETRokXFyZMn1R5TFQcOHBCOjo6iTZs2YvXq1UKSJDFjxgyxZMkSUbFiReHi4iKOHTum9piUB7Dn8thzZey5MvZcHnuujD0nc2HPlbHp8thzZey5MjZdHntO5sKeK2PPlbHpyth0eey5MjY9A3esW0ijRo1EUFCQSExMFHfv3tU71sCBAweEl5eX2LJli8pT2oaLFy+KmTNnijlz5oizZ8+qPY5qKleuLMLDw4UQhsenSEhIEOXLlxfTp09Xc0TVJCQkiNdee01IkiQcHBxE2bJlRdmyZYWDg4OQJEmEhoaKBw8eqD2mKm7duiVGjhwpKlasKJycnISTk5OoWLGiGDVqlLh586ba46lq+/btokKFCkKSJL1/5cuXFzt37lR7PMoj2HPTsecZ2HNl7Lky9lwZe07mwJ5nD5vOnhvDnhvHpstjz8kc2PPsYc8zsOnK2HRl7LkyNl0ISYgC+FkOVuDq6oqYmBiMHj0aDx48QJEiRfDbb7+hRYsWAID3338f27Ztw+HDh1WelGyFk5MTZsyYgaioKDx8+BCenp7YsmULwsPDAQDTpk3D4sWLC+THGWlt3LgRW7duxZUrVwAAZcuWRZs2bdC+fXvZY8FQwSSEwKNHj+Dg4AAnJyf8888/OH/+PNLT0xEYGIjatWvz+kImY88pu9jzrLHnZAr2nMyJPafsYs+zxp6TKdhzMif2nHKCTc8am06mYNNf4DHWLcTe3h5ubm4AAE9PTxQqVAh37tzRLS9XrhxOnTql1ng24fHjx3jw4IHscTrKlCmjwkTq8vDwQGpqKgDA3d0dLi4uuHbtmm65m5sbbt26pdZ4NuH111/H66+/rvYYZOOeP38Ob29vfPrppxg3bhxq1KiBGjVqqD0W5VHsedbYc33sedbYczIFe07mxJ6bhk1/gT3PGntOpmDPyZzYc9Ow5/rY9Kyx6WQKNv0F7li3kPLly+P8+fMAAEmSUKlSJWzYsAE9e/YEAGzevBnFixdXc0RVPHv2DDExMVi6dCni4+MV10tLS7PiVLahatWqOHbsmO7rkJAQLFiwAG3atEF6ejoWLVqEChUqqDgh2aLTp09j+fLliIuLk91oliQJ27dvV2k6dTg6OqJ48eJwdHRUexTKB9hzeey5MvaccoI9N8Sekzmx58rYdHnsOeUUm66PPSdzYs+VsefK2HTKCfbcEJv+AnesW0ibNm2wbNkyTJkyBfb29nj33XfRp08fvPLKKwCAixcvYsqUKSpPaX1RUVFYuXIlIiIi0KhRI3h5eak9ks146623sHDhQiQnJ8PR0RExMTFo3ry57pWEhQoVwg8//KDylOoQQmDx4sVYunSpLmYvkyRJ9+rDgmL16tXo06cPChUqhIoVK8rengrq0T569+6NVatWYciQIXBwcFB7HMrD2HN57Lky9lwZey6PPVfGnpO5sOfK2HR57Lky9lwZmy6PPSdzYc+VsefK2HRlbLo89lwZm56Bx1i3kJSUFDx8+BDe3t664wqsWbMGP/zwA+zs7NCuXTv07t1b3SFV4Onpie7du2PRokVqj5InxMXF4ZdffoGdnR1atmxZYF89N3bsWMyYMQM1atQwunEYHR1t5cnUFRgYCG9vb2zduhVFixZVexyb8t133+Hjjz9GcnIyevfuDX9/fzg7Oxus16lTJxWmo7yEPZfHnmcPe56BPZfHnitjz8lc2HNlbLrp2PMM7LkyNl0ee07mwp4rY8+zh03PwKbLY8+VsekZuGOdrMrLywtTp07FoEGD1B6F8pBixYohLCwM69atU3sUm+Ls7IwZM2ZgyJAhao9iczQaTZbrSJJUID8Ci8gc2HPKCfZcHnuujD0nsjw2nbKLPVfGpstjz4ksjz2nnGDT5bHnytj0DPwoeAtLS0vDkSNHcPnyZQBAQEAAatWqBTs7O3UHU8nrr7+O2NhYRj4Ljx8/lj12BwDdx9QUJE+fPkXz5s3VHsPmVK9eHTdu3FB7DJu0Y8cOtUegfIY918eem4Y918eey2PPlbHnZG7suSE2PWvsuT72XBmbLo89J3Njzw2x56Zh0/Wx6fLYc2Vsega+Y92CVqxYgffffx937tzR3VlLkgQfHx98+umn6Nu3r8oTWt/FixfRrVs31K5dG4MGDUKZMmVkN3q8vb1VmE5dz549Q0xMDJYuXYr4+HjF9fL7q33kREREwMfHB0uWLFF7FJvy559/omvXrvj+++/x6quvqj0OUb7Fnhtiz5Wx58rYc3nsOZF1sOfy2HR57Lky9lwZm05keey5PPZcGZuujE2Xx55TVrhj3UIWLVqEIUOGoEaNGhg0aJDuOB1nz57FokWLcPz4ccybNw+DBw9WeVLryvxREdpj4cgpiCHr27cvVq5ciYiICKPHNImMjLTyZOq7ceMGWrVqhR49emDQoEEoUqSI2iPZhA4dOuD8+fM4d+4cKleuLLvRLEkSNm7cqNKE6rl//z7+++8/VK9eXXb5iRMnUKpUKcXbGZEWey6PPVfGnitjz+Wx58rYczIX9lwZmy6PPVfGnitj0+Wx52Qu7Lky9lwZm66MTZfHnitj0zNwx7qFlCtXDqVLl0ZsbCwKFSqktywlJQVNmzbF9evXERcXp9KE6pg4caLRuGtFR0dbYRrb4unpie7du2PRokVqj2Jz3NzckJ6ejmfPngEAnJycZGOWmJioxniq8ff3z/L2JElSgbufATI2hs+ePYsDBw7ILn/11VcRFBSEpUuXWnkyymvYc3nsuTL2XBl7Lo89V8aek7mw58rYdHnsuTL2XBmbLo89J3Nhz5Wx58rYdGVsujz2XBmbnoHHWLeQW7duYfTo0QaRB4BChQrhjTfewLhx41SYTF0TJ05UewSbJUkSatWqpfYYNqlz584mbRwWNNpjSZGhP/74A0OGDFFc3r59eyxcuNCKE1FexZ7LY8+VsefK2HN57Lky9pzMhT1XxqbLY8+VsefK2HR57DmZC3uujD1XxqYrY9PlsefK2PQM3LFuITVr1sS5c+cUl587dw41atSw3kA26unTpwAAZ2dnlSdR3+uvv47Y2FgMGjRI7VFszooVK9QegfKYu3fvomjRoorLixQpgjt37lhxIsqr2HPTsOcvsOfK2HPKLvaczIU9Nx2bnoE9V8aeU3ax52Qu7Lnp2PMX2HRlbDplF5ueQZP1KpQTc+bMwbp16zBr1ixdyICMqH355ZdYt24d5s6dq+KE6rl69Sr69OkDX19fuLq6wtXVFb6+vujbty+uXLmi9niq+d///oe4uDgMHDgQR44cwd27d3H//n2Df0RyHj16hP/++w9Xr141+FcQlShRAn///bfi8iNHjsDHx8eKE1FexZ4rY8/lseeUG+y5PvaczIU9N45NN8SeU26x6S+w52Qu7Llx7Lk8Np1ygz3Xx6Zn4DHWzaR69eoGp92/fx83b96Evb09SpYsCQC4ceMGUlNTUaJECRQpUgTHjh2z9qiqOnPmDEJDQ5GQkIAWLVogKChId/rvv/8OLy8v7N27FxUrVlR5UuvTaF68zsXYR7CkpaVZYxyb9N9//+Hvv/9GYmIi0tPTDZa//fbbKkylrgULFmDGjBlGj+lSEK8zo0aNwrx58/D999+jQ4cOess2btyIrl27YsiQIZg1a5ZKE5KtYs9Nw54rY8+zxp4bYs/lseeUU+y56dh0eex51thzeWy6Ifaccoo9Nx17roxNzxqbbog9l8emZ+COdTMJCwvL0fEoduzYYYFpbFdERAT27duH7du3o1q1anrLTp48iWbNmuHVV1/Fhg0bVJpQPRMnTjTpOhQdHW2FaWzLs2fPEBkZiR9++AHp6emQJAnau67Ml1lBi9nChQsRFRWFVq1a4bXXXsMHH3yAUaNGwcnJCStWrICvry+GDx+O3r17qz2q1SUmJiI0NBSnTp1CcHAwqlatCiDjfubYsWMICgrC3r174enpqe6gZHPYc9Ow58rYc2XsuTz2XBl7TjnFnpuOTZfHnitjz5Wx6fLYc8op9tx07LkyNl0Zmy6PPVfGpv8/QWRFnp6e4uOPP1ZcPmnSJOHp6WnFiSgvGDVqlLC3txdTp04Vu3btEpIkiVWrVolt27aJtm3bipo1a4oTJ06oPabVVa5cWYSHhwshhLh3756QJEls375dCCFEQkKCKF++vJg+fbqaI6rq8ePH4qOPPhJVq1YVzs7OwtnZWVStWlVER0eLx48fqz0eUZ7GnlNOsOfy2HPj2HMiy2LTKbvYc2VsujL2nMiy2HPKCTZdHntuHJsuBI+xbiPu3buHcuXKYf/+/WqPYlEpKSlwdnZWXO7i4oKUlBQrTmS7nj59qne8oILs+++/R58+ffDee++hSpUqAAA/Pz80b94cmzZtgqenJ+bNm6fylNZ38eJFtG/fHgBQqFAhAMDz588BAB4eHujfvz/mz5+v2nxqK1y4MGJiYnDixAk8efIET548wYkTJzBx4kQULlxY7fEon2LPM7DnL7DnL7Dn8thz49hzUkNB6TnAppuKPX+BPVfGpitjz0kN7PkL7PkLbPoLbLo89tw4Nh3gjnUbkZaWhsuXL+f7O/WaNWviq6++QmJiosGyhw8fYunSpahVq5YKk9mGq1evok+fPvD19YWrqytcXV3h6+uLvn374sqVK2qPp5o7d+6gXr16AKDbSExKStIt79y5M3788UdVZlOTh4cHUlNTAQDu7u5wcXHBtWvXdMvd3Nxw69YttcazGTdv3sSxY8f0rjNElsKes+cAe66EPZfHnpuGPSdrKig9B9h0Y9hzeey5MjY9a+w5WRN7nqGg9xxg05Ww6fLYc9MU5KZzxzpZVUxMDC5evIhKlSphwoQJWLFiBVasWIH3338flSpVwsWLFxETE6P2mKo4c+YMatWqhdWrV6NWrVoYMWIERowYgdq1a2PVqlWoU6cOzp49q/aYqvD19UV8fDyAjFdYenl56V0WDx8+xLNnz9QaTzVVq1bFsWPHdF+HhIRgwYIFuH79Oq5du4ZFixahQoUKKk6oro0bN6JSpUooVaoUatWqhb/++gtAxiuWa9asiZ9++kndAYnyMPZcGXuujD2Xx54bx54TWRabLo89V8aeK2PTlbHnRJbFnitj05Wx6fLYc+PYdPAY67bi1q1besdqyM+2bdsmatSoISRJ0vtXs2ZNERsbq/Z4qnn99deFj4+POH78uMGyEydOiGLFiomIiAgVJlNf165dRbt27XRfR0ZGimLFiok1a9aIVatWCR8fH9GyZUsVJ1THsmXLRL169cSzZ8+EEELs3btXODk5CY1GIzQajXB0dBSbNm1SeUp1/Pzzz0Kj0YiGDRuKmJgYg/vXtm3big4dOqg4IeVX7Dl7zp4rY8/lsefK2HNSS0HquRBsuhz2XBl7roxNl8eek1rYc/ZcCDbdGDZdHnuujE3PIAkhhNo79wm4ffs2SpQogdjYWDRt2lTtcazi1q1buo9aKVu2LIoXL67yROry8vLC6NGj8eGHH8ou//jjjzFjxgw8ePDAypOpb+/evVi/fj0+++wzODo64tq1a2jevDnOnz8PAAgMDMSmTZtQsWJFlSdVX1xcHH755RfY2dmhZcuWBfbVc3Xr1oWrqyt27NiB+Ph4+Pj46N2/Tp48GYsWLcLVq1dVnpTyG/acPWfPlbHnpmPPM7DnpJaC2HOATc+MPVfGnmcPm86ek3rYc/YcYNONYdNNx55nYNMz2Ks9ABVcxYsXL/BhzywlJUV3LBM5Li4uSElJseJEtiM0NBShoaG6r0uXLo3Tp0/jxIkTsLOzQ6VKlWBvz7szAChXrhxGjBih9hiqO3nyJGbMmKG43NfXF3fu3LHiRET5F3uujz1Xxp6bjj3PwJ4TWReb/gJ7row9zx42nT0nsjb2XB+broxNNx17noFNz8BbBVnUqlWrAAC9evWCJEm6r7Py9ttvW3Ism1SzZk189dVX6N+/Pzw8PPSWPXz4EEuXLkWtWrVUms72aDQaBAcHqz0G2SgXFxckJSUpLo+Li0ORIkWsOBFR3saem449zx72nIxhz4nMj003DXuePew5GcOeE5kfe246Nj172HQyhk3PwI+CtxH59aNpNBoNJEnC06dP4eDgAI1Gk+V5JElCWlqaFaazLX/88QfCw8NRpEgR9OnTR/dxImfPnsXKlSsRHx+PX3/9FU2aNFF5UsvbvXt3js732muvmXkS26K9PWWHJElITU210ES2q0uXLjh79iz+/vtvJCYm6n0sza1bt1CtWjW0a9cOy5cvV3tUymfY8xfYc/acPZfHnpuOPSe15NeeA2y6qdjzF9hzZWy6adhzUgt7rq8g9hxg0zNj0+Wx56Zj0zPwHes2wsHBAY0bN4aXl5fao5jVpUuXAGT8fpm/JkNNmzbFli1bMHbsWEydOlVvWY0aNbB69eoCEXgACAsLy1bMhBAFYuPwo48+ynbkC6rJkycjJCQEdevWRdeuXSFJEn777Tf88ccfWLRoEYQQiI6OVntMyofYc2LPX2DP5bHnpmPPSS35tecAm24q9vwF9lwZm24a9pzUwp4TwKZnxqbLY89Nx6Zn4DvWLejOnTu4fv06nj59CldXV5QvXx4uLi5qj0V5wK1bt3DlyhUAQNmyZQvccXF27dqVo/M1btzYzJNQXvbvv/9ixIgR2LFjBzKnLiwsDPPmzUNQUJCK01Fewp5TTrHn7DnlHntO5sKeU06x5+w55R57TubCnlNusOlsOuUem84d62YXHx+PqVOnYu3atbhx44beMo1Gg5CQEIwePRoRERHqDKiycuXKYebMmejQoYPs8k2bNmH48OGIi4uz8mREece1a9eg0Wjg5+cHAHj27Bnmz59vsF6pUqXQrVs3a49ncx48eIALFy4gPT0d5cqVg4+Pj9ojUR7AnhvHnhPlHnuePew55QR7njU2nSj32HTTseeUE+x51thzotxjz7OnIDedHwVvRleuXEGjRo1w48YNBAUFoWTJkjh16hRSUlLQt29fJCQkYNeuXejcuTMGDBiAhQsXqj2y1V2+fBmPHz9WXP748WPdq8byu1WrVgEAevXqBUmSdF9n5e2337bkWGTjTpw4gZo1a2LmzJkYOnQoACApKQljxowxWNfOzg5BQUGoVq2atce0KV5eXqhbt67aY1Aewp5njT1/gT2nnGDPs489p+xiz03Dpmdgzymn2PTsYc8pu9hz07DnL7DplBPsefYV6KYLMpsuXboIHx8fcezYMd1p9+7dE82aNRPh4eFCCCHS0tLE1KlThUajEUuWLFFrVNVIkiS++eYbxeXvvvuu8Pb2tuJE6pEkSWg0GpGcnKz7Oqt/Go1G5amta82aNeLXX3/Vff3w4UPRvn17g39Dhw5VcUrreuedd0S5cuVEWlqa7rR79+7pbluXL18Wly9fFnFxcaJMmTLinXfeUXFa9cTGxorPPvtM77SlS5eK0qVLi2LFiomRI0eK1NRUlaYjW8eeZ409f4E9zxp7bog9Nw17TrnBnpuGTc/AnmeNPZfHpmeNPafcYM9Nw56/wKZnjU03xJ6bhk3PwB3rZuTl5SUmT55scPrhw4eFRqMR58+f153WrVs3UbNmTWuOp5qZM2eKgIAAERAQIDQajShWrJju68z/vL29hUajEW+99ZbaI1uF9s745a+z+ldQbN68WWg0GrFt2zbdadqYFS9eXPj7+wt/f39RtmxZodFoxJYtW1Sc1noqV64sxowZo3ea9nLZvn273unjx48XlStXtuZ4NiM0NFT07NlT9/Xx48eFvb29qFWrlujWrZvQaDRi6tSpKk5Itow9l8eey2PPjWPP5bHnpmHPKTfYc2VsuiH23Dj2XBmbnjX2nHKDPVfGnstj041j0+Wx56Zh0zPwo+DNKDk5Ge7u7gane3h4QAiB27dvo3z58gCAFi1aYNOmTdYeURXFihVDlSpVAGR8LI2fn5/uOBVakiShcOHCqF27NqKiotQY0+rKli1r9OuCbtWqVahXrx6aN29usOzrr79G06ZNdV/Xq1cPK1asQOvWra05oiouX76MSpUq6Z1mb2+P4OBguLm56Z0eEBBQYD7m6WWnT59G586ddV+vXr0a7u7u2LNnD1xcXDB48GCsWrUK7733nopTkq1iz+Wx5/LYc+PYc3nsuWnYc8oN9lwZm26IPTeOPVfGpmeNPafcYM+Vsefy2HTj2HR57Llp2PQM3LFuRjVr1sSaNWswZMgQ2NnZ6U5ftmwZ7OzsUKFCBd1pjx8/hrOzsxpjWl2PHj3Qo0cPAECTJk3w4YcfolmzZipPZXvKlSuHmTNnokOHDrLLN23ahOHDhyMuLs7Kk6njwIED6Nevn0nrdujQAV999ZWFJ7Id6enpel97eHjg77//NlhPkiQIIaw1lk1JSkrSe+D166+/Ijw8HC4uLgCAunXrYs2aNWqNRzaOPZfHnpuGPdfHnitjz7PGnlNusOfK2PSssef62HPj2HTj2HPKDfZcGXtuGjZdH5uujD3PGpuegTvWzSg6OhqtW7dG5cqV0bFjRzg7O+PPP//E9u3b0a9fP/j4+OjW3b17N6pVq6bitOrYsWOH2iPYrMuXL+Px48eKyx8/flygXgl169YtlC5dWu80Z2dnjBgxAmXKlNE73c/PD7dv37bmeKopVaoUjh07ZtK6x44dQ6lSpSw8kW0qXbo0Dh06hL59++LChQs4efIkRo8erVt+//59ODo6qjgh2TL2PGvsuTL2XB97Lo89Nw17TrnBnpuGTZfHnutjz5Wx6Vljzyk32HPTsOfK2HR9bLo89tw0bHoGjdoD5CctWrTATz/9BHt7e3z22WeIiYnB4cOHMW7cOMybN09v3c6dO+Ozzz5TaVL1rF27Fr1791Zc3qdPH6xbt856A9kYSZIUlx06dAienp7WG0ZlTk5OBhs9Li4u+PLLL3Uf8aSVlJQEBwcHa46nmhYtWuDrr7/GnTt3jK53584dfP3112jRooWVJrMtPXv2xOLFi9GhQwe0atUKXl5eeP3113XLjxw5oveqZqLM2POssefGsecvsOfy2HPTsOeUG+y5adh0Zez5C+y5MjY9a+w55QZ7bhr23Dg2/QU2XR57bho2/f+pd3j3/O3+/fvi5s2bIj09Xe1RbErdunXFwIEDFZcPGTJEhISEWHEidc2cOVMEBASIgIAAodFoRLFixXRfZ/7n7e0tNBqNeOutt9Qe2Wrq1KkjunXrZtK63bp1E7Vr17bwRLbh0qVLonDhwqJq1ari0KFDsuscOnRIVKtWTRQuXFjExcVZeULbkJKSIiZMmCBq1KghwsLCxO7du3XL4uPjha+vr/j0009VnJDyCvZcHnuujz1Xxp7LY89Nw56TubDnytj0F9hzZey5MjY9a+w5mQt7row918emK2PT5bHnpmHTM/Cj4C3Ey8sry3WuX78OPz8/K0xjO86ePYu+ffsqLg8ODsa3335rxYnUVaxYMVSpUgVAxsfS+Pn5GVwnJElC4cKFUbt2bURFRakxpioiIiIQExODAwcOICQkRHG9v/76Cz/++CMmTpxoveFU5O/vj7Vr16JHjx6oX78+ypcvj6pVq8LV1RWPHz/GyZMnceHCBTg7O+Obb75BQECA2iOrwt7eHpMnT8bkyZMNlnl7e+PWrVt6p6WkpGD//v0IDg6Gh4eHtcakPIA9l8ee62PPlbHn8thz07DnZC7suTI2/QX2XBl7roxNzxp7TubCnitjz/Wx6crYdHnsuWnY9P+n9p79/CQkJEQcP348y/XS0tLEF198Idzd3a0wlW1xc3MTU6ZMUVw+ZcoUUbhwYStOZDvCwsJEbGys2mPYjIcPH4py5coJNzc38fnnn4sbN27oLb9x44b4/PPPhbu7uyhXrpx4+PChSpOq4+LFi2LgwIHCz89PSJKk+1eyZEkxYMAAcf78ebVHzFNu3bolNBqN2L59u9qjkA1gz7PGnitjz/Wx58ax5+bFnlNm7Llp2HR57Lk+9jxrbLr5sOeUGXtuGvZcGZuuj003jj03r/zadEkIIdTeuZ9flChRAvHx8Rg5ciRiYmLg7OxssM5ff/2FwYMH49ixY2jXrh1+/vlnFSZVT+PGjZGQkIBDhw4ZHJ8jOTkZdevWhYeHB/bs2aPShGRLLly4gI4dO+Lff/+FJEnw9PTUvUosISEBQghUrlwZGzZswCuvvKL2uKp59OgRHj58CDc3N7i7u6s9Tp50+/ZtlChRArGxsWjatKna45DK2POsseeUHey5adjz3GPPKTP23DRsOpmKPTcdm5477Dllxp6bhj2n7GDTTcOe515+bbpG7QHykzNnzqBv37744osvUKVKFWzevFm3LDExEYMHD0bDhg1x//59/PjjjwUy8uPHj8fJkyfRpEkT/PLLL4iLi0NcXBx+/vlnhIWF4d9//8X48ePVHlMVa9euRe/evRWX9+nTB+vWrbPeQDagfPny+Oeff/D111/jjTfegL+/PxwcHFC2bFl0794da9aswT///FOgAw8Abm5u8PPzY+CJzIQ9zxp7row9N8Sem4Y9JzIv9tw0bLo89twQe246Np3IfNhz07Dnyth0Q2y6adhzUqTm2+Xzq3379omqVasKjUYjOnfuLBYuXCh8fX1FoUKFxJgxY0RSUpLaI6pq+fLlwt3dXWg0Gt0/SZKEu7u7WLp0qdrjqaZu3bpi4MCBisuHDBkiQkJCrDgRUcFx69YtIUlSvvtYGsod9tw49lwee06kHvac5LDnWWPTDbHnROphz0kOe5419lwem06knvzadHu1d+znRw0aNMDff/+NoUOHYvHixdiwYQOCgoIQGxuLqlWrqj2e6nr37o1OnTph27ZtuHjxIgAgMDAQLVu2hJubm8rTqefs2bPo27ev4vLg4GB8++23VpzI9iQnJ+Po0aO4c+cOGjZsiKJFi6o9EhHlY+y5cey5PPY8a+w5EVkTe541Nt0Qe5419pyIrIk9zxp7Lo9NzxqbTpQ93LFuAampqZgyZQpWrVoFLy8vuLi44Ny5c1ixYgViYmJQuHBhtUdUnbu7Ozp37qz2GDZFCIGEhATF5Q8ePEBKSor1BrIxs2fPxsSJE5GQkABJkrBt2zY0bdoU9+7dQ6VKlfDZZ58Z3UgiIsou9jxr7Lkh9tw49pyIrI09Nw2bro89N449JyJrY89Nw54bYtONY9OJso/HWDezPXv2oHr16oiOjkbHjh1x+vRpnD59GoMHD8bMmTNRuXLlAnusl8wePXqEkydPYs+ePdi9e7fBv4KoZs2a+Pbbb/H8+XODZcnJyfjmm29Qs2ZNFSZT3/LlyzFy5EiEh4dj2bJlEELolhUtWhRNmzbF2rVrVZyQiPIb9tw07Lkh9lwZe05E1saem45N18eeK2PPicja2HPTseeG2HRlbDpRDqn3KfT5T9++fYVGoxHly5cX27ZtM1h+6NAhUbNmTaHRaERERIT477//VJhSXffu3RNvvPGGKFSokN6xXl7+/4Joy5YtQqPRiFdffVX8/PPP4uLFi+LixYti48aNIiQkRGg0GrFp0ya1x1RFlSpVREREhBAi4zr08nE5pk6dKkqWLKnWeJQP5NfjvVDOsOdZY8+VsefK2HOyNPacMmPPTcOmy2PPlbHnZGnsOWXGnpuGPVfGpitj08nS8mvT+VHwZrRmzRq8//77+N///gdHR0eD5XXq1MHhw4cxa9YsREdHo3LlykhMTFRhUvUMGDAAv/zyC4YPH45GjRrBy8tL7ZFsRuvWrbF06VKMGDECERERutOFEHBzc8OSJUvQtm1b9QZU0YULFzB8+HDF5d7e3oiPj7fiRGTLnjx5grfeegudO3dGz549TTqPh4cHli9fjipVqlh4OsoL2POssefK2HNl7DllB3tOucWem4ZNl8eeK2PPKTvYc8ot9tw07LkyNl0Zm07Zwaa/wB3rZvTPP/8gKCjI6DoajQajRo1Cly5djN5p5Ve///47Ro0ahc8++0ztUWxS79690alTJ2zbtg0XL14EAAQGBqJly5Zwc3NTeTr1eHp64t69e4rLT506heLFi1txIrJlLi4uiI2NRevWrU0+j5OTEyIjIy04FeUl7HnW2HPj2HN57DllB3tOucWem4ZNV8aey2PPKTvYc8ot9tw07LlxbLo8Np2yg01/gTvWzShz5G/cuIFff/0Vp0+fxsOHD+Hm5obKlSsjPDwcJUuWROnSpbFhwwYVp1WHi4sL/P391R7Dprm7u6Nz585qj2FT2rRpg8WLFyMqKspg2b///oslS5agb9++KkxGtio0NBT79+/HgAED1B6F8iD2PGvsedbYc0PsOWUXe065wZ6bhk03jj03xJ5TdrHnlBvsuWnY86yx6YbYdMouNj2DJIQQag+Rnzx9+hRjxozBV199hdTUVLx88RYqVAj9+/fH9OnT4ezsrNKU6nn33Xdx4sQJbNu2Te1RbNajR49w5coVPHjwwOD6AwCvvfaaClOp68aNG6hfvz6EEGjfvj0WL16Mt956C2lpafjhhx9QokQJHDx4EEWLFlV7VLIRcXFxaNWqFbp3747BgwejVKlSao9EeQx7bhx7njX23BB7TtnFnlNusedZY9ONY88NseeUXew55RZ7njX2PGtsuiE2nbKLTc/AHetmlJqaipYtW2Lnzp1o0qQJ3n77bQQHB8PNzQ2PHj3CsWPHsGrVKuzYsQNNmjTB77//Djs7O7XHtqp9+/Zh2LBh8PHxwcCBA1G6dGnZy6BWrVoqTKeu+Ph4DB06FD/88APS0tIAZBzrRZIkvf/XLito7ty5gwkTJuDHH39EQkICAMDNzQ2dO3fG1KlTUaxYMXUHJJvi5uaG1NRUPH/+HABgb29vcCwuSZIK5HG3KGvsedbYc2XsuXHsOWUHe065wZ6bhk2Xx54bx55TdrDnlBvsuWnYc2VsunFsOmUHm56BO9bNaM6cORgxYgTmzZuHIUOGKK63cOFCREVFYfbs2Rg6dKgVJ1SfRqPR/b82XpkV5JB16tQJv/zyC4YPH45GjRrBy8tLdr3GjRtbeTJ1JScn47fffoO/vz+qV68OALh79y7S09Ph4+Ojd50i0urdu7fsfczLli9fboVpKK9hz7PGnitjz+Wx55QT7DnlBntuGjZdHnsujz2nnGDPKTfYc9Ow58rYdHlsOuUEm56BO9bNqF69evDz8zPpWC4RERG4fv06Dh06ZIXJbMfKlStNWi8yMtLCk9geV1dXREVF4bPPPlN7FJsihICTkxNmzZqFwYMHqz0OERUA7HnW2HNl7Lk89pyIrI09Nw2bLo89l8eeE5G1seemYc+Vseny2HSinLNXe4D85PTp0+jXr59J64aHh2Ps2LEWnsj2FMR4m8rFxQX+/v5qj2FzJEnCK6+8gnv37qk9ChEVEOx51thzZey5PPaciKyNPTcNmy6PPZfHnhORtbHnpmHPlbHp8th0opzjjnUzkiQJ6enp2VqfSOutt97Chg0bEBUVpfYoNmfChAl499130bVrV1SsWFHtcSgP+e+///D3338jMTFR9v757bffVmEqsnXsOeUGe66MPaecYs8pJ9hzyg32XBl7TjnFnlNOsOeUW2y6MjadcqqgN50fBW9G9erVQ4kSJbBx48Ys142IiMCNGzdw8OBBK0xmO/r27ZvlOpIkYenSpVaYxrbs27cPw4YNg4+PDwYOHIjSpUvDzs7OYL1atWqpMJ26hg8fju3bt+PcuXMICwuDv78/nJ2d9daRJAmzZs1SaUKyNc+ePUNkZCR++OEHpKenQ5IkaHOX+UFWQTy2FGWNPc8ae66MPVfGnlN2seeUG+y5adh0eey5Mvacsos9p9xgz03Dnitj05Wx6ZRdbHoG7lg3o9mzZ2PUqFGYO3cuhgwZorjewoUL8c477+DLL7/E8OHDrTih+vz9/Q1eOZiWloabN28iLS0NPj4+KFy4MOLi4lSaUD0ajUb3/3KvrhRCQJKkfH+nJCfzZaOkoF42JO/dd9/FnDlz8Mknn6BBgwYICwvDypUrUaJECcycORM3btzAqlWrULVqVbVHJRvEnmeNPVfGnitjzym72HPKDfbcNGy6PPZcGXtO2cWeU26w56Zhz5Wx6crYdMouNj0Dd6ybUWpqKpo3b449e/agWbNm6NWrF4KDg+Hm5oZHjx7h+PHjWL16NWJjYxEaGort27fD3p6fxg8AKSkpWLRoEWbOnIlt27YhICBA7ZGsbuXKlSatx2PmEGWtTJkyCA8Px+LFixEfHw8fHx/ExsaiadOmAICmTZuiYsWKWLBggcqTki1iz3OOPWfPicyJPafcYM9zp6A3nT0nMh/2nHKDPc+dgt5zgE0nMic2PQN3rJvZkydPMGrUKCxbtszg2AJCCNjZ2aFPnz748ssvUbhwYZWmtF1RUVG4cuUKNm/erPYoZINOnjyJLVu24PLlywCAgIAAtG7dOt+/Aoqyz8nJCXPnzkX//v3x5MkTuLq6YuPGjWjfvj0AYN68eZg0aRJu376t8qRkq9jz3GHPyRj2nEzFnlNusee5x6aTEvacTMWeU26x57nHnpMxbDqZik3PwJdvmZmLiwsWLVqEjz76CFu3bsWpU6fw6NEjuLm5ISgoCK1bt0apUqXUHtNmBQcHY/Xq1WqPQTYmOTkZgwYNwurVqyGE0H1MTXp6OsaPH4+ePXviq6++goODg8qTkq3w9fVFfHw8gIz7ZS8vL5w9e1YX+YcPH+LZs2dqjkg2jj3PHfac5LDnlF3sOeUWe557bDq9jD2n7GLPKbfY89xjz0kOm07ZxaZn4I51C/Hz80P//v11X585cwbr16/Hp59+ikqVKqF3795wd3dXcULbtG3bNri4uKg9hir69u2b5TqSJGHp0qVWmMa2vPfee1i1ahWioqIwbNgwBAYGQpIkXLhwAbNnz8aCBQvg7e2NmTNnqj0q2Yj69etj7969eO+99wAA7du3x+eff44SJUogPT0dX375JUJCQlSekvIC9jxn2HPj2HP2nEzDnpO5sOc5V1Cbzp4rY88pu9hzMhf2POcKas8BNt0YNp2yi03PwI+CN6O5c+di9uzZ2LdvH4oWLao7fdOmTejSpQtSUlKgvbjLlSuHAwcO6K1XEEyaNEn29ISEBOzevRtHjx7F+PHj8emnn1p5MvX5+/tDkiS909LS0nDz5k2kpaXBx8cHhQsXRlxcnEoTqqdo0aJo27at4jFxevXqha1bt+LevXtWnoxs1d69e7F+/Xp89tlncHR0xLVr19C8eXOcP38eABAYGIhNmzahYsWKKk9Ktog9zxp7row9V8aeU3ax55Qb7Llp2HR57Lky9pyyiz2n3GDPTcOeK2PTlbHplF1s+v8TZDYtWrQQ4eHheqelpKSIYsWKCRcXF7FixQpx8uRJMW3aNGFvby9Gjhyp0qTqkSRJ9p+3t7eoW7euWLRokUhPT1d7TJvy/PlzMWfOHBEYGCji4uLUHkcV7u7uYv78+YrL58+fLzw8PKw3EOVJaWlp4p9//hEnTpwQKSkpao9DNow9zxp7nn3sOXtO5sGek6nYc9Ow6dnDnrPnZB7sOZmKPTcNe559bDqbTuZREJvOd6ybUalSpTBgwABER0frTtu2bRtatWqFCRMm4JNPPtGd3rNnTxw+fBhnz55VY1TKg6KionDlyhVs3rxZ7VGsrlu3bnj+/Dl++ukn2eWvv/46HB0dsW7dOusORkT5EntOlsSes+dEZB3sOVkSe86eE5F1sOdkaWw6m06UXTzGuhnFx8ejdOnSeqdt374dkiShY8eOeqc3bNgQP/74ozXHU4W3tzcWL16MLl26AMj4WJpOnTqhatWqKk+W9wQHB2P16tVqj6GKjz/+GN26dUOnTp3wzjvvoHz58gCA8+fPY968ebhy5Qq+++473L9/X+983t7eaoxLKti9e3eOzvfaa6+ZeRLKD9hzQ+y5+bDn7DkpY8/JnNhzeWy6ebDn7DkpY8/JnNhzeey5+bDpbDopY9Plcce6Gfn6+uLWrVt6p+3ZswcuLi4IDg7WO93BwQEODg7WHE8Vjx8/xpMnT3RfT5w4EeXLl2fkc2Dbtm1wcXFRewxVBAUFAQBOnDiBjRs36i3TfuhG5cqVDc6XlpZm+eHIJoSFhRkcL8kYIQQkSeJ1hGSx54bYc/Nhz9lzUsaekzmx5/LYdPNgz9lzUsaekzmx5/LYc/Nh09l0Usamy+OOdTOqU6cOVq5ciWHDhsHNzQ3//vsvDh48iNdffx329voX9ZkzZ1CqVCmVJrWewMBAfP/992jUqBHc3d0BAElJSQavcnpZQXzV06RJk2RPT0hIwO7du3H06FGMHz/eylPZho8++ihbd+BU8OzYsUPtESgfYc8NseemY8+VseeUFfaczIk9l8emm4Y9V8aeU1bYczIn9lwee246Nl0Zm05ZYdPl8RjrZnTixAnUrVsXnp6eqFKlCo4cOYInT55g//79qF27tt66gYGBaNq0KZYsWaLStNaxevVq9OnTB9m9muX3V7TI0Wg0sqd7eXkhMDAQ/fv3x4ABAxg7IiILY88NseemY8+JiGwDey6PTTcNe05EZBvYc3nsuenYdCIyN75j3YyqVauGP/74A5MnT0ZcXBxCQkIwZswYg8jv3LkTLi4u6Nq1q0qTWk+vXr1Qr1497Ny5E7dv38bEiRPRsWNHVK9eXe3RbE56erraIxAREdhzOey56dhzIiLbwJ7LY9NNw54TEdkG9lwee246Np2IzI3vWCerCggIwKxZs9ChQwe1R1Gdt7c3Fi9ejC5dugDI+FiaTp068Vg4RGbQt2/fLNeRJAlLly61wjRE+Q97/gJ7TmQ57DmR5bHpGdhzIsthz4ksjz1/gU0nshw2PQPfsU5WdenSpWytf+/ePdSrVw9ff/01GjRoYKGp1PH48WM8efJE9/XEiRNRvnx5Rp7IDP744w+Dj3BKS0vDzZs3kZaWBh8fHxQuXFil6YjyPvb8BfacyHLYcyLLY9MzsOdElsOeE1kee/4Cm05kOWx6Bu5YJ5uWlpaGy5cv4+nTp2qPYnaBgYH4/vvv0ahRI7i7uwMAkpKScP/+faPn8/b2tsZ4RHna5cuXZU9PSUnBokWLMHPmTGzbts26QxEVYOy5IfacKGvsOZHtya9NZ8+JLIc9J7I9+bXnAJtOZElsegZ+FDzZtNu3b6NEiRKIjY1F06ZN1R7HrFavXo0+ffoguzfBtLQ0C01EVHBERUXhypUr2Lx5s9qjEBUI7Lkh9pwo99hzIuvLr01nz4nUw54TWV9+7TnAphOpqaA0ne9YJ1JJr169UK9ePezcuRO3b9/GxIkT0bFjR1SvXl3t0YjyveDgYKxevVrtMYgoH2DPidTDnhORubDnROphz4nInNh0IvUUlKZzxzqRiipWrIiKFSsCAJYvX47IyEh06NBB5amI8r9t27bBxcVF7TGIKJ9gz4nUwZ4TkTmx50TqYM+JyNzYdCJ1FJSmc8c6kY24dOlStta/d+8e6tWrh6+//hoNGjSw0FREedOkSZNkT09ISMDu3btx9OhRjB8/3spTEVFBwJ4TmQ97TkRqYc+JzIc9JyI1selE5sOmZ+COdaI8Ki0tDZcvX8bTp0/VHoXI5kycOFH2dC8vLwQGBmLhwoUYMGCAdYciIpLBnhMpY8+JKK9gz4mUsedElJew6UTK2PQM3LFORET5Tnp6utojEBERUS6x50RERHkfe05ERJQ/sOkZNGoPQGSMg4MDGjduDC8vL7VHISIiohxiz4mIiPIHNp2IiCjvY8+JiHKO71gnq7tz5w6uX7+Op0+fwtXVFeXLl4eLi4vsul5eXtixY4eVJySivObq1as5Ol+ZMmXMPAlRwcGeE5G5sedE6mDTicic2HMidbDnRGRubLo87lgnq4iPj8fUqVOxdu1a3LhxQ2+ZRqNBSEgIRo8ejYiICHUGJKI8zd/fH5IkZft8aWlpFpiGKP9iz4nIkthzIuth04nIUthzIuthz4nIkth0edyxThZ35coVNGrUCDdu3EBQUBBKliyJU6dOISUlBX379kVCQgJ27dqFzp07Y8CAAVi4cKHaIxNRHrNs2TK9yKenp2PWrFm4cuUKevbsiYoVKwIAzpw5g2+++Qb+/v4YPny4WuMS5UnsORFZGntOZB1sOhFZEntOZB3sORFZGpsujzvWyeLGjBmDZ8+e4ejRo6hevTqAjFfTde/eHZcuXcLWrVuRnp6Ozz//HBMmTECdOnXQv39/lacmorykd+/eel9PnjwZz549w4ULF1CkSBG9ZRMnTkRoaChu3bplxQmJ8j72nIgsjT0nsg42nYgsiT0nsg72nIgsjU2Xp1F7AMr/tm/fjpEjR+oCDwBFihTBtGnT8Pvvv+PChQvQaDR477330KVLF8yfP1/FafMOBwcHNG7cGF5eXmqPQmRzFi5ciIEDBxoEHgB8fHwwYMAALFiwQIXJiPIu9twy2HMiZew5kWWw6ebHnhMpY8+JLIM9tww2nUgZm56B71gni0tOToa7u7vB6R4eHhBC4Pbt2yhfvjwAoEWLFti0aZO1R7QZd+7cwfXr1/H06VO4urqifPnycHFxkV3Xy8sLO3bssPKERHlDfHw8njx5orj8yZMniI+Pt+JERHkfe2469pzIPNhzIstg003DnhOZB3tOZBnsuenYdCLzYNMz8B3rZHE1a9bEmjVrkJaWpnf6smXLYGdnhwoVKuhOe/z4MZydna09oqri4+MxduxYlC5dGiVKlECdOnXQqFEj1KxZEx4eHmjUqBF++ukntcckylNCQkIwc+ZMHDlyxGDZ4cOHMWvWLNSvX1+FyYjyLvbcOPacyPzYcyLLYNOVsedE5seeE1kGe24cm05kfmx6BkkIIdQegvK3bdu2oXXr1ggMDETHjh3h7OyMP//8E9u3b0e/fv2wePFi3bqdOnXCgwcPCsyrwq5cuYJGjRrhxo0bCAoKgouLC06dOoWUlBT07dsXCQkJ2LVrF27duoUBAwZg4cKFao9MlCecOnUKYWFhiI+PR0hICF555RUAwPnz53HgwAF4e3tj586dqFKlisqTEuUd7Lky9pzIMthzIstg0+Wx50SWwZ4TWQZ7roxNJ7IMNv3/CSIr+OWXX0TlypWFJElCkiTh5eUlxo8fL54/f6633po1a8TBgwdVmtL6unTpInx8fMSxY8d0p927d080a9ZMhIeHCyGESEtLE1OnThUajUYsWbJErVGJ8pxbt26JkSNHiooVKwonJyfh5OQkKlasKEaNGiVu3ryp9nhEeRJ7Lo89J7Ic9pzIMth0Q+w5keWw50SWwZ7LY9OJLIdNF4LvWCerevDgAZKTk+Hr6wtJktQeR3Xe3t4YM2YMJkyYoHf6kSNHUK9ePZw9e1Z3LJzu3bvj/PnzOHr0qBqjEhER6bDn+thzIiLKq9j0F9hzIiLKq9hzfWw6EVkSj7FOVuXl5YXixYsbDfz169etOJG6kpOT4e7ubnC6h4cHhBC4ffu27rQWLVrg7Nmz1hyPiIhIFnuujz0nIqK8ik1/gT0nIqK8ij3Xx6YTkSXZqz0A5X8NGjTA4sWLUa1aNaPrpaenY+bMmYiJiUFiYqKVplNXzZo1sWbNGgwZMgR2dna605ctWwY7OztUqFBBd9rjx4/h7OysxphEedLp06exfPlyxMXF4cGDB3j5A1okScL27dtVmo4o72HPlbHnRJbDnhOZH5sujz0nshz2nMj82HNlbDqR5bDp3LFOVnD58mXUrl0bI0eORExMjGyo/vrrLwwePBjHjh1Du3btVJhSHdHR0WjdujUqV66Mjh07wtnZGX/++Se2b9+Ofv36wcfHR7fu7t27s9xQIqIMq1evRp8+fVCoUCFUrFgRXl5eBuvwSChE2cOeK2PPiSyDPSeyDDZdHntOZBnsOZFlsOfK2HQiy2DT/58aB3angiUhIUEMGjRIaDQaERAQIDZt2mSwzM7OTpQpU0Zs2LBBvUFV8ssvv4jKlSsLSZKEJEnCy8tLjB8/Xjx//lxvvTVr1oiDBw+qNCVR3lKuXDlRp04dcffuXbVHIco32HPj2HMi82PPiSyDTVfGnhOZH3tOZBnsuXFsOpH5sekZJCEKwssHyBbs378fAwcOxKlTp9CxY0e0aNEC0dHRuH//PkaMGIGYmBi4uLioPaZqHjx4gOTkZPj6+ho9Hg4RZc3Z2RkzZszAkCFD1B6FKN9hz41jz4nMhz0nsiw2XRl7TmQ+7DmRZbHnxrHpRObDpmfgjnWyqtTUVAwdOhSLFy+GJEkICgrC2rVrUbVqVbVHyxOuX78OPz8/tccgsnn169dHy5Yt8fHHH6s9ClG+xJ7nDntOZBr2nMjy2PScY8+JTMOeE1kee547bDqRadj0DBq1B6CCIzU1FVOmTMGqVavg5eWFkiVL4ty5c1ixYgWSkpLUHk8VDRo0wIkTJ7JcLz09HTNmzEDlypWtMBVR3jdjxgwsXboU+/btU3sUonyHPTfEnhNZBntOZFlsuj72nMgy2HMiy2LPDbHpRJbBpmfgO9bJKvbs2YNBgwbhzJkz6NGjB7788ku4uLhgwoQJmD9/Pvz8/DBnzhx06NBB7VGtqkSJEoiPj8fIkSMRExMDZ2dng3X++usvDB48GMeOHUO7du3w888/qzApUd7SoUMHnD9/HufOnUPlypVRpkwZ2NnZ6a0jSRI2btyo0oREeRN7Lo89J7IM9pzIcth0Q+w5kWWw50SWw57LY9OJLINNz8Ad62Rx/fr1w4oVK1CuXDksWLAAzZs311t++PBhDBw4EMeOHUOHDh0wd+7cAvPRK4mJiXjvvfewZMkSlC1bFnPmzEHbtm31ln311Vfw8/PDrFmzEBERoe7ARHmEv79/lsdNkiQJcXFxVpqIKO9jz5Wx50SWwZ4TWQabLo89J7IM9pzIMthzZWw6kWWw6Rm4Y50sztHREWPHjsX//vc/ODo6yq6Tnp6OWbNmITo6GpIkITEx0cpTqmv//v0YOHAgTp06hY4dO6JFixaIjo7G/fv3MWLECMTExMDFxUXtMYmIqABjz7PGnhMRUV7AphvHnhMRUV7AnmeNTSciS+COdbK406dPIygoyKR1r127huHDh2PDhg0Wnsr2pKamYujQoVi8eDEkSUJQUBDWrl2LqlWrqj0aERERe6U/BDQAAQAASURBVG4i9pyIiGwdm5419pyIiGwde24aNp2IzI071smqbty4gV9//RWnT5/Gw4cP4ebmhsqVKyM8PBwlS5ZUezzVpKamYsqUKZgyZQqcnZ3h4uKC27dvY/jw4YiJiUHhwoXVHpEoz3r06BESExORnp5usKxMmTIqTESU97Hn8thzIsthz4ksg003xJ4TWQ57TmQZ7Lk8Np3Icgp00wWRFTx58kRERUUJBwcHodFohCRJev8cHBxEVFSUePLkidqjWt3u3btFUFCQkCRJvPnmm+L27dvi0aNHYtiwYcLOzk6UKVNGbNy4Ue0xifKc+fPni/LlywuNRqP4j4iyhz1Xxp4TWQZ7TmQZbLo89pzIMthzIstgz5Wx6USWwaYLoVF7xz7lf6mpqWjbti0WLFiA0NBQLFu2DEePHsX58+dx9OhRLF++HKGhoViwYAHatWuHtLQ0tUe2mn79+iEsLAwpKSn4/fff8fXXX6NYsWJwdXXF7NmzceDAARQpUgQdO3ZEx44dcf36dbVHJsoTFi5ciHfeeQfly5fHJ598AiEERo4cifHjx6N48eIIDg7G0qVL1R6TKE9hz5Wx50SWwZ4TWQabLo89J7IM9pzIMthzZWw6kWWw6f9P3f36VBDMnj1bSJIk5s+fb3S9BQsWCEmSxJw5c6w0mfocHBzEBx98IJ49e6a4TlpampgxY4Zwc3MT7u7uVpyOKO+qXLmyCA8PF0IIce/ePSFJkti+fbsQQoiEhARRvnx5MX36dDVHJMpz2HNl7DmRZbDnRJbBpstjz4ksgz0nsgz2XBmbTmQZbHoGHmOdLK5evXrw8/PDhg0bslw3IiIC169fx6FDh6wwmfpOnz6NoKAgk9a9du0ahg8fbtLlSFTQOTk5YcaMGYiKisLDhw/h6emJLVu2IDw8HAAwbdo0LF68GBcvXlR5UqK8gz1Xxp4TWQZ7TmQZbLo89pzIMthzIstgz5Wx6USWwaZnsFd7AMr/Tp8+jX79+pm0bnh4OMaOHWvhiWxH5sDfuHEDv/76K06fPo2HDx/Czc0NlStXRnh4OEqWLInSpUsz8EQm8vDwQGpqKgDA3d0dLi4uuHbtmm65m5sbbt26pdZ4RHkSe66MPSeyDPacyDLYdHnsOZFlsOdElsGeK2PTiSyDTc/AHetkcZIkIT09PVvrFyRPnz7FmDFj8NVXXyE1NRUvf4hEoUKF0L9/f0yfPh3Ozs4qTUmUt1StWhXHjh3TfR0SEoIFCxagTZs2SE9Px6JFi1ChQgUVJyTKe9hz49hzIvNjz4ksg01Xxp4TmR97TmQZ7LlxbDqR+bHpGTRqD0D5X6VKlfDrr7+atO6vv/6KSpUqWXgi25Gamoq2bdtiwYIFCA0NxbJly3D06FGcP38eR48exfLlyxEaGooFCxagXbt2SEtLU3tkojzhrbfewsmTJ5GcnAwAiImJwenTp1GmTBn4+/vj7Nmz+OSTT1SekihvYc+VsedElsGeE1kGmy6PPSeyDPacyDLYc2VsOpFlsOn/T80DvFPBMGvWLKHRaMT8+fONrrdgwQKh0WjErFmzrDSZ+mbPni0kSTLpspEkScyZM8dKkxHlPxcvXhQzZ84Uc+bMEWfPnlV7HKI8hz1Xxp4TWQ97TpR7bLo89pzIethzotxjz5Wx6UTWUxCbLgnx0mdgEJlZamoqmjdvjj179qBZs2bo1asXgoOD4ebmhkePHuH48eNYvXo1YmNjERoaiu3bt8PevmAcpaBevXrw8/Mz6TguERERuH79Og4dOmSFyYiIiPSx58rYcyIiykvYdHnsORER5SXsuTI2nYgsqWDck5Kq7O3tsWXLFowaNQrLli3D9u3b9ZYLIWBnZ4d+/frhyy+/LDCBB4DTp0+jX79+Jq0bHh6OsWPHWngiIiIieey5MvaciIjyEjZdHntORER5CXuujE0nIksqOPempCoXFxcsWrQIH330EbZu3YpTp07h0aNHcHNzQ1BQEFq3bo1SpUqpPabVSZKE9PT0bK1PRIY0Gk22bx+SJCE1NdVCExHlT+y5PPacyDzYcyLrYdMNsedE5sGeE1kPey6PTScyDzZdHj8KnlR15swZrF+/Hjdv3kSlSpXQu3dvuLu7qz2W1dSrVw8lSpTAxo0bs1w3IiICN27cwMGDB60wGVHeMnHixBxtBEdHR1tgGqKChz1nz4nMgT0nUl9Bbjp7TmQe7DmR+gpyzwE2nchc2HQF6h3enQqKOXPmiFdeeUXcvXtX7/RffvlFODo6Co1GIyRJEpIkicDAQIP18rNZs2YJjUYj5s+fb3S9BQsWCI1GI2bNmmWlyYiIiPSx58rYcyIiykvYdHnsORER5SXsuTI2nYgsie9YJ4tr2bIl7OzssHXrVt1pqamp8PPzw+PHjzF//nzUqVMHmzdvxgcffIChQ4fiyy+/VHFi60lNTUXz5s2xZ88eNGvWDL169UJwcDDc3Nzw6NEjHD9+HKtXr0ZsbCxCQ0Oxffv2AnU8HKLsunbtGjQaDfz8/AAAz549w/z58w3WK1WqFLp162bt8YjyNPZcGXtOZF7sOZFlseny2HMi82LPiSyLPVfGphOZF5v+ErX37FP+5+fnJyZOnKh32u+//y4kSRIffPCB3ulvvvmmqFChgjXHU11SUpIYOHCgsLe3FxqNRu+fJEnC3t5eDBgwQDx+/FjtUYls2vHjx4WdnZ2YM2eO7rR79+7pXp2b+Z+9vb04fvy4itMS5T3suXHsOZF5sOdElsemK2PPicyDPSeyPPbcODadyDzYdEN8GQ5ZXHx8PEqXLq132vbt2yFJEjp27Kh3esOGDfHjjz9aczzVubi4YNGiRfjoo4+wdetWnDp1Co8ePYKbmxuCgoLQunVrlCpVSu0xiWzeokWLULZsWURFRRks+/rrr/Hqq68CANLT0xEWFoZFixZh7ty51h6TKM9iz41jz4nMgz0nsjw2XRl7TmQe7DmR5bHnxrHpRObBphvijnWyOF9fX9y6dUvvtD179sDFxQXBwcF6pzs4OMDBwcGa49kMPz8/9O/fX/f1mTNnsH79enz66aeoVKkSevfuDXd3dxUnJLJtO3bsQKdOnaDRaAyW+fr6omzZsrqv33zzTfz888/WHI8oz2PPTcOeE+UOe05keWx61thzotxhz4ksjz03DZtOlDtsuiHDS4LIzOrUqYOVK1fi0aNHAIB///0XBw8eRKtWrQyOXXLmzJkC9UqxuXPnokKFCrh3757e6Zs2bUKNGjUwceJELFy4ECNHjkStWrUM1iOiFy5fvoxKlSrpnWZvb687hlJmAQEBuHLlijXHI8rz2HNl7DmR+bDnRJbHpstjz4nMhz0nsjz2XBmbTmQ+bLoh7lgni4uOjsaVK1fwyiuvoFmzZmjYsCEkScL7779vsO6GDRt0Hx1REPz8888IDAxE0aJFdaelpqaiX79+sLOzw7Jly3DixAlMnToVV65cweTJk1Wclsj2paen633t4eGBv//+G3Xr1tU7XZIkCCGsORpRnseeK2PPicyLPSeyLDZdHntOZF7sOZFlsefK2HQi82LT9XHHOllctWrV8Mcff6B27dq4ceMGQkJCsGXLFtSuXVtvvZ07d8LFxQVdu3ZVaVLrO3XqFEJCQvRO27FjB+7evYtRo0YhMjISVapUwbhx49CtWzds2bJFpUmJbF+pUqVw7Ngxk9Y9duxYgXqlLpE5sOfK2HMi82HPiSyPTZfHnhOZD3tOZHnsuTI2nch82HRDPMY6WcWrr76KzZs3G10nLCwMJ06csNJEtiE+Ph6lS5fWO2379u2QJAkdO3bUO71hw4b48ccfrTkeUZ7SokULfP311/joo49QrFgxxfXu3LmDr7/+Gj179rTidET5A3sujz0nMh/2nMg62HRD7DmR+bDnRNbBnstj04nMh003xHesE6nI19cXt27d0jttz549cHFxQXBwsN7pDg4OcHBwsOZ4RHnKmDFjkJKSgmbNmuHw4cOy6xw+fBjNmzdHSkoKRo8ebeUJiSi/Ys+JzIc9JyK1sOdE5sOeE5Ga2HQi82HTDfEd60QqqlOnDlauXIlhw4bBzc0N//77Lw4ePIjXX38d9vb6N88zZ84UiI/RIMopf39/rF27Fj169ED9+vVRvnx5VK1aFa6urnj8+DFOnjyJCxcuwNnZGd988w0CAgLUHpmI8gn2nMh82HMiUgt7TmQ+7DkRqYlNJzIfNt2QJArCkeSJbNSJEydQt25deHp6okqVKjhy5AiePHmC/fv3GxwPJzAwEE2bNsWSJUtUmpYob4iLi8O0adOwefNm3LhxQ3d6iRIl0LZtW4wbNw7ly5dXcUIiym/YcyLzY8+JyNrYcyLzY8+JSA1sOpH5sekv8B3rRCqqVq0a/vjjD0yePBlxcXEICQnBmDFjDAK/c+dOuLi4oGvXripNSpR3lCtXDosWLQIAPHr0CA8fPoSbmxvc3d1VnoyI8iv2nMj82HMisjb2nMj82HMiUgObTmR+bPoLfMc6ERERERERERERERERERGRERq1ByAiIiIiIiIiIiIiIiIiIrJl3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoREREREREREREREREREZER3LFORERERERERERERERERERkBHesExERERERERERERERERERGcEd60REREREREREREREREREREZwxzoRERHlWcHBwZAkCY6OjoiPj9dbNnHiREiSlO1/O3fuBAD07t3bYJm9vT18fHzQokULrFq1CkIIFX5rkqP9G9m6X375BY0aNYK7u7vBdY6AsLAwXia5NGvWLEiShB9++EHvdO192ooVK9QZzMbklfsMAvz9/Q167OjoiDJlyqB79+7Ys2ePwXny8/Vdu30zceJEs3w/W7msbGWO/CQ/389p7xcuX76s9igEYNSoUdBoNDh8+HCuv5e57+PIvM6fP4+hQ4eicuXKKFy4MJycnFCqVCnUrVsXQ4cONdj+tEW2fP+xYsUKSJKE3r17qz2KrH379qFly5bw9vaGRqMxudvax3iSJGH69OmK6/Xv3z/Ht38hBD7//HNUrVoVzs7O+bqBSvLK72zr1/PsuHz5MiRJgr+/v8EyW76vyY7r16+jV69eKFmyJOzt7fPN3y637NUegIiIiCgnDh06hOPHjwMAnj9/jjVr1mDEiBG65TVq1EBkZKTB+X799Vfcvn0bwcHBqFGjhsHy4sWL630dGBiI0NBQAMCzZ89w8uRJxMbGIjY2Fhs3bsS6detgZ2dnxt+MXhYWFoZdu3Zhx44dCAsLU3ucHPvnn3/QuXNnpKeno2nTpihRogQkSTK4zhHl1N27dzFx4kTUrVsXnTt3VnscIrNq2LAhypcvDwBISEjA4cOHsW7dOqxfvx7Tp0/Hu+++q/KElN9NnDgRMTExiI6O5k4/yrbLly8jICAAZcuWNfuT7GpuK58+fRpz585F586dUadOHav+bLKuH3/8EW+++SaSk5NRpEgRNGzYED4+Pnjw4AH++ecfzJs3D2vXrlV1G5T305Zz48YNtG3bFomJiQgNDYW/vz80Go1u28xUU6ZMQf/+/eHp6WnW+RYsWIBx48bBw8MDrVu3hru7u1m/P5lOu3Ofb0QxLzUuVyEEOnXqhIMHD6Jy5cpo0qQJChUqpHuOtCDjjnUiIiLKk5YuXQoA8PPzw/Xr17F06VK9HesRERGIiIgwOF9YWBhu376NiIgIkx5sh4aGGrwKe8GCBYiKisKPP/6IlStXom/fvrn5VcgMTp8+rfYIWfrpp5+QkpKCCRMmYPLkyWqPQ/lQTEwMEhIS+ESiCfLCfQbp69+/v967I549e4ZBgwZh1apVGDduHNq1a4cKFSqoNyDlypQpUzB+/HiUKFFC7VGIKBvGjh2L1NRUs217DB06FG+88QaKFi1qlu9H5nH79m1ERkYiOTkZo0ePxieffAInJye9dY4cOYLvv/9epQnzh44dOyIkJAQeHh5qj2Lg999/R0JCAt588018/fXXOfoeLi4uuH//PqZOnYqpU6eadb5169YBANavX48WLVqY9XsTKfHz88Pp06dRqFAhtUexiCtXruDgwYMoU6YMjh07Bnt77k7W4kfBExERUZ7z5MkTfPvttwCA1atXw9XVFSdOnMChQ4es8vOHDBmCxo0bA3jxAI7UValSJVSqVEntMYy6evUqAOCVV15ReRLKjxISErBixQr4+fkhPDxc7XFsXl64zyDjnJycMG/ePBQuXBhpaWn48ccf1R6JcqFEiRKoVKmSTe5MICJ5586dw5YtWxASEoIqVaqY5XsWLVoUlSpV4o51G7Np0yY8fvwYJUuWxPTp0w12qgNA7dq1MWXKFBWmyz88PDxQqVIlm3yRmTkeyw4bNgwajQazZ8/GjRs3zDUaAD7WJnUUKlQIlSpVQmBgoNqjWIT2dhUQEMCd6i/hjnUiIiLKc9avX4+HDx+iatWqaNKkCbp37w7gxbvYraF27doAkO2Pcty4cSMaNWoENzc3eHh4oHHjxti8ebPRYzMBwIMHDxAdHY0aNWrAzc0NLi4uqFatGj755BM8efLEYP3Mxye8e/cu3nnnHZQuXRoODg4oXbo0hg0bhoSEBMU5z507h0GDBiEwMBBOTk7w8PDAa6+9hjVr1siun/nY2Hv27EH79u3h4+MDjUaje8f/o0ePsGTJEnTq1AmvvPIKChcujMKFC6NatWr44IMPDObZuXMnJEnCrl27AABNmjTRO8Zu5k8SMHY8sfv372PChAmoUqUKXFxc4Obmhtq1a+Ozzz7D06dPDdbX/tywsDCkpKRg2rRpqFKlCpydnVGkSBF06tQpW+921f4tli9fDgDo06ePbl7tx3Vm/vunpaVhxowZqFmzJlxdXQ1+r99++w3t2rVDsWLF4ODggJIlS6J79+6Kx7XM/Lc5cOAA2rZtiyJFisDNzQ2NGzfWOz7xr7/+imbNmsHLywuurq5o0aIFjh49avLvmtnJkyfRuXNnFC1aVHd9nTlzJtLT07N9vLGsjr2e1fE4jxw5gsjISAQEBMDJyQne3t4IDg7G2LFjceXKFYP1Dx48iG7duqFkyZJwcHBAsWLF0L59e2zbtk32+ycnJ+Pzzz9H7dq14ebmBgcHBxQvXhx169bFuHHjcP/+fYPzPH36FF988QVCQkLg6ekJJycnVKxYEePGjUN8fLxJl0tmy5cvR1JSEnr16gWNxvjDvEuXLqFXr14oXrw4HB0dERgYiA8//BDJycl660VGRkKSJKNPkq5btw6SJKFevXq60zIfNy8+Ph7vvPMOypQpA0dHR5QtWxajRo3CgwcPFL+nqZd/cnIy6tSpA0mSMH78eIPvk5aWhsaNG0OSJAwZMkRvmbH7jNTUVCxbtgzNmzdH0aJF4ejoiFKlSqF58+aYM2eO4txkfa6urqhYsSIA5R6ben3P7MiRI+jZs6fueuvt7Y1WrVphy5Ytsutnvk/bsWMHWrZsCS8vLzg7O6NWrVpYtWqV3vrp6ekoV64cJEnC/v37FeeIioqCJEkYN25cFpcEkJKSgjVr1qBnz56oVKkS3N3d4ezsjIoVK2L48OHZfgI7u83O6WWhpXSMdWtvzyiRJAkxMTEAMj4dJPP2yMvHmczudocxmbcPUlNT8dlnn+m2SYoWLYpu3brhzJkzWX6fH374AaGhoXB3d0fhwoXRsGFDxeszkHE/+NVXXyEsLAze3t5wdHREQEAAhgwZgmvXrhmsn5Ntpx07dkCSJFSqVEnxY02fPXuGIkWKQJIknDp1Ksvf88qVK5g2bRqaNm2qu/16enoiNDQUixYtQnp6uuJ5Hzx4gEmTJqFOnTrw8PCAs7MzypUrh27dumHr1q25uox69+6NgIAA3YyZrz+ZW2TJbWXt75idxxNZmTdvHoQQBreBtLQ0lCpVCpIk4cCBA4rnHzNmDCRJwqhRo3SnKW3Tmfs+jrLn9u3bAAAfHx+Tz/Pw4UO4u7vD3t5e9n5Dq02bNpAkCfPnz9edlnnb/59//kGnTp1022SVK1fGF198YXC/kZ37aa3sdErr+++/R3h4OHx8fODg4AA/Pz+89dZbivdRR44cQffu3VGqVCk4ODjA3d0d5cqVQ+fOnbFx40a9dY0dezo2Nhbt27eHr68vChUqBC8vL7zyyit46623sHv3bqMzy1m7di2aNWumu/8qW7Ys+vbti3PnzsnOFB0dDUD/slV67kJJ1apV0atXLzx9+lT3/XJLe125dOkSgIwdgNr5Xr4fyc3jaKXnODJv92zduhVhYWHw8PCAl5cX2rVrhxMnTui+3zfffIMGDRrAzc0Nnp6e6NSpEy5evKj3M5cvXw5JktCqVSvF3/nGjRsoVKgQnJ2dTXrceOrUKURHR6Nhw4bw8/ODg4MDihQpgubNmyu+USQnXdfef2u93Dq5bfWkpCS8//77KF++PBwdHVG8eHFERkbi+vXrRmd68uQJPvroIwQFBcHFxUXvunjw4EGMGzcO9erVQ/HixeHg4ABfX1+0b98esbGxsr9v5ttedmbK6nk8OdndVsnu5WqObV/t76V9Q9GuXbtkf6YptxEA+O+//zBs2DC88sorupkaNmyIRYsWIS0tzeDnZ/57JCYm4t1334W/vz+cnJzwyiuvYNq0abrL6fr16xg0aBBKly4NR0dHVKxY0TqP2QURERFRHtOoUSMBQMyYMUMIIcSff/4pAAgPDw/x5MkTo+dt3LixACCio6ONrhcZGSkAiMjISNnl/fv3FwBE9erVTZ572rRpAoAAIOrXry969Ogh6tatKwCIcePGCQCibNmyBuf7999/RenSpQUAUaJECREeHi7at28vfH19BQBRo0YNkZCQoHee6OhoAUD07dtXlCpVSvj6+opOnTqJNm3aCA8PDwFA1K1bVzx//tzg561bt044OTkJAKJSpUqiY8eOomnTpqJw4cICgOjTp4/BebSXa1RUlNBoNKJy5crijTfeEC1bthTffPONEEKIPXv2CADCx8dHhIaGiu7du4uWLVuKIkWKCACifPny4t69e7rvefr0aREZGan7PVu1aiUiIyN1//bs2aNbV3u5vuzixYuibNmyup/buXNn0aFDB+Hm5iYAiFq1aon79+/rnWfHjh0CgHj11VdF8+bNhYuLiwgPDxedO3fW/R08PT3FpUuXjP69tTZs2CAiIyNFYGCgACAaNmyo+x2mTJkihBDi0qVLAoAoU6aM6NChg3BwcBDNmjUTPXr00LuOffjhhwKAkCRJNGzYUPTo0UPUqFFDABB2dnZi6dKlin+bMWPGCHt7e1GzZk3RvXt33fkcHR3Fn3/+KebOnSs0Go149dVXRbdu3USFChUEAOHq6irOnz9v0u+qtXPnTuHs7CwAiMDAQPHGG2+IFi1aCAcHB9G9e3fd3+Tly1A7644dO0w6XUt7fZe7XX/22WdCo9EIAKJChQqiW7duon379iIoKEgAEMuXL9dbf/Hixbr1a9asKXr06CFeffVV3XVs4sSJeuunpaWJZs2aCQDC3d1dtG7dWvTo0UM0b95c93v+/fffeue5fv26qFatmgAgvL29RfPmzUXHjh116/v7+4vLly+bcEm/8NprrwkAIjY2Vna59j5txIgRwt3dXZQtW1Z069ZNNG/eXPe3ioiI0DvPkSNHdNfL1NRUoz935cqVutOWL18uAIgOHTqIwMBA4enpKSIiIkTHjh2Fl5eXACAqVqwo7ty5Y/D9snv5X7x4UXh6egpJksSWLVv0lr3//vu67/Ps2TO9ZUr3GQkJCSI0NFQAEIUKFRKNGzcWPXr0EE2aNBE+Pj6y5yHL0t4uXr6tapUvX14AEMOHD9edlpPru9bMmTN118EaNWqILl26iNDQUOHg4CAAiJiYGMUZ//e//wlJkkTt2rXFG2+8IUJCQnTXtS+//FLvPF988YUAIN58803ZORITE4Wrq6vQaDR695VK93fXrl3TbQuFhISIrl27ijZt2oiSJUvqGih3X669rF6+fLPb7NxcFsbmsPb2jJLIyEgRHBwsAIjg4GC97ZElS5bo1svJdocx2u2DsmXLik6dOolChQqJ5s2bizfeeEOUK1dO1+l9+/YZnFd7eX/00Ue67Ybu3bvrfg9JksSPP/5ocL6HDx+KsLAw3fdu3Lix6NKli6hYsaIAIIoUKSKOHj2qd56cbjtpW/j777/L/v7Lli0TAESTJk30Tlfajvj4448FABEQECCaNWsm3njjDdG4cWPd7bdTp04iPT3d4Of8888/ws/PT3cbatOmjejevbto0KCBcHZ2Fo0bN87VZbRkyRLRuXNnAUAULlxY7/qT+bGGJbeVc/J4IitlypQRAMSFCxcMlmkbPGjQINnzpqSk6H728ePHdaeb+z6OzGP16tW6xxtK25pyhg0bJgCICRMmyC6/cOGCkCRJuLu7i0ePHulO1277jx8/Xjg4OIigoCDd7dnOzk7X+MxMvZ/OaadSUlJEt27ddI+fXn31VdG1a1fdz3R2dhZbt27VO09sbKwoVKiQbqYuXbqIjh07inr16glHR0fx+uuv662v3YZ++TmIFStWCEmShCRJon79+qJ79+6iQ4cOolatWsLOzs7gsjAmPT1dvP322wKAsLe3F02bNhVvvPGG7rGfi4uL3u+xZ88exct29OjRJv1M7d9z9erV4sqVK8LR0VHY2dmJ06dP663Xr18/xcd0SqZMmSIiIyN1be/cubNuvg0bNujWy83jaGPPcWivT+PHj9d978yPpT09PcWFCxfE2LFjdZd3ly5ddPfHJUuW1NsuePbsmfDx8RGSJImzZ8/K/s4fffSR7HaM0uMb7eVaqVIl0apVK13ftNu7o0aNMjhPTrqufd5DO8fLrbt7964Q4sX1PCIiQlSvXl14enqK9u3bi9dff10UK1ZMt93zcpO0M9WvX1/UrVtXFC5cWLRu3Vp0795dNG/eXLdes2bNhEajEdWqVRNt2rQRXbt2FbVq1dLNNXPmTIPfN6czZd5Oe5m5tlVMvVyFMN+27927d0VkZKRo1aqVACB8fX1lf6Ypt5GDBw8Kb29v3fMK3bt3F+Hh4bo5W7VqJZKTk2X/Hq+//roICgoSxYoVE507dxYtW7bUPZYbOnSouHDhgihevLgoXbq06Natm2jSpImuEVOnTjXpd80pPitAREREecrZs2d1Ozwy75SpVKmSACBWrVpl9Pzm2LGelJSkeyLp7bffNmnuo0ePCjs7O2FnZ2fwJOa6det0D2pe3iB/8uSJbofshx9+qLfBmZSUJHr06CG7gax9UgqA6N27t95OpatXr+qePNRu7GodP35cODo6CicnJ/HDDz/oLbt8+bLuCdDMO9GEeHG5AhDz5s2TvQyuXbsmYmNjRVpamt7pSUlJugf3UVFRBufLaqeqEMoPIuvXry+AjB18jx8/1p1+584d3YOrl3dqaB+wARk75G7evKlb9vTpU92Di4EDByrOI0dpp4EQLx6QARClSpWSfRC9detWAUA4OTkZPAH91Vdf6W4XJ0+e1FumvfwkSRKrV6/WW/buu+8KIGMnp6urq94TZampqbongfv372/y7/nkyRPd9Wv06NF6f+9///1X9ySq3INMc+9Y37hxo+4y++677wzO9++//4pTp07pvj5+/Liwt7cXkiQZ3Jds2bJF92A38+W/a9cu3XXl4cOHBj/j0KFDek+Cp6eni4YNGwoAol+/fnrnSUlJEaNHj5bdiWDMkydPhIODg9BoNLIzCCH0Hox/8MEHejvKT5w4oXuw/fLOGe2scjtfTpw4oXsyO/N9jPaBMAAREhIi4uPjdcsePHig21H+xhtv6H2/nFz+QmQ82QBAFC1aVFy7dk23vvaJWrkn/JXuMzp16qT7e758/UxJSRE//fSTwXnIsoztWD927Jiun8uWLdOdntPr+6+//iokSRJFixYVu3bt0lt2/PhxUapUKQFA7Ny5U3bGQoUKiV9++UVvmfb28PKL/xISEkThwoWFg4ODuHXrlsHvNmfOHAFAtG/fXu90pfu7hw8fio0bNxo8MfX8+XPdDq42bdoY/BylNuW02Tm5LIzNYe3tGWOMvYhLKyfbHcZk3j4oWrSoOHbsmG5ZamqqbqdV2bJlFV9A5OnpKQ4cOCD7u1SoUMHgZ7755psCgGjXrp24ffu23rIvv/xSABCvvPKK3u0qp9tOS5Ys0V1ecmrXri0AGPwNlZ6sPnjwoDhx4oTB97l+/bpup9C6dev0lj1+/Fi3k+Dtt9/W27knRMZtddu2bbm+jIw9+a5lqW3lnD6eMObChQu6bQA5586d013/nj59arBcu41Wu3ZtvdPNfR9H5vHo0SPd/a0kSSIsLEx8/PHHYvPmzbIvlNQ6d+6ckCRJFCtWzOA+Sgih2+4dNmyY3umZH1suXLhQb9n27duFJEnCzs5Ot92nZcr9dE47NWHCBAFk7NSLi4vTW7Z+/XphZ2cnvLy8xIMHD3SnN2nSRAAQa9asMZgjISFB7N+/X/bnv/wcREBAgACg92IZrdu3bxu82MmYBQsW6JqS+cW/6enpusvP09PT4O9qymWrJPOOdSFePAbt2LGj3no52bGupdQFIXL/ONrYcxzan+vo6GjwWLpr164CgKhataooUqSI+Oeff3TLk5KSdI+JPvnkE73v+cEHHwhA/0WjWs+fPxfFixcXAMSRI0f0lik9vtm5c6e4ePGiwelnzpzRbdv+9ddfesty85yI0hxamR8rtmrVSiQmJuqW3b9/X/eCh08//VRxpurVq+vNlNmWLVvEjRs3DE7ft2+fcHd3F4UKFRL//fefWWbKyY71nGyrCJH15WqJbV/tZf7yCwy1srqNPHv2THc5DB48WO+FsBcvXhT+/v4CMHzxVea/R/v27UVSUpJu2ZEjR4S9vb1uR/7gwYNFSkqKbvlPP/0kgIw3PWQ+n7lxxzoRERHlKe+9954AMl6JnNlnn31mdINPKzc71p8+fSoOHz4smjdvLoCMVzYfPHjQpLn79u0rAIgePXrILu/SpYvsBrn2gW+7du1kz/fo0SNRrFgxYW9vr/dKZ+0D31KlSsluTE6dOlUAGe8Ay6x79+4CgJg+fbrszzt48KDsk2Day7Vp06ay58tKUlKSsLe3l31yLqc71rXv+nFxcZHdaXH48GEBQGg0Gr0nZbQPHiRJ0nvwq3XgwAEBQJQrVy4bv6HpO9aVXhyifVf0u+++K7u8Xbt2AoAYMGCA3unay69r164G54mPj9f93LFjxxos175jOSAgwITfMMOqVat012W5dxDOnTtX9zMtvWNd+wD4iy++MGl27ZM5nTp1kl0+dOhQAUC0aNFCd9q6desUn/iQo31ip0aNGnoPALXS0tJE1apVBQDZB9xyDh06JICMV4Ar0V7/ateuLftuvcGDBwsAYtKkSXqna3+/Zs2aGZxn0KBBAoB4//339U7P/ED45XfrC5HxoF+SJIPbXk4uf61Ro0YJIOMTIeLi4nTv7lu/fr3s95K7z/jnn390T7q9/GQLqUdux3pCQoLYvHmzbkdRyZIl9XZi5vT6rt0p+v3338vOor09vLwNop1R6f5Z++K/3bt3650eFRUlAIiPP/5Y8Ty//fab3uk5fWK7ZMmSsi++MdYmJcaandPLIqsd69banjEmq8s+p9sdxmTePpB7h9WzZ890O7u+/vprvWXa882ePVv2fNp3/F+9elV3+qlTp4QkSaJkyZKKL9Rq06aNAKC3Qyqn205PnjwRRYoUERqNxuCTWvbv3y8AiNKlSxt8aoqxHShKfvvtN9ntoZkzZ+q6rPTpLJnl9DIyZce6MbnZVs7p4wlj1q9fLwCI1157TXEd7aeMvfzCFyGEiIiIEADE3Llz9U43930cmc+ZM2d0nXz5X40aNcSCBQtkb0Pa28PLL/B98uSJ8PLyEpIkiTNnzugt016nlbYJw8PDZR83ZWfHenY6FR8fL5ydnY1uI2qbPmfOHN1plStXFgBMvl0p7Vh3cXERHh4eJn2PrGi3neTakJ6eLqpXry4AiMmTJ+stM+eO9fj4eF2DMr+4wFI71nP7ONrYcxzanyv3WPro0aO624jcTscffvhBAIYvqL5+/booVKiQ8PDw0Nu+FUKIb7/9VgAQDRo0MPh+co9vsrJo0SLZ+XPznEhWc2iv54ULF5bdAb527VrZyz3zjvWXtyNNpX0h1st/j5zOlJMd68YobasIkfXlaoltX1N3rCvdRrSfdlKyZEnZF1d9//33AoBwc3PTexGe9u/h6upq8AJGIYTo0KGD7vkPuRfvaV9E8PILpc2Jx1gnIiKiPCM1NRUrV64EAPTt21dv2dtvvw17e3vs3r3b4DhVubFy5UrdcYScnZ1Rp04dxMbGws3NDatXr0bdunVN+j7aYx/27NlTdrnS6Zs3bwYA3XHkX+bq6oo6deogNTUVhw4dMljerFkzuLi4GJweFBQEAHrHiUpPT9cdQ1Lp59WpUweurq74+++/8ezZM4PlXbp0kT1fZvv27cO0adPwzjvvoE+fPujduzeioqLg4OCAu3fvGj32cnZoj8cdHh4OX19fg+W1a9dGcHAw0tPTdX+fzMqUKYPg4GCD0+UuO3Pq3LmzwWmpqan4888/AUDxGIH9+vUDkHGsQDlt2rQxOM3b2xtFihRRXP7KK68AQLaOW6m9LLt27YpChQoZLFe6rpvbrVu38M8//0Cj0egum6xorzNZXcZ79uzRHQusVq1asLOzw7JlyzBv3jzcvHnT6M/Q3qY7d+4Me3t7g+UajQavvfYagIzbiim0x73U/i2NadeuneyxxZWu1x07dkTp0qWxfft2veP4JiYmYs2aNbCzszM4frlWcHAwatSoYXB6tWrVULNmTaSnp+sdEzInl7/WtGnTEBISgj///BM1a9ZEfHw8hg0bZtJ9ktavv/4KAGjbti38/PxMPh9ZR58+fXQ99vT0RNu2bXHx4kUEBgZiy5YtKFy4sMF5snN9v3fvHg4ePAhnZ2e0b99edoawsDAAyrdNpfMp3b6GDx8OSZKwaNEipKam6k7X3t4qVqyIFi1ayH5PJceOHcOMGTMwbNgw9O3bF71790bv3r2RmpqK9PR0XLhwIVvfL6fNzu5lkRVrb8/kRG63O7ISGRlpcJqjo6Pud9T+/JfJ/S0cHR1Rrlw5APqX3ZYtWyCEQOvWreHm5ib7/YzdDrK77eTs7IyBAwciPT0dCxYs0Fs2b948AMDgwYNhZ2cnO4uc5ORk/PLLL/joo48wePBg3fV20aJFAICzZ8/qra+97+/Xr59JPye3l5EpzL2tnNvHE3JM2fbo06cPABgc6/3u3bvYvHkzHB0d8eabb5r087TMfR9HpqtYsSIOHDiAv/76Cx999BFatWqlO+b6P//8gyFDhiA8PBzPnz/XO9+IESMAAHPnztU7/ZtvvsGDBw/QvHlzVKxYUfZnmrslOf3eO3bswNOnT3XHqJYjd7uvV68egIzHP3v37tVrfXbUq1cPiYmJePvtt3HkyBGDYzCb6r///tM9VyLXFEmSdLdbpceU5uDt7Y333nsPAHT/tRRzPI425fGEscfSWS1/+bF2yZIl0aVLFyQmJmL16tV6y7RtHDp0aJYzZfb48WOsX78eEyZMwMCBA3X3nT/88AMAwzZqWfI5kTp16qBEiRLZ/t7FihVDo0aNjH7v+Ph4rFq1CuPGjcOAAQN0v692+0vp983pTNmV3W2VrKi17auldBvRbpu+8cYbcHR0NFjeqVMneHl54dGjRzhy5IjB8tq1a6NYsWIGp2tvO02aNIGTk5Pi8uw8j5Vdhs/kEBEREdmozZs349atW/Dz80OrVq30lvn6+qJNmzb4+eefsWzZMkyePNksPzMwMBChoaEAADs7O3h6eiI4OBgdOnSAp6enyd/nv//+AwD4+/vLLlc6PS4uDgDQq1cv9OrVy+jPuHv3rsFpZcqUkV3X3d0dAPQ2qOPj4/Hw4UMAQOnSpY3+LO36Lz+xoPR7AMCdO3fQuXNn7N271+j3ffjwIby8vLL8+VnRPugJCAhQXCcwMBDHjh2TfYCU1WWXnJyc6xlfVqxYMdkdB/Hx8bq/ldLvExgYCED5wZ7S7+Pq6or4+HjZ5donjLPzu2Z1Xff09ISHhwcSExNN/p45cfXqVQBAiRIl4OHhYdJ5srrOaC/jZ8+eIT4+HsWKFUNgYCC+/PJLjB07FkOHDsXQoUNRtmxZNGjQAO3atUPXrl3h4OCg+x7a2/T//vc//O9//zM6j9xtWo72stReN43Jzn0CANjb2yMqKgrvv/8+5s6dq3tSdOXKlUhKStLteJdj7LYXEBCAo0eP6q4vQM4uf61ChQph7dq1eOWVV5CYmIjg4GBMnz5d8efLuXLlCgCgUqVK2TofWUfDhg1Rvnx5AICDgwOKFSuGkJAQhIeHy75IBcje9f3SpUsQQuDp06eyT/xkpnTbzO7tq2LFimjZsiV+++03/PTTT7onpbRPmkZFRcm+MEBOUlISevXqhQ0bNhhdT9v5rOS22dm9LLJi7e2ZnMjtdocxnp6eitud2p+X+f40s+xcdtpGLV26FEuXLjU6U062O+W2J6KiovD5559j6dKlmDhxIpycnHD37l2sX78ejo6OGDBggNE5Mjtw4AC6d++u2waQ8/JtILv3/bm9jIyx1LZybh9PyDFl26Nbt24YPnw4YmNj8d9//6FUqVIAgDVr1iAlJQXdu3c3+fcw930c5Vy9evV0O4yFEPj777/x+eefY+3atYiNjcWsWbMwduxY3fotWrRAUFAQ/vrrLxw5cgS1a9cGYNoOQnO3JKffW3sb2r59e5ZdznwbmjJlCo4fP46tW7di69atcHZ2Rq1atRAWFoaePXvqdtZlZf78+WjXrh1Wr16N1atXw83NDXXr1kXTpk3Rq1cvxd/lZdr2FClSRPG2m9VjSnMZOXIk5s6di927d2PTpk1o166d7HpnzpzB1KlTDU4PDQ1F//79s/w55ngcbew5Di25v4Grq6vR5drH2nLX4+HDh+Pbb7/FvHnzMHjwYADA8ePHsXfvXvj6+mbrxcO//PIL+vTpg/j4eMV1lO47LfmcSE5v31n9PZYsWYJRo0YhKSlJcZ2c/r7m2CGdk22VrKi17aul9DfJattYkiQEBATgwYMH2XpOTnvbUlpu7LZlLtyxTkRERHmG9smrZ8+eoXHjxgbLtRtiK1aswKRJk7L17hYloaGhBu+yyA2lB+JKp2tfja707qfMypYta3CaRmP6BxRlfuW73CvYXya348HZ2Vlx/f79+2Pv3r1o0KABYmJiEBwcDC8vL927mkuWLImbN29CCGHyzJaUncvOXIxdfrmV1e9j7t/X2JNOpu4oMlVO37VhLsOGDUO3bt3w888/Y+/evdi7dy/Wrl2LtWvXIjo6Gnv27NG98l07a2hoqO5JHCVVqlQx6edrd7aY8gA8J3/nAQMGYNKkSVi1ahWmTJkCV1dXzJ8/H0D23y3xMnPe3teuXYuUlBQAGS+suHnzpuz9IuVN/fv3V3ynkZKcNNDV1VX2k0PM/fO0RowYgd9++w3z5s1Dly5dcO3aNfz8889wdXXN1u/7/vvvY8OGDahUqRKmTp2KunXromjRoroX9rz66qvYv3+/ybe53Dbb3E2x9vZMXmSOv4X2sqtRo4bsO9Qyq1+/fq5+llapUqXQqVMnrFu3Dt999x0iIyPx1VdfITk5Gb169dK9IzYrT548QUREBG7fvo0+ffpgyJAhKF++PNzd3WFnZ4dz586hYsWKue5Obi8jYyy1rZzbxxNyTNn2KFy4MLp164Zly5Zh1apVmDBhAoAX72DXvjPWFOa+jyPzkCQJtWrVwrfffosnT57g559/xk8//aS3Y12SJAwbNgxRUVGYO3culi9fjv379+Pvv/+Gv7+/4g5VwLKPx3Jy31i+fHk0bNjQ6LqZX6RTvHhxHD58GLt27UJsbCz+/PNP/PXXX/jzzz/x6aefYsqUKSa9YzsoKAhnz57F77//jj/++AP79u3Dnj178Mcff2DSpElYunQp3nrrLZN/H1vg7OyM6OhoDBo0CBMmTJB9RzeQ8Slk2k8tfJkpO9bNwZTH6OZ+rB0SEoJ69erh4MGD2LVrFxo3bqx7McrAgQP1XrhtzPXr19G9e3c8ffoU48aNQ8+ePeHv7w9XV1doNBr8/vvvaNWqldW258zxvY39PY4cOYJBgwbBzs4O06ZNQ/v27VGmTBm4uLhAkiQsXrwYgwYNUuX3BSy3raL2tq+lnsey9nNY2cEd60RERJQn3Lx5E1u2bAGQ8epK7cd5yblx4wZ+/fVXtG3b1lrjZcnPzw9xcXG4fPkyKleubLD88uXLsucrXbo0zpw5g379+mXrVck5UbRoUTg7O+Pp06eYPn06ihYtarbvnZSUhC1btkCj0WDLli0G77pKSkrCrVu3zPbzAOhegat9h4Ec7TJb/9jnIkWKwNHREcnJyYiLi0P16tUN1rGV30X785Wu04mJiUhISMjW99Q+cfDo0SPZ5dp3m2WmffXyzZs3kZiYaNK71v38/HDx4kXExcWhatWqBsu1l7GTkxO8vb31lvn6+mLAgAG6d9adOXMGffv2xf79+zF+/HjdE0LaV5C//vrrGDNmTJYzmUL7zm1j70LIjSJFiqBnz5746quvsGrVKlSoUAFnz55F5cqV0bRpU8XzXbp0SXGZ9vqhffcakLvLf+/evfjwww/h4vJ/7N13fFPV+wfwz83u3rSUDlr23sjeAiJ7FQRFXIhQHLjgx1QQVETFL4KAgAgVECh7z7Jkb2RJKdCW1U1nmpzfH+GGhCZtkia5afO8Xy9etHflyW1yz7nnueccV/Tu3RurV69GVFQUDh8+bHBKAkP4z4zukPfEefDfTY7jsHTpUrs11HTv3h3Vq1fHwYMHceXKFcTExEClUuH11183aRQK3tq1awEAa9asMVhG3Lx50+RjCVFmW5Mt6zPFsWW9Iz09Henp6QZ7rRu6nlqK/x60bt26yLDNtjRu3DisXbsW8+fPx/Dhw7Fw4UIA5j28FRcXh4cPH6Jx48ZYunRpkfXGvgNhYWH4999/ce3aNXTp0qXE17HVObLl984W9xOm1j1GjhyJpUuXYvny5Zg4cSLOnj2LixcvIiQkxKypLqx5jSO20bVrV2zevBlPnjwpsu6NN97AxIkTsXr1asyZM0f73Rk9erSgiRFT8d/7GjVqmP3QPcdx6NChg3ao+Ly8PCxfvhxjxozBxIkTMXDgwBIftgU0o0j16NFDm4DOzMzE3LlzMX36dIwaNQr9+vUzOC2OLr7s4Xu3Gqpn2POe8u2338bcuXNx6dKlIkOe8zp06FCqB2bK0n30i8aNG4fhw4fjf//7Hxo0aIBVq1ZBIpFoe7CbYsuWLcjNzUW/fv3w7bffFllf3q6df//9NxhjiI6Oxueff15kvdDv19K6SkmEqvuWxJS6Md9m4Gjfv5I4fslFCCGEEAJNzwaVSoWXXnoJjDGj//jKc0lDM9obP2dyTEyMwfXGlr/yyisAnjcm2ZJYLNY2cFn79TIyMqBSqeDp6WmwUXjlypVGb5j5pKq5c9LxjRc7d+7UzgOp69y5c9o5uPm/j6OSSCTaKQmMNebwN2YdO3a0V1gG8efy77//Nvg3M/ZZLw5/k/Xvv/8WWZeTk2NwPrygoCDtXLaGbloN4T8zJZ3jtm3bGh16mlezZk1tD5Tz589rl/Pfaf6m3xrq1KkDmUyG+/fvG334oLTGjRsHQDNsJ98YOmbMmGL3uXjxIi5evFhk+ZUrV3D27Nki3z1Lz/+TJ08wZMgQFBYW4n//+x/+/PNPtGzZEidOnDBr3sbu3bsD0Myfa8v52IhjCg4ORv369ZGVlaWdc9ke+J58ADB37lwsWbIEgPmjQaSmpgIw3Nt0165dBhMdxpSmzHYEtqrPlFQfsXW9w1DSoaCgAGvWrNF7/dLgy6jNmzfbdPjMF7Vu3RpNmjTBqVOnMGnSJNy9exfNmjXTDjdtCv47YGxY0JUrVxpczl/7ly5dCpVKVeLrWHqOSvr82LKubIv7icaNGwMwXDfT1aZNG1SvXh03b97E0aNHsWzZMgCaHnXmJFSteY0j5jPlms8Pa2zoIR83Nze8/fbbyMvLwzfffIN169ZBoVBo57a2FkvvG0vSuXNnyGQyHDx4EI8ePSrVsRQKBd5//33Ur18farXaYF3ZFJ6enpg2bRq8vb2Rk5ODGzdulLhPSEiINolvqL7NGNMut8c9pVgsxjfffAMAmDJlik2mWitL99EvGjx4MCpWrIiNGzdi5syZ2mm4goODTT5GcddOxphF9+Yl4R9qtvb30BTFvd+8vDztnPJCsbSuAhR/Xm3ZllcafN10zZo1ButMsbGxSEtLg4eHh3aakLKCEuuEEEIIKRP4m52ShjV64403AABbt241e15DWxo7dixEIhFWr16NTZs26a3bsGGD0Qr+e++9h/DwcPz999/44osvDCbNHjx4gMWLF1slzqlTp0Imk+Gzzz7DH3/8YXCI7cuXL2PDhg1mHTcwMBA+Pj5IT08v0jD8zz//YMKECUb35Rtnrly5YtZrtmnTBi+99BJyc3MxatQo5OTkaNc9efIEo0aNAgAMGTLEpHmohDZ+/HgAwIIFC7Bv3z69dcuXL8fmzZshlUrx4YcfChGe1qBBg1CxYkXcuXMH//d//6f3Gbp27Rq++uors4/J9yCbP3++3txb2dnZeO+993Dv3j2D+02dOhUA8H//938Gv2NXr17VaxD+8MMPIZFIsHHjxiI3tbt378Zvv/0GAHo9zffv34/t27drhyDnMcawdetWAPo39n369EGzZs1w8uRJjBw50uB1Ki0tDQsXLjS5McLFxQUtWrSAWq3GiRMnTNrHXPXq1UOnTp3w77//YvPmzfD09NReb41hjGH06NFIS0vTLsvIyMDo0aPBGMOAAQP0vnuWnH/GGIYPH47ExESMGDECI0eOhEQiwerVq+Hr64sff/yxyDXXmIYNG6JPnz7Izc1Fnz59isx7V1hYiM2bN5t0LFI2zZgxA4Cmh+WWLVuKrGeM4cSJE9i9e7dVX/fNN9+El5cXli5dikePHqFjx44GR7cpDj9P6y+//KK3/Pr162b1bAJKV2Y7ClvUZ0qqj9i63vH111/j8uXL2t/VajW++OIL3L9/H6GhoRZPYaCrUaNGGDBgAO7du4f+/fsbHH0mOzsbq1atMvjwQGnw9Rd+Ll1zHy7hvwP79u3D1atX9dYtWrRI+wDCi9555x2EhITg3LlzePfdd4vMyZqZmYm9e/dqf7f0HAUEBEAmk+HBgwfahnVdtqwr2+J+IjIyEmFhYXj8+DFu3bpV7Lb8kO8LFy7UJnHMndrDmtc4Yr5ff/0VI0aMwLFjx4qsY4xhw4YN2gcvhwwZYvAY/P3w3LlzUVBQgKFDh8LPz8+qcVp631iSwMBAREdHIzs7G7169cKlS5eKbJOfn4/NmzfrjXw0Z84cg/MoX7t2TdsztaTpF3JycjB37lyD9wyHDx9Geno6xGKxyaOW8PXor7/+GhcuXNAuZ4xhxowZOH/+PLy9vbWjcNla//798dJLL+Hu3btmtzGYqqzcR79IKpVi9OjRKCwsxJw5cwBYXjauW7cOycnJ2uUqlQpTpkwx+J0uLVt9D03Bv98//vhDr6zLy8vDBx98UOyIavZgaV0FKPm82qotrzQGDRqEsLAwJCUl4ZNPPtFr34iPj9d+N6Ojo6FQKOwWlzVQYp0QQgghDu/QoUO4desW5HK50Rt1Xp06ddC4cWMolUqsWLHCThGWrEmTJpgxYwZUKhX69u2Lli1bYtiwYXjppZcwYMAAfPTRRwBQZK4sNzc3bNu2DZUrV8Z3332HsLAwtG/fHsOGDUO/fv1Qp04dBAcHY/LkyVaJs3HjxtqE1ptvvonw8HB069YNw4cPR48ePRAaGop69eqZ/RSsWCzGlClTAGgefmjRogVee+01tGnTBq1atULPnj2NNirwDcWff/45evXqhbfffhvvvPOOSTeBMTExCA8Px6ZNmxAREYFBgwahb9++qFKlCk6dOoXGjRvbdajT0njllVcwadIk5OXl4eWXX0bbtm0xbNgwNGnSBCNHjoRYLMbChQtNnpfbVlxdXbFy5UooFAp89913qFGjBoYOHYpu3bqhQYMGaNu2rfYJbVPnhhs8eDCaNm2Ku3fvok6dOujZsyd69OiBiIgIHDx4EG+99ZbB/fr164eZM2ciLy8PAwcORK1atTBkyBD06dMHderUQZ06dfQS0fXq1cP8+fPBcRxef/11NGnSBMOGDUObNm3QvXt35OfnY9q0aejatat2n4sXL+LVV1+Fv78/OnbsiGHDhqF///6IiIjAkiVL4OXlpfcwgUgkwsaNG9GwYUP88ccfiIiIQOvWrTF06FAMGDAAjRo1QkBAgLYRxVR9+/YFAOzZs8fkfczF91oHNA85ubu7F7t979698eDBA0RGRqJ///4YMGAAIiMjcfjwYVSrVq3Id8+S8//NN99g165dqF27tnbed0DTC2D58uXgOA4jR440OjXBi5YtW4YWLVrg9OnTqFatmvZv2rlzZwQHB6NPnz4mHYeUTb169cLPP/+M1NRU9O7dG9WqVUPPnj0xbNgwdO3aFUFBQWjRogX2799v1dd1d3fXm2vY3EZTQNOYxnEcJk+ejPr162Po0KHo3Lkz6tWrh8jISLRq1crkY5WmzHYUtqjPdOvWDW5ubti4cSPatGmDkSNH4p133tH2wAVsV+8ICwtDmzZt0LhxY3Tt2hVDhw5F9erV8dNPP8HNzQ0xMTFWa5BctmwZOnfujB07dqBGjRpo3rw5oqKiMHjwYDRv3hy+vr4YPny43kNT1hAVFaWd/zsgIABRUVFm7d+oUSP06dMHWVlZaNSoEbp164ahQ4eiVq1aeP/997Xze7/I3d0dmzdvRlBQEJYtW4aQkBD07NkTQ4YMQevWrREUFKR96IZnyTmSSqXo3bs3VCoVGjZsiNdeew3vvPOOdo5gW9aVbXU/YWrd44033oBYLMbKlSuRmpqKdu3aoWrVqma9ljWvccR8/L1169atUaFCBXTr1g3Dhg3Dq6++isjISAwYMAA5OTkYPny40V7olStXRu/evbW/W1LWlcSU67SlZs+ejddeew0nT55Ew4YN0bhxYwwcOBBDhgxBmzZt4Ofnhz59+ujVOWfMmIHw8HDUqlUL/fv3x7Bhw9CxY0fUq1cP2dnZeOONN7SjPxhTUFCA8ePHIygoCA0bNsSgQYPw2muvoVWrVmjfvj0AzUPEAQEBJr2PUaNG4fXXX8eTJ0/QtGlTdOnSBa+99hpq1aqFKVOmwMXFBTExMSYfzxr4Icp1H0izprJyH23IqFGjtPNh169f3+wRb3r16oUmTZrg/v37qF69Onr27ImoqChUqVIF3377rVmje5mKL5O6dOmCqKgobVlnq2nLdI0cORLh4eE4d+4cIiIi0K9fPwwcOBDh4eFYt26d4A9PWFpXAUo+r7ZqyysNuVyOdevWwdfXFwsWLEDVqlUxZMgQvPrqq6hduzbi4+PRrVs3bYeIMoURQgghhDi4119/nQFgAwcONGn7n376iQFgtWrVKrKuffv2DACbOnVqsccYMWIEA8BGjBhhQcTGbdiwgbVu3Zq5ubkxDw8P1qZNG7Zx40YWFxfHALCWLVsa3C8zM5N99913rGXLlszb25tJpVJWsWJF1qxZM/bZZ5+xY8eO6W0/derUYt/ngQMHGADWvn17g+vj4+PZxx9/zOrWrcvc3NyYQqFg4eHhrEOHDmz27Nns1q1betvz5/XAgQPFvv+NGzeyVq1aMW9vb+bu7s6aNm3Kfv31V6ZWq1l4eDgDwOLj44vst3jxYta4cWPm6urKADAAbNmyZdr1/DJDUlJS2IQJE1itWrWYQqFgrq6urFGjRmz27NksJyfH7HNT0usZw3+mdOPmxcfHMwAsPDy8xOPs2LGD9ejRg/n5+TGJRMKCgoLYoEGD2IkTJwxuX9Lfprjzzphl75Uxxi5cuMD69evHfH19mUKhYLVr12bff/89y8/PZzKZjIlEIpabm2tyrGlpaWzs2LEsJCSESaVSVqlSJfbee++xhw8flvh5P378OBs6dCirVKkSk0qlzNfXlzVo0IB9/vnnLCEhocj2//zzDxs4cCALCgpiEomE+fn5sVdffZXt3r27yLa3bt1i06ZNY507d2ZhYWFMoVAwHx8fVr9+ffbll1+ye/fuGYwpLy+PLVy4kHXs2FH7t6xQoQJr2LAhGzNmDNu1a1fJJ/mF8+Pm5saCg4NZYWFhkfXFff4YY2zZsmUlXvOysrKYWCxmHMexa9euGd1O91iPHj1io0aNYiEhIUwmk7HQ0FA2btw4lpKSYnR/U8//wYMHmVgsZq6uruzKlSsGjzV+/HgGgDVv3pwVFBRolxf3uc7Pz2cLFixgbdu2Zd7e3kwmk7GQkBD28ssvs/nz5xuNm9gGf40y9tk1pLSf90uXLrH33nuPVatWTVtuREZGsm7durF58+axxMREgzEau46WFA9jmms7ABYaGmrwO8wr7noXFxfHOnfuzPz9/ZmrqyurW7cumzlzJsvPzzd6fS0uNkvKbEvPhbHl9q7PlCQuLo516dKF+fj4MJFIZPBzZG69ozi69QOlUslmzpzJatasyeRyOfP19WUDBgwwev0rqfwursxVqVQsJiaG9ejRgwUGBjKpVMr8/PxY3bp12ciRI1lsbKzeNdVadaeoqCgGgE2YMKHY7Yx9zgoKCtj333/P6tWrx1xdXZmvry/r2rUr2717d4l1rcePH7NJkyaxevXqMTc3N+bi4sIiIyNZVFQU27lzZ5HtzT1HjGk+G6NGjWJhYWFMKpUaPCe2qiszZv79REmuX7/OOI5jzZs3L3HbHj16GI1Ll7WvccQ6MjMz2caNG1l0dDRr3ry5tj7u4uLCqlSpwoYOHcp27NhR4nEWLFhQ7D0vr6S/Z0mfk+Ku06Uts7dv38769++vva/w9vZmtWrVYkOGDGExMTEsOztbu+3KlSvZyJEjWd26dZmvry+Ty+UsPDycvfLKKyw2Npap1Wq9YxuqnyiVSrZw4UI2dOhQVrNmTebl5aU97wMGDGD79u0zGGdJYmJiWIcOHbTXgtDQUPbmm28areOXVB4Xh/97/vnnn0a30b1GWPIaJf1dGbP+fbQpr1tc2WdqG8BLL73EALDffvut2O2MvVZWVhabOHEiq1GjBlMoFKxChQqsb9++7PTp00bL79KU67m5uezzzz9nVatWZTKZTLsdf45KqocbOy+mxMSYpjz/4IMPWJUqVZhcLmfBwcFs+PDh7ObNm0Zf29KYivsbWruuUtJ51Y3JWnXfks65qWXv3bt32ZgxY1hkZCSTyWTMw8ODtWzZki1YsIAplcoi25f09yjpemTKvVdpcYw58MRYhBBCCCFO4quvvsLUqVMRHR2NefPmCR0OITYTFxeH9u3bo169ehbPKUgMGzt2LObPn4/NmzejV69eVj/+kiVL8O6776Jr167YtWuX0e2WL1+OkSNHYsSIEUbnMiSE6Bs+fDhWrVqFb775pkwMtU7s486dO4iIiEB4eLjJI2+UVenp6QgJCUFeXh7i4+PLxDQ9BOjZsye2bduGixcvol69ekKHQxxcmzZtcPToUcTExGDo0KFCh0OIw7tx4wZq1qwJLy8vJCYmwtXVVeiQCCGgoeAJIYQQQuzm5s2bBofN3Lx5M2bNmgWO40qcQ56QsuDx48cG5y+7fPmydr4+3WGPiXVMnToV3t7eFs1jX5Ls7GzMmjULwPN5Cgkh1nHp0iWsWbMG7u7u2nm4CXE2s2bNQnZ2NgYPHkxJ9TLku+++g0QiwfTp04UOhTi4HTt24OjRowgLC8PAgQOFDoeQMmHKlClgjGH06NGUVCfEgUiEDoAQQgghxFnwPdEaNWqE0NBQKJVKXL9+HdevXwcATJs2DU2aNBE4SkJK78qVK+jYsSNq166NyMhIuLi4ID4+HmfPnoVarcbLL7+M6OhoocMsdwICAjBt2jR89NFHWLdunVUaLb///ntcvnwZR44cwe3bt9G9e3e9Oc4JIZZ75513kJ2djR07dqCwsBCTJk2Cr6+v0GERYjfHjh3D0qVLER8fj/3798PV1bXIfObEsdWuXRtjx47FTz/9hNOnT6Np06ZCh0QcSEpKCr744gukpaVh+/btADQPY0ilUoEjI8Rxbd68GZs2bcKVK1dw4sQJBAUF4fPPPxc6LEKIDkqsE0IIIYTYSffu3XHz5k38888/+Pfff5GXlwc/Pz/06tULH3zwAbp37y50iIRYRfXq1TFmzBgcOnQIR48eRVZWFjw8PNCqVSu89tprePfddyGR0K2ILXz44Yf48MMPrXa8bdu24dChQ/D398ebb76JuXPnWu3YhDi733//HSKRCKGhofj000+p0ZQ4nRs3buD333+Hi4sLWrRogW+//RaRkZFCh0XM9OOPP+LHH38UOgzigLKysvD7779DIpEgMjIS48ePR1RUlNBhEeLQzp49i6VLl8LDwwNdunTB3Llz4e3tLXRYhBAdNMc6IYQQQgghhBBCCCGEEEIIIYQQUgyaY50QQgghhBBCCCGEEEIIIYQQQggpBiXWCSGEEEIIIYQQQgghhBBCCCGEkGIInli/efMmxo4di9q1a8PNzQ0KhQIhISFo1qwZxo4di/Xr1wsdYokqV64MjuNw584doUMpYvny5eA4Dm+++abQoRh07NgxdO3aFb6+vhCJROA4DsuXLy9xvw4dOoDjOEybNs0qcfDHO3jwoFWORwx78803Tf4bCykxMRGvv/46goODIZFIHPo7RITnyGUAIeWdse9facr1slgnOHLkCDiOK3Nz83IcB47jhA7DLEJ+PsriZ5MQUj6U5/ouXVv1WXrPbu22J0f6u1j7vXXp0gWenp548OCBVY5HCCGEvOjgwYPgOA4dOnQQOhRCyiVBE+sbNmxAvXr1MH/+fDx69AitW7fGgAEDUL9+fSQmJmL+/PkYNWqUkCFi2rRpVk3gkueSkpLw6quvYu/evahbty6GDx+OESNGoGrVqkKHRpwYYwz9+/fHypUr4ePjg6ioKIwYMQJt2rQROjSrK4vXN6oYEkJKoyxe90zRpk0bvPrqq/j5559x8+ZNocMhhBBCiI2VxYfTiMbs2bORlZWFCRMmCB0KIYQ4rLL2UF9Z6UxGCLEOiVAv/PDhQ4wYMQL5+fkYP348ZsyYAYVCobfNmTNnsG7dOoEiLB/69euHFi1awMvLS+hQiti9ezfS09Px2muvYdWqVUKHQ+xg1qxZ+PLLL1GxYkWhQzEqISEBJ0+eRFhYGC5cuACJRLDLJCGEEAutWLECOTk5CAsLs+u+Qpo+fTq2bduGL774Ahs2bBA6HGIDZfWzSQghhJDnmjZtip49e+KPP/7ARx99hAYNGggdEiGEEEIIMYNgGaOtW7fi6dOnCA4Oxpw5cwxu06RJEzRp0sTOkZUvXl5eDplUB4C7d+8CAKpVqyZwJMReKlas6NBJdeD55zIiIoKS6oQQUkaVJvFYVpOWTZo0QYMGDbBp0ybcuXMHlStXFjokYmVl9bNJCCGEEH1vv/02tm7dip9//hlLly4VOhxCCCGEEGIGwYaCf/jwIQAgICDA5H0yMzPh6ekJiUSCe/fuGd2uR48e4DgOv/76q3aZ7vxM58+fR//+/eHv7w+5XI7atWvjhx9+AGNM7zgcx2H69OkANL2A+KG2iptb6cCBA+jatSt8fHzg4uKCxo0bY8WKFcW+r3Xr1qF79+4ICAiATCZDpUqVMHz4cFy9etXg9mfOnEFUVBRCQkIgk8ng6emJyMhIDBgwAJs2bdLbtri5oPbu3YtevXohMDAQUqkUPj4+qFatGoYPH464uLhiYzZk9erV6Ny5M3x9fSGXyxEeHo633noLN27cMBjT1KlTAeifW0dsBF62bBlkMhl8fHxw4MABtG/fHhzH4a+//jK6z3fffQeO4zB48GDtMt3hZx8/fowxY8YgNDQUMpkMoaGhiI6ORnp6epFjlTRsrbGhsXWX5+TkYMqUKahVqxZcXV2LnOczZ85g2LBhCAsLg1wuh6+vL7p164bt27cbfE3d4XjM+cwbGxbH0nPDu3HjBkaNGoUqVapAoVDAy8sL7dq1w8qVK43u86I7d+6A4zi0b98eAHDo0CG97zw/9JDuteTw4cPo1asXAgICIBKJ9N7X/fv3ER0djWrVqmljat26NX777TeoVKoir6/7Xc3IyMAnn3yCypUrQ6FQoFq1avj222+hVqsBaOaAHzVqFEJDQyGXy1GjRg388ssvJr9XwPTrm+7fetOmTejUqRN8fX2158Aa34eEhAS88cYbqFixIhQKBapXr45p06YhNzdX7zgdOnRAx44dDf59XvxMFxYWYuHChWjVqhW8vLy053HcuHFITEw0ek744RQXL16MJk2awM3NDd7e3ujRowf++eefEs+rOd+HhIQEfPvtt+jUqZP2u+ft7Y02bdrgt99+0/69dfGf08qVK4MxhkWLFmnj9PLyQteuXXH8+PES4yREV05ODn766Se0adMGPj4+2jK8V69eiImJMbj97Nmz0bhxY3h4eMDV1RV16tTBpEmTkJaWVmT70n5ur169ikGDBsHf3x8uLi6oW7cu5syZY/BayjM0L6ep173i5vQs7bVl/fr1aNOmDTw9PeHm5obWrVsbLWuTk5Px4Ycfonr16lAoFHB1dUVoaCg6d+5s9KHUN998E2q1GgsWLDB6bmxl06ZNaNu2LTw8PODl5YX27dtj27Zten//kpSmrg9opnPZsGEDevbsiaCgIMhkMgQFBaFNmzb49ttvi5QrgOn11+JYWhaq1WosWrQIrVu3hre3N6RSKSpUqIAGDRogOjq6yLCHxj6b+fn5+P7779GkSRN4eHho33ezZs3w+eefIzU11eT3Qggp3+xd3+WZ0+ZhSb3hv//+g1gsho+PD3JycozGUadOHXAcZ7Ts1fX48WPMmzcPPXr0QEREBFxcXODp6YmmTZvi22+/RV5entF9za1bmXOO+Psonm59Rve+ValUYuXKlRg2bBhq1qwJT09PuLi4oEaNGhg3bhySkpJKPAcvKiwsxE8//YR69epBoVAgICAAAwYMwKVLl0rctzT37Ka24VlaHgvx3l599VX4+/vjr7/+onKaEEJ08O2jCQkJADQdn3TLuYMHD5bY5l2aeoFuO2hsbKz2/t3DwwMdOnQoUofg6y1//PEHAGDkyJF68b7Ynl+a9uKUlBSMGTNG24YYHh6Ojz/+2GA7jC6lUolvv/0WderUgYuLC/z8/NC/f3/8+++/Brffu3cvoqOj0bBhQ23ZGxISgqioKJw6dcrgPqVt2yekzGEC+fPPPxkAJhaL2d69e03eLzo6mgFgEydONLj+1q1bjOM45unpybKysrTL27dvzwCwL7/8kslkMlarVi02ZMgQ1r59eyYWixkA9uGHH+oda8SIEaxBgwYMAGvQoAEbMWKE9t/ixYu124WHhzMAbPLkyYzjONakSRM2ZMgQ1qJFCwaAAWA//vhjkViVSiUbPHgwA8Dkcjlr1aoVGzRokPY1XVxc2I4dO/T22bt3L5NKpdqYBg4cyPr168eaN2/O5HI569Onj972y5YtYwDYiBEj9JYvX76ccRzHOI5jL730EouKimK9e/dmjRs3ZmKxuMi5KI5arWZvvPEGA8AkEgnr1KkTGzJkCKtevToDwFxdXfXex+HDh42e2/Hjx5v0mvzfc+rUqSbHacrxDhw4oLd88uTJDACrXLkyu3LlCmOMsfXr1zMArFWrVgaPpVKpWOXKlRkAdujQIe3yqVOnMgDsrbfeYiEhISwwMJD179+f9ejRg3l5eTEArFmzZqygoEDvePx+xt7rgQMHGADWvn17g8tfeukl1qxZM+bm5sZeeeUVFhUVxbp06aLd7qeffmIikYgBYA0bNmQDBw5kbdq0YTKZjAFg06dPL/Kaln7mR4wYwQCwZcuWGXyP5p4bxhhbu3YtUygUDACrWbMm69evH+vUqRNzc3NjANjIkSMNnrcXPX78mI0YMYJ169aNAWCBgYF63/nHjx8zxp5/Vj744AMmEolY7dq12ZAhQ1jXrl1ZTEwMY4yxkydPMl9fXwaAhYWFsaioKNa9e3dtnN26dWP5+fl6r89/V/v06cNq1arFKlSowAYMGMC6du3KXFxcGAA2duxYduvWLRYUFMRCQ0PZ4MGDWceOHbXXsNmzZ5v0Xvm/hTnXt7FjxzIArGnTpmzo0KGsffv2LC4urtTfhzfeeIP5+fmxwMBANmjQINazZ0/t365169YsNzdXu8+sWbOM/n10rx15eXmsS5cuDABTKBTaz31oaCgDwPz9/dmZM2eKxMp/dj/++GPGcRxr06YNGzp0KKtbt672+rZhw4Yi+1n6ffj6668ZABYREcE6d+6sLZP4717//v2ZWq3W2yc+Pp4BYOHh4WzEiBFMKpWyTp06scGDB2uvuXK5nP3zzz/G//iE6Lh79y6rXbu2trx++eWX2ZAhQ1jbtm2Zl5cXCw8P19s+JSWFNWzYkAFgnp6erHfv3mzAgAHM399f+3mOj4/X26c0n9vDhw9rrwmRkZFsyJAhrEuXLkwqlbIBAwZov38vvqahct3U656xOkFpry1TpkxhHMex1q1bs6ioKG0sHMcVubYkJyez4OBgbTnSp08fFhUVxdq2bct8fX2Zl5eXwb/n5cuXGQBWvXp1g+tt5dtvv9W+z5deeokNHTqUNWvWjAFgn3/+ufbv/yJ+H12W1vULCgpY//79GQAmEolYixYt2NChQ9nLL7/MKlWqVORzYm79lWfo82FpWThy5Ejt56lLly5s6NChrFu3bqxatWoMAIuNjS3xtVUqFevcubP2O/nKK6+woUOHsi5dumi/H+fOnTMYFyHE+di7vmtJm4el9YZevXoxAGzRokUG3/v+/fsZAFalShW9Oraxcp9vs6pUqRJr3749GzJkCOvcuTNzd3dnAFjLli1ZXl5ekdcxt25l7jmKjY3V3lfzbT2G7lvv3bvHADAvLy/WokULNmjQINajRw9t/SIgIIDdvHmzSPzG7tlVKhXr27cvA8BkMhnr2rUri4qKYpUrV2YKhYJ98MEHBtueGLPsnt2SNjxLy2N7vzfewIEDGQBtOwIhhJDneQP+OjpgwAC9cu7ff/81qc3b0noBX+f5+OOP9dpBmzdvri17582bp92eb0+uUqWKti1TN17de7rStBf37t2bValShXl7e7O+ffuyfv36MR8fHwaA1ahRgz169EhvP/4ctWrVinXp0oW5urqy7t27swEDBmjbMLy9vYu0pTDGWJUqVZhMJmONGjVivXv3Zv3799fWbSQSCVu3bl2RfUrTtk9IWSRYYj0rK0vbyMVxHOvQoQP7+uuv2bZt24pcCHTduHGDcRzHKlSoYPAmZvz48QwAi46O1lvOV8oBsIULF+qt27dvH+M4jonFYnbv3j29dSUlNRl7fsGVSqVsy5Yteuv4i5+XlxfLycnRWzdx4kRtIXD79m29dX///TcTi8XMx8eHpaWlaZd37NiRAWArV64sEkd6ejo7fvy4wdd/8QYgIiKCAWCHDx8ucpyHDx+ys2fPGn2/L1qwYIG2QVm34U6tVmvPn7e3d5G/qynn1hhbJ9bz8/PZsGHDtAXogwcPtNsWFhZq/+aGztOWLVsYAFa/fn295fz7BcDefPNNvc/v3bt3td+HF2+qSptY52NJTk4usu/OnTsZx3HM399f78aSMcYuXrzIQkJCGAB28OBBvXWWfuZLSqybe24uXrzI5HI5UygUbP369Xrr7ty5w+rVq8cAsD/++KPoiTPC2Pnk6V5L5s+fX2R9Xl6e9vy8//77ehWG//77T3sj/2LCgD9vAFivXr1Ydna2dt2ZM2eYRCLRJvLff/99plQqtes3btyobVDX3a8k5lzfxGIx27RpU5H11vg+9OnTR++zcu/ePW2j2Zdffqm3X0l/H8YY++KLL7SVY90KYkFBAXv77bcZoEn+vVhZ5eNxcXFh+/bt01v33XffaT/XDx8+NHiOzP0+nDx5kl26dKlI/ImJidqGtLVr1+qt4xsa+cbG69eva9cVFhayt956iwFgXbt2NXp+COGpVCrWtGlT7WfmxXI6NzeXbdu2TW9ZVFSUtu7y5MkT7fKsrCz2yiuvGGzMtPRzm5ubq73h++ijj1hhYaF23YULF7TJfMC0xDpjpl33jO1b2muLt7d3kSQAH8+LifDp06czAOy9994r8oBNQUGB0YdS1Wo18/b2ZgCK1Glt5ezZs0wsFjOxWFwkGbN27Vrtw3umJtYtret/8sknDNA8DHn+/Hm9dWq1mu3du5elp6drl1lafzX0+bCkLExISGAAWEhIiME62tWrV1lCQkKJr33o0CEGgDVq1IhlZmYWOc6pU6f0vquEEOdm7/quJW0eltYb9uzZwwDNw3OGDBgwgAFgP/zwg95yY+X+1atXi7SvMMZYamoq69q1KwPAvvvuO711ltStLDlHjBkuQ3VlZmayTZs2FamXFBQUsAkTJjAArEePHkX2M3bP/r///Y8Bmgecr169ql2uVCrZ6NGjtfG82PZk6T27JW14lt6b2vu98ebOncsAsLffftvgekIIcWbGHqJnzLQ2b0vrBfzrchxXJP+yevVqxnEck0gkRdrzjJWfPGu0F7do0YKlpKRo16WlpbFWrVoxAGzIkCFGz1GjRo30zlFubq6249J7771XJNbY2FiWmppqcLlEImF+fn5F6nyWtu0TUlYJllhnjLFr166xl156Sful0/3XsGFDtmDBAr1GVF6PHj0YAPbnn3/qLc/JyWE+Pj6M4zh27do1vXV8pbx///4GY+nevTsDwFasWKG33JzE0yeffGJwfc2aNRkAFhcXp12WkpLCXFxcmEKhYPfv3ze4H/9U7C+//KJdxj8dZOjiZoixxLqrq6vR3k7m4p/I0n1ai6dWq1n9+vUZADZz5ky9dY6aWE9NTdX+3rt3b4OJSr7RwdANEF8w/fbbb3rL+fcbEhJi8JizZ89mgObJLkP7lSaxrvvZ08V//ww9acaYpkEc0DwdqMuSzzxjJSfWzT03fIJnzpw5BuM4efIkA8CaNGlicL0hpibWO3XqZHA937MhODjYYEJg3bp1DADz8PDQ643Nf1fd3d2LNGQxxljv3r0ZoHmiUXc/Hn/T/uIDEsUx5/r24rnXVZrvg4uLi8EKMN/o4enpqfd+S/r75ObmanuRbN68ucj67OxsFhgYyACwVatW6a3jvy8fffSRwWPzjWQvXsss/T4UZ9euXQwAGzRokN5y3YZGQ+8vOTmZAZreLvQUKCkJ/1BOxYoV9Xr+GpOQkMBEIhHjOI5duHChyPr79+9rn7Q+evSodrmln9uVK1cyACw0NNTg5/nHH3/UHtfWiXVrXFsM1ZPy8vK0T2/fvXtXu5yvAxrqNViSli1bMgAGH4ayBT7BMXToUIPr+d5YpibWGTO/rv/w4UPtaB+nT582KW5L66/GPlvmloV8HaV3794mxWvstfm62rhx40w+DiHEedmzvmtpm0dp6rt16tRhQNEOBPfu3WMSiYS5uroWSVAbu64X5/r16wzQ9LzSZW7dytJzxFjJifWSBAcHM5FIVOShLGP37FWrVmUA2IIFC4ocKzc3lwUFBRlse7L0nt3SNjxL7k3t/d54/D1fo0aNDK4nhBBnZmpivbi2NkvqBfzr9u3b1+Ax+YT8u+++q7e8pMR6aduLAcMjkV28eJFxHMdEIpHew2b8OeI4rsiD54wx9s8//zBAMzKgOYYOHcoAFHlQ0NK2fULKKsHmWAeAGjVq4J9//sGJEycwZcoUdOvWTTvn+vnz5zF69Gh0794dBQUFevt9+OGHAID//e9/estjYmKQlpaGLl26oEaNGgZfs1evXgaX16pVCwCMzo9pCnOOfeDAAeTm5qJ169aoVKmSwf34ObOPHTumXda8eXMAwLBhw3DkyBEUFhZaFGvz5s2RkZGBN954A2fOnDE4l68p7t+/j//++w8AMGLEiCLrOY7DyJEjAWjes6OLj49Hq1atcOjQIYwdOxaxsbFwdXUtst0777wDV1dX7WeOd+vWLezevRve3t4YPny4wdfo3LmzwWNa4zNoSIUKFdC2bdsiy588eYKTJ0/CxcXF6GfX0GdQl7W/T+acG7VajR07dgAAoqKiDB6vadOmcHd3x7lz54qdA88SAwcONLicn/d0yJAhkMvlRdb3798fPj4+yMrKwpkzZ4qsb9KkCSpUqFBkebVq1QAAHTt2hEKhMLrekrnyTGHs/QKl+z507doVQUFBRZb37NkTfn5+yMzMxNmzZ02O8/Tp03j69Cl8fX0Nfj5dXV0xZMgQAMavSYauZQDwxhtvAIDBeZcBy74P+fn52LJlC6ZMmYL3338fI0eOxJtvvonffvsNAHD9+nWDx5RIJOjevXuR5UFBQfDx8UF+fj5SUlIM7ksIb+fOnQCA1157De7u7iVuHxcXB7VajUaNGqF+/fpF1leqVAndunUDYPj7Ze7nlv+uDR48GFKptMh+xr6rtmCNa4uh/eRyOSIjIwHoXyP4+t6XX36JDRs24OnTpybH6ufnBwB4+PChyfuUxqFDhwBo6qaGGFteHHPr+gcOHEBBQQGaNGmCJk2alHh8W9RfzS0La9asCQ8PD2zfvh0zZ85EfHy8Sa/zosaNG0MsFmPp0qWYP38+kpOTLToOIcS52KO+a2mbB8+S+u64ceMAFC0/fvvtNxQWFmLYsGHw9vY2GIshKpUK+/btw9dff40PPvhAW1efOXMmgKJ1dXPrVqU9R6a4cOEC5s6di+joaLz11lt488038eabb6KwsBBqtRq3bt0q8RiJiYna7Qzd1ykUiiJzlgPWuWc39x7L3PJYyPdm7zobIYSUN8bavHmlqRcYqyvxy43VlYwpbXtxgwYN0LBhwyLL69Wrh0aNGkGtViMuLq7I+rCwMDRo0KDI8pLa7pOSkrB48WKMHz8e77zzjrb+cOXKFQDG2yvtnfcgRCgSoQMANI2HfAMiYwznzp3D999/j9WrV2Pv3r34+eef8dlnn2m3f/nll1GrVi2cOHECZ86c0TagzZ8/HwAwduxYo68VFhZmcLmnpycAlCr5Zs6xb9++DQDYt28fOI4r9riPHz/W/jxr1ixcvHgRO3bswI4dO+Di4oLGjRujQ4cOGDZsmPYiVZJff/0VPXv2xJ9//ok///wTHh4eaNasGTp16oTXX3/d6Ht5EX8x9PPz077PF1WpUkVvW3ubPXs2rl27VmT5nDlz4O/vr7fsvffeQ2FhId555x388ssvRo/p4+OD119/Hb/99ht+//13fPrppwA055UxhpEjRxosRADbfgYNqVy5ssHl8fHxYIwhNzfXYIGuS/czqMva78Wc46WkpCAzMxMAEBoaWuKxU1JSjDZWWMLYeeU/5xEREQbXcxyHiIgIpKWlGfxOGDsHfMOMsfUeHh4ArP/54Rl7v0Dpvg/GzhP/mikpKbh//77JcZZ0/oGSr0nG9uWXG4vH3O/DP//8g6ioKNy9e9dorPxn/EUVK1Y0mGjkXy8tLc1mnwVSfiQkJADQJPhMUdrvl7mfW/67Zuz1fHx84OXlhYyMDJPiLw1rXFvMuUa8/vrr2LNnD1atWoUBAwZALBajdu3aaNOmDQYOHIhOnToZjYM/nm5jcnGOHDmCJUuWFFnet29f9O3bt8T9+b+TsXKiuPLDGHPr+pZ+lq1ZfzW3LPTw8MCyZcswcuRITJo0CZMmTULFihXRokULdO/e3eSkTJUqVfDjjz/is88+w9ixYzF27FiEh4ejZcuW6NmzJwYNGgSZTGbSeyCEOA971HctbfPgWVLfHT58uPahtOTkZFSsWBEFBQVYvHgxgOLbil508+ZN9OvXT9uAbMiLdXVzy6PSnqPiZGdn4/XXX0dsbGyx2xm739DFfx78/f2Nlk2GPlPWuGc39x7L3PJYyPdmbp2NEEKIvpLuNUtTL7C0rmRMaduLi2uLiIiIwNmzZw3GVFI5mp+fX2Td9OnTMXPmTCiVSqOvaaz+YO+8ByFCcYjEui6O49C4cWP89ddfyMnJwebNm7Fx40a9xDrHcYiOjsYHH3yA//3vf1i2bBmOHz+Oc+fOoXLlyujZs6fR44tEtuukb86x+R7iVatWRevWrYvdVvemLCgoCKdPn8ahQ4ewd+9eHD16FCdOnMDRo0fxzTffYNasWfjiiy9KfP1atWrh+vXr2L17N/bv349jx47h8OHD2L9/P7766iv8/vvvRnuYljU7d+7U9qbSNW3atCKJ9eHDh2PFihVYtWoV+vfvj1deecXocceNG4fffvsNCxYswCeffIK8vDwsW7YMHMdhzJgxRvez9mewpNEGXFxcit3P3d0dAwYMsOi1rf1eLPkOAab1Wizp4QFzGTuvpVXSObDlNaw4Jb1fS78PpmCMlWp/azMWjzl/m5ycHPTt2xcPHz7EyJEjMXr0aFStWhWenp4Qi8W4ceMGatSoYZXXIsRROPvn1pz3LxKJsHLlSkycOBHbtm3D0aNHcfToUSxYsAALFixAr169EBsbC7FYXGRf/kEDHx8fk17r1q1b+OOPP4osr1y5skmJdZ6xhEBJiQJj+1ha1xeSuWXhgAED0KVLF2zevBmHDx/G0aNHERsbi9jYWEyZMgV79uxBvXr1Snzd6OhoDB48GJs3b8aRI0dw5MgRrF69GqtXr8bUqVNx+PBhVKxY0RZvmRBSTlmjDmppm4clr8VzdXXFu+++i++++w6LFi3C1KlTsX79ejx8+BBt27Y1OOKOMQMHDsSVK1fQs2dPfP7556hduzY8PT0hlUpRUFBglfvL0p6j4kyYMAGxsbGoWbMmZs+ejWbNmsHf31/7sFWrVq1w/Phxm95rWeOe3ZLPgS3vTXnWeG/m1tkIIYToK6mt0pr1ghc5WlslYDgmc8vRDRs2YNq0aXB3d8f//vc/dOrUCcHBwXBxcQHHcZg4cSJmzZpF7ZXE6TlcYl1X165dsXnzZjx58qTIujfeeAMTJ07E6tWrMWfOHO2QHqNHjy4TX2D+idYaNWpg+fLlZu3LcRw6dOigHRIsLy8Py5cvx5gxYzBx4kQMHDhQ28umOBKJBD169ECPHj0AaJ40mjt3LqZPn45Ro0ahX79+cHNzK/YY/BO3/NO6hnr98E9hW7O3sDnMGZplxIgReOWVVzB8+HD07dsXMTExRpPOtWvXRpcuXbB3717s2LEDSUlJSE9PxyuvvGLS+TcVf+OblZVlcD3/VLy5+M8gx3FYunRpmfje6PL394eLiwtyc3MNjj4gFP5zzn/uDeGHexXqO2ELln4fihv69s6dOwCAkJAQk+Pgz2lxxy3pmhQfH29weCVL4jEmLi4ODx8+ROPGjbF06dIi62/evFnq1yCkJPyTxIZGdTHElOubNct8/hj8d+9F6enpdumtrhtLaa4tlqhduzZq166Nzz77DIwx7N+/H6+99hq2bNmCFStWaIcr18UPixsYGGjSa/BDulmqUqVKuH37Nu7cuYPatWsXWW/s71cSc+r6ln6WrV1/taQs9PLywuuvv47XX38dAHDv3j1ER0dj06ZNGDt2rMGHQw0JDAzEu+++i3fffReA5ly89dZbOH78OL788kuDD08QQpyXPeq7pWnzKI0xY8bghx9+wKJFizBx4kRt+WFOb/Vr167h4sWLqFChAmJjYyGR6DedGaurm1se2fIcrV27FgCwZs0ag4kDc+43+LLwyZMnePr0qcGe3YbKe6Hu2c0pj4V8b+bW2QghhJjP0npBfHy8wSHULa0rlba92Nrtp8bw9YeZM2fivffeK7Ke2isJ0RAsk2bKUz388LiGLgpubm54++23kZeXh2+++Qbr1q2DQqHA22+/bdU4+aSmpXOZG9O5c2fIZDIcPHgQjx49KtWxFAoF3n//fdSvXx9qtRoXL1606Dienp6YNm0avL29kZOTgxs3bpS4T0hIiPbGxNCNIGNMu7xjx44WxWVvgwcPRmxsLEQiEaKiorBixQqj2+rOAWrKVASW4AvTf//91+D6bdu2WXTc4OBg1K9fH1lZWdq54MoSsViMl19+GcDzQt8R8A+8rFmzxuDwNrGxsUhLS4OHh4dJ88DakrWvb5Z8H3bv3m3wGrh9+3akpKQUOU8lxczPY5eamorNmzcXWZ+bm4vVq1cDMH5N+vPPP4tdzv+NSyM1NRWA8SGSVq5cWerXIKQk/Lylf/31F7Kzs0vcvl27dhCJRDh//jwuXLhQZH1ycrK2PLFGmd++fXsAmmu8oSHIiiufjbH0umeNa0tpcRyHzp0747XXXgMAnD9/vsg2arVaW1+wVxnTrl07AJr5zw0xtrwk5tT1O3XqBJlMhjNnzuDs2bMlHtuW9dfS1g1DQ0Mxffp0AIb/xqaqWbOmdhSr0hyHEFI+2aO+a802D3OEhYWhb9++SEpKwpQpU3Ds2DEEBwejf//+Jh+Dr6sHBwcXSaoDxuvq5tatSnOO+GHyjdVp+PcQHh5eZN2uXbsMdmAxJiQkBJGRkQAMl+v5+fn4+++/iywX8p7d1PJYyPd2+fJlAParsxFCSFlirTZLS+sFxupKfDvEi3WlkuItbXvxxYsXDeZ8rly5grNnz0IkEmnvzUujuPrDo0ePsGfPnlK/BiHlgWCJ9V9//RUjRozAsWPHiqxjjGHDhg3aJ4iGDBli8Bhjx46FSCTC3LlzUVBQgKFDh8LPz8+qcfJJ/eLm1bJEYGAgoqOjkZ2djV69euHSpUtFtsnPz8fmzZv1nnaeM2eOwfl4r127pn1iyNCFT1dOTg7mzp1rcI6uw4cPIz09HWKx2OSnnPg5q77++mu9hnbGGGbMmIHz58/D29tb24OmLHj11Vexfft2uLi44M0338Svv/5qcLsePXqgatWq2LlzJy5cuIAqVaoUO3y8JTp16gSRSIRdu3bp9VpijGHevHlYv369xceeMWMGAGDkyJHYsmVLkfWMMZw4cQK7d++2+DVsaerUqZDJZPjss8/wxx9/GBwW//Lly9iwYYPdYho0aBDCwsKQlJSETz75RK9CFR8fj/HjxwPQDNuqUCjsFpch1r6+WfJ9yM3NxejRo5Gbm6tdlpSUpD1P77//vt554mO+efOmwUSbQqHQDu83fvx4vREdlEolPvzwQzx48AAREREYOHCgwZgWLFhQZKSLH3/8ESdPnoSHh4dVHuCqVasWAM18ilevXtVbt2jRIqxZs6bUr0FISXr37o1GjRohKSkJgwYN0vaa4eXl5WHHjh3a38PCwjBo0CAwxjBq1Ci97bOzs/Hee+8hLy8PrVq1QqtWrUod38CBA1GpUiXcvXsXEyZM0LvGX758WVuGmcPS6541ri3mWLFiBc6cOVNkeVZWlvb6ZKi+d+XKFWRkZKB69ep2GxWFr4+vXr0amzZt0lu3YcOGUtVTTK3rV6hQAaNHjwagKYf5hmoe39tfd4QDW9VfTS0Lz507hzVr1uiVfzy+TlZSnR4A9u/fj+3btxcpExlj2Lp1q8nHIYQ4F3vUdy1t87AGPqk6e/ZsAMCoUaMMJsiNqV69OsRiMS5dulTkPG3ZsgU//vijwf3MrVuV5hyVVKfh7zd++eUXveXXr1/H+++/b3Cf4nz00UcANFPq6caiUqnw6aefIikpyeB+Qt2zm3NvKtR749tDO3XqZM5bI4QQp2DNNktL6gWxsbHah+d569atw/r16yGRSBAdHW1WvKVtL2aMYfTo0UhLS9Muy8jIwOjRo8EYw4ABA7Qj4ZQGX39YtGgRCgoK9F5rxIgRdhs1kBBHJ9hQ8EqlEitWrMCKFSsQEBCARo0awd/fH+np6bh69ap2CIvhw4cbvamrXLkyevfujY0bNwKwfk9hAOjWrRvc3NywceNGtGnTBtWqVYNYLEbr1q0NDr9pjtmzZyM5ORkxMTFo2LAhGjRogMjISEgkEty/fx/nz59HdnY2duzYoZ1Pa8aMGfjss89Qs2ZN1KpVCy4uLkhKSsKRI0dQWFiIN954A40bNy72dQsKCjB+/Hh89tlnqFevHqpVqwapVIo7d+7gn3/+AQD83//9HwICAkx6H6NGjcKxY8fw559/omnTpmjfvj0qVKiAs2fP4vr163BxcUFMTIzJxzPHkiVLiu1tPXnyZLz66qsWHbtjx47Yu3cvXnnlFYwZMwZZWVlF5q8XiUQYO3as9kbsgw8+sGgu0eKEhoYiOjoaP//8Mzp37oy2bdvC19cXFy5cwN27d/Hll19qKwbm6tWrF37++WeMHz8evXv3RtWqVVGjRg14eXnh8ePHuHDhAh49eoQvvvgCXbt2ter7sobGjRtj5cqV2mFsJ02ahNq1ayMgIACpqam4dOkS7t+/j6ioKLN6KJSGXC7HunXr0L17dyxYsADbt29HixYtkJWVhf379yMvLw/dunXD1KlT7RJPcax9fbPk+/DGG29g69atiIyMRNu2bZGXl4f9+/cjOzsbLVu21Pba44WFhaFp06Y4ffo06tWrh6ZNm0KhUMDf31/7PZg+fTpOnz6Nffv2oVatWujYsSM8PDxw/Phx3L17F35+fvj777+1T5O+aNSoUejUqRPatm2LSpUq4fLly7h06RLEYjGWLl2KoKAgs8/Nixo1aoQ+ffpg06ZNaNSoETp06ABfX1+cP38e169fx8SJEzFz5sxSvw4hxRGJRIiNjUW3bt2wY8cOhIWFoU2bNvDz80NiYiIuXLgAb29vveEv58+fj2vXruHEiROoUqUKOnbsCIlEgkOHDuHx48eIiIjAqlWrrBKfi4sLVq1ahR49euCHH37Axo0b0axZM6SkpODgwYPo1asXzpw5Y9aUKKW57pX22mKODRs2YMSIEQgODkbDhg3h4+ODtLQ0HD16FBkZGahbt67BhO/evXsBwKy50UurSZMmmDFjBiZOnIi+ffuiRYsWiIyMxK1bt3Dy5EmMHz8eP/zwg0XnxZy6/nfffYf4+Hhs3rwZDRo0wEsvvYSIiAg8efIEV65cQWJiIuLj4+Hl5QXAdvVXU8vChIQEDBkyBC4uLmjcuDFCQ0NRWFiIS5cu4fr165DJZPjuu+9KfL2LFy/i448/hqenJxo3bozg4GDk5ubi7NmzSEhIgJeXF7766iuz3gMhpPyzR30XsKzNwxratm2LRo0a4dy5c5BKpQaHMi2Ov78/xo4dq3cPHhwcjOvXr+Ps2bOYNGmSwQf8LKlbWXqOBgwYgDlz5qBLly7o1KkTPDw8AADffvst/Pz8MHXqVAwcOBCTJ0/G2rVrUadOHTx69AiHDx/Wvh9DHV2MGTNmDPbs2YMtW7agQYMG6NixI3x8fHDixAkkJydj9OjRWLBgQZH9hLpnN+feVIj3plQqERcXB4VCgW7dulntfRNCSHkxYMAAHDhwAMOHD0fXrl3h4+MDAPjss8/MPpYl9YIPP/wQQ4cOxdy5c1GtWjX8999/OHHiBABNx8cXp1np27cvpk+fjnnz5uHy5csIDQ2FSCRC79690bt371K3F/fu3RuXL19GZGQkOnbsCI7jcPDgQaSmpqJatWraDqql9dFHH2HFihXYvn07IiMj0aJFCyiVShw6dAiurq546623DE5pSYjTYQLJzMxkGzduZNHR0ax58+YsJCSESaVS5uLiwqpUqcKGDh3KduzYUeJxFixYwACwli1bFrtd+/btGQB24MABg+unTp3KALCpU6cWWRcXF8e6dOnCfHx8mEgkYgDYiBEjtOvDw8MZABYfH2/w2CNGjGAA2LJlywyu3759O+vfvz+rVKkSk0qlzNvbm9WqVYsNGTKExcTEsOzsbO22K1euZCNHjmR169Zlvr6+TC6Xs/DwcPbKK6+w2NhYplar9Y69bNmyIvEqlUq2cOFCNnToUFazZk3m5eWlPe8DBgxg+/btMxhnSWJiYliHDh2Yt7c3k0qlLDQ0lL355pvs2rVrBrcv7pyXhP97lvTP2Dk3djxDn48LFy6wChUqMADs//7v/4qs//fffxkA5urqytLS0oy+Rknv98CBAwwAa9++fZF1arWa/fDDD6xWrVpMJpMxX19f1qtXL3bmzBmj+xV3vBddunSJvffee6xatWpMoVAwV1dXFhkZybp168bmzZvHEhMT9ba39DNvbHlpzg1jjMXHx7OPP/6Y1a1bl7m5uTGFQsHCw8NZhw4d2OzZs9mtW7dKPAemvlZJ1xLe3bt32ZgxY1hkZCSTyWTMw8ODtWzZki1YsIAplcoi2xv6ruoq6RyVdJ0xprTXtxdZ8n24ffs2Gzp0KAsMDGQymYxVrVqVTZkyRe/apyshIYG99tprrGLFikwikTAALDw8XG8bpVLJfv31V9aiRQvm4eHBZDIZq1KlCouOjmb37983eFz+usGYpmxp2LAhc3FxYZ6enqx79+7s6NGjBvez9PtQUFDAvv/+e1avXj3m6urKfH19WdeuXdnu3btZfHy8wfdlbLk58RDyoqysLPbtt9+yZs2aMQ8PD23donfv3mz16tVFts/OzmazZs1iDRs2ZK6urkyhULBatWqxiRMnstTU1CLbl/Zze+nSJda/f39tvadWrVps1qxZTKlUGt2vuGt1Sde94vYt7bXFEEOvFxcXxz766CPWvHlzFhQUxGQyGQsKCmItW7Zkv/zyC3v69KnBYzVo0ICJRCJBvv8bNmxgrVu3Zm5ubszDw4O1adOGbdy4kcXFxRmtq5d0bhgzva7PmKa+FBMTw7p27cr8/PyYVCplQUFBrG3btuz7779nubm5RfYxt/5qSj3AlLIwOTmZzZ49m/Xo0YNFREQwV1dX5unpyWrXrs3GjBlj8PUNvfatW7fYtGnTWOfOnVlYWBhTKBTMx8eH1a9fn3355Zfs3r17xZ4zQohzsXd9l2dOm4e16rtffPEFA8CGDh1qdBvGjF/X1Wo1+/3331mTJk2Yu7s78/LyYm3atNHWjYorw8ytWzFm3jlijLHc3Fz2+eefs6pVqzKZTKaNR/ecxMXFsc6dOzN/f3/m6urK6taty2bOnMny8/ONvu/i/pZKpZL98MMPrHbt2kwulzM/Pz/Wp08fdv78+RLvZ829Zy9NGx7P1HtTe783xjT1JgBs5MiRxcZFCCHOSqVSsVmzZrE6deowhUKhLecOHDhgVps3z9R6gW4dY+3ataxly5bM3d2dubm5sbZt27ItW7YY3Tc2Npa1bt2aeXh4MI7jDJZTpWkvfvToERs1ahQLCQlhMpmMhYaGsnHjxrGUlJQi+5lyjozVZeLj49mwYcNYWFiYtg7z/vvvswcPHhgtf0vbtk9IWcMxZsJk5w6sTZs2OHr0KGJiYjB06FChwyFOaNKkSZg5cybee+89/Pbbb0KHQ4igTP0+TJs2DdOnT8fUqVMxbdo0+wVYDL4HQxkvFgkhTurMmTNo2rQp+vXrZ9cpUEry1VdfYerUqYiOjsa8efPM3r8s1vWpbkgIcVTOUt9VqVSoUqUKEhIScOzYMbRs2VLokIgAHLk87tWrF7Zt24azZ8+iYcOGQodDCCHlmjn1gsqVKyMhIQHx8fGoXLmy/YI0Yvny5Rg5ciRGjBiB5cuXCx0OIeQZweZYt4YdO3bg6NGjCAsLs8p8loSYKzk5GfPnz4dIJNIOMUaIs6LvAyGECGfKlCmQyWT49ttv7f7aN2/e1Jvrjbd582bMmjULHMdhxIgRZh+3LNb1qSwkhBDhLVq0CAkJCWjZsiUl1Z2UI5fHp06dwtatWzFixAhKqhNCiB1QvYAQYm2CzbFuqZSUFHzxxRdIS0vD9u3bAWjmVJRKpQJHRpzJl19+icTEROzduxfp6el4//33UatWLaHDIkQQ9H0ghBBhHTlyBNu3b8dnn32GatWq2f31V61ahW+++QaNGjVCaGgolEolrl+/juvXrwPQjFLSpEkTk45VVuv6VBYSQoiwrl+/ju+//x4PHjzAzp07IRKJMGfOHKHDInZWFsrjCRMmwMPDA7NmzRI6FEIIKbeoXkAIsaUyl1jPysrC77//DolEgsjISIwfPx5RUVFCh0WczOrVq3H37l0EBQXho48+wuzZs4UOiRDB0PeBEEKE1aZNG0GH9e3evTtu3ryJf/75B//++y/y8vLg5+eHXr164YMPPkD37t1NPlZZretTWUgIIcJKTk7G77//DplMhjp16mDatGlo1aqV0GEROysL5fHevXuFDoEQQso9qhcQQmypzM+xTgghhBBCCCGEEEIIIYQQQgghhNhSmZ5jnRBCCCGEEEIIIYQQQgghhBBCCLG1MjcUvKnUajWSkpLg4eEBjuOEDocQQggBADDGkJWVheDgYIhE9HxbSag8J4QQ4qicoUw/ePAgOnbsaHDd8ePH0aJFC5OOQ+U5IYQQR+UM5bm1UHlOCCHEUdmzPC+3ifWkpCSEhoYKHQYhhBBi0L179xASEiJ0GDZ39uxZTJs2DUeOHEFeXh4iIyPx3nvvYdy4cSbtT+U5IYQQR+cMZfq4cePQrFkzvWVVq1Y1eX8qzwkhhDg6ZyjPS4vKc0IIIY7OHuV5uU2se3h4ANCcRE9PT4GjIYQQQjQyMzMRGhqqLafKs927d6NXr15o1KgRJk+eDHd3d/z333+4f/++yceg8pwQQoijcqYyvW3bthg4cKDF+1N5TgghxFE5U3leWlSeE0IIcVT2LM/LbWKdH47G09OTCnpCCCEOp7wPm5aZmYk33ngDr776KtatW2fxEDxUnhNCCHF05b1M52VlZcHFxQUSifnNCFSeE0IIcXTOUp6XBpXnhBBCHJ09ynOaOIYQQgghVhcTE4OHDx9i5syZEIlEyM7OhlqtFjosQgghhFhg5MiR8PT0hEKhQMeOHXH69Olit8/Pz0dmZqbeP0IIIYQQQgghpKyjxDohhBBCrG7v3r3w9PREYmIiatSoAXd3d3h6emL06NHIy8szuh81xBNCCCGOQyaTYcCAAfj555+xadMmzJgxA5cuXULbtm1x7tw5o/vNmjULXl5e2n80HyshhBBCCCGEkPKAY4wxoYOwhczMTHh5eSEjI4OGpiGEEOIwnKV8atCgAW7dugUAePvtt9GhQwccPHgQv/zyC4YMGYK//vrL4H7Tpk3D9OnTiywv7+eLEEJI2eMsZfqLbt26hfr166Ndu3bYuXOnwW3y8/ORn5+v/Z2f787ZzhUhhBDH56zluSXoXBFCCHFU9iyjyu0c64QQQggRztOnT5GTk4P3338f8+bNAwD0798fBQUF+O233/DVV1+hWrVqRfabMGECPvnkE+3vfEM8IYQQQhxD1apV0adPH2zYsAEqlQpisbjINnK5HHK5XIDoCCGEEEIIIYQQ26Gh4AkhhBBidS4uLgCAoUOH6i1/7bXXAADHjx83uJ9cLoenp6feP0IIIYQ4ltDQUBQUFCA7O1voUAghhBBCCCGEELuhxDohhBBCrC44OBgAEBgYqLe8QoUKAIC0tDS7x0QIIYQQ67h9+zYUCgXc3d2FDoUQQgghhBBCCLEbSqwTQghxCg8ePABjTOgwnEaTJk0AAImJiXrLk5KSAAABAQF2j4kQQsqKnJwc5OTkCB0GIXj8+HGRZRcuXMDmzZvRtWtXiETUpEAIIcR8GRkZVNchhBBCyri8vDyn7DxFd8GEEELKvU2bNuHll1/GwoULhQ7FaQwePBgA8Pvvv+stX7JkCSQSCTp06CBAVIQQUjYMHDgQUVFRQodBCKKiovDqq69i5syZWLx4MT7++GO0atUKrq6umD17ttDhEUIIKYMKCgrQsWNH9O/fX+hQCCGEEFIKw4YNQ7t27ZCVlSV0KHYlEToAQgghxNZOnToFANi2bRtGjx4tcDTOoVGjRnjrrbewdOlSFBYWon379jh48CD+/vtvTJgwQTtUPCGEkKLu3bsndAiEAAD69u2LVatWYe7cucjMzERAQAD69++PqVOnomrVqkKHRwghpAx6+vQplEplkdHNCCGEEFK23LhxA4BmJBoPDw+Bo7EfSqwTQghxGjQUvH0tXLgQYWFhWLZsGWJjYxEeHo4ff/wRH330kdChEUIIIcQE48aNw7hx44QOgxBCSDmiUqmEDoEQQgghVqRWq4UOwa4osU4IIeXIli1bULNmTVSrVk3oUBwSx3FCh+BUpFIppk6diqlTpwodCiGEEEIIIYQQB1BYWCh0CIQQQgixImfrzEaJdUIIKScePXqEiRMnokKFCti3b5/Q4TgkZyvkCSGEEEIIIYQQR6KbWGeM0QPwhBBCSBnnbD3WRUIHQAghxDoKCgoAaBLsRB+fUKcbdkIIIYQQQgghRDhKpVL7M/VeJ4QQQso+Z+vMRol1Qggh5R6fUHe2Qp4QQgghhBBCCHEkusl03SQ7IYQQQkhZQIl1QggpJyhpXDLqsU4IIYSUXbm5uRgxYgQ2bdokdCiEEEIIsZBuMp0S64QQQkjZ52x5CUqsE0IIcRrOVsgTQggh5cmVK1dw9uxZTJo0SehQCCGEEGIhfhq7F38mhBBCSNnkbHOsS4QOgBBCCLEX6rFOCCGO4cmTJ4iJiUFBQQEaNWqEzp07Cx0SKQPoATlCCCGk7KPEOiGEEFK+ONu9OiXWCSGEOA1nK+QJIcRRbdiwAYsXLwYA/P333/jnn3/o4SdCCCGEECeQn59v8GdCCCGElE3O1mOdhoInhJByghISJaNzRAixtydPnmDhwoVIS0sTOhSHcvfuXQCAX7A3cnJykJKSInBEhBBCCCHEHiixTgghhJQvztaZjRLrhBBSTjhbAWYO/tzQOSKE2NvPP/+M+fPnY9GiRUKH4lDu3bsHkUiE0BoVtb8TQgghhJDyLy8vT/szJdYJIYSQso96rBNCCCHljFKpBEA91gkh9nf79m0AwP379wWOxLEkJCTA088NPoGeAIA7d+4IGxAhhBBCCLEL3cS67s+EEEIIKZtUKpXQIdgVJdYJIYSUewUFBQCoxzohhDiCzMxMpKSkwCvAA94VKLFOCCGEEOJMKLFOCCGkLNmxYwe6dutGI+0VgxLrhBBCSDnD36xTj3VCCBEe34vfN8gLPs8S6/wyQgghhBBSvuXm5hr8mRBCCHFE06dPR3JSEnbv3i10KA5FtwMbJdYJIYSQcoZPrPNDwhNCCBHOzZs3AQC+Qd5QuMnh6umiXUYIIYQQQso3SqwTQggpS7KzswHQSKgv0k2mU2KdEEJImUSFu3H8zTrdtBNCiPCuX78OAPAL9tb+n5iYiMzMTAGjImUBjTxDCCGElH05OTkGfyaEEEIcGd2P6qPEOiGEkDJPrVYLHYLDosQ6IYQ4jsuXL0MiFcMnyAsAUCHUFwBw9epVIcMiZQA9REgIIYSUfXzPvxd/JoQQQhwZ3Y/q002mFxYWChiJ/VFinRBCyglnezLMHHxCPS8vjx5AIIQQAeXk5ODatWvwr+QDsVhzKxIY7g8AOHfunJChEUIIIYQQO6DEOiGEkLKIeqzr002mO1teghLrhBBSTjhbAWaO7JznN+v8fOuEEELs79y5c1CpVKhULVC7rGJkADiOw6lTpwSMjJQF1JBBCCGElH1ZWVnan2kqIEIIIWUF9VjXp5uLcLaObJRYJ4SQcoIS68bl5T5PptMcboQQIpzDhw8DAEKqBWmXyV1kqBDmi3Pnzuk1tBJCCCGEkPJHN5lOiXVCCCFlBT3orU+3x7pSqRQwEvujxDohhJQTzvZkmKmUSqVe4U7zrBNCiDDUajX2798HhascFSMD9NZVrhuCwsJCxMXFCRQdIYQQQgixh7T0NIgUml5/6enpwgZDCCGEmIh6rOujxDohhJAyT7cwI8+9mEinHuuEECKMM2fOIDn5ASLqhUAk1r8NqdIgFACwZcsWIUIjhBBCCCF2wBhDSkoKOCnAyYCUlBShQyKEEEJMQj3W9enmIpwtL0GJdUIIKSeox7phLybWqcc6IUQI9GQzsH79egBAzeaRRdZ5B3iiYkQAjh07hqSkJHuHRsoI+h4RQgghZVtmZiaUBUqI5AyclOHx48dCh0QIIYSYhO5H9en2Uqce64QQQsokmmPdsLy8vGJ/J4QQYnuPHj3Czp074VvRC0ER/ga3qdO6KhhjiImJsXN0hBBCCCHEHh48eAAA4OSASM6QlZWF7OxsgaMihBBCiLmcObEuKc3OBQUF2Lt3L65du4bs7GxMnjwZgCZpkZmZCX9/f4hElLsnhBB7oB7rhuXn5+v9XlBQIFAkjovKc0Jsz9mHDFu5ciVUKhUatK9p9FxUaRCG41su4O91f+Pdd9+Fl5eXnaMkpGyj8pwQQoiju3//PgBo5lhnnHZZjRo1hAzL4VCZTgghxNHpJtNpKHgTbd68GWFhYejVqxc+/fRTTJs2Tbvu4sWLqFixIlavXm2NGAkhhJiAeqwbpk2kizXD9VCPdX1UnhNiH848ZFhKSgr++usvuHu7onqTyka3E0vEaNixJnKyc7By5Ur7BUhIOUDlOSGEOJ709HQkJSU5dT3wRQkJCQAAsQIQuzC9ZUSDynRCCHFMzt5h4kXO3GPdosT60aNHMXDgQMjlcvz888947bXX9NY3b94cVatW1c6jSAghxPaox7phfGKdE2t+d7Yn6IpD5TkhxB6WLFmCvLw8NO5SB2KJuNht67SsCjdPF6z4cwVSU1PtFCEhZRuV54QQ4nhu376Ntm3bolu3bliwYIHQ4TiM//77DwAgcmMQueovI1SmE0KII6MH5fQ5c2LdoqHgv/76a3h7e+PMmTPw9/dHSkpKkW2aNm2KEydOlDpAQgghpqEe64bxBTsnBhhoKHhdVJ4TYj/O+mRzUlIS1qxZAy9/d9RqUaXE7SUyCZp0rYu4daewePFifPHFF3aIkpCyjcpzQghxPHfu3NH+TInj565fvw5ODIgUAPdsVLnr168LHJXjoDKdEEIcl7O26xjjzIl1i3qsnzhxAn369IG/v7/RbUJDQ/HgwQOLAyOEEGIeemrOML6HOt9jnR5AeI7Kc0KIrf3yyy9QKpVo/kp9iMWm3XrUalEFXv4eWLNmjXYeTkKIcfYuz2fOnAmO41C3bl2rHI8QQsqjzMxMgz87s9zcXNy8eRMidwaOA0QyQCQHLl++LHRoDoPu0QkhxHFR27s+3VFhnW2EWIsS6/n5+fD09Cx2m/T0dIhEFk/hTgghxEyUMDZMe164F34nVJ4TQmzq33//xdatWxEQ4ouqDcNN3k8sFuGlVxtAqVRi3rx5NoyQkPLBnuX5/fv38c0338DNza3UxyKEkPJMN5melZUlYCSO4/Lly1Cr1ZB4PE9MiD3UePjwISWKn6F7dEIIIWWF7qiwzjZCrEWlcGRkJE6dOlXsNsePH0fNmjUtCooQQoj5aI51w7SJdNELvxMqzwkhNsMYww8//AAAaNW7ETiReUOmVWkQisBwP+zYsYN6MRFSAnuW559++ilatGiBpk2blvpYhBBSnukm06nHusbp06cBABIvncT6s5/5dc6O7tEJIYSUFTQUvJkGDBiAo0ePYtmyZQbXz5kzB5cvX0ZUVFSpgiOEEGI6Shgbxj9wwHGaG3Y6T89ReU4IsZXjx4/jxIkTCK8djErVAs3en+M4tOzVCAAwd+5cGnKNkGLYqzyPi4vDunXr8NNPP5XqOIQQ4gy0yXQOyMjMEDYYB8HPCy72fl6vkz77+Z9//hEkJkdD9+iEEELKCmfusS6xZKfPPvsM69evxzvvvIOYmBjk5+cDAD7//HMcP34cx44dQ8OGDTF27FirBksIIcQ46rFu2Is91uk8PUflOSHEFtRqtSbxxgEtXm1o8XGCq1RA5TqVcOrUKRw/fhytWrWyWoyElCf2KM9VKhWio6PxzjvvoF69eiVun5+fr40DoN6ahBDnw/dYl7jL8TTrKRhj4DjzRvApT7KysnD+/HmIPRlEOq3RIjfNXOtHjhxx+nME0D06IYSQssOZE+sW9Vh3d3fH4cOHMWTIEBw8eFBb+ZkzZw6OHTuGwYMHY+/evZDL5daOlxBCiBHUE9swbSKde+F3QuU5IcQm9u7di3///RfVm1SGX7B3qY71Uo8GAAf8/PPP1Gud0GfACHuU5wsXLkRCQgK+/vprk7afNWsWvLy8tP9CQ0Mtfm1CCCmL+MS61F0GtVqNnJwcgSMS1tGjR6FSqSD11b8f5zhA4qtGSkoKrl69KlB0joPu0QkhhJQVzjwUvEU91gHAx8cHq1atwrx583Dq1CmkpqbC09MTzZo1Q2Cg+cM9EkIIKZ3CwkKhQ3BI/AMH3LNHyeg86aPynBBiTSqVCvPnz4dIJEKzbvVLfTy/YG9UaxSOq2ev4sCBA+jUqZMVoiRllW4PaKLPluV5SkoKpkyZgsmTJyMgIMCkfSZMmIBPPvlE+3tmZiYl1wkhTiUrKwvgOIhdZdrf3dzcBI5KOPv37wcASP2KPiQn9WMoeADs27cPderUsXdoDofu0QkhxDE5+6gqL8rLy9P+7Gz36hYl1jt16oTWrVvj66+/hp+fH7p3727tuAghhJhJ98kwGkLtOT6Rzok1vzvbE3TFofKcEGJtu3btwu3bt1G7RRV4+btb5ZjNutXDrXN38euvv6JDhw4QiSwadIuUA7m5uUKH4JBsXZ5PmjQJvr6+iI6ONnkfuVxOvekIIU4tKysLYpkYYplY+3tQUJDAUQkjLy8Phw4dgsiFQWTg2QKJDwMnBvbs2YPo6Ginbsuge3RCCCFlhW4yXTfJ7gwsapU6ceIEDTlMCCEOxpmHXykOX8hTYr0oKs8JIdakUqmwcOFCiMQiNOla12rH9a7giepNKuP69eva3k7EOWVnZwsdgkOyZXl+8+ZNLFq0COPGjUNSUhLu3LmDO3fuIC8vD0qlEnfu3EFqaqpNXpsQQsqyrKdPwUlFEEk1N6JPnz4VOCLhHDlyBDk5OZAGMBjKmXNizXDwd+7cwY0bN+wfoAOhe3RCCCFlhe6D75RYN0HNmjWRkJBg7VgIIYSUQkFBgcGfnR1/LjgJ0/udUHlOCLGu7du3Iz4+HjWbR8LDx7pDnTbtWheciMP8+fOhVqtL3oGUS86clCiOLcvzxMREqNVqjBs3DhEREdp/J06cwI0bNxAREYGvvvrKJq9NCCFl2dOnTyGSisHp9Fh3Vtu3bwcAyAKM1+FkFZjets6K7tEJIcRxMVZ0OhNnpptYz8nJETAS+7MosR4dHY1Nmzbh6tWr1o6HEEKIhZx5+JXi8IU8J9P/nVB5TgixHqVSiQULF0AsEaHJy9afG9MrwAM1mkXg1q1b2LVrl9WPT8oG3aQENWo8Z8vyvG7duoiNjS3yr06dOggLC0NsbCzefvttq78uIYSUZYwxZGc/hUgmhvhZj3Vna3DmZWVl4eChgxC7MYiLmSVI4svASYBt27c59UOUdI9OCCGOy5mnKjFE++C7iDnd6HIWzbEeGRmJDh06oEWLFhg1ahSaNWuGwMBAgx+sdu3alTpIQgghJdNNpusm2Z0dX7DziXXq7fYcleeEEGvZsGED7t29h3ptq1u9tzqvade6uHnmDn755Rd06dIFUqnUJq9DHJduYj07Oxvu7sW00DsRW5bn/v7+6Nu3b5HlP/30EwAYXEcIIc4uLy8PapVa02NdqunT5GwNzry9e/dCWaCEolLxyXJOBEgD1HiY/BCnT59G8+bN7RShY6F7dEIIcVz0cLc+vm4jkjvfyDwWJdY7dOgAjuPAGMMPP/xQ7JMaNC8MIYTYh25inXplP5eZmQkAECs0lR9nK+iLQ+U5IfZTnm/AsrOzsWDBAsgUUjS14tzqL/L0dUedVtVwMe461q5di2HDhtnstYhj0i3Dnz59Son1Z6g8J4QQx6JtaJaKnX6O9c2bNwMAZIEl14VlgWoUJIuwZcsWp02sU5lOCCGkrMjKygI4TWe27MxsqNVqiEQWDZJe5liUWJ8yZQoNe0AIIQ5GN5lOQ8E/l5GRAQDg5Jqn4PnfCZXnhNgDn1AvLCwUOBLb+e2335CSkoKXXm0AF3eFTV+rade6uH4qHr/++it69OgBHx8fm74ecSy6SYmsrCwEBQUJGI3jEKI8P3jwoF1fjxBCyhK+vNJNrDtjj/XExEScPn0aEm81RPKStxd7AiIFsHv3bkycOBEuLi62D9LB0D06IYQ4rvLcYcISWVlZ4CSASMKgYprh4D08PIQOyy4sSqxPmzbNymEQQggpLd3EOvVYfy41NRUAwEkATsq0vxMqzwmxBz6hXl6vy7dv38aff/4JTz93NGhf0+avp3CTo1n3ejgSewY///wzXcecjG5i3RkTFMbQ94AQQhyLdjoyqcipe6xv3boVgGm91QGA4wBpoBo5CTk4cOAAevToYcvwHBKV6YQQ4rhopBB9GRkZ4CQM3LNZ+jIzM50mse4c/fIJIcQJ0FDwhqWkpICTam7SORnw5MkTesKQEGI3BQUFev+XJ4wxfPXVVygsLESbfk0gedZwbGt1W1eDX7A31q9fj3PnztnlNYljyMnJ0f5MiXVCCCGOik+ii2ViiGTOmVhnjGHr1q3gxIA0wPT7b1mgZi52fgh5QgghxFGo1WqhQ3AomsS6pjMb/7uzsKjHOi87OxsbN27E+fPnkZmZCU9PTzRs2BB9+/aFm5ubtWIkhBBiAt3EOg0F/9yjR48gkmtu5DkZQ15WHp4+feo0T9CZgspzQmxHqVTq/V+erFu3DmfOnEGVBqGoXKeS3V5XJBahw+DmWP/zbkybNg1r166FXG7C+KKkzNNNrNNDhEVReU4IIY5BOxS8EyfWr169ijt37kBaQQ3OjGcvxS6A2JPh+PHjePLkCfz9/W0XpAOjMp0QQhwPJdafy8/PR15eHiSuDJxU0+6enp4ubFB2ZHFiff369XjvvfeQnp6u1/OP4zh4e3tj8eLF6N+/v1WCJIQQUrL8/HyDPzuznJwcZGdnQ+KrKaf4ed0ePXpEifVnqDwnxLb4oeDL25BhDx48wJwf5kDuKkObfk3t/vqB4f6o37YGLsZdx8KFC/Hhhx/aPQZb0f2sMMZonk0dNO2NcVSeE0KI48jMzAQAiGQS7VDwztSLC9AZBr6C+aPFySqokZvJYdeuXRg2bJi1Q3N4VKYTQohjKm/tOqXBJ9E1U69qlqWlpQkXkJ1ZNBT8sWPHMGTIEGRnZ+Odd95BTEwMDhw4gL/++gvvvvsucnJyMGTIEBw/ftza8RJCCDFCd5jh8jjksCUePHgA4HlCXSRjesudHZXnhNge3xhWnp5sZoxh6tSpyMnOQes+jeHm5SJIHC/1aAAvfw8sXbYUly9fFiQGW9B9OK48jnRQGnn5z0fkoYcIn6PynBBCHAvfsCySS8CJOIhkEqdKrKtUKuzYsQMiKSDxMT+xLg1gAAfs2LHDBtE5NirTCSHEcVFi/bnU1FQAgEjqnIl1i3qsf/PNN5DL5Th69CgaNGigty4qKgoffPABWrVqhW+++QZbtmyxSqCEEEKKx/eKfPFnZ/Y8sf6sx7pCf7mzo/KcENvjb7zKU2L977//xrFjxxBeOxg1mkUIFodULkHHIS9h4/y9mDhxItauXQuFQiFYPNbyYq9smUwmYDSOpSD/+YODlFh/jspzQghxLHzDslghefa/WNsA7QzOnDmDlJQUyILV4Czo0iWSARJvNS5cuICkpCQEBwdbP0gHRWU6IYQ4rvLUrlNafL2GkzGIng0Fn5KSImRIdmVRj/Xjx48jKiqqSAHPq1+/PgYPHoxjx46VKjhCCCGm0+3VRj3cNJKSkgA8T6jzCfbk5GShQnIoVJ4TYnv8jVd5ebL53r17mDNHMwR8h8EvCT5MeXCVCmjQrgbi4+Pxyy+/CBqLtdA84sblFzxPptPoPM9ReU4IIY7lyZMnAACJi6YLl9hFipTUFKdpkN+9ezcAQBZg+fuVPhtCfs+ePVaJqaygMp0QQhxXeWnXsYbHjx8D0DwMxz3rC8DXf5yBRYn1nJwcBAYGFrtNYGCgXqMQIYQQ29It3Kmg19Am1l/osc4vd3ZUnhNie/yDTuXhgSeVSoVJkyYhNzcX7QY0FWwI+Be91KMBfCp44s8//8Tp06eFDqfUdK+52dnZAkbiWFQqFZQFSoDTlOl5eXkl7OE8qDwnhBDHwjc2i100PdYlLlKoVWqn6MmlUqmwe89uiGSA2Mvy40j9NMPB79y503rBlQFUphNCiONylgfkTMHXdTjZ8ylYHz16JGBE9mVRYr1y5colPjG4b98+VK5c2ZLD65k5cyY4jkPdunVLfSxCCCnPdAt3Kug1EhMTAQCiZ7kfTg6Ae77c2dmzPCfEWfHJv9xykARcuXIlzp49iyoNw1CtcWWhw9GSyCToNKwlwAH/93//V+aT0VlZWQZ/dnb80O/8/G3Um/85Ks8JIcSxJCYmQuImAyfSNLtK3DUtzs4wctrZs2eRlpoGib8apRnYSCTVDAd/+fJlp3owntrcCSHEcdHUq889HyWWgRNp7tOdaepVixLrgwcPxpkzZzBixIgilZvk5GS8+eabOHPmDKKiokoV3P379/HNN9/Azc2tVMchhBBnQIn1ou7fvw+Ing9Jw3Ga3uv37t0TNjAHYa/ynBBnpVQqtcnAnDKe7L116xbmzZsHV08XtB/YTOhwiggM80OTLnWQlJSE77//XuhwSoUS64ZlZmYCeP40PJ2b56g8J4QQx6FUKpGcnAyJu0y7TOKhKbzu3r0rVFh2wyeFpf6s1MeSBjjfcPDU5k4IIY6LEuvPPR8lVvO7SMGQmJgIxkpf/pcFEkt2+uKLL7Bz5078+eefWLNmDapWrYrAwEA8fPgQt27dQkFBAZo3b44vvviiVMF9+umnaNGiBVQqlVONz08IIZbQHf6dEusaCQkJmifndJ6UF7kwPH78GDk5OXB1dRUuOAdgr/KcEGfFJwIBTSNrWb3uKJVK/N///R8KCgrw8oj2ULjJhQ7JoCZd6yLhaiLWr1+Pzp07o23btkKHZBFKrBuWnp4OABC5MqiyOKSlpQkbkAOh8pwQQhzHvXv3oFKp4Oqp0C6TeWrqTvHx8UKFZRcqlQq7d2uGgZd4WyGx7s+Qe1OTWB8xYoQVInR81OZOCCGOqzxM8WctCQkJmvnVxZrfRQqG/Kx8PHr0qMQpTcoDi3qsu7q6Ii4uDtOmTUNISAiuXr2KAwcO4OrVqwgJCcH06dNx6NAhuLhYPu9iXFwc1q1bh59++sniYxBCiDPRTabTHOtAWloaMjIyIHLRv6EXPctpJSQkCBCVY7FHeU6IM+MTgcZ+LyuWLFmCq1evolaLKgivXUnocIwSi0Xo9FpLiCUiTJkyBRkZGUKHZBHdZLruwxnOjh8+V+zGwImda5i5klB5TgghjuPmzZsAAJn382uuzEfz861btwSJyV7Onj2LlJQUSPxKNww8jx8O/sKFC04xjD5Abe6EEOLIKLGuUVBQgOTkZHA6be58e/udO3eECcrOLOqxDgByuRxTpkzBlClTkJWVhczMTHh6esLDw6PUQalUKkRHR+Odd95BvXr1TNonPz9fO9QmQI1QhBDnoyx8XrhTQf+80UL8wshmYlemXV+rVi17h+VwbFmeE+LsUlJS9H5PTU1FcHCwQNFY5tq1a/jtt9/g4eOG1n0aCx1OifwqeqN59/o4vvU8vv32W3zzzTdCh2Q23cT606dPBYzEsfDTuIhcAE7OcPfuXTDGwFmj5b4coPKcEEIcw7Vr1wAAct/niU+xQgqJqwxX/70qVFh2sW3bNgCArIL1RtCTVmAoTAO2b9+Ot99+22rHdWSO1OZO7e2EEPKc7vXQmd2+fRtqtRoyt+eJdd329pdeekmo0OzGoh7rL/Lw8EClSpWsdtO+cOFCJCQk4OuvvzZ5n1mzZsHLy0v7LzQ01CqxEEJIWVGQX6D9mRLrwI0bNwBoerbp4n+/fv263WNydNYuzwlxdo8ePQIA+FUK1/u9rCgsLMTkyZOhUqnQIeolyBRSoUMySYOONREY7octW7bg8OHDQodjNt1kOiXWn+MTFWI3BrE7Q1ZWVpG5R4kGleeEECKcS5cuAQDkfvpPeMv8XPEg+UG5HXY7Ly8Pu3fvgkgOiL2sd1yZPwMnAjZv3uw087bqErrNndrbCSHOTrfsKSgoKGZL52Gozd3Z2tstSqwfPXoUn3zyidHh95KTk/HJJ5/gn3/+MfvYKSkpmDJlCiZPnoyAgACT95swYQIyMjK0//geDYQQ4izy8vIgkom1Pzs7vkFD7PFCYt0dAPd8vTOzZXlOCHmeSK9YpZbe72XFypUrce3aNdRqUQWhNYKEDsdkIpEIHYe0gEgswldff4WcnByhQzKLbrxlLXZbOn/+PDgJIFI8L9svXLggcFSOgcpzQghxDEqlEhcvXYLMx0V7b85TVNAk2s+dOydEaDa3Z88eZGU9hTTQOsPA8zgJIPFX4/bt205R7jtamzu1txNCnF1hYaH259zcXAEjcRyXL18GoN/mLnLVzLd+5coVocKyK4sS63PnzsWWLVsQFGS4ga1ixYrYunUrfvzxR7OPPWnSJPj6+iI6Otqs/eRyOTw9PfX+EUKIs1AqlVAqlRC7anoTZmdnCxyR8M6fPw9OqmmA18WJNU/RXblyxel79tuyPDdk5syZ4DgOdevWtcrxCHF0/FyQlarX0/u9LHj48CF+/fVXuHoo0LJXI6HDMZtvkBcad6mNB8kPsHjxYqHDMYtuMp3Kc4179+7h3r17kHhrGuul3pob+GPHjgkcmWOwd3lOCCHEsCtXriA3JweKwKK9i12eLTt16pS9w7I5xhhWr14NAJBXtN4w8Dz+mPxrlGeO1uZO7e2EEGene39OiXWNS5cuASL96Vc5DhC5M9y6dcspOghYlFg/deoU2rRpU+w27dq1M/vpuZs3b2LRokUYN24ckpKScOfOHdy5cwd5eXlQKpW4c+cOUlNTLQmZEELKNb7hXeIqA0BDx96/fx+JiYmQeBl+Wl7ixZCfn+8UT7wXx1bluSH379/HN998Azc3t5I3JqSc4IepDq3VUO/3smDevHnIzc1Fi54NoXhWtpQ1jTvVhqevG/744w/cv39f6HBMpnuzTiPQaMTFxQEAJL6ahLrIDRDJNct1exA4K3uW54QQQozjr7MuFYsm1uX+bhBJxTh+/Li9w7K5c+fO4eLFi5D6q4s82G4NYi/Nw/E7duwoU/VpS1CbOyGEOBZKrOvLycnBv//+C7G7ZqoWXRJPBrVa7RTt7RYl1h89eoRKlSoVu01QUJDZw10mJiZCrVZj3LhxiIiI0P47ceIEbty4gYiICHz11VeWhEwIIeVaRkYGAEDiLgM4IDMzU+CIhMU3Vkh8DM/Bxi939p5utirPDfn000/RokULNG3atNTHIqSsuH//PlzcveATFAqJTF5mkrvx8fHYunUrAkJ8UKNphNDhWEwik6BFz4ZQKpVlqte6bjKdbtw1tm/fDnCA1E9TfnOcZljYtLS0ctnzz1z2LM8JIYQYd/ToUXAizmBinV9+584dJCYmChCd7SxatAgAIA+1fm91QFPuy0PVUKvVWLp0qU1ew1FQmzshhDgW3VHknL0jG6AZIValUkHiVbTNXfJsZDlnuEe3KLHu7e2Nu3fvFrtNQkIC3N3dzTpu3bp1ERsbW+RfnTp1EBYWhtjYWLz99tuWhEwIIeUan1gXyyUQyyRIT08XNiCB7d+/HwAg9TWSWPfWPFXHb+esbFWevyguLg7r1q3DTz/9VKrjEFKWqFQq3Lt3Dz4VQ8FxHHwCK+HOnQQwZvi65EiWLFkCtVqNZt3qgRNZcZJMAVRpEAbfil7YtGmT0bkqHU1eXp72vOfn5wscjfDu3LmDixcvQuKthkhn8ARZgKbxfvPmzQJF5jjsVZ4TQggxLjU1FRcvXoQ8wA1imcTgNq4hXgCej8RSHpw6dQpHjx6FxEcNiQ1HCZdWYBC5Mqxbt65cz/NNbe6EEOJYdJPplFjXPEQIGO7MJvHStLc7Q0c2ixLrLVq0QGxsrNGKzN27d7Fx40a0atXKrOP6+/ujb9++Rf75+/vDw8MDffv2Rb169SwJmRBCyjV+yC6xQgKRQoK0tDSBIxJOVlYWTpw4AbE7MzoMHScGJL5q/Pfff4iPj7dvgA7EVuW5LpVKhejoaLzzzjsmleH5+fnIzMzU+0dIWXT//n0olUr4VgwFAPgGh+Pp0yw8efJE4MiKl5GRgZ27dsIn0BPhdYrvLVMWcCIODTvUgkqlwoYNG4QOxyS5ubngJCJwEhENBQ8gNjYWACCrqH/jLvYERK4Mu3fvdvqywh7lOSGEkOLFxcVBrVbDLczb6DZ8Yv3AgQN2isq2VCoVfvjhBwCAIsI2vdV5HAcoKquhUqksml+8rKA2d0IIcSxZWVnan/Py8px+KrLDhw9r2tUN9FjnxIDYU40rV644fNtXaVmUWP/kk0+Qk5OD1q1bY8WKFUhOTgYAJCcn448//kDr1q2Rm5uL8ePHWzVYQgghhj1PrEshVkiQlp4Gtdq2N7aOas+ePVAqlZAGFP/+pQGaCsCOHTvsEZZDskd5vnDhQiQkJODrr782aftZs2bBy8tL+y80NNTi1yZESDdv3gQABIRGAgD8QzVDqt+4cUOwmEyxa9cuFOQXoHaLquC4st1bnVelYRjkLjJs3LSxTIwYkJeXB04sgkhMiXWlUonYjbEQSZ8PA8/jOEAWpEZBQQG2bt0qUISOge7PCSFEePv27QMAuIV6G91G4iqDPMANJ0+e1I46V5b9/fffuHLlCqSBakiKjn5vdVJ/BrEXw549e3DkyBHbv6AAqEwnhBDH8mIvdWfutX7r1i3Ex8dD4qsuMr86T+qvuW8v76PEWpRYb9euHebOnYukpCSMHDkSISEhkEgkCAkJwVtvvYUHDx7g559/Rrt27awS5MGDB3H58mWrHIsQQsojbWLdRQKxQgq1Sl0ubtQtsWXLFgCALLD45InUj4ETA1u2bC4TiRZbsHV5npKSgilTpmDy5MkICAgwaZ8JEyYgIyND+688D/NHyrerV68CAAIjamj+r1xdb7mjOnjwIABNMrq8kMokCK8djOSkZO0DD44sLy9P22Pd2edY37dvH9JS0yANMnzjLgvSDDW3Zs0apy3LAfvfnxNCCNGXnZ2No8eOQebjAqmnkWHTnnEL94FKpdLWucoqTdnyE0RSwCXSPg/1cxzgWk0FcMCMGTOQk5Njl9e1J2pzJ4QQx8L3WBdJxQDg1KOl7dq1C8DzzmqG8In1nTt32iUmoViUWAeADz/8EGfPnsWoUaPQuHFjREZGokmTJhg9ejTOnTuHMWPGWDNOQgghxUhJSQHwrMe6i0RvmTNJSEjA6dOnNfOwyovflhMD0gA17t9PxOnTp+0ToAOyZXk+adIk+Pr6Ijo62uR95HI5PD099f4Rx8cYQ2FhodOOlGHI+fPnwYlE2oR6xSq1AAAXLlwQMqxiKZVKnDx5En7B3nD3dhU6HKsKrx0MADh+/LjAkZQsJycHIokInJgS66tXrwYAyCoavraIpIDEX43bt2/jzJkz9gzN4dD9OSGECCcuLg7KggK4V/YpcVv3cM02e/bssXVYNsMYw9SpU/H0aTbkESqIZPZ7bbEbIA9VIzExEXPnzrXfC9sRlemEEOI4+ES6xENT2OkODe9M1Go1Nm3aBE4CSH2NJ9ZFckDircapU6fKdWcpSWl2rl+/Pn799VdrxUIIIcRC+ol1KYDnvdidCT9/7ovzsBojC1Kj4IEI69atQ7NmzWwZmkOzRXl+8+ZNLFq0CD/99BOSkpK0y/Py8qBUKnHnzh14enrC19fXqq9LhPHJJ59g7969cHV1xapVq1C1alWhQxJUXl4eLl68iICwKpC7ugEA3Lx84VsxDKfPnEFhYSEkklJVw23iv//+Q35+PqpGlJ/e6rygyppRM8pCj5zc3FxwXlJwIoac3PLXE8tUt27dwpkzZyDxUUPsYnw7ebAaykcirFmzBk2bNrVfgA6I7s8JIUQYu3fvBqDpjV4SqYccMj9XHDt2DFlZWfDwsMMY6lb2999/49ixY5D4qiELsv+IMYpwNQpTOKxZswYdO3ZE69at7R6DrVGZTgghjoFPrEvd5ShIzXXaHuvHjx9HcnIyZBXV4MTFbysLYihMB2JjYzFu3Di7xGdvFvdYJ4QQ4jiez7GuGQped5mzKCgoQGzss3lY/U27uRd7AiI3zRxtzna+bC0xMRFqtRrjxo1DRESE9t+JEydw48YNRERE4KuvvhI6TGIlx4//A0DT0/bixYsCRyO8kydPIj8/HxH1m+str1y/GbKfPsX58+eFCawE169fBwAEhJTcKFzWuPu4QuEmx7Vr14QOpVhKpRIFBQUQScXgpGLk5uQ67RDnfG91eXDx71/sCYjdNWX548eP7REaIYQQopWTk4O4uDjIvF0g8y7mSTAd7uE+UCqVOHDggI2js77//vsP3333HURSwLW6Ghxn/xg4EeBaUwVOBEycONEpR+sjhBBiH897rMv1fnc2K1euBADIgkseqVIawCCSah7Ey8vLs3VogjArsW5seM/09HR8/PHHaNCgARo0aICxY8fi0aNHVgmQEEJIyVJTUyGWS8CJOIgVEu0yZ7J3716kpRmfh9UQjgPkFdVQKpXYuHGjTeNzJPYoz+vWrYvY2Ngi/+rUqYOwsDDExsbi7bffLs3bIA4iKysL2dlPIZZpbjKSk5MFjkh4e/fuBQBUbazfe6Zq4zYAHHfoz4SEBACAd0D5m4KB4zh4B3jg/v37KCwsFDoco/i5QkVSMURSERhj5XL+0JI8ffoUm7dshkgBSPyKT6xznObmXqVSYd26dXaK0DHQ/TkhhAjv0KFDKCgogJsJw8Dz3CM0o3bxPd3Livz8fHz+xefIz8+HS3VVidOv2ZLYHVBEqJCamorJkyeX+QcRqUwnhBDHpNtjXfd3Z/Lff//hyJEjkHipIXEveXtOBEiD1UhPT8fmzZttH6AATE6sz5s3D1KptEhDYF5eHtq1a4d58+bh0qVLuHTpEn799Ve0bt3aaecbIIQQe0tLS4NIrhmHxVkT6yXNw2qMLJCBEwNr1qyBSqWyRWgOxV7lub+/P/r27Vvkn7+/Pzw8PNC3b1/Uq1fPWm+LCIgf6t8npIre784qNzcXe/bshad/IIKr1tFbF1a7IVw9fbBz504olUqBIjSOfyjC08+EO6UyyMPPHYWFhQ7dq4m/SRfJxBDJNOW5M95Tbdq0Cbk5uZBVVJnUE05WgYGTAmvXrnXI75Yt0P05IYQ4hl27dgGASfOr86Qecsj9XHH06NEy1UD/448/4sb1G5AFq00eJc6WZJUYJL5qHD58GDExMUKHYzEq0wkhxHFpe6y7y/R+dyZLliwBAMhDTS/75cGajm9Lly516M4NljI5sX7o0CFUqFABL7/8st7yxYsX4/Lly6hduzb279+PkydPYuDAgbh9+zbmzZtn9YAJIYToY4whPT1dOwS8WK5piE9PTxcwKvu6fv06zp07B4lv8fOwGsJJAGkFNZKSknD48GHbBOhAqDwn1nb//n0AgHelyuBEIu3vzmrPnj14+jQLddu9Ak6kX9UWiSWo07YbUlNTHXLoz8ePHwMc4OKhEDoUm3Dz1BQQjjxcuG5iXfzsgbmMjAwhQ7I7tVqNmJgYcCJAVtG0G3dODMgC1Xjy5InDjghhbVSeE0KI8HJycnD48GHIfEwfBp7nVtkHhYWFOHjwoG2Cs7K4uDisWrUKYjcGl0jzHma3FY4DXGuoIZIBc36Yo53WqKyhMp0QQhxXZmam5v78WUc2Z0us37t3D9u3b4fYnUHia3piXSQDpBXVSExMxPbt220YoTBMTqxfvHgR7du3L7J8zZo14DgOq1atQocOHdC0aVP89ddfqFSpUrnt5k8IIY4kKysLhYWF2gLeGXus//XXXwBKnofVGHkltd5xyjOhy/ODBw/i8uXLVjseER6fSHf1DoDCwwd3794TOCJhrV69GpxIhPodehpcX79jLwCa75yjSU9Ph9xFBrHYrNmiygwXN83QbY784Bnfm16skGgflHOm8hzQNNzfvXsX0kA1RFLT95NVUgMcsGLFijI/HKwp7FWeX7lyBYMGDUJkZCRcXV3h7++Pdu3aYcuWLdZ4G4QQUqYdPHjQ7GHgee6VNcPB8z3eHVlaWhomT56smdu8lgqcWOiInhPJAJcaKhQqCzFhwgQUFBQIHZLZhL5HJ4QQYlxWVpZmqjaZcybWFy1aBLVaDXmY2qTR5HQpQjW91hctWlTueq2b3Gr2+PFjVKlSRW+ZUqnE6dOnUbVqVdSvX1+7XCwWo1u3brhx44b1IiWEEGJQWloaAED0LKEukksA7vny8i4jIwNbt26FyMW8J+d0id0AiZcax44dQ3x8vJUjdCxUnhNru3v3LgDAxdsXLt5+ePLksVPOCQ0Aly9fxqVLl1C1cWt4+gca3MYvOAyV6zXDyZMn8d9//9k5wuJlZWVBrjAjk1nGyFw0Q7c58tCZfGJd4iKF2EXzt3jy5ImQIdndH3/8AeD5Q2+mEisAqb8aV65cwZkzZ2wRmkOxV3mekJCArKwsjBgxAj///DMmT54MAOjduzcWLVpUujdBCCFl3PNh4H3N3pcfDv7YsWMO30g/Y8YMpKamQh6hgthN6GiKkvoyyILVuHnzJv73v/8JHY7Z6B6dEEIcVwbfY/3ZiHKOXmZb071797B582aI3ZhFU8CI5IA0SI2EhATs2LHDBhEKx+TEel5eHvLy8vSWXbp0CQUFBWjRokWR7QMDA522UZUQQuyJT6DzPdU5joNYLnWaHm4bNmxAfn4+ZMHmPzmnS1ZJU0Hg52ovr6g8J9aWkJAAgIOLlx9cvf0AaCrfzogf9aJx1/7Fbtfo2XpHGyUjLy8PkmdPYZdHEqnmRvjFa6Aj4YepF7tKIXaV6i1zBpcuXcLp06c1U7tY0HAvD9Ek45cvX27dwByQvcrzHj16YOfOnZg6dSreffddfPjhhzhw4AAaNGiAuXPnWhw/IYSUdU+fPsXhI0c0w8B7WTaNDj8c/P79+60cnfUcPHgQu3fvhtiLQV7JcUeEcYlUQ+SieUDv2rVrQodjFrpHJ4QQx6RSqfA0KwtihcQpe6z//vvvmt7q4Za3uStCNSPLLV68CCqVyroBCsjkxHrFihVx9epVvWXHjh0Dx3Fo3rx5ke0zMzPh5+dX+ggJIYQU6/mwsc97GYoVEu3y8qywsBB//fUXODEgDyrdTb7Un0GkAGJjYx26N2NpUXlOrO3u3btQeHhBLJHCxdsfgHMm1jMyMrBr1y74VgxDWJ0mxW5bpVFLePoHYuvWrQ7VKKZUKiEqp8PAA4BIonlvSqVS4EiMe/DgAQBA4iqDxE2mt8wZLFu2DMCzm28LSDwBsRfDoUOHcPPmTWuG5nCELM/FYjFCQ0MdeloFQgixtQMHDkBZUAD3CPN7q/P4nu47d+60VlhWlZOTgxkzZmiGgK+uKtWD7LbGiQGXaiqo1WpMnz4darVjzANvCrpHJ4QQx5SVlQXGGEQyCTgRB5FMjIyMDKHDsosHDx5g06ZNELta1ludJ1IAsiA14uPvYO/evVaMUFgmt5y1a9cOe/fuRVxcHAAgNzcXixcvBgB07969yPYXLlxASEiIlcIkhBBijO6wsTyxiwQZGRkOnTywhn379iE5ORnSQDW4Unay5DhAFqxCbm4uNmzYYJ0AHRCV58SaCgoK8ODBA7h4aRp2XJ/9zw8P70y2b9+O/Px8NOjUC1wJrY4ikRj1O/REdnY2du/ebacISyYSld+kui5Hfp/JyckAAInb88Q6v6y8i4+Px969eyH2YBB7WX4cPim/dOlSK0XmmOxdnmdnZ+PJkyf477//8OOPP2LHjh3o3Lmz0e3z8/ORmZmp948QQsoTfkhTdwvmV+dJPeSQB7jhn3/+ccip3P788088fPgQshA1xK5CR1MyqQ+DtIIaly9fdtiHFQyhe3RCCHFM/IPE/DDwIpkE6U6SWF+1ahUKCwstmlv9RfJnvdaXL18Oxhx39BtzmNyq9Pnnn0MsFqNLly5o3LgxIiMjcenSJfTq1avIPDApKSk4fvw42rRpY/WACSGE6Hv06BEAaIeMBaCdl7U891pnjGHFihUAng/9WlqyigycGFi5cmW5fSiBynNiTcnJyWCMwcVL09vGxVvz//3794UMSxCbNm2CSCxGnbbdTNq+brtXwHEcNm3aZOPITCcSiaBWlZ3ePeZiz3ouicVigSMxLikpSTPMnFQMsUwz3FxiYqLQYdnFsmXLwBgr9Y27xJdB7Mawfft2JCUlWS9AB2Pv8nz8+PEICAhA1apV8emnn6Jfv37FzmM7a9YseHl5af+FhoZa/NqEEOJo0tPTcezYMcj9XSH1tGwYeJ57hC9UKpVDPWwJAKmpqfj9998hkgGKsLJTP3SJUIMTAT///HOZuaene3RCCHFMfGJdJNf05hLLxUhPd7wH4awtOzsbf//9t2aO9IDSJ8LFLoDUT/Pg27lz56wQofBMTqzXqVMHW7ZsQVhYGM6fP4+UlBT079/fYE+A3377DYWFhejWzbSGRUIIIZbj517V7bEucdX0cuOT7uXR2bNncfHiRUj91RC7WOeYIgkgDVLjwYMH2LVrl3UO6mCoPCfWxCetFJ6anjoKD83/ztLDlvfff//hypUriGzYEq6epvVa8vQPRFjtxjh9+rTDPIjg4uKCQmX5mfPqRcp8zXtTKErXAG4rjDEkJiZqe6oDgMRdiqSkpHLzVLcxDx8+xJYtWyByZZD6le69chwgD1NDrVaX67nW7V2ef/TRR9izZw/++OMPvPLKK1CpVCgoKDC6/YQJE5CRkaH954xThBBCyq9du3ZBpVLBPaL0w3G7V/YFOGDbtm1WiMx6/vrrL+Tm5kIepgLnuM8kFiFSALJgNZKSkrB9+3ahwzEJ3aMTQohj4od9Fz9LrIvkEuTm5JaZB7cstX37dmRnZ0NWUQXOSgP+8Z3i1q5da50DCsys0/Lyyy/j1q1bePjwIZ4+fYp169bB17foXEIfffQR0tLS0LVrV6sFSgghgGZe1L59++Llri/TkJLPPHz4EAAgdn3eEM/3Xi/PifUlS5YAeDacjBUpQjTD0/z+++9lal42c1B5TqyFv/7I3TXjNoulMkgVLtrlziI2NhaAphe6Oeq20wzt6Ci91l1dXaHMLxQ6DJtR5mtuft3c3ASOxLCUlBTk5+dD4iHXLpO6y5GdnV3u6zx//vknCgsLoQgt/TBzgOapepEC2LBhA1JTU0t/QAdlz/K8Zs2a6NKlC9544w1s3boVT58+Ra9evYw+9CGXy+Hp6an3jxBCyostW7YAHEo1vzpP4iqFS0VPnDt3zmEeQsrNzUVMTAxEUkAWVPYe7pM/u6dfunRpmXk4ke7RCSHE8fCJ9ec91jX/8z3Zy6t169YBnGZkV2sRewJiV4bde3aXi3nqLXreICAgADKZzOh6V1dXeHl5lTi/JCGEmCslJQX//fcfHiQ/cJibTqE9fPgQYrkEIsnzSzrf2628JrcuX76MI0eOQOKthsTK7bQiBSCroMatW7ewf/9+6x7cwVB5TkrryZMnAAC5m4d2mdTVQ7vcGeTn52Pjpk1w8/ZFlUatzNq3evMOULi5Y/36DQ7xxLOnpyfycwrKTAOkufJzNb1rHTXBpzu/Os8Z5lnPyMjA2rVrNcPMVbDOZ4/jAHmICvn5+fjrr7+sckxHJkR5PnDgQJw6dQo3btyw2jEJIaQsuHPnDi5cuADXYE9IdKZjKw2PKpqe71u3brXK8Upr9+7dyMzMhDRYXaZ6q/M0Q9eqcfv2bZw9e1bocMxC9+iEEOI4ns+x/rzHOoBykRg25tatW7h69SokvmqIjBdHZuM4zSixygJluRgl1kod+QkhxD50h5zMz88XMBLH8fDhQ7351QFob/DLa2L9119/BQAowm3To1wernnC/ddffy23vdYJsQb+ZkLq8rwHsMzFHRkZGU7z3YmNjUVGejrqd+wFsURi1r5SuQJ12/XA48ePsGPHDhtFaDovLy+o1epy22s9L1tTb/Dy8hI4EsMePHgAAJC66w4Fr/m5PM8VvmbNGuTm5kIWYr1h5gBNDzuRFIiJiUFOTo71DkwAaHozAuW7UYkQQgzhRxryqOpvtWO6hXtDJBVj48aNDlGH3rBhAwBAHiR8LJaSV9TEzr8XQgghxFzPe6xrnjITP/u/PN8D8VPTyAKt3+FCVoEBnOM8SFgalFgnhJQpuj36HKF3n9BycnKQlZWl17sNeN7DjW+kL0/Onj2Lw4cPa3qre9vmNcQugCxQjZs3b2Lnzp22eRFCygF+eGqpwlW7TKpwgVqtRnZ2tlBh2U1OTg4WL14MiUyOJt0GWHSMZj2iIBZLsHDhwmLnK7YHfrjJ3Kw8QeOwldynmsS6t7e3sIEYwZfZulO78OV5eZ3aJS8vDytXroRICsitOMwcAHBiQFZJhczMTKxfv96qx3Ymhj57SqUSK1asgIuLC2rXri1AVITYn1KpxOLFixEfHy90KERAhYWF2LRpE8RyCVxDva12XJFEDLcIHyQlJeHkyZNWO64lkpOTcfbsWUi81RApBA2lVMRegMiFYe/evdQpgxBCiEX4Ni+x7FmP9Wf/l9ep2tRqNbZt2wZOAkj9rJ9YF8kBibca586dQ2JiotWPb0+UWCeElCmFhc970VFi/XmP9BeHoBMrpADHlbuGeMYYfvrpJwCAIsK2T88rwtXgRMC8X+bRZ40QI7KysgAAEtnzOaHFz352hsT6kiVL8OjRIzTvORSunj4WHcPDrwIadx+Ie/fuYfny5dYN0Ex8Yj2nvCbWs/IgFosdtsc6X2brluliF6neuvJmw4YNSEtLs9lQs7JgBk4MLF++nMpyC40aNQqdO3fG9OnTsWTJEsyYMQP169fH2bNnMWPGDLi7uwsdIiF2sX//fsybNw8TJkwQOhQioCNHjuDx48dwj/TVm4rNGjyraXrAC/0w2O7duwFYb3oWoXAcIPVnyMnJwZEjR4QOhxBCSBnEt3mJZGK9//nl5c358+eRnJwMqb/aqqPJ6ZI9q184wqiNpUGJdUJImaKbWHeEIdKExje06/ZuAwBOxEHiKi13DfF79uzBuXPnIPW3/tzqLxIpAFmwGon3ExETE2PbFyOkjOKHVxbrJNYlTpJYP3/+PH7//Xd4VaiIl3oNK9WxWvUbAXcffyxYsAD//vuvlSI0n7+/pkE3JzNXsBhsKTsrF/7+/hCJHPMW6HliXafHumv57bGuVCqxbNkycGJAXsk2dTqRVFOWP3r0CJs3b7bJa5R3UVFREIlEWLBgAUaPHo25c+ciJCQEmzZtwieffCJ0eITYDT/k55UrVwSOhAhp3bp1AACP6gFWP7bc3w0yHxfs3bsXKSkpVj++qfbv3w88S0qXddIATf1i//79AkdCCCGkLHr69CmA5wl1cTlPrPP3zLZ8uE7qz8CJNFPrMFZ26xqO2apECCFG6CbTKbEOPH78GEDRHuuAppfbo0ePynQhpSs/Px8//PADOBGgiLTP314epoZICixcuBCpqal2eU1CypKcnBxwIjFE4udzi4ulcu268urx48f49NNPwQC8OnoSpPLSjZMpd3VHj/cnorCwEJ988gnS0tKsE6iZAgMDAQBPM8rf344xhuz0XO17dERPnjwBAIhddL5Pz3qs8+vKk61bt+LBgweQBWnKWluRh2ietl/y+xK9BzSJaYYMGYI9e/bgwYMHUCqVSE1NxZ49e9C7d2+hQyOEELtKSkpCXFwcFBXcIPdxsfrxOY6DZ40A7XDzQkhLS8P58+ch9mQ2LZvtReyuGXb20KFDUKlUQodDCCGkjMnKygI4DtyzUWr4BDufcC9PcnJysH3HdogUgMTbdrkETgJIAtS4c+cOzp8/b7PXsTVKrBNCyhTdZDrdGD1PrPMN77rELlLk5+eXm6foli5diqSkJMgqqSG2fjuGQSIpIA9X4enTp5g3b559XpSQMiQnJwcSmf6IGeV9KPjs7GyMGzcODx8+RPsh7yOkRn2rHLdyvWZoPeAt3L9/Hx9++CFyc+3fa7xixYoAgKzU8ve3y32aB1WhSvseHdHjx48hVkjA6fSoF0lEEMnE2vK+vFCpVFiyZAk4ESAPte3DciIZIA1S4/69+9i5c6dNX4sQQkj5tW7dOjDG4Fmjgs1ewyPSDyKJGGvXrhWkI8HRo0ehVqsh9SsfnRg4DpD4qZGRkYFLly4JHQ4hhJAy5unTpxDLxOA4DgAgkpbfHutbtmxBbk4uZEEqPHu7NiMP0tQz/vrrL9u+kA1ZlFjPyMjAxYsXjfZEys7OxsWLF5GZmVmq4Agh5EW6va/LS0/s0njeY11WZB3fi7089HK7d+8elixZApFcM/e5PcmCGcTuDOvXr8eFCxfs+tq2RuU5Ka2srCyIZfq9tfmh4MvrE7yjR4/G5cuXUb9jLzR7dYhVj9+q/5uo3fplnDt3DuPGjUNenn3nOg8LCwMApD8ufzeJ6Y8074l/j47o8eP/Z+++w6Oo1geOf2e2JZveCQkh9BK6WBALigg27O0qoghWsOu9FuyK/ESvonjtigULiAIqKB1EqhQhlFBDCOm9b5n5/bFsAGkpuzuzm/N5njwkm90574Rkz8wp71twwoVygTaxPm/ePPbv34+plYJsOfXzmyuojQISfPjRhwGZ8Uj054IgCN5lt9v54YcfMFiMhLSN8lo7stlAaPtosrOzNakL7m7TFB04Yy2mKNe5+EudddGnC4Ig6EdFRQXSoV3qELg11p1OJ1988QWSDOZE718DGCLAEKry22+/kZOT4/X2vKFJE+svvvgiAwcOPOFuUafTycCBA3nllVeaFZwgtGTp6en897//FSkr/0FMrB/tcI314wzEH3rM3+uyqqrKyy+/jM1mI6iDE8lw6td4kiRBcCdXf/fCCy9gt9t9G4AXif5caK6ysjJMlqNTSJiCrAABN9hTUlLC6NGj2bBhA93OHsLFdz5av2rZUyRJ4pK7n6LT6eexatUq7rnnHp/+HENDQ4mPj6c4p9RnbfqK+5zatWunbSAnUFNTQ2Vl5XFLuxitJkpKSgKm/1EUhY8++gikQxPePiAHgbmVwt49e1m4cKFP2vQl0Z8LgiB41/z58ykuLiasUwyy0bvJP8O7uuq3f/fdd15t559UVWXlyj+RLSBbfdq0VxmjVJBg5cqVWofSIKJPFwRBKzU1NXz88cdkZ2drHYpulJaV1ddVB5AtrrJtZWVlWoXkFXPnznUtfE9QkI/du+dxkuQq2VY/NuCHmnQ1OG/ePIYMGUJYWNhxvx8eHs7QoUP59ddfmxWcILRkDz30EJ9++imrV6/WOhRdETXWj5afn48kSxiCjMd8z72L3d8n1ufOncuff/6JMVrBFKvNYgpjOJgTFXbu3MnUqVM1icEbRH8uNEddXR1VVVWYrKFHPW4KDgGgqKhIi7C8IjMzkxEjRrB582Z6nn8pl937FLLsnVU+BqOR4eOep9vZF/HXX38xcuRIn97YpqWlUVlaTXWF71PRe1N+VjHgOj89OrxQ7ngZaFyPBcqu9SVLlrB7927MCQpy0Kmf7ymWQ7vWP/7444BbnCn6c0EQBO/69ttvAbyaBt7NEm0lKD6U5cuXc+DAAa+357Zr1y6Ki0swRCpeTwHrS5IBDOEqW7Zs8YuMWqJPFwRBKz/99BNvv/02L730ktah6EJtbS011dXIR4y5y2YDkixRUlKiYWSeZbfbmTJlCpIMQSm+m2sxxasYrCozZ85k//79PmvXU5o0sb5//346dep00ud06NDBL38ggqAXubm5AJrUWNWzIyfTA21QtClycnMwWE3H3TVpDHHtevPXlCrg2iE6YcIEJANYO2l7gx/U3rVq73//+x+ZmZnaBeJBoj8XmsPdTwWFRRz1uPtrf37vOdKff/7JzTffTGZmJmddOYJhd/0H2XDsYiZPMhhNXH7fePpfcgO7du3ipptvZu3atV5t061Pnz4AZO/M80l7vqCqKtk784iIjCA1NVXrcI7L/fdkDDl2Yt1w6DH3c/zdZ599Bni/tvo/GYLBFKuwdevWgFu4KvpzQRC0VFZWxpw5c6irq9M6FK/YsWMHGzZswJocgSnMB/VLcO1aV1WV6dOn+6Q9gPXr1wNgjAy8cRZjhIqiKH5R2k306YIgaMW9kNvdH7R0xcWuxfmGoMNZ5SRJQrYYKSzy/7KrbtOmTePAgQOYE3278F2SwNJOwel08vrrr/uuYQ9p0sS6JEmnvGCuq6s7YdoaQRCEpjryfaWlv8fY7Xby8/Ixhh7/5t79+MGDB30ZlkdNnDiR0tJSgto5fdq5H49shKBOTmw2G88//3xAZEwQ/bnQHHv37gUgOCL2qMeDwqORJIl9+/ZpEJXnKIrC+++/zz333ENNbR2X3Tee8268y+Pp309EkmUuHDGOoWOeoKKigjFjxvDZZ595fVHZwIEDAdi31X/7jn8qzi2joqSKswecjSx7N31rU7mzEphCj51Ydz8WCCn5/v77bzZu3IgxRsGgQZpZ92T+F1984fvGvUj054IgaOn111/nqaee4ocfftA6FK/4/vvvgcMp2n0htG0UhiATM2fOxGaz+aTNv//+GwBjeGBOrMPhc9Qz0acLgiDow4kWvxtDTOTl5gXEhr/8/HymTJmCbAZLqu/HuU0xKsZIhSVLlrBs2TKft98cTRpZ6tq1K/PmzTvhL4+iKMydO5cuXbo0KzhBEIR/OnIys6XXn8/OzkZRlBOumjeFmEHCb3dXL1++nF9++QVDuIq5tT4uVsyxKqY4hXXr1gXEwJHoz4Xm2LZtGwBhcYlHPW4wmgiOimPbtm1+uwCluLiYe++9lylTphAe24p/PfceaedcrEksvS+4gpueeQdrRAxvvvkmDzzwgFfreXXu3JmUlBT2bj6ArTYwanrvWOtaBDJkyBCNIzkxd19tPE6f7u7n/bU/P9JXX30FQFCyNu8NxjAwRKgsX768fnFQIBD9uSAIWnLXrt6zZ4/GkXhedXU1P//8M8ZQM9bWEad+gYdIBpmwTjGUlpaycOFCn7S5bds2JCPIwT5pzqcMYa7+cevWrRpHcmqiTxcEQdCH+sXvx0ysW6irq6vf0e7PJkyYQE1NDZZ2TmTvJmY8LkmC4I6ukm0vv/wy1dXVvg+iiZo0sX7zzTeTkZHBqFGjjhnYKysrY9SoUezatYtbb73VI0EKgiC42e32437eErkHLswRx9/KLRlkTGFBfjnAUV1dzYsvvogkgbWzU1c13oI7KkhGeOONN8jL8+9UyaI/F5pjzZo1SJJEeKuUY74X2botlZWV9ZPv/mTdunVcd931/Pnnn3ToN5CRr3xCq/baDlwlde7ByFc/JrXn6SxZsoTrb7jBaztuJEni6quvxmFzsGOd/0882m0OdqzdS2RkJIMGDdI6nBPavXs3AObIY0ezTYcecz/HXxUUFPD7778jh6gYfDc3cQxLkmtS310vNxCI/lwQBME75s6dS3V1NeGdYpFk396Uhnd27ZD3RTp4p9PJvsx9yMGqru69PUU2gWTCLzJqiT5dEARBH9wlN0zhRy9+d3/t7yU55s+fz4IFCzBGKpgTtNvQZggBS4pCTk4OkydP1iyOxmrSxPrYsWM5++yzmTp1Ku3atWPo0KGMGjWKoUOH0q5dO7744gvOPfdcxo4d6+l4BUFo4Y5MieWrlGh6tWPHDuD4g/Bu5sggSkpK6uvk+Iv33nuP3NxczCkKhhCtozmabIag9k6qqqr4v//7P63DaRbRnwtNVVhYyPr16wlPbIsp6Nj3oJjUrgAsWLDA16E1maqqfPbZZ4wePZqi4iIuuOV+rnl0AkGhYVqHBoA1PIrr/z2Jc28YQ25uLiNHjmTatGleST92zTXXEBQUxMZF23A6/DvN5NaVu6iprOWmm27CZDKd+gUaUFWVbdu2YbSaMViOXSZuDDZhCDaRnp6uQXSeM336dJxOJ5YkRdNBe1OsimyBn376iaqqKu0C8SDRnwuCIHjHjBkzQJII6xR76id7mCnMQnDrcNauXev1rDUFBQXYbXbkYH1kivMGOVglOztb96l7RZ8uCIKgD+7FWKbwoze0ub/25wxopaWlvPTyS0gyBHfW9v4cIChFwWBVmTZtGuvXr9c2mAZq0gZ/k8nEggULeOaZZ/joo4+YP39+/ffCw8N5/PHHefHFF3U7eCUIgv+qra2t//xUdacCnXuA3RJ74iKlltgQqvaXkp6eruudekfasWMHX375JXKwq2PVI3MrFVuuyu+//86KFSvqaxL7m5bUn//444+6W03aqVMnLr30Uq3DaJIff/wRRVFo1bXvcb8fk9oFoyWYH3/6ifvuu0/3v0M1NTU8/fTTzJ8/n7DoeK588EVad0rTOqxjSLLMgKtuI6lLT+ZMfp4JEyawefNmnn/+eSyW45cFaYro6GhuvPFGpk6dypYVO+l9flePHduX6mpsrF+4FWuIVde7eg4ePEh+fj4hqVEnfE5QXAg5+3PIy8sjISHBh9F5ht1u5/vvv0cygTle2wFtSQJzayfVe6uZPXs2N998s6bxeEJL6s8FQRB8ZefOnWzZsgVrmwiMVvOpX+AF4Z1iqTlYzo8//shDDz3ktXbcO6PlAO4mJJOKvdxOTU0NVuuJx1C0Jvp0QRAEfdi5cyey2YDBevT7rTnKtblk165dWoTlERMmTKCkuISgDk4MOigBI8kQ3MVJ5UaJ8ePHM2PGDIKDdRDYSTQ5c35QUBCTJk1i4sSJbN++nbKyMiIjI+nSpQsGg8GTMQqCINSrqak57uctjaIobNq0CWOoBUPQiW+oLLGu7d4bN270i4l1VVWZOHEiiqIQ0tGJ1KS8Kt7nTlFf8ZeR1157jZkzZ/rtjW1L6M83btzIs88+q3UYx9WlSxc6dOigdRiNUlVVxZdffonJEnzCiXWD0URSjzPI/GspM2fO5MYbb/RxlA1XWFjIuHHj2LJlCylp/Rg+7nms4See4NSDlG59GfnqJ8x6+1l+/vlnsrOzefvtt4mK8lzcY8aM4adZP7F23mY69WuLNUzfNzXHs3beZmoqannkkUeIiNAw9/gprFmzBoDghNATPicoIZSq/aWsWbOGK664wlehecy8efMoKirC0kZB0kHXYm6lUpfpqvl+4403Iss6veBohJbQnwuCoG+S1tudPOynn34CXJPbWrG2icRgMTJ79mzGjRvntffz+pqmAdxduK8/qqqqdD2xDqJPFwRB0JrNZmPfvn2YY4KPub4xR7p2rGdkZGgRWrMtWbKEX3/9FUO4iiVJP1lcjOFgSVbYv38///vf/3jkkUe0Dumkml2S3mAwkJamvx09ghAo9J6myteOnEyvv/lrgXbv3k1paSlhHWNO+ryguBAkWWLdunU+iqx5Fi1axNq1azHGKJii9f27bwgBc6LCvn37+P7777nlllu0DqlZArk//+KLLwBwpPRFDdNuYOxIculBDNnpfPnllzz//PNah9MoH3zwASUlJXQ4exhG84l3Saecdh7Zm1cxZcoUhg0bpsuJzcLCQu644w727dtHrwuuYMgdj2AwNvvy2CdCo2K58em3mPfha2xYMZ/Ro0fzySefEBkZ6ZHjR0RE8OADD/Liiy+ybMY6ht5+jl8NmOfuK2Tz8gxSU1N1vVsdYOnSpQBYk078N2JNiqBo7QGWLVvmdxPrqqoydepUkA7XN9eabAZTguumfenSpVxwwQVah+QxgdyfC4Kgb4E0duFwOPj5l58xBJmwJmt3DSsbZULaRVGwvYBVq1Z5LVNa/WRt4PwXHuvQufnTxLTo0wVBELSxa9cunE4nIdHHLsSSjQZM4UFs274NVVX9apyksrKSl156EUkGaxen5ing/ymorYK9UGbq1KkMHTpU132g/y/NFwShRTlyMr0lT6yvXr0agKBWJ6/9K5sMWGJD2LxlMxUVFb4IrcmcTieTJ08GCYLb62Pg/VSCUhUkI3zwwfst+vdRz3Jycli4cCFKSDRKq86ooTG6+HAm9UANCmPOnDmUlpZq/WNqsI0bNzL1iy+wRsbQpt85J32u2RpKu7OGUFJSwiuvvKK7wdaqqirGjBnDvn37OGv4rQwd/bjfTKq7GU1mLrtvPKcNu56MjAzuuusuj2Zzufbaazn99NPZ83cWO9d7t7anJ9ltDhZ9swqAl156SdcZRaqrq1mxYgWmiKBjarcdyRQRhCnMwrLly/yuFM7y5cvZsWMHpjgF2XMVC5rNkuy61vjoo4909/4kCIIgaOvPP/+kuKiY0HZRSBpnNQnr4FpMP3v2bK+1ERTkugZR/eM2vElUp+tf97kKgiAIwons2LEDAEv08TP3WaKDKS8rJy8vz5dhNduUKVPIzy/AkuLEoMPkLZIBrJ0dKIrCiy++iNPp1DqkE2rQ6OGFF16IJElMnTqV5ORkLrzwwgYdXJIkFi5c2KwABaGl86dVT74gJtZd/vjjDwCsrcNP+VxrUji1+ZWsXr2aiy66yNuhNdn8+fPZs2cP5kRFl5378cgmsCQ7KdlXynfffccdd9yhdUgn1RL78+nTp6MoCkpCJ3S1FFOScCZ0wpa5nh9//FH3vzsAxcXFPP74E6iKQreLb8BgPPVkZZs+AynYnc7cuXPp378/N9xwgw8iPTVVVXnhhRfYtWsXpw27nnNvvMtv+1tJkrhwxDjsdTX8vfhnXnvtNV544QWPHFuWZV588UWuve5als1YS6vUWMJjTpyuXC9W/PgXpfnl3HHHHfTp00frcE5q2bJl1NbWEtUl8aTPkySJkNQoSjfn8scffzB48GAfRdg8qqry/vvvAxCUoq/ReoMVTHEKmzdvZuXKlZx99tlah9RgLbE/FwRB8KWff/4Z4JQZ4nzBEhuCKSKIhQsXUlVVRUhIiMfbSEhIAED1r7V7jaLUSYSFhekuDbzo0wVBEPRn+/btAFiOs2MdwBxthX0l7Nixg1atWvkytCbbsWMH06ZNw2BVsbTR78JyYySYExS2bt3KjBkzdFtaskET60uWLEGSpPpJrCVLljTo4P46QCkIgn7V1tbWf95Sa6xXV1ezdu1azNHBGK3mUz4/OCkCNhxk2bJlup1YV1WVjz/+2JUmVmcD76diSVKpOwCfT/2cW2+9Vdc7I1taf26325nxww9gtKDEpGgdzjGU2FTI+pvvp09n5MiRuq7xW1dXx0MPPURubg4dBg4jsnVqg14nyTJpw25i7Tfv8Oqrr5KSksJZZ53l3WAbYOnSpcydO5fkrr254Jb7/PZ33E2SJC66/WHyM3cxc+ZMLr30Us4880yPHDs5OZlnxz/Lf/7zH+Z/uYKrxl6EwajfFJq7NmSyddVuunfvzrhx47QO55Tmzp0LQGi76FM+N7RdNKWbc5k7d67fTKwvXbqUzZs3Y4pTMHh+HqDZglIU7AUykydPZsCAAX7zXtDS+nNBEARfqqysZOHChZgiglwD5xqTJImw9tEUbzjIggULuPLKKz3eRnh4OFarldqaKo8fWw9UFdRaidYdW2sdyjFEny4IgqA/GRkZIIEp8vg71s2HdrJnZGRw/vnn+zK0JlFVlYkTJ6IoCiEdFST9Dj8CENRewV4k884773DJJZcQHn7qjYW+1qAfoaIoOJ1OOnfuXP91Qz70vFVfEAT/dOTEur+lQvWUlStXYrPZCGkT2aDnW2KsGK0mli5diqLoc9J6w4YNrjSxsQoGP8vMJhnB3EqhuKiY33//XetwTqql9eeLFy+mpLgYZ1w7kHU4EWg044xJ4UBWVn15Bz1yOBz8+9//ZsOGDbTq2pe2/Qc16vVBYZH0vHwEKhIPPfQQ6enp3gm0gVRV5Z133kGWDQwd/QSywb/Sv5+I0WRm6OgnAHjnnXc8euzLLruMK6+8krzMIlb9vNGjx/ak0oJylny/BqvVyv/93//peqETQFlZGcuXL8ccHYz5BDfsRzJHuZ63ePFiKisrfRBh8zidTtfvouQqnaJHhlDXrvX09HQWLVqkdTgN1tL6c0EQBF+aP38+NpuNsA4xupm8DG3v2jk/Z84crxxfkiS6du2KUi0FZDp4pcaVCr5Lly5ah3IM0acLgiDoi6qqZOzMwBQehGw8/vSpJcq18C4jI8OXoTXZ8uXLWbt2LcYYBVOUfneru8lmsKQ4KSsr49NPP9U6nOPS+doEQRCEo9lstuN+3pIsXrwYAGsDJ9YlScKaHElxcTF///23FyNruunTpwNgSfLPu3jzobhnzJihcSTCkX744QcAnHHtNY7kxJT4DgDMnDlT40iOz+l08vTTT7Nw4UKiUzrSbch1TRpgjGydStolN1NdXc3dd99dX69KCzt27CAjI4POZ5xPTGv9ZTJojoTUTnToN5BNmzaRmenZmuhPPfUUHTp0YNPSHezZnOXRY3uCw+7k96krsNXaeeGFF2jbtq3WIZ3S/PnzsdvthLVvWJpZSZIIbR+NzWbzi9Sfc+bMISMjA3OCvku8BLVTQIL//ve/2O12rcMRBEEQNOauZe6uba4HpjALQQmhrFmzhtzcXK+0kZaWBio4K7xyeE05y133L2lpaRpHIgiCIOhdYWEh5WXlJ138brCakM0Gdu3a5cPImkZVVSZPngwSBLfzn3F3S5KKHARffvklhYWFWodzDDGxLgg6p5cV0nrhcDiO+3lL4XA4WLJkCcYQM5aYho9Sh6REAuhyN1ZNTQ0LFy5ADlYx6C+zS4MYgsAYqbBu3TqvDXQIjXPw4EFWrlyJEhYHwfr9xVJDolGtESxYuJCSkhKtwzmKw+Hgqaee4tdffyWidSq9rhjZrN3d8R170O3iGygrL+fOO+/UbHL9jz/+AKDLWRdo0r63dT1zEHD4PD3FarXy5ptvEhQUxOJvVlNerK8d0ytmracwu4Qbb7yRYcOGaR1Og/z6669Aw9LAu7mf+8svv3glJk+prq7mnXcmIxn0u1vdzRAM5tYKmZmZ9Qv9BEEQhJbpwIEDrFu3juDEMIwhpy675kthHWNRVbV+4t/T3GWE7CWBN1RsL3GNq3mqVJIgCIIQuHbv3g2AOfLEKVUlScIUEcS+fft0vzh70aJFriyx8fosz3YikuzatW6z2XS5a71Bo6PLli1rcgPnnXdek18rCIJrVZFw2JGpzFti6qv169dTVlZGRLf4Ri26CE4MQzYbWLBgAQ8//LCuFmysWLGCmppaLG1VdBRWo5niVRylrh2II0aM0Dqc42pJ/flPP/2EqqooOt6tDoAk4Yxrj5S5gV9++YVbb71V64gAV336f//738yfP5/IpHb0vvIODKbmDy4mdusHqsrW+dO5Y9QoPvzgA3r06OGBiBtu7969ACSkdvZpu74Sf+i83OfpSe3bt2f8+PE8/fTTzP9iBVeNG4LBoP3g6+6N+0lfsZOuXbvy+OOPax1Og+Tm5rJu3TqCWjVu4N4UZiEoPpTVq1dTUFBAXFycF6NsuqlTp5KfX4ClrYJs0TqaUwtqq2DPk5kyZQqXXXYZERERWod0Ui2pPxcEQfClH3/8EXBNYutNaGoURauz+PGnnxg9ejSy7NlrsP79+2M0GnEU2SHVo4fWlKqCs0QmPj6e9u31d28o+nRBEAR9OTyxfvJybebIYCoKqsjKytJl/wKueaUPP/zQVZ4tRd8L3o/HnKBStx++//57xowZQ1RUlNYh1WvQxPqgQYOaPAnTEie+BEHwHr3WCPeVBQsWAId3oDeUZJCxJkWQtTeLjIwMXdUWc++qNMX49/+tKUalBtf56HVivaX054qi8NNPP4HBhBLdRutwTkmJSYX9m/jxxx+55ZZbNF/4UldXxyOPPMKyZcuIatOR3sNHemRS3S2x+2lIsoGtv33HnXeO5n//e49+/fp57PinUl5eDkBwWKTP2vQla3gkcPg8PW348OGsXr2a2bNns3beZs66rLdX2mmoiuIqlny/huDgYCZNmoTF4gezuLh2nKuq2qQ0s6HtoynMr+TXX39l5MiRXoiueXJzc/nkk0+QLRDUxj/6dtkElrZOyneX8/777/Pvf/9b65BOqqX054IgCL7kcDj46aefkM1GQtrqZ+DWTTYZCGkXxYGdWaxZs4azzjrLo8cPCQlhwIABLF++HGeNK6NLIHCUSih2GDx4sOb3Wccj+nRBEAR9cad3b8jEuvv5ep1YX7NmDVu3bsUUp+/ybCciyWBJdlKzq45vv/2We++9V+uQ6jVoYv3ZZ5/V5cWHIAgtz5E7+Fvabn5FUVi4cCGGICNBCWGNfn1I20gq9xazcOFCXU2sr1ixAskEhlCtI2ke2QyGUJW169Zis9kwm/WVOhBaTn++atUqcnJycMZ3gGakLvcZkwVnVBIZGRls3bpV09p/NTU1PPDAA6xatYqYdl3pedmtGIwmj7fTqmsfZKOR9F+ncffdd/Puu+/6LDWj0ej6nVAc+k7X1VTOQ2nI3OfpDU8//TQbN25k/cJ02nZLJLF9vNfaOhlVUVk4bSV1NTZefvllv6irDq7rl1mzZiEZZELaRjb69aHtoilak8VPP/3Ebbfdprv39TfffJO6ujqsXZ1IBq2jaThLaxVbDkybNo3rrruODh06aB3SCbWU/lzQxl9//UVYWBidOwdmZhdBOJElS5aQn59PRLd4ZKP2GXmOJ7xzLBU7C/n+++89PrEOcPHFF7N8+XLsBRKGlMAYb7Hnu/rLiy++WONIjk/06YIgCPqSkZGBJEuYIk6+aN8cFVz/fL32MVOnTgXA4icL3o/H3EqlLtN1nz5q1CjdbKZo0Ijb888/7+UwBEEQGsY9mS7LUotbnbtlyxby8/MJ6xSLJDf+xsuaFIFkkJk/fz733XefFyJsvNzcXHJzczHFKn6dBt7NGKFSl21n69at9OnTR+twjtFS+vOffvoJQP9p4I+gxLXHUJzFzJkzNZtYr66uZuzYsaxdu5a4jj3occnNzaqpfirxHXsgX3Ebm3/+kvvuu593332HAQMGeK09t+TkZABKcg8QHKbvdM9NUZKbBRw+T2+wWq1MmDCBEbeNYOG0Vdzw2CWYgzy/AONUNi3bzsHd+QwZMoThw4f7vP2mWrduHXv37iW0QwwGc+P/xgwW1066Xbt2sWnTJl31N3/99Rdz587FEK5iivevAXlJhuD2Tqq2wMSJE/nggw90O9DdUvpzwfdKS0u5/fbbAdi8ebO2wQiCj02bNg2A8K7aLBhsiKC4UCwxVhYuXEhOTg6JiYkePf6QIUN45ZVXsOXWYGnj9Pt7dNUJ9gKZpKQkn2bIagzRpwuCIOiH0+lk586dmCKDkE5RcsUS7ZpY37Fjhy9Ca7TMzEyWL1+OIULF2Pj9ebohGcCUqFC6v5S5c+dy1VVXaR0SAPpcgikIgnACDocDAIPR0OIm1uvTwDdhdxu4UsdZk8LZtWsXmZmZHoys6TZu3AiAIdy/Bt9PxBDhOg/3eQm+V1ZWxoIFC1CDI1BDorUOp8HUiARUi5VffvmFmpoan7dfV1fHgw8+yNq1a4nv1Isel/7Lq5PqbrHtutJr+EgcisK4ceNYu3at19t013Tfu3mN19vSwt6/Xefl7dr1vXr1YszoMZQXVbL6l01ebet4SgvKWfPr38TExPjdTh/3wH1El6bXRw8/9NqvvvrKIzF5gtPpZOLEiQAEd/TPwXhTjIoxWmHlypUsWbJE63AEwefq6uq0DkEQNLF9+3bWrl1LcFI45oggrcM5qYjuCSiKUn894UkhISEMGzYMpUbCUeqHHfk/2PIlVCdcddVVHq9JLwiCIASe3bt3U1NTgyU25JTPNQSZMIaa2bx5sy6z6n7//fcAWJL8d7e6m6W1AhJ8++23WodSr9lXFStWrGDKlClMmDCBKVOmsGLFCk/EJQiCcFx1dXUggSnI1KIGflRVZcGCBchmA9bE8CYfx10rbv78+Z4KrVm2bt0KgDFAJtaNYa7zSE9P1ziSxguU/vznn3/GbrfjjG+PX83qSDJKbDuqqqp8/vfpcDh44oknWLVqFXEde5B2yU3Isu/yN8e07UyvK27D7nBy/9ix9e8L3nLuuecSFBTE5iW/4rDbvNqWr9nrakn/4zfCwsK8kh70n+6++246duzI5j8yyNmT7/X23FRFZcl3a3DYnYwfP57IyEiftd1cu3fvZsGCBVjiQrDEnfpm/USCElw71n7//Xf27dvnuQCb4eeff2bbtm2YExS/XhEf3MF10/7GG29gt/tfyYhA6c8FbRgMflS/QRA86PPPPwcgMi1B20AaIDQ1CqPVzPTp06moqPD48W+66SYA6g740b3Ucagq2A7ImExGrrvuOq3DaRLRpwuCIPiWe6NUUAMm1sGVSaaoqIgDBw54MarGs9lszJ49G9nsWjzu72QLmKIV0tPTdZMhoMkT63/++SddunThvPPO44EHHuDpp5/mgQce4LzzzqNr166sXLnSk3EKgiAArlTFZosJk8VIVVWV1uH4zI4dO8jKysKa7Ern3lTW5AgkWWLhwoUejK7p3BNo/l5f3U2ygGTyr4n1QOrPVVXlhx9+cE1Sx6RqHU6jOQ+lrv/hhx982u7EiRNZtGgR0SmdXOnffTip7hbTtjM9LvkXtTU13HvffeTk5HitLavVyk033UR5YS7rf5/ptXa0sPaXb6kqLWbEiBGYzWavt2cymXjppZeQJIkl09fidPgmk8z2tXvqU8APHjzYJ216yv/+9z8AonolNmuXvSRJRPZKRFVV3n//fU+F12R1dXVMfmcykgGC2vn3iniDFcyJCpmZmfz4449ah9NggdSfC9oxmXxf1kMQtJadnc3cuXMxR1sJbsYidl+RDDIR3eOpqqqq343mSWlpafTr1w9HsYzTj4dcHMUSzmqJSy65lNjYWK3DaRTRpwuCIGhj9erVAA2+HghqFXbU6/Ri+fLllJaWYkpQkAIkYYs50bVAYNasWRpH4tKkH2t6ejoXX3wxO3fu5KKLLuKVV17hs88+49VXX2XIkCFkZGQwdOhQr+84EoSWwJ9Sm/pCaWkpFquZIKuZsrIyXaZa8Ybff/8dgNBDO86bymAxEpwYxpYtW8jOzvZEaE2mqipbt25FtqpIAbI5RpLAEKqQlZXlld0DnhZo/fnmzZvZuXMnzqhkMFm0DqfxLCEoEa1Yv349e/bs8UmT33//Pd9++y2hca3pefkIn6R/P5G4jml0HnQlxUVFjB03zqsp8e+8806io6NZ/t2H5O3b6bV2fOngznT+/PFzEhISGDFihM/a7dGjBzfddBMluWVsXLLd6+3VVNaycvZGQkJC+M9//uP19jxp48aN/PbbbwTFh2BNjmj28UJSIrHEukpIbNmyxQMRNt3MmTPJz8vH3FpB9sO3338KaqsgGeDDDz/0i13rgdafC9rxxaIsQdCbzz77DEVRiOzRym/GX8I7x2EwG/niiy+ora31+PHvvPNOAGr3++dovKpCbaaMJEmMGjVK63AaRfTpgiBoxV1+taVyOBysWrUKU6gFU1jDbmqtia6J9T///NOboTXab7/9BoA53r8XvR/JGKUimVxzJIqi/Xk16QrpxRdfxGaz8euvv/Lbb7/xn//8h5EjR/Lvf/+befPm8euvv1JbW8uLL77o6XgFocVpKRPHDaEoCgUFBVjDgrCGB2Oz2SgvL9c6LK9TVZXff/8d2SgTnOSBgfhUV91prdPBZ2dnU1FRgSE0sH7H3bvvt2/3/gRTcwVaf+7e6a3Et9c4kqZzxncA8MkuyfT0dCZMmIDZGkrv4SMxmrWfDUvuPYDk3gPI2LGDl19+2WvtREZGMmHCBJwOOzMn/YfywjyvteULxTlZ/Pjmk6CqTJw4kdBQ36YBGTduHDExMayfn05FiXe3Nq3+ZRO11XWMGzeO+Ph4r7blSU6nk9deew2AmNPbeGTgXpIkYk5vA8CECRM0u7l0OBx88sknSAawtNH+BtcTZLNr13peXh5z5szROpxTCrT+XNCO2LEutDT5+fnMnDkTU7iF0NTmLWL3JdlsILxbHMXFxV7JdnXuueeSlpaGPd8/d607iiWcFRIXX3wxHTp00DqcRhF9uiAIWvHGQi1/sn79esrLy7G2afjYuyk8CFNEEH/88YduStba7XaWLF2CHKwiN736nO5IMphiXPfoesgU26SJ9SVLlnDdddcxbNiw435/2LBhXHfddSxevLhZwQmCIBypqKgIu91OWHQIYVGunuHgwYMaR+V9GRkZZGZmYm0TiWxs/orxkJRIJFli3rx5Hoiu6Q6ngQ+wifVDddb9YQV5IPXn1dXV/Dp3LqolFDVc/7URT0SNbA0mC7NmzfLqLsnq6moef/xxHE4naZfcTFBYpNfaaqxO511OeKs2zJ49m19++cVr7Zx99tk88cQTVBTn892rD1Fe5J+T6yV52Xw/4RGqykp45plnOO2003weQ1hYGI8++ih2m4M/Z2/wWjv5WcVsXb2bzp07c+ONN3qtHW/45ptvSE9PJ6xjDEFxnlv4EJwQRmj7aP7++2+mT5/useM2xvLly8nLy8PUSkEOoDk5SxtXrXVvpNn1NG/252vXrmXs2LGkpaUREhJCSkoKN9xwAxkZGc0NW9AhUWNdaGmmTp2K3W4nsmcikuwfu9XdIrolIBsNfPbZZx6/b5AkibFjxwJQs9e/dq2rKtTulZFlmXvvvVfrcBotkO7RBUHwL3qZGNaKe5d3SEpko14X0jaSmpoa/vjjDy9E1Xjp6enUVNdgjFbxk0Q8DWaMdo256yH1fpOujsrKymjXrt1Jn9OuXTvKysqaFJQgCMLxZGZmAhARG0ZEbOhRjwWyX3/9FcBjK+gNFiPBrcNJT09n//79HjlmU7gnno1hATaxHuo/E+uB1J//9ttv1NbU4Ixrh19fOcoGnDGplJSUsGzZMq81M2nSJLKyskjtP4joNh291k5TyAYjPS75F0azhVdeeYW8PO9NeI8YMYJ7772XktwDTHv+PooOavee2BQF+3cz7YX7KC/M5ZFHHuH666/XLJbLL7+cPn36sHvjfrJ3ef7/TFVV/pi5DlR46qmnMBq1K1vQWAcOHGDy5MkYgozE9G/j8ePHnN4Gg8XIm2++SU5OjsePfyo//fQTAJbEwNit7iabXSvi09PT2blT3yUjvNmfT5w4kR9++IHBgwfz9ttvc9ddd7Fs2TL69euneQkCwfP8JQ22IHhCaWkp33//PcYQM2EdorUOp9EMQUbCusR6LbvKwIEDOeOMM3AUyThKPX54r7HlSTirJK6++mq/260OgXWPLgiCf/GHEljeYrfbmffbPIxWM0EJYY16bVi7GADdZDpbt24dAMbIwBpvh8Pn5D5HLTVpYr1169asWrXqpM9ZvXo1rVu3blJQgiAIx+OuORwZH05UQsRRjwUqRVH45ddfkc1Gj9RjdQtt5xo48OZu0FNxp20x+DZjsdfJQSAZ/WNiPZD6c/fEjhJ78kEIf+CMc6Wyd5+Tp61cuZLp06cTGteadmdd5JU2mis4IppO519BRUUFL7zwglfLotx33308+uijlBflM+35e8navslrbXnSvs3rmPbC/VQf2ql+xx13aBqPJEk8+eSTSJLEip/Wezwt+a4NmeTuK2TYsGGa7MpvKkVRePbZZ6mpqSHmjDYYgjy/IMAYbCLm9GSqq6t57rnnfFpGyG63s3LVSmSriiGA0sy5meJcP8sVK1ZoHMnJebM/f+SRR8jMzGTy5MmMHj2aZ555huXLl+NwOOrLGwiCIID/Lcz45ptvqK2tddVWl/1rV7ZbZFoCkizV14n3JEmSePTRR5EkiZrdBvyhSqHqhLq9BoKCgrjvvvu0DqdJAukeXRAE/+Kusd4Sy9IuXLiQ8rJyQttHNzqDjTkqGHO0laVLl1JUVOSlCBvOnVnMGGAZYgFkE8hBqi4WvjfpynH48OEsWbKE8ePHH1N7oba2lueee47Fixdz5ZVXeiRIQRAEONwxxCRGEt0q4qjHAtXatWvJy80lpG0kksFzN/shKZHIRgNz5szR5IJJVVW2btuKHKwi+c+mwwaRJDCEKezbt4/KykqtwzmpQOnPs7OzWb9+PUp4AlisWofTfNYIlJBoli9fTklJiUcPXV1dzfPPP48ky3Qfcj2yQb9/gInd+xPdtjPLly/n559/9mpbt99+O6+++ir22mq+f/Vhtv65wKvtNdfmJb8w4/8eQ3Xaef3113WTFr179+5cddVVFGaXsH3NXo8d12FzsHLORswWMw8//LDHjusL33zzDWvXriWkbVT9ojZvCO0Qg7VNJCtXrvRp6vKMjAxXmrkAXA0Ph1fE//XXXxpHcnLe7M/PPvtszGbzUY916tSJtLQ0tm3b1qy4BUEILP40EF9XV8c333yDwWIkrFOM1uE0mdFqJrRDDPv27fNKtqvu3btz5ZVX4qyUsOXqf+FE7X4ZxQajR48mPj5e63CaJFDu0QVB8D/+1I972g8//ABAWOfYJr0+vHMsDoeDWbNmeTKsJtmzZw+SASSL1pF4h2xVyc/Pp6KiQts4mvKi8ePH065dO1599VVSUlK4/PLLufPOO7n88stp27YtL730Eu3atWP8+PGejlcQWpyW3Kn9U0ZGBrIsE5UQTnBYEMGhQbpYoeRN7g45vFPTOvYTkU0GQlIjycrKYv369R49dkPk5eVRXlYecPXV3dy79nbt2qVtIKcQKP353LlzAVBiU7UNxIOU2LY4nU7mz5/v0eO+8847HDx4kLb9BxEWr+9dDpIk0XXwNRjNFl6bOJHCwkKvtnfFFVfwwQcfYA0O4ud3X+DPmZ/rrg9WFYVl337A3A9fIzw8nI8//pihQ4dqHdZRxo0bR3BwMGvm/o29zjOp5DYt3UFlaTUjbxvpV7tz9uzZw5tvvokh2ETsWSle3cknSRJxA9piCDIyadIkn5V6OXjwIAAGq77+VjxFNruy0LjPU6983Z+rqkpeXh6xsSe+Pq2rq6O8vPyoD0EQBL2YO3cuJSUlhHWJQzYatA6nWSK7JwDw9ddfe+X4DzzwAMHBwdTtM6A6vNKERzhrwHZAplWrVowcOVLrcJosUO7RBUHwP/6WecZT9u3bx6pVqwhqFYY5PKhJxwhrH4NsNDB9+nScTqeHI2yc/Px8JEvg1Vd3kw8tGCgoKNA2jqa8KCYmhlWrVjFy5EgqKyv59ddf+eyzz/j111+pqKjgjjvuYNWqVURH+1+NIkHQm5baqf2Tqqpk7MwgMiEMg9GAJEnEtHZNDFdXV2sdnleUlZXx22+/YYoIwhLn+fyqYYcm692r8nzJvSDCEBKYA/HuBQN6z6gQKP3577//DpKMEpWkdSgeo0SnAIfOzUPWr1/P119/TUh0PO3OGOyx43pTcHgUHQZeQnlZGa+++qrXJ7rPOOMMvvrqK5KTk/ljxifM+/A1nA59jCA67DbmTHmRVbO/IjU1lWlff03fvn21DusYcXFxjBo1iuryGjYsav5u1uqKGtYv3EpMTAx33nmnByL0DYfDwdNPP43NZiNuQArGYJPX2zRaTcSe1Zba2lqefvppn9zQu7NqSN4/Pc1IJpXi4mKtwzgpX/fnX3/9NdnZ2SfNljFhwgQiIiLqP9q0aeORtgVB0B/39ZneFiSezA8//AASRHSJ0zqUZjNHBROcGMaqVavIysry+PHj4uK4++67UWxQm6nflPm1e2RUBR577DGCgpo2MaIHgXKPLgiC/zEa9ZvR0Ju+++47ACK6Nj3TiWw2ENo+mgMHDmhaRkxRFEpLS5FN/nNN1ljSoWRqWt+jN/mKKDY2lk8//ZSysjI2bdrE8uXL2bRpE2VlZXzyyScnXb0uCELDiYl1l4MHD1JVWUVMYmT9YzGtI1FVVfe7gptq1qxZ2Gw2wjvHeeX3ICg+FHNkEPPmzfN4uulTOXDgAAByAGTtPh452HUB4z5PPfP3/jw3N5dt27a50sAbzad+gb8wB6OExrB27TqP7PKrrq7mmWeeAaDbxTcg+9ENU1KvM4lK7sD8+fPrsxN4U/v27fn6669gnzHXAAEAAElEQVTp2bMnm5f+ysxJ/8ZWq+0CrtqqCqa/9ijbVy7ktNNO46uvvtL1JNXIkSOJj49n45LtVJXVNOtY637bgr3Ozv33309IiP8U8f7888/ZsmULYR1iCEmJ8lm7oalRhLaPZuPGjXz11Vfeby80FHDVNA1YTomwsDCtozglX/Xn27dv5/7772fAgAEn3RH45JNPUlZWVv/hjckeQRD0wT2hXlVVpXEkDbNv3z42btyINSkCY0hg3D+4F83Pnj3bK8cfMWIESUlJ2A7KOJt3aecVjjKwF8r07duXiy++WOtwms3f79EFQfBP7on1ljQXUV1dzU8//YTRaiYkJbJZxwrv5lqs980333ggsqZxOByu6zL/TsZzUpLsuu602WyaxtHspYYmk4mePXsycOBAevbsickUwNsVBEED/rTq25vck+fRR06sH/o8ECfWnU4n33zzDbJRJqyjd2q+SZJEeJd47HY7M2bM8EobJ5KTkwOAbAnM3293Wprc3FxtA2kEf+3PV65cCYAS6T/poRtKiWyNojhZu3Zts4/15ptvkpWVRdv+g4hopd8J2eORJJluQ67DaLbw8iuvkJ+f7/U2o6Oj+eSTT7jgggvY+/cavn35IarLfbsAya2ypJBvXhpH1raNDBs2jA8//JCIiAhNYmmo4OBgxo4di8PmYO1vm5t8nNKCctJX7qJdu3ZcffXVHozQu/bs2cN7772H0Wom5gzf/73FnuHaIT958mSvp4SPi3MNHig1gTn4ojpAsR0+T3/gzf48NzeXyy67jIiICGbMmIHBcOIRG4vFQnh4+FEfgv4piqJ1CIIfchzK7uMv9z7ujFCh7QNnx29ISiSyUfZotqsjmc1mHnroIVQFavfpa9e6qkLNbld/9MQTTwTUhJC/3qMLguCfWuKO9Z9//pnKykrCu8Qiyc3rPyxRVoISQlmxYoVmi4rd12QETld4rEPn5tA4u2Szrobq6ur48ccfefbZZ3nwwQd59tln+fHHH6mrq/NUfIIgCADs3r0bgJhWhycTohLCj/peIFmyZAkHDhwgtEMMBov3LmzCOsYgmw1MmzYNu90ztXAbwp2+XwrQazb3eflLmQJ/7s/XrVsHgBLe9JRNeqWGu+olus+xqf7880++++47QmMTaXfmRZ4IzeeCI6LpeN7lVJSX89xzz/lk0VlwcDBvvvkm1113Hbl7tjHthbGUF+V5vd0jleRl8/UL91Gwfze33norEydOxGz2j51Vw4cPp3379mxbvZvSgqZlXVgz929UReXBBx/0m5t8RVF48cUXsdvtxA5I8WoffiKGICMxZ6Vgs9l48cUXvfr30qNHD0xmE46SwLxzd5S6zqt///4aR9Iw3uzPy8rKuOSSSygtLWXevHm0bh14C9oEfHo/4G+0HrzTq9raWkpLS4HDi6f1bvHixUiyREhypNaheIxsNBCcFMGePXu8tqhu6NChpKWlYc+XceooOYGjWMJZITF06FB69OihdTge48/36IIg+KeTLZoNRKqq8v333yPJEmGdPLOQOrxLPKqqMn36dI8cr7HqF2AF8lrZQ+em9dhYk0d6Zs+ezV133UVBQcFRgzWSJBEfH8+HH37IFVdc4ZEgBUEQ9u7dC0BkwuHdLlEJrkn2PXv2aBKTt6iqyqeffgpARPcEr7YlmwyEd46lcEsev/zyC1dddZVX23ML+BV0h87LHwYn/b0/37IlHQwmCA68nXBqSBRIMunp6U0+RlVVFc899zyybKD7UP9KAf9PrdNOp2DXFv744w9mzZrlk/cro9HIs88+S1RUFB999BHTXrifG578L9GJ3t+FXJC1h+8nPExVaTHjxo1jzJgxfrUDx2Aw8MADD/DQQw+xdt5mhowY2KjXF2aXsGvDfnr16sWFF17opSg9b9asWfz111+EtI0ipE2kZnGEpERibRPJ6tWr+eWXX7j88su90k5wcDBnnnEmf/zxB85KMIR6pRnN2PJcf3PnnXeexpGcmjf789raWq644goyMjJYsGAB3bt391TYgs5onVJRz2pqdJj/WgeOnEzPy8vD6XTqemC+oqKCrVu3YokLQTbrN86msCaFU5VZwurVq0lJSfH48SVJYuzYsdx7773UZsqEdNd+1F5VXTvoZVnmvvvu0zocj/H3e3RBEPyTP403eMLmzZvZsWMHIalRGK2eyQgS2jaSoiATP/74I2PHjvX55K/JZMJoNKI69T8e3VSq0/V7GhwcrGkcTdqxvnDhQq699lpKS0sZNWoUn3/+OXPnzuXzzz/njjvuoKSkhGuuuYZFixZ5Ol5BaHFEKniXzMxMZINMePThEVtzkAlreDCZmZkaRuZ5f/31F3///TchbSMxRwR5vb2I7glIssSnn37qs/SP7lqlaoBu/HCfl95rsvp7f+5wOMjM3IdijYBAvAGQDShBYexqRlaO9957j9zcHNqecQFhcf69u1CSJLoOvgaj2cLrkybV747yRbsPPPAAjz76KOWFeXzz4lgK9ns3U0rOnu18+9I4qstKeOaZZ7jrrrv88ib3wgsvJC0tjZ0bMinOK2vUa90p5MeNG+c3515eXs5///tfZJOB2DO1LbkgSRKxZ7ZBNspMmjSJyspKr7V1yy23AFCbpa/UsM3lrHLVa+3du7fuJ5K92Z87nU5uvPFGVq5cyfTp0xkwYIAXzkDQC39YFKoVf6kf7msFBQX1nzudTkpKtCmd01B///03iqIQ3Erf92lNEXTonDZs2OC1NgYOHEiPHj2wF+qj1rqjVMJZKXHxxRfTvn17rcPxCH+/RxcEwX/5y323p8ycOROA8M6eK/slGVwlXUtLS1m8eLHHjtsYkZGRqAF8Se8+t+hobUv6NGn047nnniM4OJh169bx0UcfcdtttzF06FBuu+02Pv74Y9asWUNQUBDPPfdco4+9du1axo4dS1paGiEhIaSkpHDDDTeQkZHRlFAFwe+1tE7tRPbt20d4TCiy4ei3rcj4MLKzswNqEOijjz4CILJnok/aM1rNhHaMYe/evSxYsMAnbdbXZK0LzN9v5VB2Nr3XZPVmf+4LBQUFOJ1OVHOI1qF4jyWEivLyJg3mZmVl8fXXX2ONjCW1/wVeCM73gsIiaTfgYsrLyvjggw982vbtt9/Os88+S3V5Cd++/AC5e3d4pZ3sjM18/8pD2GqqeOWVV7jxxhu90o4vSJLk2j2kwvoFDc+8UJRTyt7NB+jbty9nnnmmFyP0rPfee4+SkhKieiditGqfst8UaiGyVyJFRUW8//77Xmtn4MCBdOvWDXu+jKNx6yd0S1WhZpfrmtMfFrZ4sz9/9NFHmT17NpdccgnFxcV89dVXR30IgSWQ7qk8zZsLlPyZe6Gjuy6p3ifWt23bBoAlNvDuH0xhFmSzof4cvUGSJG6//XZQoS5b+wV1dQdcv3e33367toF4kLf6dDHeLgjCqbSkzX3V1dX8OvdXTKEWghM9u9gurFMsAD/88INHj9tQcXFxqDaJQP3vVA4l2IqNjdU0jiZdBW3YsIEbb7zxhLVrevXqxQ033MD69esbfeyJEyfyww8/MHjwYN5++23uuusuli1bRr9+/diyZUtTwhUEwc+VlZVRUlJCZNyxHV1kXDiKopCVlaVBZJ63efNm/vzzT4JbhxPkw5v9qJ6JIEl88OGHPrmQ6tSpEwDOcn0PVDeVs8J1Xp07d9Y4kpPzZn/uC+Xlh+o2myzaBuJFqtE1OVdW1vjZqk8//RSn00mHgcP8OgX8PyX3GkBwRDTffz+d4uJin7Z9/fXX88orr1BXXcl3rzxEzp7tHj3+ge1/8/2ER3Ha63j99dcDIsXjueeeS+fOndm1fj8VJQ1bILJxkWtA2J/S3+/YsYNvvvkGU0QQEd3itQ6nXmRaAqbwIL766iuvlc6RJInx48cjyxI1GQZUp1ea8SlbroSjVOaCCy7g3HPP1TqcU/Jmf75x40YA5syZw4gRI475EAJLSxpQbaz6607Ez+lItbW1AASHuK7H9V7/2V1izhypbfpQb5AkCVNEEJmZmV7NRDd48GASEuKx58ma9vlKLTiKZfr06UNaWpp2gXiYt/p0Md4uCMKp+CqLqR7MmzePmuoawjrFeHzMwRwRRFBCKKtWrSI7O9ujx26INm3aoDpBDdAKT0q1RExMjH+mgrdarafchRcfH4/Vam30sR955BEyMzOZPHkyo0eP5plnnmH58uU4HA5ee+21poQrCIKf27dvHwCR8cfWUHZPtruf4+/cOzCjevtmt7qbKcxCaPtoMnbsYOnSpV5vr3fv3siyjKPMPyZMGstR6jqvfv36aRzJyXmzP/cFh+NQzn1J+90SXnPo3JzOxo1a1dbW8uvcuQSHRxPXMXAGmgBkg4E2fc7BZqtj3rx5Pm//iiuuYOLEiTjqapg+4WHy9nlml8fBnenM+L/HUJ12/vvf/zJkyBCPHFdr7p1NiqKwZcXOUz6/uqKWnRsy6dixI+ecc44PImw+VVWZMGECiqIQe2YbJIN+3pMkg0zMGW1wOp288sorXpsM6tmzJ7fdNhJntUR1huzXq+MdFVC7y0BYWBhPPfWUXyzu8GZ/vmTJElRVPeGHILQUR5agqa6u1i4QvTr0Xqn394Xs7GyQJIwh2meW8QZTmAW73U5+fr7X2jAajVx99TWoDrAXaNdH2nJdbV9//fWaxeAN3urTxXi7IAinUj/G1gJMnz4dJAjr6J1dz+Gd41BVVZNd6x07dgTAWaX/+9jGUp2g1Er156ilJo36XHTRRadMF7xgwYImDQieffbZmM1HX+B26tSJtLQ0r6YzEgRBv9w7rKISjjOxfmiy3Vu7sHxp27ZtLF26lKCEUIITfF/zLapnKwDe/+B9rw+IWK1W+vfvj7NcQqn1alM+pzpdK+fbtEkmJSVF63BOypv9uS8EBQW5PnEG8MW/4ppQrz/XBtqyZQvVVVXEdeqBFIALD+I79wRg1apVmrQ/bNgwJkyYgK2mmhkTH6Mkr3mrkAsP7GXG/z2O027njTfeYNCgQZ4JVCeGDh1KVFQU21fvwek4+SKR7Wv2oDgVbr75Zr+Y0AT4+eef+euvvwhpG4W1dYTW4RwjJDkCa5tI1qxZ49XFKA888AD9+vXDni9Tl+Uf/3f/pNRB9VYjqBKvv/46rVq10jqkBvH3/lzQD2MAZbjxtCNTnOs93bkvucfv6qrrjvpar4qKijAEG+tT1wcaQ7AJcJ2nN1111VUA2PK0+TmqKtjyDFit1oDr27zVp4vxdkEQTsWdhUbvi+SaKz09nS1bthDSJtJrC+1C2kZhsBj5YeYPPi+15M7i4iw/xRP9kONQ5tvu3btrHEkTJ9YnTZpEfn4+t9122zHpl7OyshgxYgSFhYVMmjTJI0GqqkpeXt5J8+bX1dVRXl5+1IcgBIJA78waYudO1w636FbHDlbHJEYe9Rx/5q6tHtW7tSbtmyODCUmNIn1LOitXrvR6e5dffjkAtvzAGtSwF0qoTrj88it0Pynkzf7cFzXc3NcFkq3GY8fUG8lWjSwbiIqKatTr3Gkuw+K0eT/xNktIOGZrqKbZSi655BLGjx9PVVkJMyY+Rm1VRZOOU11ewoz/e5zaqgpefvklLrzwQg9Hqj2z2czw4cOpqawla3vOCZ+nqio71u7FYrFw2WWX+TDCpisrK+P1SZOQjQZiz2ijdTgnFHtGG2SjzMSJE6moaNrv6qmYTCbeeOMNEhMTqd1roC5H333gPyl2qNpsQKmFhx56iIEDB2odUoP5+v5cCFxiYv3ExMT68YWGhgLgdChHfa1XpaWlGMyB+3tusLjO7cgMC96QlJREv379cJTKKBpk/3eWu1LBDxkyRPNUsJ7myz5djLcLgnCklpKR5+uvvwYgvOvJs4M0h2yUCesUQ3FRsc8zLfbt2xdZlrGXBt4mG3eG2P79+2scCTToavJ4A3xRUVF8/fXXfPvtt6SkpJCQkEBeXh779+/H6XTSq1cvRowYwcKFC5sd5Ndff012djYvvvjiCZ8zYcIEXnjhhWa3JQiC/mzbtg1JkohuFXnM90KjrFisZrZu3er7wDxoz549LFiwAEtsCMGJvt+t7hbVK5GqfSV8+OGHnH322V5ta8iQIbz22mvUHqzGkuwIiGzeqgp12TKyLDN8+HCtwzmGL/vziRMnsmLFCq6//np69epFbm4u7777Lv369WPVqlUnrBnXGBEREUTHxFBUWdrsY+mSqiLXlJGS0qbRA902m6uYkmw0eSMyXZCNZs3reF533XXk5OTw4Ycf8st7L3PNoxOQ5Ia/mSmKk9nvPE95YR4PPvhg/YKjQHTZZZcxdepUdm7IJLVH8nGfU5xbRkleGcOGDSMkJMTHETbN22+/TUlxMTH9k3WdVtYUZiGyVyJF67N55513eOqpp7zSTmxsLB9++CEjbhtB6c5SJIMTc7z+F4mqDtekurNKYsSIEdxxxx1ah3RSWt+fC4FL77uNtXTkZLq3Jy39yT8n0vU+sV5TU4MUFrgT67LJdR3q3nXoTZdffjnr16/HViARlOzbvt6W7zpPf1mIeTJa9ulivF0QhCOVlZUBYLfbUVVV95uFmiInJ4dff/0Vc1QwwYnHZsb1pIhuCZRtzefTTz/lsssuQ27EWFFzhIWF0atXLzZu2ohiBzmAhgXtRRJms9l/JtaXLFlywu85HA727NlzTBrmTZs2eeSPb/v27dx///0MGDCAkSNHnvB5Tz75JI888kj91+Xl5bRpo99dI4LQUIqiaB2Cpux2O+np6UQnRmCyHPuWJUkS8Skx7Nu+j9LSUiIjI30fpAd8+umnqKpKVO9ETS9cLNFWrMkR/PXXX2zYsIG+fft6ra3Q0FCuueYavvrqK+wFEuYE/Q+8n4qzHJwVEkOGDCY5+fgTR1ryZX/+yCOPMG3atKMGaG+88UZ69uzJa6+9xldffdXoYx5P7169WLx4MdRVgcU/JuIaSqopA4eN3r17N/q17h3udVWBuaNAVRTsNZVEtUnUOhTuv/9+tm7dyh9//MHGhbPoO+TqBr927S/fsT99PRdddBF33nmnF6PUXteuXUlKSmL/9hwURTnuTWVmuiul/uDBg30dXpNs2rSJ6dOnY44KJqJ7vNbhnFJkWgKVe4r59ttvufLKK+tTxHlaamoqH7z/AaNHj6ZiewWg78l1xQFVfxtwVkhcffXVPPbYY7ofRNLy/lwIbCZTAI28eZDdbj9qotI98CxwzG7hxtZ99jW73Y7ZELi/5+4Fnu5Ftt40ZMgQXn31Vex5KkHJJy/140mqAo4CmeiYaM444wyftestWvXpYrxdEIR/Ki4uBsDpdFJeXk5EhP7KnDXXhx9+iNPpJKZnK6/fGxlDzIS2i2bXrl0sWLCAiy++2KvtHeniiy9m48aN2AskLK31ey/eGM4qUKokzh18ri6uNxu0TEJRlCZ9OJ3Nu7DKzc3lsssuIyIighkzZmAwGE74XIvFQnh4+FEfghAItN6Np7UtW7ZQU1ND6w4nHrBu3d71vbVr1/oqLI/Kzc3l559/xhwZjDVZ+4uWqF6uiapPPvnE623deuutGAwG6vbLBELVg9pMV7d6shtTLfmyP/dVDbdzzjkHALmkeTWu9UgqOQAcPsfG6Nq1KwDlOfs9GpNeVBTm4LTb6s9TS7Is89JLLxEREcGSaf+joii/Qa8ryctmxYxPiI2L4/nnnw/4CS9JkjjnnHOoq7aRv7/4uM/ZvyMHWZYZMGCAj6NrPIfDwUsvvQRA3IC2jcpUoBXJIBM7IAVVVXnppZeafa92Mt27d+fjjz8mLDSM6u0GbLn6/P1W7FC1yTWpfu211/L888/7bCdBc2h1fy4EPpEK/vj+mRq1paRKbYh/jtPp/T1UVVUI5GsuH55aZGQk55xzDs5KCWeV79p1lEgodrj0kktPOk7sL7To08V4uyAIx1NQkH/E5wUaRuId+/btY+bMmZgjgwlNjfZJm1F9WiPJEpMnT/ZprfVhw4ZhMBiw5QTGeDtAXY7rGlMvmR51e8VbVlbGJZdcQmlpKfPmzaN168CsESoIp9LSb9qXLVsGQErXE+9KbHPoe0uXLvVJTJ42depUnE4nkT5YLdcQQfGhBCWEsnTpUq/Xrk9KSuLKK6/EWS1hL9D+3JvDUQ6OEpmzzz67STuMWwJv1HAbPHgwBoMBuWAPAXO1CKAqGAr2ERQczHnnndfol7dr147ExNYU7t2O0+G7i3dfKdi5GUA3NZBjY2N5/PHHsdfVsHz6Rw16zbJvP8Bht/Hkf/4TkCvBj8e9qyhn77E36U6Hk/zMIrp27eoXP4/vvvuOHTt2ENYplqB4fae9PVJwQhhhHWJIT09nxowZXm2re/fufPrpp0SER1C9w0DdQX3184rt0KR6pcT111/Ps88+q/sJIUHwNj3ci+iRe7G7Kci1O9sXu4H9xT8HiX05aNwUkiQF1j3DPx06NV/9LV955ZUA2PJ813+6F+u52xYaR4y3C4JwPBUVFVRVHZ6DyMvL0zAaz1NVlYkTJ6IoCtGnJSHJvuknTWEWwjrHkZmZyTfffOOTNgHi4uK46KKLXIvfAiCRpeoAe55MQkI8gwYN0jocQKcT67W1tVxxxRVkZGTw888/0717d61DEgTN1NTUaB2CZlRVZd68eZiDTCR1anXC58UlRxEWFcKiRYv8bpCjrKyMH374AWOomdB2UVqHUy+yp+vn/fnnn3u9rTFjxmA0GqnN9N9VdKoKtXtdXeq9996rcTT65a7hduONN57wORMmTCAiIqL+41Rp5mJiYhgyZAhydSlSeeBc+Esl2Uh1lQy/4oompTiSJInLL78MR10NBbu2eCFC7ShOJznb/iIsLIxzzz1X63DqXXHFFXTt2pX05b9RnJN10ufm79/FjtWL6dOnD0OGDPFRhNpzlxfJPc7EemF2CQ67kz59+vg4qsYrLi5mypQpGCxGYk5L0jqcRovun4xsNjD5ncleT2fctWtXPv/8c6Kjo6nZaaAuWx+TdkodVG0y4qySuPXWWxk/fryYVBcE4YTUQzcpsmw46mvBNRB/pFMtitWa0WhEVQL3/8/9u+mrndznnXcekZGR2PNkVB9UMVRsYC+S6dKlC126dPF+gwFGjLcLgnAi7ol0c5DpqK8DxaJFi/jjjz8ITgr3ebbY6L6tMViMTJkyhfz8hmU49IRbb70VgNr9/n+fW3dQQnXAzTf/SzcZtpoVxYEDB1i8eDEHDx48brpqSZIYP358o47pdDq58cYbWblyJbNmzfKLVJCC4E0tecf6unXrOHDgAF3PaI/RdOIbQ0mS6NivLRsWbmXRokUMGzbMh1E2z7Rp06ipqSHmjDa6SiNrTYrAHBXML7/8wtixY0lM9F4d4+TkZK699lq+++47bLkSlkT/G+hwlEg4SmUGDRrkF5NC/+SN/vyfvFnDbdSoUcybNw9D5gYcnc/BpzkQvUFVMGZuRJblZpUVuPbaa/nkk0/Yv+EPErr0CZhdaPk7/6auspwbRowgKChI63DqybLMPffcw0MPPcTaX75l6OjHT/jcNXOmAa6FOIHy/9IQcXFxxMXFUXig5JjvFRx6rEePHr4Oq9GmTJlCRUUFsWemYAjyvzqtxmATUb1bU7Q2iylTpvDUU095tb2OHTvy+eefc+edd1KwqwBVcRLURru+XqmDqr+NOKtd/cdDDz0UMH+HvujPBaElck9SKoor9bJYiHOYO1WsyWLEXuegoKCA+PgTl3HTmslkwhHIE+tO1+y2xWLxSXtms5mrrrqKzz//HHuhhDneuz9bW64EKtxwww0B03efiKf7dDHeLgjCybgnfONTYjiQkRtQE+vl5eW88sorrtJoZ6b4vP8wWIxE90+mYMU+XnnlFd566y2fxNCnTx/OPPNMVq9ejaNcweinlTxUB9gOGAgPD+emm27SOpx6TZ5Yf/zxx3n77bePqumiqmr9L4X788beuD/66KPMnj2bK664guLiYr766qujvu9eaSEILUVLnlh3p0jpdlaHUz63+1kd2LBwK9OmTfObifXq6mq++vorDEFGwjufODW2FiRJIrJnK/KX7eXzzz/nySef9Gp7d999N7NmzaIusxZzvAPJj0qluXerS5LE2LFjtQ6n0bzVnx+psTXcGjsQ1K1bN4YNG+bKcLHplybHqTfXXn89KSkpTX59UlISQ4YM4bfffqMkaxfRKZ08GJ02VFUlc91SZIOBW265RetwjnHBBRfQpk0btq74nUH/uheL9dgU4VVlJexYtZhOnTq1yAGtbt26sWzZMmqr6ggKOfy3XphdUv99Pdu9ezczZszAHBlMeJc4rcNpsohucZRnFPD9999z8803065dO6+2165dOz7//HNGjRpF3p48JNmJJcn3ExuKDSr/NqJUw1133cXYsWMDZmDeF/25ILRU7mtXx6HJLb3slNGDgwcPApDYPo7923LIyckhLS1N46hOzGKxYHMG7hiL6nT1rSaT7xb+XX/99UydOpW6bBlzfNPrfp+KqoDtoAGr1cqll17qtXb0wBt9uhhvFwThZIqKigCITYriQEZu/deB4M0336SgoIDofkmYw7XZnBHWMYaK3UUsWrSI3377zWdzF2PHjmX16tXU7DEQ2tuJP9761mbJKHa44747CAkJ0Tqcek26G/joo4944403GDJkCPfccw/XXnstt99+O0OHDmXZsmV8/PHHXHXVVdx3332NPvbGjRsBmDNnDnPmzDnm+6KjF1qCI+uStdSJ9aysLBYuXEh8SjStUk896RwRG0ZqWhIbNmxg48aNfrFrePr06ZSXlRPVtzWyUX8zyaGp0ZSsP8iMGTMYM2bMSetiN1dcXBy3334777//PnUHJILa+s8uAnu+hLNS4sorh/tdOjpv9uduR9ZwW758uddquP373/8mKirK78pBnEhwcDD33HNPs48zevRofvvtN/auXkhUm45+P4FUuHsrlYU5DB8+nKQk/aXglmWZ66+/njfffJNtKxfSZ/CxtR+3/vEbTqejRey0OZ4uXbqwbNkyinJKSeqYUP940cESTCYTqamp2gXXAG+++SaKohDTP9lnddm8QZJlYk5LJnfRLt566y3efvttr7eZkpLCZ599xsiRIynYVQCy06dZahQ7VP1tQKl2vTcG0qS6L/pzQWjJ3BPr6qFc175Ks+0PMjMzAWjTJZH923Lqv9ar4OBgyksrtQ7DaxSHayI2ODjYZ22mpKRw/vnns2TJEhzleG1HnL1QQqmDa269htDQYxevBgpv9elivF0QhJNxlwiLjAsD9F/apaH++OMPfvjhByzRViJ7JJz6BV4iSRLxA1M5MCudl19+mf79+3t1nN3NXX5w/vz52IskzLH+M94OoNSC7YBMQkKC7vqpJk2sf/jhh6SmpjJ37tz6FFipqanceOON3Hjjjdxwww0MGTKE66+/vtHHXrJkSVNCEoSAcmRd9ZZaY/3jjz9GURT6XtC9wYOefS/szr70bD766COmTJni5Qibp6amhk8/+xTZbCCiqz5T5UmyRGSvVhT8mcnnn3/OY4895tX2br/9dqZPn05xVhHmVg5k32SvaxbVCbV7DQQFWfxyt7o3+3M4uobbggULvFrDLTY21uvpjP1R165dueCCC1i8eDHF+3cS07az1iE1maoo7Fn1O7LBwF133aV1OCd0+eWX89Zbb7H1j9+PP7G+Yj4mk4lLLrlEg+i017mz63ew6ODhiXVVUSnOKaNDh4663gW4YcMGli1bRnBimM/rsnmDtU0EQQmhLFq0iL///ptevXp5vc02bdrw8ccfM/L2kZTuLEU2OzHFeP/mXlWgaouhvqb6Aw88EDCT6uD9/lwQWrp/vl8E0vtHc+3duxeDUSalW2tW/LSevXv3ah3SSVmtVtRCHxQD14hqd52b1Wr1abu33XYbS5YsoS5Lxpjm+Z+vqkJdlowsy/zrX//y+PH1xFt9uhhvF4TDFixYQJs2bfxuc4w3VVa6Fp2FRFqP+tqflZeX89xzzyEZJOLObad5CVZTmIXo/skUrtrPyy+/zH//+1+fXFM+9NBDLFmymNrdYIryryyxNXtkVAUefPBBXZWCBGjSb9P27dsZNmzYUXWlHA5H/efnn38+l112GZMmTWp+hILQAh1ZP6m2tlbDSLRx4MABZs2aRXSrCNr3Onlt5SMlto8jqWMCy5YtY8uWLV6MsPm+/fZbiouKiegWj8Gi30mEsA4xGEPNfPvtt/X1drwlJCSEhx56CNUJNXv9o25h7X4ZpQ7uvHM0rVq10jqcRvNmf35kDbfp06e3yJTXejFu3DhkWWbX8l/ra4P6o5xtf1FZmMvVV11F27ZttQ7nhOLi4ujfvz/ZGZupKis+6nul+QfJ25fB2WefTUSE/0/MNkX9xHpOaf1j5cWV2G2O+u/p1TvvvANAdD/9ZUtoCkmS6s/l3Xff9Vm77du35/3/vU9QUBDV2ww4vLwZQlWhepuMs1zisssu4/HHHw+4STFxfy4I3lX/93TovePIv6+WTFVV9u7dS0RsGBGxoRiMBt1PrIeFhaHYnaiqf+3YaijF5rrWDwsL82m7/fv3p2fPntgLZbyRad9R4soSN3ToUNq0afgYkT8SfbogeFdtbS0PP/wwo0eP1joUXXHPPwRZzSAFxnzExIkTyc/PJ6p3ayxRvsvkcjLhXeIITgxj4cKF/PKLb0pZpqSkMHLk7Si1rrTq/sJeImEvkOnbty+XX3651uEco8k/ycjIyPrPQ0JCjqm70KVLF9LT05scmCC0ZEemMg6UtMaN8eGHH+J0Oul/cY9Gp1k9fVhPAN577z1vhOYRlZWVfPzJxxgsRiLStEtD0xCSQSaqd2vq6ur48MMPvd7e8OHD6dGjB/Y8GUep15trFme1Kx1NYmIit99+u9bhNJm3+nN3DbdLLrmkvobbkR+C73Tq1Imrr76aysIcDmz8U+twmsReU8XuP+YSHBzM/fffr3U4p3ThhReiqiq7N6w86vE9h74ePHiwFmHpQkpKCmazmeIjJtbdk+ydOnXSJqgG2LFjB2vXrsWaFE5QXOCkHw1OCCM4MYyVK1eya9cun7WblpbGf9/8L5IqU73ViFJ36tc0VW2mjL1Q5owzzuCll146aqA6kIj7c0HwHncWuZDwKKDllmv7p6KiIioqKohKCEeWZSJiQ9m7d6+uJ63Dw8NRFbV+Z3egcdpcE7C+XsApSRJ33nkn4NpZ7ml1Wa5xoVGjRnn82Hok+nRB8B53H1VaWqptIDrj3uRnMBkwGg1HbfrzR0uWLGH27NlYYkOI7KGfjVCSJBE3MBXZZODVV1+loKDAJ+2OGTOGhIQEbFneWQDnaaoCNTsNyLLM008/rcuF8U262klKSuLAgQP1X3fo0IHVq1cf9ZwtW7boqpi8oE+1tbX8+eefVFVVaR2Krjidh3cUtrTV8AcOHGD27FlEJ0bQoXdKo1/fukM8yZ0SWL58OZs3b/ZChM336aefUl5WTkTPVhjM+t2t7hbWIQZTRBAzZsxg3759Xm1LlmWeeeYZZFmiZqcBVafjHaoKNTtd6Wieeuop3aWjaShv9udH1nAbMWLEMR+Cbz300ENERkay58/fqCr2bvYJT1NVlW0LZ2KrqeKBBx4gLi5O65BO6eyzzwYgc8tfRz2+b8s6gBadwcFoNNK+fXuKc8pQFdegRtHBUgBd71j/7rvvAAjvpu8FcU0Rceic3OfoK+eccw6PPfYYSh1UbfVOn28vlKjLlElOTubNN9/EZDJ5vhEdEPfnguBd7sH3yARXlg93HdKWbv/+/QBExLmKakfGh1NRUaHryYro6GgAnLV2jSPxDmeNA7PF7NMa624XXHAB7dq3w5Yne3TBnKMcHKUy5557Ll27dvXcgXVK9OmC4F2Busi2udyLCE0mI0az0a93rJeXl/Piiy8iGWTiz0lt9KY9bzOFWog5PZmKigpefvllnyxItFqtPPXUU4cmrGV0vAYSgLr9EkoN3Hrrrbot2dCkd5KBAweyatWq+q+vvPJKNmzYwN13380vv/zCk08+ydy5cznvvPM8FqgQmN5//33uvvtuJkyYoHUounLkG6qeV3t7w6efforTqdB/SON3q7v1H+rate6LHdaNlZuby9SpUzGGmHVbW/2fJFki5rQknE4nb731ltfbS0tL48Ybb8JZLdWvTNcbe76Eo1Rm0KBBDBo0SOtwmsyb/fmSJUtQVfWEH4JvRUZG8sILL+B02Nn885c46vznJilrwx8U7NrC6aef7jc1FVNTU4mKiiJn1+GdJKqqkrNrK61bt/bL0hGe1LlzZ+w2B+XFrrptxX6wY33FihUYgkxYk8K1DsXjrMkRGCxG/vzT9xktbr31Vi6//HKc5RK1+zw7yKXUQU2GgaAgC5MnTw7o8gvi/lwQvKuwsBCAuDbtAY7ZPdpSZWdnAxAeE3Lo39CjHtejhATXYjJHVWBmBnRU22jVqpUmO7tkWebOUXeCCnUHPNenu3fAu3fEBzrRpwuCoAX3pkdTkAmzxejXNdbfeustCgoKiOqdiDlSHyng/ymsUyzBiWEsWrSIBQsW+KTNCy+8kAsuuABHqYw9T5/j7eDKEFuXZSA+IV7XGSubdKUzYsQIOnToQGZmJgCPP/44ffr04aOPPmL48OFMnDiRtm3b8vrrr3s0WCHwuFc4b9++XeNI9OXIm6CWtJKuqKiIWbNmEREXRvveTa+b1bpDPK3axbFkyRLd1XibPHkyNpuN6H5JyEb/+b+1tokkKCGUhQsXsm7dOq+398ADDxAXH0fdfoPuUtQodqjdbSA4OJinn35a63CaRfTnLcuFF17I7bffTlVxPn///CWKH2REyd+5mV3LfyEuLo6JEyf6TZ8oSRLdu3enNP8gddWuG9SqsmKqyopJS0vTODrtuSfQ3Sngi3LKiIyMIDY2VsOoTqy0tJSDBw9iibXqMgVZc0myhDnGyv79+6moqPBt25LEM888Q0pKCnVZnisDo6pQvV1GscOTTz6l60UbniD6c0HwLvfEekJ7144ZX6Xt1LucnBwAwqJcE+uhkVbAtZhcr5KSXFkH7BX+neL2eBSHE2e1naTWSZrFcOmllxKfEI8tV0bxwK2GsxrshTK9e/emX79+zT+gHxB9uiB4VyDez3lCcXExSK4a60GhQRQVFfrlppiNGzcyffp0zFHBukoB/0+SJBE3oC2SQea1117zWTbnp556iuDgYGr3GFB0mLxHVaE6w5XNbvwz47FarVqHdEJNGp0cNGgQc+fOpW3btgCEhoayatUqvv/+e1599VWmTZvG5s2b678vCKciOrWjGQyG434e6H744QdsNhu9z+/a7MmTPoNcKcK++eYbT4TmEenp6cyZMwdLjJXQ9tFah9MokiQRc7prscPrr7+Oong3R3toaCjPPP0MqnKoQ9XRtVzNLtdA/YMPPuj3u05Ff97yPPTQQ1x00UWUZO1i8y9f6XpyvWD3VtLnfoPVamXKlCl+kQL+SO3atQOgJM+VyrE017V7KzU1VauQdKNDhw4AlOSW43Q4KSusoEOHjrq9HnRPNhuCAzONOIAhyFWaxtcT6+CqHfp///d/yLJM9U6jR1LC23JdmWUGDx7M1Vdf3fwD6pzozwXBu/Ly8gCIjG9NcGh4/dctXX6+q7xQSIRr0DPk0MS6nn8+HTt2BMBWUqNxJJ7nPif3dZYWTCYTt424DdUBtpzmX9fVZbvGhe644w7dXid6mujTBcG7Wsp7SWNlZ2cTEm5FNsiERlqpq7O5Jtv9iNPp5NVXXwVwTVrrLAX8P5nCg4jq1Yr8/Hw++OADn7TZqlUrHnzwwUObxvS3ccWWK+EskxgyZIjuM8R6rLivyWTiuuuu89ThBKFFO7L+o9ls1jAS33E6nUyfMR1zkIku/VObfbzUtCRCI63Mnj2bhx9+WJMaY0dSVZVJkyYBEHNGG7+8kAuKDSG0Qwxbt27l119/5fLLL/dqexdeeCFDhgxh/vz52HIkLK21n123F0nY810r5m+66Satw/EK0Z8HNoPBwGuvvcbDDz/M8uXL2TT7c3pePgKj2aJ1aEfJ3b6Brb9/T1BQEO+99x7dunXTOqRGcy+8qSwphHZdqCgpOOrxlqx9e1cq3ZK8MsoKK1EVtf4xPUpISECSpIDc3ebmqLIhG2Ti47UpU5OWlsatt97KF198QV2WRFDbpvf5ih1q9xoICQnhqaee8strLk8Q/bkgeI479XtIRDQhkTEiFfwhhyfWXffaIeGuf/W8o79Dhw4YjUZqC32zM8yX6gpdqd60vm6++uqreXfKu9gO1mJJdtDUblhxgD1PJikpSfeD694m+nRB8Bx/3IXtbTU1NRw8eJDWHV33gpHxrvJne/bsISYmRsvQGmXWrFls27aNsI4xBMWHah1Og0T2aEXFriK+/PJLrrvuOlJSUrze5k033cScOXNIT0/HlKBiitLH34RiO3wf/5///EfrcE6pScsS2rdvz+TJk0/6nClTpuh6gEzQF9GpHe3IiXWj0WPrX3Rt9erV5Obk0qlfW0yW5u8Ikw0y3c7sQFVVFfPnz/dAhM2zZMkS1q1bR0hKJMEJYVqH02Qx/ZKQDDJvv/02dXXen2B46qmnCA0LpXavAUXj+QzVCTW7DBiNRl544YWAyCYh+vOWyWKx8NZbbzF48GCK9+9k/YwPqKvy/S7V41FVlcy/lpE+71tCQ0P58IMP/DbtY1RUFAA1FWVH/RsZGalVSLqRmJiI2WymtKCC0oJyQN87+c1mMx07dqQuvxJ7ZeBNrtsr6qgrqKJzp86aXnfef//9REdHU3egeWnp6rJkVDvce++9mi0U8DXRnwuCd5WVufrw4LAIgkLDKSsrE2MYuHamG81GzEGu+3f3BLued6wHBQXRq1cv6gqrUexOrcPxqJoc1zXV6aefrmkc4eHhXDn8SpRacBQ3fXGbPU9CdbomAALh3ruhRJ8uCN4l+u9j7dq1C1VViUmMBCAmMQKAjIwMDaNqnOrqaiZPnoxsNBDdT7uSKI0lGWSi+yfjcDh46623fNKmwWDg+eefR5ZlanYaPJIxzhNq9rju4x966CG/uI9v0sT6vn37KC0tPelzSktL6+vBCILQOEcOaraUG4iffvoJgK5nei5tWZczXGl4Z82a5bFjNoWiKLw7ZQpIEtGnJWsaS3MZQ8xEdIsnNzeXH374wevtxcbG8vhjj6M6XCnYtVS7V0aphTFjxmiaXs+TRH/ecpnNZt544w2uv/56KvKzWffdFCqLtB0EVRQnGUtmsWv5LyS0asUXU6fSp08fTWNqjtBQ1wppd431uupKAMLC/HdxlafIskxycjLlRZWUF7p+Lm3atNE4qpO78847URWVkk05WoficcUbD6IqKqNHj9Y0DqvVyr333ovqcE2ON4ViB1u2TKtWrQI2s8zxiP5c8BQx2Hx87rqX5uAQzMFWFEWhpibwUok3Vk5ODqGR1vrMICHhwUiyxMGDBzWO7OQGDBgAqkpVVpnWoXiM4nBSc7CC1NRUEhMTtQ6nfne1LbfpE+t1OTJGo5Hhw4d7Kiy/IPp0QRB8bdu2bQDEJrs2B8Qlu8qXbt26VbOYGmvatGkUFRUR0SMBo9W/sv+GpEQSFB/K/Pnz2bJli0/a7Nq1K7fccgtKDdTt1z7Dm71Ewp4n06NHD66//nqtw2kQr81SlJWVYbHoK62ooD92ezO2owSwllZjvby8nIWLFhKVEEF8G8/VHg+PDiWpUwJr1qzhwIEDHjtuYy1evJiMHTsIax+NOSJIszg8JbJHK2STgY8//tgnu9avvvpqTj/9dOyFMvYibTp7RwXUHZRJTU3VfOLB10R/HrgMBgPjx4/nwQcfpLa8hL++f4/irN2axOKw1fH3nC84sGklXbp0ZdrXX9fXwPRX7gn0uhrXxLF7Yj08PFyzmPSkdevW1FbVUZxXVv+1ng0bNozOnTtTsbOQ8p2FWofjMeU7CqjcXUS3bt0YMmSI1uFw7bXXEhMTgy1HRm3CRkLbQQlVgVGjRom+6x9Efy40hLg/Pz6bzQaA0WTGaHIN1rb0n1VVVRUlJSWEx4TUPyYbZMIiQzS9926IoUOHAlC5N3BS+ldnlaE4nPXnprWuXbvStWtX7EVyk7LQOKtAqZK44IILiI723BhRoBB9utAQTz31FNdffz0Oh0PrUHTF6QysbCWe4J5Ajz80oR4RG4Y5yOQ3E+sVFRV8+umnGIKMRKYlaB1Oo0mSRPRprl327777rs/aHTt2LPEJ8dRlGXBquF5UVaB2l4wsyzz77LN+MxfW4Fx/y5YtO+rrffv2HfMYuN6csrKy+Prrr+ncuXPzIxQCWnW1qwaUougk54ROHLlToCXsGpg7dy62OhunndHd43Uwu57RnuydecyaNYv777/fo8duqKlTpwIQ1Vv7leOeYAgyEt41joLNufz2229eX0EuSRLPPPMM1157LTW7wBjpQPJhH6uqULPTACqMHz8es9m/Vj7+k+jPhSNJksTo0aNJTk7mqaeeYtOPn9Bt6A206tLHZzHYqivZNOszyvMOcO655zJp0iSsVqvP2veWiAhX+rT6VPCVrvScYmLdxV1rPj+z6Kiv9cpgMPDWW29x8803U7gyE2OIGWtr//6/rD5QRsGq/URFR/Hf//4XWdY2Mwy4yiHdfPPNvPvuu9jyJSyJDb8OVlWw5RgICwvlyiuv9GKU+iD6c8Eb3PfnwtGcTieSJCNJErJsqH+sJXPvlo2MO7ovjIgLJWtHLtXV1bq9nmvXrh09e/Zk85bN2CvrMIX6/wRl2Q5XXfsrrrhC40gOu/TSS9m+fTv2IglLq8aNa9nyXdckl1xyiTdC0x3RpwveMGfOHMC1EMp9byogFhocx44dOzCaDPW11SVZIqZ1JHv37sVms+l+HPTrr7+moqKCmP7JyCb/mJT9p+CEMIJbh7NixQo2bdpE7969vd6m1Wrl30/8m0cffZSaXTKhPbWZn6vLlnBWS/zrXzfRrVs3TWJoigZPrA8aNKh+wkuSJKZOnVo/WfRPqqoiSRKvvfaaZ6IUAlZFhaumqzu1muDiXhEPgb8SXlVVZsyYgSzLdDm9nceP375XG/6Y+RczZ87k7rvv9nnt0IyMDDZs2IA1OQJTuP/vVncL7xJH6ZZcvv32W5+kZmvfvj133HEHH330EbVZMsGpvuvsbbkSzgqJyy67jDPOOMNn7XqL6M+F4xk2bBhxcXGMGzeO9Hnf4qitIbn3AK+3W1tRyoaZH1FdUsi1117LM888o2mNZ0+Ki4sDoLKk8Kh/3Y+3dO6fQ3FuGWaz2S8WHLRp04bJkyczesxochfuIuH8doSkRGkdVpNU7ishf9kezCYT70x+h6Qk/dShu+qqq3jvvfew5apYEhs+ceUokVDq4NKrLtPtZI4nif5c8Ab3/blwNEVRwP33dmgRUkvfHLBr1y4AohKOnqyJbhVB1o5cdu3aRa9evbQIrUFuuukmNj+9mfLt+cT013c5mlOpK66mNreCgQMH0rZtW63DqXfxxRfz5ptvYi9s/MS6vUgiKCiIc88910vR6Yvo0wVvEtkNjtYSNrA1hqIo7N69m8j4cGTD4YXW0YmR5OwpYO/evXTp0kXDCE+uurqaL7/60rUJrIt/j7VE9U6k5mA5H3/8Me+8845P2hwyZAgDBgxg5cqV2ItUTDG+/ftQ6qAu00BUdJRmGyKbqsEjl88++yySJKGqKi+++CLnn38+gwYNOuZ5BoOB6OhoLrjgAr9aYSBoo6zMtYvrVPWDWprKysrjfh6INm3axPbt2+nQOwVrWLDHj28yG+ncP5XNyzNYsmQJF110kcfbOJm5c+cCEN451qftepsp1II1KYLNmzeTnZ3tkwH50aNHM2v2LAqy8jG3UjD4YJ2C4oC6fQaCg4N59NFHvd+gD4j+XDiR0047jc8//5y7776bHYt/QlUU2vQd6LX2astLWP/Dh9SUFTNmzBjGjRvn8awlWoqMjMRqtVKalw1ASW42MTExBAUFziKr5jgyrWd0dLTf/N/369eP/733P8Y9MI68xXuIHdiW8I7+1ceXZxRQsDKTEGsI7777rk9WwzdGQkICZ555JitXrkSpBbmBfzL2AtfvkJ5263mT6M8Fbzjyvryurk4Mxh+iKEp9Vg93f9XSJ9bT09OBw/VY3WKTDtdl1fPE+rBhw3j77bcp3FFIZM9EDBb/XdhZ8ncOACNHjtQ4kqMlJSXRvn179mbuQVUUpAYmxlFqXWngzzz/zBZz3Sz6dMGbRF9+NH+57/SV/Px8ampqSE6IP+rxqEO71/ft26frifUff/yR8rJyovq29tvd6m7BCWEEJYSyZMkSdu/eTYcOHbzepiRJ/Oc//+Gaa66hZjcYoxwN7q89oWavqwTcww897BebLY7U4CvH559/vv7zpUuXcscdd3Dbbbd5IyahhVBVleLiYsC1ukjcuB/m/rn88/NA9MUXXwDQ6zzvpbHqeU5nNi/P4IsvvvD5xPrSpUuRjDLBrQMv7VJISiTVB8pYsmQJt9xyi9fbs1qtPP7Y4zz++OPU7pEJ6e79way6TBnFBvc8fE/A7DIV/blwMp07d+bzzz9n1KhRZCydjWwykdTD85ka6qoq2DDzY2rKinnggQcYM2aMx9vQmiRJtGvXjh0ZO7HVVlOWf5D+/U/TOizdiIyMPO7n/uDMM8/k008+5Z577qHgj304Km1E9U7U/SCNqqqUbDhIyd85REVF8f7779O9e3etwzquIUOGHFo1L2FJOvWqeVUFe5FMQqsEXU/keJLozwVv+Od9aGJiYJSyaq66urr62uqGQ//W1dVpGZLmNmzYgNFkILZ15FGPJ6TG1H//pptu0iCyhjGbzYwaNYrXXnuNsvQ8ovvpJ3NLY9SV1FC1r4QePXpw1llnaR3OMQYOHMiePXtwVoCxgUMijlLX9dSAAd7PnqUXok8XvEnv9yi+FigZ8jxl3759wLGlXSLjw4DDpV/0SFEUvvr6a2SjTETX+FO/wA9E9mhFbt4upk2bxvjx433SZvv27fnXv/7Fl19+SV22RFAb3+xad5SDPU8mLS3NL0u5NWn9weLFi0UHLzRbRUUFtbW19V8XFBRoGI2+5OTkHP48N1fDSLxr9+7dLFiwgIS2MbRq571Jy8j4cNr1TGbDhg2sXbvWa+38U1VVFTt37iQoPhTZqH3dUk8LPlRbdtOmTT5rc+jQofTp0wd7gYyjzLttOWvAdlAmOTmZW2+91buNaUT058LxtG3blk8++YSo6Gh2LJxJ4Z5tHj2+025j06zPqC4t5J577gnISXW3rl274rDb2LFqMaqq6Hqlt68duRrZH2v+9ejRg6+++orkNsmUbDxIwYp9qDrevag6FfKX76Xk7xxSUlL4+uuvdTupDnDBBRcgSRL2woYNBDrLQLXDhRdc2CIHD0V/LnhKSUlJ/eeBvsC7McrLy7GEhAIQFBJW/1hLVVpayvbt20lIjcVgPHp3WERsGCERwaxZs0b36Xavu+464uPjKduWj7PWP0vwlWxwZUYaO3asLvu/fv36AeAoa3hsjnLXc087rWUuSBV9uiB4l8Hg37uaPc09cR4RF3bU4+6Jdj1PrP/5558cyMoipH20X2eeOZI1KQJTqIXZs2f7NIvxPffcQ0REBHX7DSi2Uz+/uVQVana7/hb/85//1GeG8ifNinjDhg088cQTDB8+/KhdoJmZmXz//ffiRkw4qSMnj4/3dUu2f/9+AGSDkdKSkoC9aX/77bdRVZX+F/fw+k3gaUN6HNWmL+zZswcAc5TnU9zrgTHEjGwy1J+nL0iSxOOPPw5AzR4D3vyvrN0royrwyCOPYDabvdeQDoj+XPin1NRU/vfee1gsFtLnfUNlkWcWeamqSvpv31GRn811113Hfffd55Hj6pV74nLT4p+P+lo4emI9LCzsJM/Ur9TUVL7+6mt69+5Nxa4ichfuQrE3vCa4ryh2JzkLd1G5p5i+ffvy9ddf06aNvuvJxsbGkpaWhqNMRnWc+vn2Ytdt7fnnn+/lyPRN9OdCc7lLtUHLnjg+kqIoFBQUEBrpKvsRGunakZ2Xl6dlWJpasWIFqqqS0uXYjAaSJNGmSyKFhYVs2+bZxZmeZrFYuPvuu1HsTko2+9+GhtrCKqr2l9K3b1/OPvtsrcM5Lne5GWdFw8d7nBWu+uqdOnXyVlh+QfTpgiD4wt69e4HDqd/dQqOsGE2G+u/r0Zw5cwCI6BwYGUYBJFkirHMstbW1LFy40GfthoeHM3bsWFQH1GZ6f5LbXiDhLJcYNmwYffr08Xp73tDkn9ITTzxB//79mTRpEj///DOLFy+u/56qqvXpAwThRLKysgAIi2sNHJ5MFg53ap1OO+eorwPJ8uXLWbx4Ma07xJPSrbXX24tvE03Hvm3ZtGkTP/30k9fbg8ODLabQwCxxIEkSxlAzObm+XRTTq1cvLr74YpzlEvYi7yzIcJSDvUCmd+/ePi8f4GuiPxdOJC0tjddeew2HrY7Nc77EUVd76hedwv6/llGwawtnnHEGTz/9tC531niSu/bhwZ1bjvpagJCQkPrPQ0NDNYykeaKjo/noo48YNGgQ1dnlHPwtA2dtA2aCfcRZa+fgvB3UHCxn8ODBfPTRR36Tev+8884DFewlp36fsBe5BuH79+/vg8j0SfTngidUV1fXf15VVaVhJPqRnZ2NzWYjqlUyAFGtXAuTfLm4WG+WLFkCQNu046dPdz++dOlSX4XUZFdffTWtW7emfHsBjmofbNHyoOJDu9XHjRun22vquLg4oqOjcVY1LD5VAWe1ROfOnVv0rlLRpwuC4Cu7du0CyZXt9UiyLBMRF8bu3bt1mYGmtraWRYsWYQoPwhxj1TocjwptFw3A3LlzfdruddddR7v27bDlyDi9eBugKlC714DJZOLhhx/2XkNe1qSJ9c8++4xJkyZx+eWX8/fff/Pkk08e9f3U1FTOOOMMZs+e7ZEghcDkruER277bUV8Lrk7NZAmmXR9Xjazdu3drHJFnlZaW8txzzyEbZM67tr/PbgLPHt4Xc5CJiRMnkp2d7fX23HX3JIM+b3I9QTLI2G2+T5v34IMPYjAYqNsre2XXeu0+V/f48MMP63aQwhNEfy6cyuDBgxkzZgzVpYXsWPxTs45VlrOf3SvmkZCQwKRJk1pEbbOOHTvWv4eYTCZSU1O1DUhHjpxMP3KS3R8FBwfz3//+l2uuuYa6wioO/r5DF5Przlo7B3/LoK6omuuvv5433ngDi8V/Fvudd955ADiKT94PK7WgVEucddZZfnV+nuTt/ryyspLnnnuOYcOGER0djSRJfP755x6IXNCbI0u1Hfl5S7Zli2txXEK7zkf96368pbHb7SxfvpyI2DCiEsKP+5yULq0wGA0sWrTIx9E1nslk4t5770V1KpRu8Z9d67UFldRkl3PmmWdy+umnax3OSXXq1AmlRkJtQFIfpRZQaNG71cU9uiAIvqKqKhkZGUTEhGI6Tir1mMRIqqurfTKG3libNm2itraWkJTIgBu3NYVZMEdbWbduHTab7xb9GY1GHnv0MVChZq/3dq3XZUsotXDrrbfSurX3N1t6S5N+Qu+99x7dunXjhx9+oEePHsdNkdu1a1d27tzZ7ACFwOXehR3XIe2or1s6p9PJ3r17iUlKJa5Ne4CA+luy2+089thjFBQUcOalvYhOjPRZ26GRVs69pj9VVVU8/PDDR+3I8Ab3CmtV0d/KPo9RVSTZ9xcwKSkpXH311TirJez5nm3fUQqOEplzzjkn4Ou6if5caIj77ruPvn37krt9A7k7NjbpGE67jfR53yJJMHHiRKKiojwbpE4FBweTmOhKk5qSktIiFhM01JGT6Var/68wNxqNPP/889x8883Yims4+NsOnHXaTa67J9VtJTWMGDGC8ePH+93Or27duhEVFYWj5OSL6OyHJt7POeccH0WmP97uzwsLC3nxxRfZtm1bfVpfITDZ7fbjft6SrVmzBoCkLr0ACIuOIyIukb/++gunU3/lP7xt7dq1VFVVkdoj6YQD2SaLieTOCWzfvp2DBw/6OMLGu+yyy0hKSqJ8R6Hf1FovPZS6/p577tE4klNr27YtcGjS/BSUatfvVEtejCru0QXBu1pi330iBQUFlJSUENP6+OMzMUmux7dv3+7LsBrkr7/+AiColX+WlTuV4FZh1NXVkZ6e7tN2zz33XE4//XQcRTKOUs8fX7GDLctAeHg4o0eP9nwDPtSkifWtW7cyZMiQkw4OJiQkkJ+f3+TAhMC3b98+ZIOB0NhEzNZQ9ood64Ar1VxdXR2xyanEJLluQAIlzZyiKDz//POsXr2a9r3a0GeQ71Pidu6fStrATmzbto0nnnjCqwNGMTGu+nt62LXmLc4aO7GxsZq0fffdd2MymajN9Oyudfdu9bFjx3ruoDol+nOhIYxGI6+++irBVisZi2dhq65s9DF2r5hHTVkRd9xxR8AvWPmn6GhXGq9WrVppHIm+BAUF1X8eCBPr4CqR8uSTT7om10tqyF24E8Wh+DwOxeEkZ8EubCU13HrrrTz++ON+uYpflmUGDhyIUgfKSVLROcTEutf788TERHJycsjMzOT1119vapiCHzhyV4yYWHft5FqxYgXBYREkpB7eQZva83RKS0t9PtipB+5d6O16JJ/0ealpru8fmcJar0wmEyNHjkR1KpTvKNA6nFOyl9dStb+U3r17+0UJlJSUFACUmlNfizgPTb63adPGmyHpmrhHFwTvcjgCd4y2sdzXMfFtoo/7/fhk1+N6zNLjznxsiQ7WNhAvMUe5zsvXGZ4lSeKRRx4BoGavweNZYuuyZBS7a0w/PPz4mY/8RZMm1o1G4ynTEBw8eNCv6yUK3peVlUVQeDSSLBMcEcPB7GyxaozDO/djktpiDrISHpsQELv5FUXhpZdeYvbs2SSkxjL4XwM02eksSRLnXH0abbu3ZunSpfz73//22qCRe5eivTww0ygqdieOajuJrRI1ab9Vq1Zcc801KDUS9gLP/C45ysBRJnP++eeTlpbmkWPqmejPhYZKTk7m0UcewV5bTcbSOY16bVluFlmb/qR9+/bcd999XopQ//xtt7C3yfLh25BAmViHw5Prl19+ObX5VeQv3+PTzDWqopK3dC91hVVcddVVPPHEE345qe7mniw/UZ11VQFHqUxqaipJScev9dsSeLs/t1gsYnFQC3FkRi9vZ/fyBxkZGeTk5NC+91nI8uF+vH3fAcDhWuMthdPpZOGihQSHBtGq3ckXV7frmQQSzJ8/30fRNc+VV15JaGgo5RmFus84V57hmvy/9dZbNY6kYdzjIkrdqZ+r1rr6e39ODdtc4h5dELxLj/XCtbJp0yYAEtoev0+Pa+MqAfX333/7MqwGycnJQZIlDMEmrUPxClOoK1uJFpl/evTowZAhQ3CWS6csy9YYSh3YDsq0atWKm266yWPH1UqTJtZ79uzJokWLTjgJWl1dzYIFC1rcjiSh4WpqaigpKSE4wrXyKTgiGofDQUGB/lcHe1tmZiYA0a1cK3SjEpLJzc2tr9ftj+x2O8888wwzZswgLjmay+8adNzaLb5iMMgMHXkOyZ1bMX/+fB588EFqamo83k5SUhKhoaHUFQXmoJStxPUz69bN95kH3EaNGuWqtb7fM7vWa/e7usW77rqr+QfzA6I/Fxrj+uuvp3fv3uTt2EhJ1u4GvUZVFXYs+hFUleeff/64qQwDnXsC+ciJZOFogTSxDq7J9RdffJGzzjqLqsxSijf67ma4eH021VmlnH322Tz77LN+PakOcPbZZyNJJ76hd5aD6mzZu9VBn/15XV0d5eXlR30I+ldWVnbcz1sq9+7sjqcNPOrx1B79MZotLW5ifeXKlRQWFNKhd5tTXtdYw4JJ6pjAX3/9RVZWlo8ibDqr1cqll16Ko8pGTY5+369URaVidzGRkZEMHjxY63AaxD1JrtSe+prEPfnunoxvifTYpwtCIPH3+yNPWrduHbIsE59y/B3r5iATMa0j+fvvTT6t9d0QNpsNySgH7P+nZHRdZ2mVQWrs2LHIskztPs9lia3NklGdrnKTgTA22KQRvlGjRpGRkcE999xzzGRfeXk5t99+O7m5uYwZM8YjQQqBx73aJijMVavDEhZ51OMtWXZ2NgAR8a3r/1VV1W9/NrW1tTz88MPMmTOHVqmxDL/vQizB2r95Gs1GLh19Pm27t2b58uXcfffdHh88kiSJnj17Yi+r9ZtabY1Rk1cBoOnO7tatW3PppZfirJJwnGA3W0M5q8BRLHP66afTq1cvD0Wob6I/FxpDlmWefvppJEkiY9nPqOqpU1znbttARX42w4cPp2/fvj6IUn/cg55nn322xpHo15Fp4QOFyWTijTfeICUlhdK/c6jMLPF6m5V7iyndkktqu3a88cYbmEz+v3o/KiqK7t274yh33YT/k73EdTvb0v++9NifT5gwgYiIiPqPlpzW158cudBdpBl27Ug3GE2k9jrzqMdNliBSe57Ozp07OXDggEbR+d6XX34JQPcBHRv0/O5ndQDg66+/9lpMnnTFFVcA+KTPbqravAqcNXaGDRvmN/18QkIC0LAd60qdhMViISIiwstR6Zce+3RBCCQnK7PQkpSVlbFlyxYS2sZgspy4P0nu3Iq6Olt9TXNdCeTkAxqfW/v27V3j7ZUSjqLmL15Q6sCeI5OUnFR/veXvmjyxftNNN/HJJ58QFxfHJ598AsAZZ5xBUlISM2bMYOTIkVx33XUeDVYIHPv37weo37FuPfSv+/GWLCcnB4Dw2ISj/nU/7k8qKiq4++67Wbp0KSndErniXn1MqrsZTQaGjTqPzqelsmHDBkaNGkVhYaFH2zjrrLMAqD5Y4dHj6kHNwXIkSao/R62MHDkSgLoDzevo67Llo47XEoj+XGisbt26ceWVV1JZcJC8HSdPB6Y4HOxZ+TsWi4UHHnjARxHqzx133MHChQu5+eabtQ5Ft4KDA7MuWnh4OJMnTybYGkzBH/uwlXmvNIyttIaCPzMJCQlh8ttvB1R60LPOOgsUcJQd2887SiSMRmOL37Wlx/78ySefpKysrP7DH3astnSqqpKVlYU1Kg5JklvUhPHx5ObmsnXrVlLS+mEJPjazSsfTXJky/KGGuCf8+eef/Pnnn7Tp0orYpKgGvaZ97xQiYkP5/vvv/WKsp1evXsTGxlKdVabbVMFVWaUAXHTRRdoG0ggxMTEYDAaUulPfr6t1EgkJCQG7A7Eh9NinC0IgESXaXJYsWYLT6aRt95OX3nB/f8GCBb4Iq8GioqJQ7E4Ux6k3fPgjZ60DgOjo42cT8IW77roLSZKo9UCW2LoDMqoCd425K2AWtzQ5J+W0adP44IMPaNeuHdnZ2aiqyrp160hJSeF///sfn376qSfjFALM9u3bAQiJSTj0r6tm344dOzSLSS9yc3MxBwVjsboGRMMP/Yxyc3O1DKvRysrKGDNmDOvXr6dTv7Zccuf5mMz6e+M0GGQG/2sAPc/tTEZGBiNHjiQvL89jxx840JU2sPpAqceOqQfOOge1eZWkpaURGRmpaSxdunThtNNOw1Ei42xiRn/FAfZ8meTkJM4991zPBqhzoj8XGuvee+/FaDSyb83Ck+5az9n2F7UVpdx88831O1VaIkmSiI+P1zoMXbNYLFqH4DUdOnTg5ZdeRrE7yVuyG8Vx/LSezeE69h4Uu5NXXnmFdu3aebwNLZ15pmunqKP06EF21QHOSonevXsHXDmBptBbf26xWAgPDz/qQ9C3wsJCKisrCY1tRXBkNHv27NHt5KIvuNO8d+w38Ljf79DXVaqiJaSDr6io4IUXXkCWZQYMb3gGIoNBZsAVfbHb7YwfPx6Hw+HFKJtPlmXOOeccnDV2bKWeLxXnCTU5FQQHB9OvXz+tQ2kwWZaJi4tDPUUWYVUFxYa4bkZ/fbogCIHnp59+AqBj37YnfV5i+zhCIqzMnTvXK2VUm6pVK9dckqNKXynqPcV9Xlr2ie3atWPw4ME4KySczaiSozrAluu6FgiU3erQjIl1gDFjxrBp0yYqKys5cOAA5eXlpKenc/fdd3sqPiFArVq1CkmSiEhMASAsrjUGk5k//1ypcWTaO3jwIOGxrepX6IbHJdQ/7i+qq6u5++67SU9Pp/uAjlx0y9kYDPqtLSvJEudcfRqnDUlj//793HnnnRQXF3vk2J07d6ZNmzauVe/OwFlFV32gDFVRdbNS/qabbgLAdrBpv2f2PAnVCTfeeFOLrIMs+nOhMVq3bs0VV1xBVXE+RfsyjvscVVXZv2E5ZrO5RWWBEJomEFPBH+niiy/mlltuwVZSQ+Fqz+7aVVWVglX7sZXWcNttt/lNvdXG6NOnDyaT6ZiJdUeZBCqcfvrpGkWmP6I/F5pj27ZtAITGJhIam0hpaalHFxz7m4ULFwKHd6b/U0hEFK079eCvv/6itLTUh5H5ltPp5KmnnuLgwYOcNiSN2NYN263u1r5XGzr2bcv69et56623vBOkB51xxhkA1ObqL+Ocs9aBraSGfv36+U0aeLf4+HhUm3TSHW/uiXcxse4i+nRB8A69L/LyhS1btrgW63RNJDzm5JnOZFmm+1kdqKioYObMmT6K8NTat28PgK1EP5P9nuQ+rw4dOmgax+EssU0fK7flSqgOuOWWW/zu+uVkPDJ7EBwcTOvWrQMq5aDgPfv372f9+vVEJrfHFOTaXSIbjcSkdmHPnt2kp6drHKF23KkSIxOS6h+Lind97g+p08B14/3II4+Qnp5Ot7M6cP51pyPJ+k/jJUkSZ1zSi34XpZGZmcn9999/TD2rph53yJAhKHYn1Qc8W8NdS5V7XQsPLr74Yo0jcRk8eDCRkZHY812pZRrLlitjNBoZPny454PzI6I/FxrqlltuASB786rjfr/0wB6qiwsYNmwYsbGxvgxN8EOBkgrsZB599FHSeqRRsbOQit1FHjtuxa4iKncX0atXLx566CGPHVdPgoODSUtLw1kpHVVn3Z0avqWngT8e0Z8LTbFx40YAwlu1IbxVylGPtTSlpaWsXbuW1h3TCIuOO+HzOp9+Hk6nk0WLFvkwOt9RVZX/+7//Y8mSJaR0TeS0i9OadJxBN5xBVKsIpk6dyrfffuvhKD2rT58+ANQWVGkbyHHUFbpi6t27t8aRNF58fDyq4tq1diLuGuwtOdPV8Yg+XRA8yxNjvf5MVVXefPNNAPpd1LB+vee5nTFZTHz00UdUVOhj4VmXLl0AqCuu1jgS76grrsZkMpGamqppHH369KF79+7Yi+T6froxVBXqcmRMZhPXXnut5wPUUMvblidozl0fKKnn0XWZk3q5vv7www99HpNeZGS4dv7FJB1OwxIaHYfFGlr/Pb17//33WbFiBW27t/abSXU3SZI489JedD2jPVu2bOG1117zyHEvvfRSACr2eGYXvNactXaqs8vp2bMnbdq00TocAEwmE1dccQWKDRzFjfudc1a6UslecMEFmtauEQR/0qVLF3r06EHR3h3Yao4deMzZvh4g4C6cBe9oCXU0TSYTk16fRGhoKIUr93uk3rqtpIai1fsJCw9j0qRJAbX6+5/69u0LKjiPGMdxlEvIskyvXr20C0wQAsjKlSuRZAMRiW2JSnbtAlq16vgL6ALd/PnzcTqddDlz0Emf1+XMCwCYO3euD6Lyvffff59p06YR0zqSi0ee0+TMXuYgE5eNOR9reDCvvvoqv/76q4cj9Zzk5GTCw8OpK9LfQH1dkeuaOy2taQsctOTeha6eZFBesbmuB8WiXEHwrJZc1uV47Ha71iFo6ueff2bt2rW065lM6w4NyxASFGLhtIu6U1RUxNtvv+3lCBume/fuSJJEXUGl1qF4nOpUsBVX061bN13c4994442gunaeN5azDJRqiWFDh2leStbTGrw9xJ1eoTEkSWL37t2Nfp0QuNLT0/nxxx8JjU0kvmOPo74XldyByKR2LFq0iJUrVzJgwACNotTO5s2bAUhs363+MUmSSGjXmT1bN1BRUUFYWJhW4Z1Seno6H374IRGxoVx069nIOk7/fiKSJHHedadTdLCUGTNmcNFFF9XXSW+qLl260KlTJ3bt2Y2zzoHB4t878yr2FIOqctlll2kdylGuvPJKvvzyS2x5EqbYht842PJcv6ctZbe66M8FT7nkkkvYsmULhXu20jrtcCpmVVEo3LOVVq1auSbDBOEUWspgT3JyMi+99BIPP/wweUt2k3RZN2Rj066VFLuTvKV7UBwKr77yKomJiR6OVl/ck+eOcgljpOqqw1op0aVz5xZbX93X/fm7775LaWlpfXmqOXPmcODAAQDGjRtHREREk44r6ENubi5btmwhOqUjRrOFsPjWWELCWLx4Mc8880yLyCxypLlz5yJJEl3PuvCkzwuPTSCpc0/WrFlDYWFhQE0ITp06lffee4+I2DCuuPsCzEHNG9gNjw7lirsv4KcpC3jyqSexWCy6LF8iSRJdunRh7bq1KA6lyf20N9QdSgvbtWtXjSNpvLg4V+YHxSZh4PjXfS05Fby4Rxe8yWazYbFYtA5DNwwGg9YhaCY7O5tXX30Vc5CJc65uXNav3oO6snNDJt999x2DBg3inHOOXyrHV8LCwmjfvj179+9DVVS/2th3KnVF1aiKqpsF5MOGDeO11yZQl1eDJcVJY/ZFuMfcr7nmGi9Fp50GXyHu27ePrKwscnJyGvzhTzWhBe+z2+08++yzqKpK50HDkf6x2lmSJDoPuhJJknnhhReortbfCmFvW7NmDQBJXY5+40zu0htVVVm/fr0WYTWIqqpMmDABRVG44KazsASbtQ6pyYwmA4P/dRayQeaVV1/xyGrGK6+8EtWpULmvxAMRaqtidxEGg4FLLrlE61CO0qVLFzp37oyjWEZpYMkkVQV7vkxkZGSzF1D4C9GfC54yaNAgAIoyj86oUp53AHtNNeedd16L2IksCI1x0UUXcfPNN7t2m69rer31wrVZ9XXV3X+LgaxHD9eCXGel6z1FqQbV6Z+79jzF1/35pEmTGD9+PP/73/8AmDlzJuPHj2f8+PGUlPj/9W1L595xHd/JdR8qSTJxHXtSXFzc4nat5+fns27dOpK69CIs5tQTfN3OvghFUfj99999EJ1vfPfdd0yaNInQSCvD770Qa3iwR44b0zqSy+8ahMFo4PHHH2fFihUeOa6nderUCVSweyC7jCfZSmsIDw+vn6T2J+5FJ4rtxM9pyTvWxT264E21tfp6L9OaHnYAa8Fms/HYY49RWVnJOdecRlhUSKNebzAaGHzLAAxGA08++SS5ubleirTh+vbti2J3Blyd9dp81y58vWxUsVqtXHzxUJQaCWd5w1+nOsFeIJOcnBSQ5dsavfRy0KBBfPHFF5SXl1NTU3PKD0Fwe///2bvvuKbu/Y/j7xP23nsPRRAF96ziwL2tWutCW3fd6/7co1qr1llntXW21llH1br3VtyKAxCVJXtDxvn9kYJSEghwkpOQz/Px8F4hIedjqrxIzjnfs2kTXr58CZfaTYqWlvsvMzsneDQIwYcPH4qu96Et8vPzcffePdi5+8DEwqrYbZ6B9QFAbV94AtLZHj58CJ9gd4WXklFn1k6WqNnUF+9i3uH48eOVfrzOnTtDIBAg83USB9PxJz81BwXJ0h1m6rhseufOncFKAGGSYjvzRGkMJAXSo++07Ydr6jmpLDc3Nzg4OCDtQ1SxM47TPkQBABo1asTXaETDaNsBGFOmTEH16tWR8eIjst+llfvrs96mIvNlEvz9/avsddX/y8HBAZaWlkU71gv/PyAggM+x1IKqeh4dHQ2WZWX+4vvaf6RyWJbFoUOHoKOrV7RjHQCcAqRvgB06dIiv0Xhx5swZsCwL/yaKnU3t1ygEjECAf/75R8mTqcaRI0fw/fffw9jcCN3GtIaZdfneeC+Lg4ctOg9vCRYsJkyYgLt373L6+Fzw8fEBIN2RrS5YiQSijHz4+vpq5M9NhQcDsKXsWC+8TRMPHOAKvUYnyiAWi/keQa1o23t/hX744Qc8efIENRp5o0aD8q+SAQC2zlb4olc9pKWlYdKkSbxfr75wx3NeFVsOvnDHenBwML+DfKZLly4AgIJExXcnC5MZsGKgS5euGvmzS1kUfiaePXuGCRMm4MGDB/jqq6/g7OyMSZMmFS1dTUhpXr16hW3btsHIwhq+zUs/y9WrURuY2jrizz//RHh4uIom5N+dO3eQn5cH76DGJW5zrhYAQxNTXLlyRW2XS921axcAoH5oYBn31Bx12wRAIBBg9+7dlX7ebW1t0axZM+R/zObkuqp8yXydDEB6Br46KjyLXpioWLAL79epUyelzaRuqOeEKwzDICgoCAXZmcjPSi/6fEaC9CxcdVm2iqg/df3ZRlkMDAywbNkyGBgY4OO1txDnKb4yjihXiKQbb2FoaIhly5ZpzRtDDMOgRo0akORKX5wX7livXr06z5Pxh3pOuHL//n1ER0fDrlot6Bl+OjPZzN4FpnbOuHDhApKTk3mcULXOnj0rXU2vYUuF7m9iYQ3XGkEIDw9HUpJmH0R96tQpzJ07F4YmBug2qhUs7cyVsh1nH3t0/OYLiMRCjB07Fg8fPlTKdirKy8sLANTqdbswIx+shK3QkuHqwMbGBgDAFsh/nV54NnvhfbUJNZ0ok7ZdzqUs2nigwb59+3DgwAHYuVmjRe8GZX9BKfwb+8C/kQ+ePHmChQsX8vpavnDHc+GO6KqAZVnkfcyGs7OzWl0apUGDBrC1tYXoowCK/icvqOLvuSu8Y71GjRpYsWIF3r9/j4MHD6JJkyZYv349goODUb9+fWzcuBHp6ellPxDRSsuWLYNYLIZf657Q1S/9ui4CHV3UaPslwDBYunSp1rzZevnyZQCAd52S15YX6OjCs1ZDvH//HlFRUaoerUzx8fG4ceMGnH3sYeNsyfc4nDGxMIZXbVe8fPkSz58/r/TjFe6MznyjmW+4sBIWWZEpsLCwQIsWLfgeRyYnJycEBQVBlCaApIz9FKwEECYL4ODggKCgINUMqAao54RL/v7+AICsj3FFn8v6GAdLS0s4ODjwNRbRMFXx6OWy+Pj4YNKkSRDnCZF0S/El4ZNuxkCcJ8KUKVO07ixhX19fAIA4R/rr889pI+o54UrhGenOgQ2LfZ5hGLgENoRIJOJkBS9NkJKSgvv378PFrzZMLBRfnat6gxZgWRbnz59X4nTKde7cOfzvf/+DnoEuuo5sBWsnS6Vuz83PCe2GNEdeXh5GjRqFJ0+eKHV75VF4xrpQjc5YL0iT7uTX9B3rpb1GZwsY6Ovrw8SE21USNAE1nSiTNl9TXBa+z7JWtXv37mHJkiUwNjNEh6FfQFevcn8fGIZBiy/rw8HDBkePHsWePXs4mrT83NzcYGlpifyP2bzNwDVRVgHEuUK1e59aR0cH7du3h0QoXf21LKwIEKcKUKNGjaIDFquaci8Fr6Ojgx49euDo0aN49+4dlixZguzsbIwdOxbOzs4YOHAgYmJilDEr0VAPHjzAzZs3YePpBxsPxc4qsXB0g6NfMJ49e1a0w7kqY1kWly5dgqGJGVyqyb5WpE/dpgCgls/HuXPnwLIsqtfz5HsUzvn9+2c6c+ZMpR8rJCQEpqamyHqTopEHjOTEZkCcK0Tnzp3V+gy59u3bS6+JV8Zy8KI0BqwQCA0NhUBQ7hxqPOo54UK1atUAAFnJCQAAsUiI3PRkVKtWTSt3lpKK0cQmcqF///6oU6cOsqJSkPO+7DdLs9+lIfttKho0aIC+ffuqYEL1UviCXJLLQJLDwNbWFqampjxPxT/qOamMrKwsnD59GsZWtrB09ixxu4NfMAS6ejh06LBWfK++cOECJBIJqjco30HE1etL73/u3DlljKV0p0+fxpQpU6CjJ0DnESGwc1PNJb+8Al3RdlBTZOdkY/jw4Wpz5rq1tTUsLS3Vain4wlkKd/prGktLSwBlLAUvlD732vwagppOlEEbz9AujTZdRiExMRFTpkwBy7JoN6R5ua+rLo+Org46DP0CJuZGWLFiOe7cucPJ45YXwzCoVasWhJn5EOeJeJmBa3n/HiSgjitAtm/fHgAg/Fh2p4XJDFjJp6+piiq1J8HBwQEzZszA8+fPcebMGVhbW+OPP/7AgwcPOBqPVAV79+4FAHg0aFWur/NoEFLs66uyN2/eIDY2Fl5BjSDQkb1Ej1ftRmAEArXcsX716lUAgGdNF54n4Z5rdUfo6ukU/Rkrw8DAAB06dIAouwB58ZkcTKdaWW+kS0B269aN50lK16aN9HqIZe1YFyZLbw8NDVX6TOqOek4qqnBHV05qIgAgNy0JLKu5y1QSfmjrG6gCgQBz586FQCBA8t13YCXyd1qxYgmS77yHjo4O5syZo5UHhLm5uQEAxDkMJPkM3N3deZ5I/VDPSXkdP34ceXl5cApoIPN7sZ6hEex8aiIy8o1WXKat8GDqavXLt2PdzMYeTr4BuHXrtsadVfrXX39h2rRp0NXXQZeRreDoaavS7fsGuyN0UDPk5ORgxIgRuHXrlkq3L0+1atUgzMyHRCThexQAmr9jXU9PD+bm5pAI5f/MxwoZrVwGXh5qOuGKSFQ1djhyRdM6XVFCoRCTJ09GcnIymvWsC2cfbpcVN7EwRvuhzcECmDp1KhISEjh9fEUFBkovSZufXDXOWi/8cxT+udRJUFAQ7OzsIEoqezn4gn93vrdr104Fk/Gj0u/I3LlzB6NHj8aXX36JDx8+wNnZGa6urlzMRqqA3NxcnDt3Tu4R8KUxtXGEhZM7rt+4gZSUFOUMqCaKloEPLrkMfCFjc0s4+QTg/v1wZGaqz05ZlmXx6NEjWNqbw9jcqOwv0DC6+rqwd7fBy5cvOTmqsWvXrgCAzDeadZ1CiVCMnHdp8Pb2RkBAAN/jlMrZ2Rl+fn4QpwnAyjkwl2UBUbIAlpaWare8Dl+o56QinJ2doauri5zUjwCA7H//X9uWqCaVow1nQcrj6+uLPn36oCAtD5mv5V8qJuNVEoQZeejfv3+VXUqtLC4u0gM4xekAWFCj5KCeE0UJhULs3LkTAh1dONesL/d+rkHS16i//vqrqkbjRXJyMm7evAln35qwsHMs99fXaNQKYrEIp0+fVsJ03GNZFlu3bsWcOXNgYKyPbqNbq3yneiHfYHd0GNoc+QX5GD16NE6dOsXLHJ+rXr06wEJtzlovSMmFhYWFWl1vtbxsbGwAOTvWWbH0l7W1alZL0BTUdMIFWgq+uKysqnM97tIsX74cDx8+RPX6nghsVk0p23D0tEPzHnWRkpKCKVOmQCgs45qcSlCzpnTl3/zkHJVvWxnyk7IhEAjg5+fH9yglCAQCtGnTRrocfHopB8qJpcvAV69evUofDF+hHetJSUlYtWoVateujcaNG+PXX39FmzZt8Pfff+Pt27eoW7cu13MSDXXt2jXk5eXBvlrtCp2NZF+tNiRiMS5cuKCE6dTHlStXwAgE8A5qVOr9fOo0gVgswvXr11U0WdnS09ORkZEBKwdzvkdRGitHC0gkEnz48KHSj1WnTh24uroiOyZNbY5+V0RWdCokIgm6dOmiEWcWtmjRAqxEfuglOYAkH2jevLlWv8ignpPK0tXVhbu7O3JSpTsEC/+fdqwTorhRo0ZBT08PaU8TZB5kwEpYpD9NgIGBAYYPH87DhOrBwcEBACDKYIp9TKjnpGJ+//13vHv3Di61GkHfWP5lFSydPWHl6oNLly6p1etQrv39998Qi8UIaF6xM2v8m7YFIxDgr7/+4nYwJRAKhViwYAHWrFkDM2sT9BzXVmXLv8vjGeiKrqNaQaDLYNq0adi6dSuvB97VqFEDAFCgBm/USwrEEGbkoUaNGhrxWlweGxsbSISQeZabpODTfbQdNZ1wzdDQkO8R1Io2rPx1+PBh/PHHH7BxtkLLPg2V2o6azaqhen1PPHz4EEuWLFF5u/39/QFUjR3rLMuiIDUXXl5eMDJSz5MXC1eJFSXL/zslTJUuA19436pK4e8kEokEx48fR69eveDi4oIpU6YAAH766Sd8+PABBw4cQMeOHbXimxNRXOE1xux9K7Z8hd2/X6ep1ypTRFZWFsLDH8DJJwBGZhal3tc7uDEA6Y54dZGTIw2XgbE+z5Moj4GR9HrihX/WymAYBp07d4akQHoGuKbIipSeYd+5c2eeJ1FM06ZNAQCiFNmhF6Yyxe6nTajnhGteXl4Q5uXgXfg1xNy9VPQ5QohibG1t0a1bNwjT82Reaz3nXRqEmfno2bOnVp/JZWBgAAsLC0Aibbgmn7XHBeo5qYyIiAisWbsW+sam8Grctsz7Vw/pCkYgwJw5c5CamqqCCVVLIpFg//790NXTR0DTil0mytTKFt5BjfHo0SNERERwPCF30tPTMWrUKBw8eBB2btboPaEdLO3V4yB5Zx979BwfCjNrE6xZswZz5sxBQUEpF+VWosIlWPOS+F9atnBZ2Fq1avE8SeXY2NgArPRa6v/FavmOdWo6USZ9/ar7fm1FmJhwc51xdXXr1i0sXLgQRqaG6DjsC+jpy77kLFcYhkFIn4awc7XGgQMHsHPnTqVu77/s7OxgbW1dJXasi7IKICkQFx0soI7q1asHU1PTosurylK4071ly5aqGosXChfZ1dUV3bt3x8WLF/HNN9/g9u3bePToESZOnAhbW36WiyLqLT8/HxcuXICRuTVM7Zwr9BhG5lYws3fBjRs3quw1UG7dugWxWFTm2eoAYO9RDSaW1rh+/braLJtaeORjfg4/L3hVIT9X+sqPq6PFNG05eFF2AXLjMlG/fn04O1fs37Kq1a5dG3r6ekVntf2X+N8z2Rs0aKDKsdQC9ZxwrXCJqpeXjkJUkAdTU1M4OTnxPBUhmqVfv34AZP9sUPi5wvtos887pa1vvheinpOKSk1NxYQJEyAsKIB/aB/oGRqX+TWmtk7wadoBiYmJvC31qUw3btxAdHQ0/Ju2haGpWYUfJ7htDwDS1QDUUVRUFL7++mvcvn0bPkFu6PFdW7W7nJu1owV6T2wHBw8bHDlyBN9++y2Sk1X/utnLywtmZmbIS+B/yeDcf2fQ9EuY2dnZAfh0dvrnJAVMsftoG2o6USZNXulCGQwMDPgeQWmePn2K8RPGgwWL9mHNYW4jf0UiLunq66LjNy1gammMFStW4OjRoyrZLiD9++3v7w9RVj7E+SKVbVcZCg+kU+cd63p6emjSpAkkuQzEMq6Ww7KAKFUAaxtrtf5zcEHhQ1bi4+Ohp6eHoKAgREdHY+7cuWV+DcMw+Pvvvys1INFcly9fRnZ2NjzqN6hUxB38gvD6ygmcOXMGX375JYcTqofC5fS8ajcs874Mw8AzsAGeXv0Hr169kl73i2dWVlawtLRE0vtUsCxbJX9g+/guBTo6OpxdF8TDwwNBQUF4+OghRDkF0FXzs/0L39Dv1q0bz5MoTl9fH7UCa+F++H2wYoD5bLV3lgXEGQI4OjrC0bH810/UdNRzwrVBgwbBxcWl6E32GjVq0NkUpFyq4s8O5VWjRg34+PggMjoKEqEYAj1puCQFYuS8T4efnx98fX15npJ/lpaWRb+3srLibxA1QD0nFSEUCjFlyhR8+PAB3k3awdarhsJf616vBTIS3uHOnTv48ccfMXv2bCVOqlq//fYbAKBexz6VehzvoEawdnLHsWPH8N1336nVTsLbt29j4sSJyMzMRP12gWjQvhYYgXr219jMCN3HtsWFP28h/F44BgwYgPXr18PHx0dlM+jo6KBevXq4ePEiRNkF0DXh7zV7bnwmGIbR+CXACy/hwuYzgGnxE0XYfOn/a+tqNNR0QlQnPz+f7xGU4unTpxg+fDhyc3PRfkhzOPuo9vupqaUxuoxshb9+Pos5c+aAYZiiE8uULSAgANeuXUN+Sg6MndRjFZ6KKDzrXt13SDdt2hRnzpyBKJWBjlHxnhdeerVJ2yZV/n3Bcq0FIRQKcenSJYXvT2+SabcDBw4AAJwC6lXqcRz96uDN1VPYv38/evfuXaX+XrEsiytXrsDI1BwO3n4KfY1XUCM8vfoPrl69qhY71hmGQYsWLXD06FHERyfByUt93jzgQnpSJhJjktGkSRNOj2rs3r07Hj58iMzXybCqrb5ndrIsi8xXSTA0NES7dhW73iBfatasifv370OcDeh+9nMVWyA9Sl7Tl9KrDOo54ZKpqalGHXhD1I+6rMLDJ4Zh0KZNG7zZsgW58ZkwcbMEAOTEZYCVsGjbtuylmrXB5zvWLSxKv4SSNqCek/Jau3Yt7ty5A/tqteHZsHW5vpZhGAS064vctGT8+eefCAoKUtkbpsr04MED3Lp1C95BjWHvXrkDmBiBAA27fo1TW5bit99+w/Tp0zmasnJOnDiBWbNmgYUEbQc2RfV6nnyPVCZdPR20HdAEVg7muH3iEQYNGoR169ahXr3Kvb9UHk2bNsXFixeR8yEd5tX5eZ9DnC9CfmIWgoKCYG6uuTsLABQd1C7JK3mbJF/aJ01ZIU8ZqOlEWarqSVAVxcVlPtXNvXv3MHbsWOTk5KDtwCbwru3GyxzWjhboNro1jm44j1mzZiE3Nxd9+/ZV+nYL39/N/5it0TvW8z5mQyAQoGbNmnyPUqqGDaUnh4rSGRg4F38vR/TvCrGNGpW9MrOmU3jHelRUlDLnIFXM8+fPcf36dVi5+cDEunJHSBmYmsPOtyaePXuM27dvV6l/mBEREYiLi0NAs1AIBDplfwGkZ7YLBDo4f/48hg0bpuQJFdOrVy8cPXoU4eefwembqnX9jPDzzwFI/4xc6tixI5YvX47MV0mwrOWotj/k5sZlQpiZj669emncdYgKj/ATZzHQNf8UenGW9LmuUUPxM3SqEuo5IUTdqGsDVa1Zs2bYsmULcmMzinas58ZmFN1GpAfyFDIzq/hyzVUB9ZyU14MHD7Bjxw6YWNvDP/TLCn3v1dHTR60ug3D79zVYvHgxGjVqpNFnmLIsi7Vr1wIAmvYK4+QxazZvjxuHd+DPffswePBg3lfIOnDgABYsWAADI310+jYETt6a89+LYRjUDw2EhY0pzv1+EyNGjsDP635GkyZNVLL9li1bYsmSJch6m8rbjvWc9+lgJWyVuE6pi4sLAECSxwD4zxlu/+5s19bLSVHTiTLl5ubC2Ljsy75oi6p2xvqFCxcwddpUiEQitBvSDD5B3Ky2WlG2LlboPrYNjm06j0WLFiE5ORmjRo1S6mv+2rVrAwDyEvm/fEtFsWIJCpKy4evrq/b/Xt3c3GBra4uU9CQAkmK3Fe5Y1/RVdhSh8I51Dw8PZc5BqhCWZbFy5UoAgGfDNpw8pmeDVkh89RirVq3Cnj17oKOj2E5odXf8+HEAgF+jVgp/jZGZBdxr1sXDh3fw7t07uLnxcxTa5+rWrYv69evj7t27+PA6AS6+DnyPxImUuDQ8v/kG3t7eCA0N5fSxTU1N0alTJxw8eBA5H9Jh4mrJ6eNzJf15IgCgT5/KLYvIh8Ilc8U5xV+4i/89OLVatWo8TMU/6jkhRN3QGetStWrVgr6+frE3BPISs2BkZISAgAAeJ1Mfnx/kp2kH/HGNek7Ka+XKlWBZFv6hX0JXv+IrcRlZWMP3i854cfYgtmzZotFLwl+9ehV37tyBT52mcK7GzdlBOrq6aNZ7GE5sWoz169dj0aJFnDxuRfz9999YsGABjM0M0XV0a9g4WfI2S2VUq+sJA2MDnPr1Mr4b9x22bd2G4OBgpW/X2dkZtWvXxqPHjyHKFULXSE/p2/yvzEjpZdnat2+v8m1zzdXVFYDsM9bFuQxMTEy09jIv1HSiTGlpaWq/o06VCi9hBwAikQi6uuVa0FmtHDx4EAsXLoSOng46Dw+Bm596XO7SxtkSPceH4timC9iwYQOSk5Pxf//3f0rbn2NjYwMfHx9ExUSDlUjAaOAS5HlJ2ZCIJEVng6szhmEQFBSEc+fOQVIACD67Wo44k4GlpSVnl9NVZ5r3t4yovSNHjuDmzZuw9faHtRs318Ays3eBk389PH36FLt37+bkMfmWk5ODv44cgbG5FbyDy3fEda2WnQAA+/btU8Zo5cYwDKZNmwaGYXD5wF2IRWK+R6o0lmVx+eBdsCyL6dOnKyX+AwYMAACkP03g/LG5IMzIQ867NAQFBSEwMJDvccqt8MWp5D+rPElypEfPeXp6qngiQgghstAZ61J6enqoWbMmClJzIRFJIBGKUZCWi8DAwCpzUGllGRkZFf3e0NCQx0kI0SzPnj1DeHg4bL0DYOFU+R04zgH1YWRpg7/++guZmZkcTKh6BQUFWLZsGQQCHbTsP4rTxw5oHgp7z2o4cuQInjx5wuljK+rZs2eYO3cuDIz00U2Dd6oXcq/hhA7DWkAkFGH8+PH4+PGjSrbbtWtXgGWR+TpJJdv7nDAzH7kfMhAcHKwWJ1RUlqWlJczMzP498P0TlgXYPAYeHh70MyEhHCkoKCj6vaq+X2qKtLS0ot+np6fzN0glbd26FfPnz4eBsT66j22jNjvVC1nYmqHXhFDYuVrhzz//xIwZM4od1MC1Jk2aQCIUIzdeM38uzY5JAyC9DI0mKNxPIM781G2JCJDkMqhZs6ZW9Jx2rBNORUZGYskPP0DXwBB+rXpw+ti+LTrDwMQMa9aswePHjzl9bD4cPHgQ6WlpqNOuJ3TKeXRc9YYtYWZtj3379iE1NVVJE5ZPQEAA+vXrh9SEdDy6FMH3OJUWcScKsW8SERoaqrTlV6tVq4amTZsiNy4TeR/Vb7ma1MfxAIDBgwfzPEnFGBsbw97eHpLc4jGX5AICgaDoiHlCCCH8ojPWP/H39wcrYSFMz0VBai7Ags5W/4yBwaezbPX0VH/mICGa6tSpUwAAl1rcnAXDCARwDmiA/Px8XLx4kZPHVLVt27YhOjoaddr1gq2rF6ePLRDooO2QiWBZFvMXLFDqG8myiEQizJ49GwUFBWg3pBmsNXyneiH3Gk5o1rMuUlNTsWTJEpVss0uXLjAyMkJmRBJYiWp/XsmIkO4MU8X1aVWBYRh4eXmBzWPw+Y9+bD7AiunAd0K4FB8fX/T7uLg4HidRP59feiEyMpLHSSqGZVmsX78ea9asgbm1CXqND4W9mzXfY8lkbGaE7mPbwsXXAf/88w8mTZpU7KAPLhWu7JIVmaKUx1cmlmWRHZ0Kc3NzNG7cmO9xFOLn5wcAEH+2O0Py7++15dKrtGOdcCYtLQ3jxo1Dbk4O/EP7wNDMktPH1zcyQUCHryASiTBhwgQkJKjnWb6KyMzMxJZffoGBsSnqtutd7q/X0dVDo24DkJOTgy1btihhwooZN24cbGxscOf0E2Sn55T9BWqqIE+IG8cfwNjYGNOnT1fqtkaOHAkASH0Qq9TtlJcwMx9Zb5Lh7e2Ntm3b8j1Ohbm5uUGSz4D97JIvkjwBnJyc6A15QghRE9pwNLOiCi9Tkp+ai4K03GKfI7QznZCKunDhAnT1DWDtzt33Eztf6Zkqmrhj/enTp9i8eQvMbR3Q/MtvlLINV7/aCGrdDREvXmDjxo1K2YY8Fy9exKtXr+Df2AduflXrmtWBzarBydsOZ8+exevXr5W+PVNTU/Ts2RPCrHxkRavuzXpxgQgZLz/CxsYG7dq1U9l2lc3LywuspPhy8IVnsHt5cXuACyHa7P379zJ/T4BXr14V/V4VHeHa1q1bsWnTJljYmaHHuFBY2JnxPVKp9A310HlECDwCnHHp0iVMmzYNYjH3K90GBQXBzc0N2dGpEOep9oDGysqJSYMouwAdOnTQmNe7he9RiLM/vZdT2PPCS7NWdbRjnXAiJycH48aNQ0xMDLwatYG9r3KWjbZ284Vviy74+PEjRo8erbFLtmzYsAFpqalo3H0gjEzNK/QYQa27wsrRFX/8sRdv3rzheMKKMTc3x4QJEyAqEOHOKc1dVeDBhefIzczD8OHD4eio3KV06tati8aNGyPnQwZy4jKUuq3ySLn/AayExZgxYyDQwGvTFHJ1dQVYQJIv/ZiVSH9PZ6sTQoj6oDPWP/H29gYACNPzUJCeV+xzBBp9DURC+PLu3TtER0fDys0XAh3u/g0ZW9nCyMIG165dV/kZ2ZWRmpqKSZMnQyIRo+PImTAwNlHatkIGjIWlgwu2bt2Ky5cvK207/3Xp0iUAQO0v/FS2TVVhGAa1/v1zqeo5HTJkCHR0dJD2KF5lZ61nPE+EpECMsLCwYqu1aDofH+nlIiXF3ogvfhshpPI+f59YE8/KVqaUlE8HSSUnJ/M4SfkdO3YMa9euhbmNKXqMbQNTS2O+R1KIrp4OOgz9Am5+jjh//jx+/PFHzrfBMAyGDBkCiUiCtGeJnD++srAsi9TH8WAYBoMGDeJ7HIU5ODjAyNio2CqxhTvWtaXnWvvOxL1793DhwgW+x1AaMzMz9O/fH+bmFdtpWx65ubmYMGECHjx4ACf/evBqHKrU7bkFN0NeRipehV/F6NGjsXnzZpiZqffRWZ979OgRfv/9d1g7uaNehz4VfhwdXT20HjQeB5dPx/z587F9+3a1uP5mt27dsH37dry4E4X67WtpTOQLFeQJ8fjKS9jY2GDgwIEq2ebUqVPRp08fJN9+B6OuAWAE/J65l5uQiayoFAQFBWn80fEuLi4AAEkeAx0jtujIeNqxTggh6oN2rH/i7u4OABBm5Be9eV/4OQKNPtiPEL4Uvu9h6+3P6eMyDANbb3+8C7+Ke/fuacTSlTk5ORg7diziYmPxRd/h8KhZV6nbMzAyRo+J32P3vFGYOnUqfvvtN9SsWVOp2wQ+XU/X3MZU6dvig8W/fy5VXTfY2dkZPXr0wMGDB5EVlQIzHxulbk+cL0La0wRYWVtXmWXgCxW+2S7OAQrPyZNo2RvxhKjCw4cPi37/4LPfk+I0aeW0ly9fYv4C6TXVu4wIgYmFZr3frqMr3bl+eN1Z/PHHH6hVqxa6du3K6Ta6d++OzZs3I/lZAsyr20LPVP0PTMuKSkF+Ujbat2+vUZdEYRgGXp5eeB7xDCwLMAwg+fdAOQ8PD36HUxGt3LGekpKCUaNGIS8vr+w7a7C4uDjMnz9fqdvIyMjA+PHjce/ePdj5BqJGaG+lR4lhGFRr0Rmigjw8fnwX33zzDTZs2ABbW1ulbpcL6enpmD59OliWRYfhM6Crp1+px/Op0wQBzULx4NoZbNiwAePGjeNo0orT0dFBWFgY5s6di+e33qBB+1p8j1Qur+5HIz+3ACOHD4ChoaFKtunn54fevXvjwIEDSH+WAMtA5Z4lXxpWLEHSjRgwDIPp06dr1A+ZshTuQC/coS7Jk/55Cne4E0II4Z+mt4ZLNjY2MDAwgChbumPdxMQEFhYWfI+lNmjHOiHld/bsWelOcC9ud6wDgJ1PTbwLv4qzZ8+q/Y71rKwsjB07Fo8fP0atkM5o3F01ZwXZe/ii27gF+GvVLAwfPhybNm1C7dq1lbpNd3d3XLt2DbGRifDwd1bqtvgQ+0Z6JpoqDzwbNWoUjh47ipTwDzDxsIJAV3k9Sn0UB0mBGCOGD4exsWbtOClL4So80rPapAcQirMZ6Orq0oGEhHCkoKAA165dg5G5NUztnfHu9RNERkbSKlgaTCgUYuasmSjIL0Cnb1vC0l75J1Iqg56BHtqHfYEDK09h8eLFaNCgAacrxRoaGmLatGmYPn06km7GwLGNr1q/1yDOFyHl7nsYGBhgypQpfI9Tbu7u7nj27BnYAoAxkL7nbmtrW+V+dpFHK3es37p1C3l5eRDb+0JiXxWPiGSh+/wCLly8iPlK3EpkZCQmTJiA6OhoOPgFI6BdXwgEqjljmmEE8G/bGwIdXTx/dBP9+3+N1atXqeTo74rKzc3FuHHj8OHDBzTtNRSuNbh5MR06dDJiXz/Dli1b4Orqip49e3LyuJXRvn17LF26FC/vRaN+u0C1jth/vbwXDYFAgB49eqh0uxMnTsT5C+eR+iAWJu6W0DNXzU79/0p9HI+CtFz0799f6W/4qIKzs/SNJOkOdbZoSXgnp6p1rUFCCCFVA8MwcHR0xPvEWEDCws1VO472VhTtWK+67t+/jy1btiA3N5fvUZRCIBCgTZs2GDBggEpfG8XExCA8PBzW7r7QN+b+7GVLZ08YmJrj5KlTmDp1qsoOTC6v2NhYjBs3Di9fvkRAs1C0/2aqSv87+NZrhs5jZuPvDd/j22+/xQ8//IA2bdoobXt9+vTB3r17ceXgXdhNCIWxmZHStqVqybFpuHv6CUxMTNCpUyeVbdfR0RGDBg7Cr7/+ivQXibBS0sHwwsx8ZLxIhKubK/r166eUbfDJ2dlZegBhzqcTnSS5DLw9POhyL4Rw5Ny5c8jKyoJH/RCY2bvg4+snOHz4sEbuuOMay7JISEgo+vjz36uzP//8ExEvIhDQ2AeeNTX7RCELW1M07V4HF/bewsqVK7Fs2TJOH79Dhw7466+/cP36dWS8SISFvwOnj88VlmWReDUaohwhxk+erJHvU39+MhujL33v3a2GG89TqY5W/tRS+AKKNTABa2LF8zRKwLKAjh4ESnqhyLIs9u/fj+XLlyMvLw8e9UPg06w9GEa1b3QxjAB+rXrA0MwSb67/g0GDBmHChAkYOHCgWiyJ/rmkpCSMGzcOT548QUCzUDTrFcbZYxsYm+LL6cvw+/wxmDt3LtLS0hAWFsbrzmxjY2O0bNkSJ0+eRGp8OqydLHmbpTxyMvMQF/UR9evVh52dnUq3bWFhgdmzZmPy5MlIuBIFl441VL4kfF5SNtIexcHB0QETJkxQ6baV5dNS8Pj3/6XPKS0FTwgh6kOTDsBTBXt7e7x9+xaA9Npl5BN1+xmfcINlWYwdOxZZWVnST1S17wms9H/u3r0LPz8/NGjQQGWb3rx5MwDAuVYjpTw+IxDAqWYDRN86hz///BNDhgxRynYq48qVK5g5axbSUlNRr0MftB74HRgeDtLxb9oW+kbGOLZuPiZOnIhvv/0WY8aMgZ6eXtlfXE7VqlXD2LFj8fPPP+PwurPoEPYFbJwtOd+Oqr2LiMfZ3ddQkCfE98sWq3xFl2+//RaHDh1C2qM4mPnYQNeI+/92yXffgxWzmDRxklL+bvBNIBDAw8MDryJfgmUBVgSwQu1ZNpYQZROLxfjll1/AMAxcajWEgakFDEzNsW//fgwdOhTW1tZ8j8ir9+/fIzk5GdUbtMTbJ3cRHh7O90hlysrKwqZNG2FgrI/GXYL5HocTNRp649mN1zh58iSGDBnC6YmSDMNg8eLF6P1lbyTf/QADGxMY2qvfpXHSnsQj510amjZtqpY/Pyui6GS2fAaCAhZgP31OG2jljvXGjRvD0NAQeQkvUWDnBeip51HVFSVIeAWmIAetWnXm/LHfvHmD77//Hnfv3oWeoTFqdRkEe99AzrejKIZh4NmgFczsXfD89D6sWLECp06dwpw5cxAQEMDbXJ+7dOkS5s2bh+TkZNQK6Yx2w6Zy/kLe2skd/WavxcHl07Fy5Urcv38f8+bN43V5/JCQEJw8eRJRTz9ozI71t88+ACzQqlUrXrYfGhqKLl264Pjx40h9HAfrINXFSCIUI/FKFMACSxYvgYmJicq2rUx2dnbQ0dGBJE+6zFzhDnZtCj0hhKgrHx8fvHnzBkZGVedMOi58/vObjY1yr+OqaeiM9aqJYRj4+fnh3r17gI4uxFaukFi7g7VwAFS0IhrnWBZMThoEKe8gSH4LJj8bJiYmKv0Z9NixYzh69CjM7F1g76O81+zuwc0Q+/gWfv75ZwQHByMoKEhp2yqP3NxcrFmzBnv27IGunj46DJ+B2q268DqTT52mGLBgE/5aNQtbt27FrVu3sHjxYnh5eXG+rREjRkAkEmHTpk04uPo06rcPRFBIDejoaN730YI8IW6ffITHV15CV1cXCxcuRIcOHVQ+h5mZGcaNG4dFixYhNTwWdk253Rmcm5CJ7LepqFu3LkJDQzl9bHXi4eGBly9fghV+en1OO9YJ4cbx48fx6tUrONdsACML6esIzwatEXHhL2zevBn/93//x/OE/Dpz5gwA6aVVwQAvb1/C69ev4evry/Nk8v35559IT89A4y7BMDRR/2uGK4JhGDTuEowj689hy5YtWLNmDaePb2tri+XLlmPEiBGIP/8aLp1q8LYyrCxZ0SlIufcBjo6OWLJkica+xi08y16SB0gMin9OG2jmf7VKsrS0lF6LuiAXuhFXAFEB3yNxhkn9AN2YB7C0ssKYMWM4e9y0tDQsXboUvXr3xt27d2HnG4hGgybxulP9czYe1dFo4CQ4+tfFkydP8NVXX2HevHlISkribaZ3795h0qRJ+O6775CekYnWg8ajw/AZ0FHS8lZ2bt4YuGATPGs1wMWLF9GtWzfs2rULQqFQKdsrS/PmzaGrq4s3D2J42X5FvHkonbV169a8zTBz5kw4OTkh9UEschMyVbbdpFsxEKbnYciQIWjYsKHKtqtsOjo6cHBwAPvvEvCSPOn12/g86IQQQojUzz//jFWrVtH1/v7DyurTilraflbJf9EysVXXpk2bMGXKFLg6O0EnKRp6Ly9D//5f0Hl9HUzKe0Ai5nvEsrEsmKxk6LwNh97D49B78g90Yp/BVBfo27cvDh06VLSakjIVnq02e/Zs6Bkao2aHfko9Q1vPyAT+oX2QX1CA4SNG4OTJk0rblqLu3LmD3r17Y8+ePbB19cKgRVt436leyM7NG4MXb0Ngiw54/PgxvuzTB7/99htEIhGn22EYpuisdQsLC9w8/gD7lp9A1OP3YFmW020pi0QswbMbr/HHD8fx6HIEPD09sWvXLpVftu1zvXr1gq+vLzJefURBKneXrmBZFsm33wEApk+fXqVX8/l86VhaUY4Q7rAsi99++w0CHV14N2lX9HnnwIYwsrDBgYMHkZGRweOE/GJZFseOHYOOnj6qNWiBgKbSA5iOHTvG82TyicVi7N27F/qGeghsVo3vcTjl4usABw8bXLhwAbGxsZw/fsOGDTFv3jyI80SIO/MKohz12P+XE5eBxCvRMDU1xcaNGzX6QHp7e3sA0jPWJQVMsc9pA63csQ4AgwYNQq9evSDITobeiwtAgeZfy02QFA29V9dgaGiAdWvXcrLjSCgUYufOnejUqRP27NkDQ3MrBHUfitpdBsHAxJyDqbmjZ2SCmu37oe6XI2Fi44hDhw6hU+fO2Lx5M/Ly8sp+AI4kJCRg8eLF6N69O86ePQvXGkEYvHgr6nfso/QXR6ZWtujzv58QOmwKRCyDZcuWoXv37jhy5AjnL9TLYm5ujlatWiHpQyrioj6qdNsVkf4xEzEv4hAcHMzrizozMzMsX74cOgIdJF6KgjhP+QdGZL5OQubrZAQGBmL8+PFK356qOTk5QVLASJeay5cuq6upRwMSQkhV4urqirZt2/I9htr5fGlbVS9zq+6q4rK4RMrQ0BBhYWE48fff+P333zF06FC4uzhBJzkGeq+uQj/8KHTehgP52XyPWpJYBEH8K+g9Pgm9p2egEx8Bc30BunbtitWrV+PixYuYM2eO0s9WZ1kWt27dQr9+X2Ht2rXQNzFHcK9vYWKt/EtK2HrVQK3OAyEUSzB9+nSMGjUKr169Uvp2/ysjIwMLFizAsGHD8P7DBzTuNhCDv/8Fdu4+Kp+lNAZGxug0ahZ6Tl4CPSMTrFy5EgMGDMDz588531bLli1x7Ogx9O/fHxlJWTj562UcWnsGMS/i1HYHu0Qiwct70di77AQu7rsNcQGLcePG4eDBg5wuF1sRurq6mDp1KsACyXffcfa4WZEpyE/OQdeuXXn/MyrbpzPcGFpRjhAOJSQk4M2bN7Dx9IOB6af37AU6OnCqWR8F+fm4c+cOjxPy69WrV9Kz0+s0haGJGbzrNIGhiSlOnjwJiUTC93gyPXjwAPHx8fCt4wF9w6r3Oqhm02pgWRanTp1SyuP37NkTY8aMgTAzH3GnX6nkPfbS5CVmIeH8G+jq6GDNmjVqvVKCIgovo8sWSH8BtGNdKzAMg3nz5qFfv35gslOh/+wsmJw0vseqGJaF4P0T6L65CTNTU/yyZQuCg4Mr/bA3b95Ez549pddSF4pRvWU3NBo4GbZeNSo/sxJZuXqj4dfjUaNtb0gYHfz888/o1q0bLly4oNTtfvjwAd9//z06duyIvXv3wtTaAd0nLkL/Oetg68r90m7yMAyDOm174NuVf6Behz6IjYvH7Nmz0a17dxw6dEilZ7APGjQIAHDjaDhYiXq+aC90/Vg4wEItrmsSFBSEiRMnQpRTgITLUUp9w6MgNRdJN2NgamaGFStWVMk3rB0cHABWGnlJAUPXqyWEEKLWzM3NZf6e0I51bcAwDGrVqoXJkyfj+PHjOHDgAIYNGwZbSzPoxEdA/+Fx6ETeAvJUt7KTXGIhBLHPof/wGHTf3oO+KA8dO3bE+vXrcenSJSxZsgRt2rSBoaFyl54UiUQ4d+4cBg4ciG+//RYRLyPgFFAfDQdMgLm98s+QL2TnUxMN+o+DtUd1XLt2Db169cK4ceNw9+5dlezAvXz5Mnr27IkDBw7A3qMaBi3cghZfjYSuvvoum1qt/hf4Ztlu1ArpjGfPnqF//6/x888/c/6a3czMDDNnzsRffx1Bu3btkBCdhOObL+DQmtN4++yD2uxgl4gliLgThb0/nsDZ3deRlZKDfv364eTJkxgxYoTaNKBZs2Zo0qQJcj5kIDeu8md/smIJUsNjoaevJ11ds4orfD0uKZCe5QZo1xvxhCjL06dPAQDmju4lbjN3kJ5A9OzZM5XOpE4OHz4MAPBvJj1TXVdPH9UbhiAuLg43b97kczS5Cq8B7xlQNQ8+8vj3z6XMa92PGjUKYWFhKEjLle5cz1ftiYeF8pKyEX/2NRgJsHrV6iqxWqyFhQUEAgEkwk9nrGvTintavZaeQCDArFmz4OTkhNWrV0Pv2TkIfZuCtdSgawFIxNCJugOdpGi4uLpiw/r1lV5OUygUYtmyZdi7dy8YRgC3Os3h1agN9AyNORpa+RiBAC6BDeFQPQhv71xAzP3LGD9+PLp164bZs2dzei3PyMhIbN26FSdOnIBYLIaVgyua9ByMgGahEOjw90/MyNQcbQaPR8POX+Hm0T14dOEY5s2bhw0bNiAsLAy9e/dW+jVN69Spg44dO+LkyZO4889jNOxYW6nbq6gnV18i6vF7NGjQAG3atOF7HADSHfz37t3DxYsXkfYoDlZKuN66RChGwsU3kIgkWLJ4sUqWpuRD4Yt0cRYDsPSinRBCiHozNTWV+XsivaQXQEvGaovCa6/7+fnhu+++w+nTp7F161a8fv0aOh+jwRqYADwul8wIcwGxCGZm5hg0/Bt89dVXxS7loGzx8fE4fPgwDhw4gMTERIBhYOcbCM+GrVW6Q/1zxpa2qNPzGyRHRyD6zgVcvHgRFy9ehI+PD/r27YsuXbpwfsBQQUEBVqxYgT/++AM6unpo0W8EGnTur7RLsHHN0NQMHUf8DwFN2+LUL8uwefNmXLlyBT/99BPn3+s8PT3x008/ISIiAps3b8aZM2fw9y+XYOtihXrtAuEd6ApGoPp/U2KRGC9uRyH8/DNkJGdBV1cXffv2xTfffKO2ZzJPnDgRN27cQEr4Bzg7mlVqdcKMV0kQZuUjLCxMK65N+ukMN6boDLfCzxFCKu748eMAACu3kqu0WDh5QKCrh5MnT2LUqFFqc6CSqiQmJuLAgQMwt7GHT52mRZ8PbtsDjy4cx6ZNm9CkSRO1uwxHSkoKAMDUyoTnSZTDyNQQOro6SE1NVdo2GIbB5MmTkZeXh7179yLu9Es4tasOHQPV/ZyYn5SN+NOvwIokWL58OVq0aKGybSuTQCCAhYUF0vNTwP57vELh63VtoBmvNJSIYRh888038PDwwIwZM4CXlyHyagiJnerOMK4wsRC6L69CkJGAOnXqYM2aNZV+IS8SiTBhwgRcuXIFprZOCGjfF2Z26vlCRhG6+gbwadYBjv718Oz0Phw9ehTv3r3Dli1bKn3mwOvXr7FhwwacPXsWLMvCzt0HjbsNhF+jEF53qP+XmY09QodOQpMeg3D35D48OPsXfvzxR2zZsgVDhgzB119/rdQd7LNmzcLTp09x9/QT6BvqIbiVv9K2VREvbkfiyuF7sLG1wZIlS9TmhyiGYfD999+jT58+iHsQC0N7Uxg5cfcmFMuy+HgzBgXpeQgLC0OrVq04e2x1U3hZDPG/K4dq8vVrCCGEVH3GxsYyf0+A+vXr4+eff9b4ZfNI+enp6aFz587o2LEjzp07h127dinleozlYWRkg+7du+Orr75S2UEwEokE165dw759+3D58mVIJBLoGhjCLbgZXIOawNhKPXZO2Xj6wcbTD+lxb/H+4Q1EvXqEH374AStXrkSHDh3w1VdfITAwsNLbycnJwahRoxAeHg47dx90/W6eSleL45JHYH0M/XE7zu/6GY8uHEPfvn2xefNm1KpVi/Nt+fn5YeXKlXj9+jW2bt2KkydP4p/frsDa0QL1QmvCJ9hdJZfOEgnFeH7zNcLPP0dWWg70DfQxYMAAhIWFwdHRUenbr4yAgACEhobizJkzyI3NgLFLxS7dIhFJkPYoHkbGRhg2bBjHU6qnwvctWaH0V+Eb84SQirt16xbOnj0LC2fPorPTP6erbwDnwAZ49+A6fv/9d7VYrVNVJBIJFi5ciLy8PLQaPLHYgXeOXn7waxiC8NsXsW/fPvTr14/HSUsqPDEoKTYVti6qO3hTVdISMyAWiZV+cBXDMJg5cyZYlsWff/6p0p3r+cnZiDsj3an+448/ol27dkrfpiqZm5sjPS4VrFC6+pGZmRnPE6mO+uz941nbtm3x66+/YsyYMciIvAURK4HEXr2uw1WMSAjdiIsQZCUjNDQUP/zwAwwMKr/E2V9//YUrV67AxrMGanUZCB3dqnEEm4m1Her1GYlnp/cjPDwce/bswTfffFOhx0pKSsLq1atx9OhRsCwL52qBaNJjELyD1e/Its+ZWtki5OsxaNRtIO7/cxD3Tu3H6tWrsWfP7xg7dgx69eqllPktLCywYcMGfPvtN7h+NBwZyVlo2r0udPV0ON9WeUjEEtz55zHunXkKC0sLbFi/Qe1evFtYWOCnn37C4MGDkXg5Ci7dAqBrxM2/yczXych6k4ygoKAqeV31zxUuQyPO1r5laQghhGiezw94VPbqQppGR0cHLVu25HsMwiOBQIDQ0FCEhobyPYpKSSQSHDlyBL/99huioqIASJd6daklXaVNR0+f5wlls3DygIWTB6q17Iq4Z/cQ++QWjhw5giNHjqBOnToYMWIEmjdvXqHHZlkWkyZNQnh4OPybtkWHEf+Dnhov+64IfUNjdBg+Ha5+tXBqy48YNWoU9u7dCzc3N6Vsz9fXF0uXLsWYMWOwdetWHD16FGd2Xcfd00/RoEMt+NR2U8oZ7GKRGM9vvsG9s8+QnZ4DIyMjDB06FIMHDy46KFoTjB49GmfOnEHqw7gK71jPfJ0EUU4Bhg4frtIVL/hUeDabRAhIRAzMzMxUciAHIVVVVFQU/u///g+MQAC/kG5y39v1bhyKxJePsGbtWlSrVg1NmzaVeb+qJCcnB99//z0uXboEr9oNEdiiY4n7tB4yAe+eh+OHH34AwzDo06eP2ry/37ZtW6xevQq3/n4Edz8nGJtXndeGYpEYlw7cAQB06NBB6dsr3LkuFotx4MABxJ35d+e6vvJ2j+Yn5yDu9CuwQgmWLl2qkj+nqpmZmYF9D7DiTx9rC/rJ5TNBQUHYuXMnrK2toRt1B4LkGL5Hkk0ihu7LKxBkJaNbt25Yvnw5JzvVAeDJkycAAK9GravMTvVCAh1deDduC+DTn7O8zp8/jx49e+LIkSOwc/dFnxkrMGD+BvjUaao20S2Lkak5mvUeipFrD6Bpr6FIy8jE/PnzMWrUKCQlJSllmx4eHti1azeqV6+OJ9de4dCa00iOS1PKthSRnpSFoxvP496Zp3B1dcXOHTsREBDA2zylKbzGpChXiMSr3FxvvSAtF8m3YmBubo7ly5dX+SWgil645zDFPiaEEELU0eerKnH1Mz4hRHNFR0djwIABmDt3Lt7GxBRdP73BV2PhXLOB2u5U/5y+kQk86rVA48FTUaf3cNh6+yM8PByjR4/GhAkTkJ6eXu7HvHfvHq5fvw7voMboPGa2xu9U/1xgi44I/WYKMjIysGvXLqVvz93dHQsXLsTff/+N3r17IyMpC6d3XMWBVf/g/ct4zrbDSli8uh+NP5b+jcsH70JSwOLbb7/F6dOnMXnyZI3aqQ4A1apVQ6tWrZCXmIXchMxyfz0rYZH+JAEGBgYYOHCgEiZUTyYmJhAIBGBFDCAC55eHIEQbiMVihIeH4/vvv0fvL7/Ex48fUa1FF5iVcikYPUNj1OzYH2KxBCNHjsTkyZNx6dIl5Ofnq3By1cjMzMQff/yB7t2749ixY3CuFoiu4+aDkXEQj5mVLXpPXw59Y1MsWrQIYWFhuHz5MkQifq7F/TlXV1dMnDgJ2ek5OLDqH8RFfuR7JE5kpmTjr5/P4sOrBISGhqrsLG6BQIA5c+agd+/eyE/KQdyZV5AIxUrZVn5qLuLOvAQrlGDJkiXo2LHkQR1VgYmJCVgxwIoY6OjoQF9f/V+XcEUtd6zn5+djxowZcHZ2hpGRERo1aoQzZ86oZNs+Pj745ZdfYGpmBt3IW2CyklWyXYWxLHSi7kKQmYj27dtj4cKF0NHh7szfhg0bAgCenz2ErCTuXkCpg5y0ZDz9508AQIMGDcr99Xfu3MHEiRORnZOL0KGTMWTxVngFNdKYHer/ZWBkjOZfDsPwVX/Ap05TXL9+HaNHj4ZQKFTK9hwdHbFnzx707dsXSR9Ssf+nU7h98hFEBar7QUUsluDBhef4c9kJxL6R/hvat28fvL29VTZDRQwcOBAtW7ZE7ocMpD9NqNRjSUQSJFyKhEQkwaJFi7TiGm6FL9QlecU/JsrHZ88JIURTff5ilHasE3VAPedPXl4exo0bhydPnsDRvy6aDvsfAtr10djLtTEMA2s3XwR1C0OjQZNg5eqD8+fPY/bs2eV+rBcvXgAA/Bq3gkDA72poyuDfpA0A4Pnz5yrbpouLC+bPn49jx46ha9euSPqQiqMbz+PktstIT8qq1GMnxiTj0NrTOLPrOnIz8jF48GCcOnUKEyZM0OgDnwtXQkx7Uv7X6dlvUyHMykevXr20alU1hmFgYmIMiAFWzKjsUhqEeq6pRCIRIiMj8c8//+Dnn3/GqFGj0LRZMwwePBh//vkndI1MUavLILgFNyvzsazdfFGvzyiYO7rjzJkz+O6779CkSRMMHjwYP/30E44ePYrnz59r3M723Nxc3Lt3D9u3b8eoUaPQokULLFmyBEkpqWjScwi+mr0Ghibyz6R18vFH2JJfUa1BC9y/fx9jx45FSKtWmDVrFg4ePIiXL19CLFbODtiyDBkyBJMnT0ZOZh4OrzuDf3ZcRVpiBi+zVFZedj6uHw3HH0uPI+Gt9ITRxYsXq3TfikAgwNy5c9GtWzfkf8xG3NlXkIi4/W9bkJ6H+NMvIckXY+HChejcuTOnj69OClfYY4XS32vqfrKKUMul4MPCwnDgwAFMnDgR1apVw/bt29GpUydcuHChwsuElUf16tWxauVKjBw5EnqvrqEgsD2gpx5vagk+voFOUhRq166NxYsXc7pTHQA6duyIp0+fYufOnbi9ZzUc/ILhGtwM5g6uGvsPI/NjLN4/uI645/fASiTo2bMnvvrqq3I/zoYNG8CyLL6avQbOvjWVMCk/TC1t0GvqUpzcvARPLp/C+fPn0b59e6Vsy9DQEHPmzEHr1q2xYMEC3D39BBF3o9Cse1141VLu37H3L+Nx5dA9pCakw9raGjNnzkS7du004u81wzBYtGgRevXqheTwWBg5m8PAumLXXE0J/4CC1Fz069cPrVu35nhS9VS4I50VMcU+JsrHd88JIUQTfb5jvaqvKkM0A/WcP2/fvkV0dDTMHd3hH/plldqBbGrjiNpdB+P69mW4cuUKCgoKynWWS+HB8tcPb4dL9UBYO7kra1SVEwkLcOqXZQCARo0aqXz7bm5uWLJkCYYMGYJly5bh9u3biHkRhwbtAxHcyh8CHcXP0RHmC3Hz74d4fPUlwErfc5o4cSKcnTXz4JD/CgoKQp06dRAeHo6C9DzoWxiW/UWQXsog7WkCBAIBBg0apOQp1Y+JiSmy0zLBikA71lWIeq6+8vPz8eHDB7x//x7v37/Hu3fvEBMTg7dv3+LDhw8lzp42sbaHi3ct2FcLhKWrd7l+PjB3dEP9fmOQmfgBia8eI/XdGzx4+BDh4eFF92EYBo5OTvBwd4f7v79cXV2LfpmYmHD2Z1eURCJBYmIiPnz4gJiYGERFRSEqKgrR0dF49+5dsR3fjt41UL1hS9QO6QJjc0uFHt/Mxh49Jy3Gx3eReHj+KF7fvYKjR4/i6NGjAKQ7DT09PeHl5QUvLy94enrCzc0Nrq6uMDc3V9r7ywzDYOjQoahTpw6WL1+ORw8e4c3DGLj7OaFm02rwCHAuV5dVjWVZJLxNwtPrr/HmQQxEQjGcnZ0xfvx4dOrUiZf35QUCARYuXIiCggKcOnUKCRcj4djKBwwHz6MwKx9xp19ClCvE3Llz0b17dw4mVl+Fq+1JCgAj66pzqQJFMCwX6wpz6Pbt22jUqBGWL1+OqVOnApAepR0YGAh7e3tcv35docfJyMiAhYUF0tPTK7wT5ddff8WqVasgsXSGqPoXAM874JicNOg9PQNLczPs379fqdeDvnbtGlatWo2ICOlR4CbW9nDwC4Z9tUCYWDsobbtcyUlLwsfXTxAf8RBZH2MBAF5eXpgwYQLatGlTocecPn06Tp48icbdB6FpzyHQrUJLzSW9j8LfG75HQvRL7Ny5E3Xq1FH6NnNycrBlyxbs2LEDIpEIrtUc0KxHPdg4W3K6nfSPmbh+LBxRj99DIBCgT58+GDduHCwsKnYNND5dv34dI0eOhL6lEVy6+EOgW77g58RmIO70S3h5e2Hfn/uKLTVblSUlJaFVq1ZFH//++++oVasWb/Nw0SdNoE49J4QQTfL69Wv07NkTAHD8+HF4eHjwPBGRRxsaRT3nF8uy6NevH54/fw5TWyd41G8JO5+aGrH8e2mEudmIf/kQb+9cRH5WetEZS+X1yy+/YO3atdAzMESddr3QsHN/hd9AV0cSsQjPrp3FjcM7kJrwHvXr18e6det43fHIsizOnDmDpUuX4uPHj7Bzs0a7wc1gYVv2NTQT3ibhzK7ryEjOgo+PD+bMmYN69eqpYGrVOnfuHCZOnAjzGnawa6xYs/MSs/DhxAuEhoZi5cqVSp5Q/fTo0QOR0W/AioGQkBCsW7eO13m0oVHq3HN12DWhqp17BQUFePr0KZ49e4aoqKiiA+gSEhJkPg/6xqYwsrCBibU9TGwdYWrjCDM7J+gZcbtjWywsQObHWGQnJyArKR7ZKQnITUtGXmaazPtbW1vD09MTHh4e8PT0hJ+fH4KDgyu9w72goKDowIK3b98iJiam6GCD2NhYmausGptbwsbFE04+/nDyDYBztUCYWVX+0iIsyyLpfRTiXj9D7OtnSIiKQEpcDIT5eSXua2JqClcXF7i6usLd3R1ubm5wd3eHh4cH7O3tIZCxBH1FZ7p48SJ27NiBe/fuAQCMzQzhE+wO3zoecPSwBSNQjxPIUhMz8Pr+W7x+8BapCdIz7D09PTFgwAD07t1bLQ4gFwqFmDRpEi5dugRTL2vYt/Cq1PcCUa4QsScjIMzIw9SpUzFkyBAOp1VPc+fOxeHDhwFILwV8/PhxXudRZc/V7oz1AwcOQEdHByNGjCj6nKGhIb755hvMnDkT7969g5ubm0pmCQsLw+3bt3Ht2jUI4l9C4uSnku3KJBZC9/V1QCLG4sWLlbpTHQCaNWuGpk2b4s6dOzhw4ADOnT+PyBunEXnjNIytbGHr5Q8brxqwdPKEQJf/v0YSsRjp8TFIjnqBpKgXyE6WLmOvp6eHtm3bolevXmjWrFmlQjZy5EiEP3iAm0d24dm1Mwhs0QH+TdrAxsWToz+FaomEBYh6dBsvbpxDxK0LkIjF6NatG4KDg1WyfWNjY0ycOBE9e/bEihUrcPHiRexbcRIBTX3RqGNtGJpU7sAFYb4Qd08/xaPLLyAWSVC/fn3MmDEDNWrU4OhPoHpNmzbFwIEDsXv3bqSEf4BtA8W/F4oLRPh4LRq6urr4cemPWrNTHUCJH+z5OLJWG6lTzwkhRJN8/iaDrhr8nE20G/WcXwzD4JdffsGaNWuwf/9+PD21F7r6hrDzrQkbzxqwdq8GPUPNODskPysDyW8jkBT5HElRL8BKxNDX18d3332HoUOHVugxhw8fDjc3N6xYsQK3j/2O8NOH4dcoBDW/aA93/zoyr6WqjlLj3+Pp1X/w9Mo/SP8YBz09PQwcOBCTJk3i/VqVDMOgXbt2aNy4MZYvX46//voL+386hbYDm8Kzpvxr+T65+hJXD98HIH0vZcSIEbz/WZQlJCQEzs7OiH8dD+s6LtAxKLvdac+kS8dr07XVP1d4TVZA+t4QUT5V9pxlWWRnZyM1NRXp6elFv9LS0pCWlobk5GQkJiYiISEBcXFxSEtL42S7FSXQ0YG9nR0cHR3h4OAAOzs7WFtbw9LSEpaWlrCwsCj6ZW1tXeHvZYsWLcK+ffv+81kGhuaWsHLzgaG5NYwsbGBkYQ0jC2sYW9pA10A1jdfR04elsycsnT2LfV4sEiI3PRm5acnITU9BbkYKctNSkJuehPAHD3D//v1i969Tpw62bt1arudIIpFgz549OHjwIGJiYmTuPDextIadpx8s7Jxgae8ES3tnWDt7wNrZHUamytl5xjAM7Ny8YefmjdqtugAAWIkEmakfkRIbg5TYGKR9jEN6YizSP8YhOuY9IiIiSjyOpaUlXFxcMG3atEofXMYwDFq1aoVWrVrhzZs3OHDgAE6cOIHHV17i8ZWXMLMygVctV3jXdoOjly1nO/QVwbIsUuLTEfnoHSIfvUNybBoA6feZjh07ok+fPqhfv75arRyrp6eHFStWYPTo0bh79y50DHVh09CtQjNKhGLEn30FYUYehg8frhU71YHiDdemfQ2AGu5YDw8PR/Xq1UscUVB47e8HDx7IDH1+fn6x639kZFT+WhMCgQCLFy/Gl19+iaSYcEhS3gE8vTBjcjPACPMQFhaGFi1aqGabDIOGDRuiYcOGyMrKwpUrV3D27FlcvXYNMfevIOb+Fejo6cPM3oXXF6yshEXWx1iICqRHjBkaGqJVq1Zo27YtWrZsydmZyT4+Pjjy11/YtGkT9u7di+uHtuP6oe2wdfWCsYUVJ9tQGRZIfPsSednS66R5+/hg2tSpvCz95OHhgXXr1uH69ev48ccf8fTaK7wJj4GNi2WlHjc1IQM5GblwdnbG1KlT0bZtW7WKd0VNmDABV69eRfTTaOQn5yi8kIYoRwhRdgHGjRsHf39/5Q6pZgwNDSHQEUAilgCgpeZURZ16TgghmuTzHevqcCQ/0W7Uc/5ZWFhg7ty5GDp0aNGSpLHP7iHu2T0wjABm9i7QUfMdlsLcbGQlxRd97FejBnp0746OHTvCxsamUo/doUMHtG7dGocPH8aOHTvw5PJJPLl8EmbW9rBycq3s6EqXl52JxOhXAKRvTn711Vf45ptvlH4yRXmZm5tj0aJFaNKkCebNm4eT2y7DycdO5mtskVCMhOgk2NjaYMXyFahfvz4PE6uOjo4OBgwYgOXLlyP2VAR0DMt+qzU3Pgs1a9ZUyWqB6ujzN+LpwHfVUFXP161bhy1btnAwsepIxGLEx8cjPj6+7DtD2uWVK1cWPXeKEIvFOHDgQLHP6RmbwtjCGgJdPbAsK92BnZ5crtn5YmBqAX1jM+RmpCA/M73o8+Hh4Xjz5k253nd8/Pgxli1bJvM2Y3MrWNo7Q9dAevJVdnoystOT8eHVk8r9AThmaGoOQxNz5GVnIDXhA4R5uUW3FR5QUvh+Lld8fHwwY8YMTJkyBXfu3MGpU6dw9uxZPLocgUeXI2BkaghrRwtARW+FZ6XmID0pE4D00matWrVCp06d0KJFC7U+gMrQ0BBr165FWFgYXj5/ibyk7HKvEAsAouwCCDPy8eWXX2LcuHFKmFQ9aXPP1W7HelxcHJycnEp8vvBzsbGxMr/uhx9+wIIFCzifx8bGBqtWrcL48ROQmprE+eOXR5s2bTB+/Hhetm1qaoqOHTuiY8eOEAqFuHfvHq5evYorV64iMvINLzN9zt3dHV988QWaNWuGhg0bwsBAOcu0GxsbY/LkyRg9ejQuXbqEkydP4urVq0h6H6WU7SmTk5MTOvTtg44dO6JGjRq873Ru2rQpDhw4gD///BObNm3Ch1cJlXo8Y2NjjB07FmFhYVXqiClDQ0P88MMPGDt2LFLiU8r1tV+0+ALDhg1T0mTqi2EYNKjfALdu3YKXlxesrDTsQBgNpW49J4QQTWFjYwMXFxcIBAJqFuEd9Vx9uLm5YezYsRg9ejQiIiL+fT1+BY8ePSp2XVF1pG9ggObNm6N58+b44osv4O7O7fXQ9fX10a9fP/Tt2xcPHz7EsWPHcOrUKcQ8vV/2F/NMR0cHzZo1Q7du3dCqVSsYGan3CgSdOnWCh4cHJk+ejNjXsv/9A0BAQABWrVpVZa6lXpaePXvi999/x4cPHxS6v56eHkaMGMH7+zB8qV+/Pm7evAmBjkBlqyZqO1X1PDU1tWIDapCMjAxkZ2eX62t0dHSwcuVK/PPPP7h79y4+fvwIYU4W0nOylDSlapmZmaFu3bpo3rw5qlevXq6vrV27NmbOnIl9+/YhKjoa4s+uJ5+TkYqcDM3+O2VqagYvL0/MmDFDKY+vq6uLJk2aoEmTJpg9ezbu3buHs2fP4vz58/jwunLvrZeHiakJOnbsiDZt2uCLL75Q653p/2VmZoaNGzdi1KhRePXqVYUfp0uXLpg1a5ZWtb127drQ09ODUCiskpf7KY3aXWPdx8cHfn5+OHHiRLHPR0ZGwsfHB6tWrcLEiRNLfJ2sI+jc3Nyq9PVxCCGEaB5tuH4bQD0nhBBS9WlD06nnhBBCqjrqOfWcEEKI5tPqa6wbGRkVC3ahvLy8ottlMTAwUNpZyoQQQggpH+o5IYQQovmo54QQQojmo54TQggh3OHvwthyODk5IS4ursTnCz+nLctIEUIIIZqMek4IIYRoPuo5IYQQovmo54QQQgh31G7HenBwMF6+fImMjIxin79161bR7YQQQghRb9RzQgghRPNRzwkhhBDNRz0nhBBCuKN2O9a//PJLiMVibNmypehz+fn5+O2339CoUSO4ubnxOB0hhBBCFEE9J4QQQjQf9ZwQQgjRfNRzQgghhDtqd431Ro0aoU+fPvi///s/JCYmwtfXFzt27EB0dDS2bdvG93iEEEIIUQD1nBBCCNF81HNCCCFE81HPCSGEEO6o3Y51ANi5cyfmzJmDXbt2ITU1FbVr18bx48fRokULvkcjhBBCiIKo54QQQojmo54TQgghmo96TgghhHCDYVmW5XsIZcjIyICFhQXS09Nhbm7O9ziEEEIIAOpTedHzRQghRF1RoxRHzxUhhBB1RY1SHD1XhBBC1JUqG6WWZ6xzofB4gYyMDJ4nIYQQQj4p7FIVPa6Nc9RzQggh6oqarjjqOSGEEHVFPVcc9ZwQQoi6UmXPq+yO9czMTACAm5sbz5MQQgghJWVmZsLCwoLvMdQe9ZwQQoi6o6aXjXpOCCFE3VHPy0Y9J4QQou5U0fMquxS8RCJBbGwszMzMwDAMr7NkZGTAzc0N7969o2Vy/oOeG/nouZGPnhvZ6HmRT52eG5ZlkZmZCWdnZwgEAl5n0QTUc81Az4189NzIR8+NbPS8yKduzw01XXHq1HNA/f4uqQt6XuSj50Y+em7ko+dGNnV7XqjniqOeaw56bmSj50U+em7ko+dGPnV6blTZ8yp7xrpAIICrqyvfYxRjbm7O+18udUXPjXz03MhHz41s9LzIpy7PDR0FrzjquWah50Y+em7ko+dGNnpe5FOn54aarhh17DmgXn+X1Ak9L/LRcyMfPTfy0XMjmzo9L9RzxVDPNQ89N7LR8yIfPTfy0XMjn7o8N6rqOR2GRwghhBBCCCGEEEIIIYQQQgghhJSCdqwTQgghhBBCCCGEEEIIIYQQQgghpaAd6ypgYGCAefPmwcDAgO9R1A49N/LRcyMfPTey0fMiHz03hAv090g+em7ko+dGPnpuZKPnRT56bghX6O+SbPS8yEfPjXz03MhHz41s9LwQrtDfJfnouZGNnhf56LmRj54b+bT1uWFYlmX5HoIQQgghhBBCCCGEEEIIIYQQQghRV3TGOiGEEEIIIYQQQgghhBBCCCGEEFIK2rFOCCGEEEIIIYQQQgghhBBCCCGElIJ2rBNCCCGEEEIIIYQQQgghhBBCCCGloB3rhBBCCCGEEEIIIYQQQgghhBBCSCloxzohhBBCCCGEEEIIIYQQQgghhBBSCtqxTgghGignJwe9evXCnj17+B6FEEIIIRVEPSeEEEKqBmo6IYQQovmo50QRtGOdEKLWcnJyYGNjg+XLl/M9iloxNjbG2bNnkZOTw/coaqljx474/fffkZuby/cohBBCQD2Xh3peOuo5IYSoF+q5fNR0+ajnhBCifqjpslHPS0dNl9LlewBtwrIsLly4gPz8fDRv3hxmZmZ8j6QS9+/fL/fX1K1bVwmTqJ+UlJRyf421tbUSJlFfxsbG0NXVhYmJCd+jqJ3mzZvjxo0bGD58ON+jqJ3IyEgMHDgQpqam6NmzJwYNGoQ2bdqAYRi+RyNVAPVccdRz+ajnpBD1XD7qOVEmbe05QE2Xh3peNup56ajpslHPiTJRz8tHG3oOUNMVQU2Xj3ouHzVdimFZluV7iKpo1qxZuH79Oi5cuABAGvl27drh/PnzYFkW7u7uOHfuHHx8fHieVPkEAoHC/7BYlgXDMBCLxUqeSj2U57kppC3PzefGjBmDFy9e4Ny5c1r3Tbo0kZGRaN++Pfr164dRo0bB1dWV75HUyp07d7B7927s27cPiYmJcHR0xNdff40BAwYgODiY7/GIhqCef0I9l496rhjquWzU89JRzwkXqOfFUdNlo54rhnouHzVdPuo54QL1vDjquXzUdMVQ02WjnpeOmk471pWmRo0a6N69O3788UcAwP79+9GvXz8sXrwYQUFBGDlyJEJCQrBr1y6eJ1W+HTt2lPtrhgwZooRJ1M/8+fPLHa158+YpaRr1dfnyZYwZMwa2trYYPnw4PD09YWRkVOJ+2nLUZSEzMzOIRCIUFBQAAHR1dWFgYFDsPgzDID09nY/x1IZEIsGZM2ewe/duHDlyBNnZ2fD398fgwYPx9ddf0w9HpFTU80+o5/JRzxVDPZeNeq4Y6jmpDOp5cdR02ajniqGey0dNLxv1nFQG9bw46rl81HTFUNNlo54rRpubTjvWlcTMzAwrV64sWi6if//+ePz4MZ48eQIAWLJkCTZu3Ih3797xOabak0gkeP/+PRwdHaGvr8/3OIQnAoGg6PeyfijStqMuC4WFhSn0Q+Jvv/2mgmk0Q1paGkaOHIn9+/cDkP7dCgkJwaRJk9C5c2eepyPqiHrODeo5Aajn8lDPy496TsqLes4dajqhnstHTS8f6jkpL+o5d6jnBKCmy0M9Lz9tazpdY11JdHV1kZ+fD0D6DejcuXMYPHhw0e0ODg5ISkriazyN8fHjR3h5eeHMmTNo3bo13+MQnlCkZNu+fTvfI2iMq1evYvfu3Thw4ABSUlIQGBiIwYMHQ09PD7/++iu6deuGWbNmYeHChXyPStQM9Zwb1HMCUM/loZ4rjnpOKop6zh1qOqGey0dNVwz1nFQU9Zw71HMCUNPloZ4rTlubTjvWlSQwMBC7d+/GgAEDcPjwYSQnJxc7MuPt27ewtbXlcULNUZUXVbh//365v0bbll4BtGeZIsKtZ8+eYffu3fjjjz8QExMDe3t7DBkyBIMGDSp2vZcJEyZgxIgRWL9+fZWLPKk86jl3qOfFUc8JUQz1nHCBes6tqtp06rliqOekIqjnhAvUc25V1Z4D1HRFUdNJRVDTace60sydOxddu3YtinmzZs3QqlWrotv//vtvNGjQgK/xiJqoX7++wtd70dalV0jZ3r9/j/DwcKSnp0MikZS4/fOjd7VFcHAwHj9+DAMDA3Tv3h0bNmxA+/btiy1x9LlWrVph69atKp6SaALqOVEE9ZxwgXpeEvWccIV6ThRBPSdcoaYXRz0nXKGeE0VR0wkXqOclUdOlaMe6koSGhuL+/fs4c+YMLC0t0a9fv6LbUlNT0aJFC3Tv3p3HCYk6oOVWFJeXl4eDBw/i/v37MmPGMAy2bdvG03T8yMvLw5AhQ3Dw4EFIJBIwDFN0tOnnPzxqY+QtLS2xZcsW9OnTB+bm5mXev3v37oiKilLBZETTUM+JIqjniqOel0Q9l496TrhCPSeKoJ4rjnouGzVdNuo54Qr1nCiKmq44anpJ1HP5qOlStGNdiQICAhAQEFDi81ZWVli1ahUPExF1U5nlViQSCd6/fw9HR0fo6+tzOJX6efv2LVq1aoXo6GhYWloiPT0d1tbWSEtLg1gshq2tLUxNTfkeU+VmzpyJQ4cOYfHixWjSpAlCQkKwY8cOODk5YfXq1YiNjcXOnTv5HlPl8vLy0KtXL1SrVk2hwAOAsbExPDw8lDwZ0VTUc1IW6rliqOeyUc9lo54TrlHPSVmo54qhnstHTS+Jek64Rj0niqCmK4aaLhv1XDZq+ieyz88nhKi9jx8/wsvLC1evXuV7FKWbNm0a0tPTcfPmTbx8+RIsy+LPP/9EVlYWfvzxRxgZGeGff/7he0yVO3DgAIYOHYoZM2agZs2aAAAXFxe0bdsWx48fh6WlJdavX8/zlKpnaGiI//3vf4iIiOB7FEIIKRP1nHpOPZeNek4I0STUc+o5QE2XhXpOCNE01HRqOvVcNmr6J7RjnSMCgQC6urooKCgo+lhHR6fUX7q6tGAAqZzCJUiquvPnz2PMmDFo2LBh0fU6WJaFgYEBpk2bhjZt2mDixIn8DsmDxMRENGzYEABgZGQEAMjOzi66vXfv3jh06BAvs/GtZs2aiI6O5nsMooGo54QP1HPqOfVcNuo5qSjqOeED9Vy7ew5Q0+WhnpOKop4TvlDTtbvp1HP5qOlSVBqOzJ07FwzDFMW78GNCSOXl5OTA09MTAGBubg6GYZCenl50e5MmTTB16lSepuOPg4MDkpOTAUiXVbGyskJERAS6du0KAMjIyEBeXh6fI/Jm8eLF+Prrr9GqVSu0bduW73GIBqGeE6I81HPZqOfyUc9JRVHPCVEe6rl81HTZqOekoqjnhCgXNV026rl81HQp2rHOkfnz55f6MakYU1NTzJs3D97e3nyPQnjk7u6O9+/fAwB0dXXh4uKCmzdvolevXgCAZ8+ewdDQkM8RedGoUSNcvXoVM2bMAAB07doVy5cvh5OTEyQSCVatWoXGjRvzPCU/fv75Z1hbW6N9+/bw8vKCl5dX0RGGhRiGwZEjR3iakKgr6rlyUM8JQD2Xh3ouH/WcVBT1XHmo6YR6Lh81XTbqOako6rnyUM8JQE2Xh3ouHzVdinasE5WKiYlBTEwMmjdvXvS5hw8f4qeffkJ+fj769++PHj16FN1mYmKCefPm8TApUSetW7fGkSNHiv4uhIWF4YcffkBqaiokEgl27dqFwYMH8zyl6o0fPx779+9Hfn4+DAwMsGjRIty4cQODBg0CAPj4+GDt2rU8T8mPR48egWEYuLu7QywW4/Xr1yXuQ0c5E1Jx1HNSEdRz2ajn8lHPCVE+ajopL+q5fNR02ajnhCgf9ZxUBDVdNuq5fNR0KYbVlgtGKNnOnTsr9HXa9o2pR48eyMrKwtmzZwEACQkJ8Pf3R0FBAczMzJCYmIj9+/cXHRVF5EtISICTkxPOnj2L1q1b8z2OUsXExODOnTvo0qULDAwMkJeXh++++w4HDx6Ejo4OunTpgrVr18Lc3JzvUXknkUjw+PFj6OjooEaNGnRtKULKiXquGOo5d6jn1HNZqOeEVA71XHHUdG5Qz6nn8lDTCak46rniqOfcoaZT02WhnpPP0Y51jggEgnJ/DcMwEIvFSphGfTk7O2PChAlFy2gsX74cc+fOxZMnT+Dl5YUOHTogKysL169f53lS9adNkSeEEFWhniuGes4d6jkhhHCPeq44ajo3qOeEEMI96rniqOfcoaYTQspCh1VwJCoqiu8RNEJKSgrs7e2LPj5+/DhatmwJHx8fAECvXr0wc+ZMvsYjRK1dvny5Ql/XokULjifRHJcuXcLff/+Nt2/fAgA8PDzQuXNntGzZkufJiLqiniuGek5IxVHPy496TsqLeq44ajohFUdNLx/qOSkv6rniqOeEVBz1vPy0vem0Y50jHh4efI+gEezs7Ir+saWlpeHmzZtYunRp0e0ikQgikYiv8TSKqakp5s2bB29vb75H4dzChQvL/TUMw2DOnDlKmEZ9hISElOsaJSzLau2RugUFBejfvz/++usvsCwLS0tLANLvOz/99BN69uyJP/74A3p6evwOStQO9Vwx1HPuUM+Lo56XRD2nnpPyo54rjprODep5cdrQc4CarijqOako6rniqOfcoaYXpw1Np54rjpouRTvWiUq1bdu26LocFy9ehEQiQY8ePYpuf/bsGdzc3PgbkEcxMTGIiYlB8+bNiz738OFD/PTTT8jPz0f//v2LPVcmJiaYN28eD5Mq3/z588v9NdoQ+QsXLvA9gsZYsGABDh8+jKlTp2LKlClwcHAAACQmJuKnn37C8uXLsXDhQixatIjnSQnRTNRz+ajnn1DPZaOeK456TojyUdNlo55/Qj2Xj5quGOo5IcpHPZePmv4JNV026rniqOlSdI11JYqPj8e2bdtw//59pKenQyKRFLudYRicO3eOp+n4kZCQgF69euHGjRvQ19fHjz/+iAkTJgAA8vPz4eLigq+//hpr167leVLV69GjB7KysnD27FkA0ufK398fBQUFMDMzQ2JiIvbv349evXrxPCkh6s/LywshISH47bffZN4eFhaGixcvIjo6WrWDEY1EPS+Jei4f9ZwQ7lDPCZeo57JR02WjnhPCHeo54RL1XDbquXzUdEK4Q02XojPWleTRo0cICQlBbm4u/Pz88PjxYwQEBCAtLQ0fPnyAj4+PVh4l5uDggGvXriE9PR1GRkbQ19cvuk0ikeDcuXNa+bwAwO3bt4t+4AGAnTt3Ijc3F0+ePIGXlxc6dOiAFStWUOQJUUBcXBwaNWok9/ZGjRph7969KpyIaCrquWzUc/mo54Rwh3pOuEI9l4+aLhv1nBDuUM8JV6jn8lHP5aOmE8IdaroU7VhXkv/9738wNTXFgwcPYGxsDHt7e6xZswatW7fG/v37MXr0aOzZs4fvMXljYWFR4nNGRkYICgriYRr1kJKSAnt7+6KPjx8/jpYtW8LHxwcA0KtXL8ycOZOv8YgaGjZsWJn3YRgG27ZtU8E06sXV1RUXL17EqFGjZN5+6dIluLq6qngqoomo56WjnpdEPSflRT2Xj3pOuEI9Lxs1vTjqOakIarps1HPCFep52ajnJVHTSXlRz+WjpkvRjnUluXbtGqZPnw53d3ekpKQAQNHSNH369MHVq1cxbdo0XLp0ic8xlW7nzp0AgEGDBoFhmKKPyzJ48GBljqWW7Ozs8PbtWwBAWloabt68iaVLlxbdLhKJIBKJ+BqPV15eXmAYptT7MAyDN2/eqGgi9XD+/PkSz4tYLEZcXBzEYjHs7OxgYmLC03T8GjJkCObNmwdLS0tMmjQJvr6+YBgGr169wurVq7F//34sWLCA7zGJBqCeS1HPFUc9l496Lhv1XD7qOeEK9fwTarpiqOfyUc/lo6bLRj0nXKGef0I9Vxw1XT5qumzUc/mo6VK0Y11JJBIJHBwcAACWlpbQ0dEpCj4A1KpVSyuOaAkLCwPDMPjqq6+gr6+PsLCwMr+GYRitjHzbtm2xdu1amJub4+LFi5BIJOjRo0fR7c+ePdPaJXtatmwpM2Zv377FtWvXEBgYiDp16vA0HX/kXatEKBRi8+bNWL16Nc6cOaPaodTEzJkz8ebNG2zZsgW//PILBAIBAOn3ZpZlMWTIEDoalSiEei5FPVcc9Vw+6rls1HP5qOeEK9TzT6jpiqGey0c9l4+aLhv1nHCFev4J9Vxx1HT5qOmyUc/lo6ZL0Y51JfHy8kJUVBQAQCAQwMvLC2fPnkXfvn0BANevX4elpSWPE6pG4XNQeF2Xwo9JSUuXLsXLly8xdepU6OvrY8WKFfDy8gIA5OfnY9++ffj66695npIf27dvl3vbw4cP0b59ewwYMEB1A6k5PT09fPfdd3j27Bm+++47/P3333yPpHI6OjrYvn07Jk+ejBMnThQdmerh4YFOnTqhdu3aPE9INAX1XIp6rjjquXzU8/KhnlPPCXeo559Q0xVDPZePel5+2t506jnhCvX8E+q54qjp8lHTy0fbew5Q0wsxLMuyfA9RFU2dOhVHjx7Fy5cvAQCrVq3ClClT0Lp1a7Asi4sXL2LKlClYtmwZz5MSdZOeng4jI6OiH4wAIDc3Fy9fvoSbmxusra15nE49zZs3D8ePH8e9e/f4HkWtbN68GVOnTkVmZibfo6hcTEwM7OzsYGRkJPP23NxcfPz4Ee7u7iqejGga6jmpKOp5+VHPZaOeU89J5VHPSUVRz8uPei6ftjadek64Qj0nlUFNLz9qumza2nOAml5IwPcAVdWsWbPwxx9/QCgUAgAmTpyIhQsXIjk5Genp6ZgzZw6+//57nqck6sjCwqJY4AHAyMgIQUFBFHg5HBwc8OzZM77HUDtnzpyBsbEx32PwwsvLC4cPH5Z7+9GjR4uOTiWkNNRzUlHU8/KjnstGPaeek8qjnpOKop6XH/VcPm1tOvWccIV6TiqDml5+1HTZtLXnADW9EC0FryRWVlaoV69e0ccMw2D27NmYPXs2j1OpXuvWrcv9NQzD4Ny5c0qYRr3s3LkTADBo0CAwDFP0cVm08Vo4pUlOTsa2bdvg6urK9ygqt3DhQpmfT0tLw+XLl3H//n3873//U/FU6qGsxViEQmHRNWAIKQ31XIp6Lh/1nBvU85Ko59Rzwh3q+SfUdNmo59zQ5p4D1HR5qOeEK9TzT6jn8lHTuaHNTaeey0dNl6Kl4FUsMjIS+fn58Pf353sUlQgJCQHDMOX+ugsXLihhGvUiEAjAMAxyc3Ohr6+v0DcchmEgFotVMJ16kffDYlpaGl68eIGCggLs2rUL/fv3V/Fk/JL3d8bKygo+Pj749ttvMXz48Ar9G9REGRkZSEtLAwB4enpizZo16N69e4n7paWlYebMmXj06BFiYmJUPCWpKqjniqGey0Y9L456Tj3/HPWcqJK29RygpstDPVcc9Vw+avon1HOiStRzxVX1ngPU9PKgpstGPS+Oml4S7VhXkrVr1+L69evYu3dv0eeGDh1adIRUnTp1cOLECdjb2/M1IuHZ27dvAQAeHh7FPi5L4f21iawfFhmGKYrZsGHDUKNGDZ6mI+piwYIFco8o/C+WZfH9999j5syZSp6KaDrqOSkL9Vxx1HOiCOo5UQbqOSkL9Vxx1HOiCOo5UQbqOVEENV1x1HSiCGp6SbRjXUlq166NVq1aYc2aNQCAf/75Bx07dsTIkSNRq1YtzJ49G/3798f69et5npQQQqqGGzdu4Pr162BZFtOnT0f//v1Rt27dYvdhGAYmJiaoV68e6tevz9OkRJNQzwkhRLWo50QZqOeEEKJa1HOiDNRzQghRPWp6SXSNdSV5+/ZtseVn9u3bBy8vL2zcuBEAEB8fj127dvE1nlrIzMxEeno6JBJJidvc3d15mIgQ9VbRJVS05d9TkyZN0KRJEwBAdnY2evfujcDAQJ6nIpqOel426jkh5UM9Lx31nCgD9Vwx1HRCyoeaLh/1nCgD9Vwx1HNCyod6Xjpqekm0Y11J/rsQwOnTp4tdd8DT0xPx8fGqHkstbNy4EStXrkRkZKTc+2jDNU3kXcOkNAzD4Ny5c0qYRr0ULuFUXoMHD+Z4EvXi6elZoWu3aMO/p/+aN29esY/T09NhamoKHR0dniYimop6Lh/1XIp6Lh/1XDbqueKo54Qr1PPSUdOp56WhnstHTVcM9ZxwhXpeOuq5FDVdPmq6bNRzxVHTpWjHupJUr14dhw8fxqhRo/DPP/8gNjYWHTt2LLr9/fv3sLS05G9AnmzatAljx45F+/btMWzYMMyaNQuTJk2CoaEhtm/fDgcHB4wfP57vMVVCIpGU+xu2tly5ISwsrMTnCp+r/z4Hnz+HVT3yv/76a7E/r0QiwZo1a/D27VsMGDAAfn5+AIAXL17g999/h6enp9b8e5Ll7t27mD17Ni5fvoyCggKcPn0arVu3RlJSEr755htMmjQJISEhfI9J1Bz1XDbq+SfUc/mo57JRz8uHek64QD2Xj5ouRT2Xj3ouHzVdcdRzwgXquXzU80+o6fJR02WjnpcPNR0AS5Tijz/+YBmGYS0tLVk9PT22Zs2arFAoLLq9ZcuWbIcOHXickB8BAQFFf+6kpCSWYRj23LlzLMuybFpaGuvr68uuWLGCzxGJGoiOji7268GDB2xQUBDbokULdv/+/eyjR4/YR48esfv27WO/+OILNjg4mH348CHfY6vc999/z/r5+bFJSUklbktMTGSrV6/OLlmyhIfJ+Hft2jXWwMCA9fb2ZocPH17sew3LSr8Hf/XVVzxOSDQF9Vw26jlRBPVcMdRz+ajnhCvUc/mo6aQs1HPFUdNlo54TrlDP5aOeE0VQ0xVDPZePmi5FO9aV6PTp0+zEiRPZ+fPns4mJiUWfT05OZnv27MkeOnSIx+n4YWBgwK5fv55lWZZNT09nGYZhT548WXT70qVLWW9vb77GI2oqLCyMbdu2LSuRSErcJhaL2TZt2rBhYWE8TMYvV1dX9qeffpJ7+/Lly1k3NzcVTqQ+WrZsyQYHB7N5eXnsx48fS0R+/vz5rJeXF48TEk1CPS+Jek4qgnouG/VcPuo54RL1XDZqOikv6rl81HTZqOeES9Rz2ajnpCKo6bJRz+WjpkvRUvBKFBoaitDQ0BKft7a2xqFDh3iYiH8WFhYQiUQAAHNzcxgbG+Pdu3dFt5uZmWn1tXAKZWZmIj09HRKJpMRt7u7uPEzEr7/++guLFy+WuYyPQCBAr169MHv2bB4m41dycjJycnLk3p6Tk4Pk5GQVTqQ+7ty5gx9++AEGBgbIysoqcbuLiwt9ryEKo56XRD1XDPW8OOq5bNRz+ajnhEvUc9mo6WWjnhdHPZePmi4b9ZxwiXouG/VcMdT04qjpslHP5aOmS9GOdaJSgYGBePjwYdHHjRs3xsaNG9GpUydIJBJs3rwZ1atX53FCfm3cuBErV65EZGSk3PuIxWIVTqQeWJbFixcv5N7+7NkzrbkWzucaN26M1atXo2PHjqhXr16x2+7evYs1a9agUaNGPE3HLz09PZk/JBf68OEDTE1NVTgRIVUL9bx01HPZqOeyUc/lo54TonzUdPmo57JRz+WjpstGPSdE+ajnpaOmy0ZNl416Lh81XYp2rCvRo0ePsG7dOty/f1/mkVAMw+DNmzc8TcePgQMHYtOmTcjPz4eBgQEWLFiAtm3bFh0Rpqenh4MHD/I8JT82bdqEsWPHon379hg2bBhmzZqFSZMmwdDQENu3b4eDgwPGjx/P95i86NGjBzZu3AhPT0+MGjUKxsbGAKRHh23cuBGbN2/GgAEDeJ5S9X7++WeEhISgYcOGaNy4MapVqwYAePXqFW7evAlra2usW7eO5yn50bhxYxw4cAATJ04scVt2djZ+++03tGzZUvWDEY1EPS+Jei4f9Vw+6rls1HP5qOeES9Rz2ajpslHP5aOey0dNl416TrhEPZeNei4fNV0+arps1HP5qOn/4m8V+qrtwoULrIGBAevo6Mh26dKFZRiGbdOmDdusWTNWIBCwtWrV0srrU8jy5s0bdvXq1ey6devYiIgIvsfhTUBAANuhQweWZVk2KSmp2PUp0tLSWF9fX3bFihV8jsibtLQ0tkWLFizDMKy+vj7r4eHBenh4sPr6+izDMGzz5s3Z1NRUvsfkRXx8PDtx4kTWz8+PNTQ0ZA0NDVk/Pz920qRJbFxcHN/j8ebmzZusgYEB26lTJ3bXrl0swzDsypUr2V9++YX18/NjjY2N2YcPH/I9JtEA1HPFUc+lqOfyUc/lo57LRj0nXKGelw81nXpeGup56ajpJVHPCVeo5+VDPZeipstHTZePei4bNV2KYVktXMtBBVq0aIGkpCTcvHkTBQUFsLe3x9mzZ9G6dWvcunULHTt2xJ49e9CxY0e+RyVqwtDQECtXrsSYMWOQkZEBS0tLnDhxAh06dAAA/Pjjj9iyZYtWHnVZ6MiRIzh58iTevn0LAPDw8ECnTp3QtWtXmdeCIdrt/PnzGD16NF69elXs8z4+Pti6dat2HD1HKo16TsqLel426jkpD+o54QL1nJQX9bxs1HNSHtRzwgXqOakIanrZqOmkPKjptBS80ty/fx8LFiyAubk5UlNTAXy6TkejRo0wcuRIzJkzR6tDn5WVhdTUVJnX6ShcpkabWFhYQCQSAQDMzc1hbGyMd+/eFd1uZmaG+Ph4vsZTC927d0f37t35HoOoOZZlkZmZiaZNmyIiIgIPHjzAq1evIJFI4OPjg3r16tEPhURh1POyUc+Lo56XjXpOFEE9J1yiniuGmv4J9bxs1HOiCOo54RL1XDHU8+Ko6WWjphNFUNM/oR3rSqKrqwszMzMAgKWlJfT09JCYmFh0u7e3N549e8bXeLzJy8vDggULsG3bNiQnJ8u9X+EPRdokMDAQDx8+LPq4cePG2LhxIzp16gSJRILNmzejevXqPE5I1NHz58/x22+/ITIyUuYPzQzD4Ny5czxNx4+CggJYW1tjyZIlmD59OoKDgxEcHMz3WERDUc9lo57LRz0nFUE9L4l6TrhEPZePmi4b9ZxUFDW9OOo54RL1XD7quXzUdFIR1POSqOmf0I51JfH19S1aCoFhGNSoUQOHDx/GgAEDAAB///03HB0d+RyRF2PGjMGOHTvQo0cPfPHFF7CysuJ7JLUxcOBAbNq0Cfn5+TAwMMCCBQvQtm3boiMJ9fT0cPDgQZ6n5AfLstiyZQu2bdtWFLP/Yhim6OhDbbFr1y4MHToUenp68PPzk/nvSRuv9mFgYABHR0cYGBjwPQqpAqjnslHP5aOey0c9l416Lhv1nHCJei4fNV026rl81HP5qOklUc8Jl6jn8lHP5aOmy0dNl416Lhs1/RO6xrqSzJ07F7/++iuio6Ohq6uLHTt2YOjQofDx8QEAvHnzBj/88ANmzJjB86SqZWlpiX79+mHz5s18j6IRIiMjcezYMejo6KBdu3Zae/TctGnTsHLlSgQHB5f6w+G8efNUPBm/fHx8YG1tjZMnT8LW1pbvcdTK7NmzcfLkSdy4cQP6+vp8j0M0GPVcNup5+VDPpajnslHP5aOeE65Qz+WjpiuOei5FPZePmi4b9ZxwhXouH/W8fKjpUtR02ajn8lHTpWjHupIIhUJkZGTA2tq66LoCu3fvxsGDB6Gjo4MuXbogLCyM3yF5YGVlhaVLl2LkyJF8j0I0iL29PUJCQrBv3z6+R1ErRkZGWLlyJUaPHs33KGrnzz//xKJFi5Cfn4+wsDB4enrCyMioxP169erFw3REk1DPZaOek4qgnstGPZePek64Qj2Xj5pOyot6Lh81XTbqOeEK9Vw+6jmpCGq6bNRz+ajpUrRjnahUWFgYsrOzsX//fr5HUWtZWVkyr90BoGiZGm1iZmaGn376CSNGjOB7FLXSqFEjtGvXDosWLeJ7FLUjEAjKvA/DMFp5bSlCuEA9Vwz1vDjquWzUc/mo54QoHzW9bNTz4qjn8lHTZaOeE6J81HPFUNOLo6bLRj2Xj5ouRTvWlUwsFuPevXuIjo4GAHh5eaFu3brQ0dHhdzCevHnzBn379kW9evUwcuRIuLu7y3wurK2teZiOX3l5eViwYAG2bduG5ORkufer6t+UZOnRowfs7Ozwyy+/8D2KWrl27Rr69OmDAwcOoGnTpnyPo1YuXbqk0P1atmyp5ElIVUE9L456Lh/1XD7quWzUc/mo54Rr1POSqOmyUc/lo57LR02XjXpOuEY9L4l6Lh81XT5qumzUc/mo6VK0Y12Jtm/fjv/7v/9DYmJi0VFQDMPAzs4OS5YswbBhw3ieUPU+P6KlcMkeWbQxZMOGDcOOHTvQo0ePUq9pMmTIEBVPxr/Y2Fi0b98e/fv3x8iRI2FjY8P3SGqhW7duePXqFV6+fImAgACZPzQzDIMjR47wNCEhVQP1vCTquXzUc/mo57JRzwlRDeq5bNR02ajn8lHP5aOmE6J81HPZqOfyUdPlo6bLRj0nZaEd60qyefNmjB49GsHBwRg5ciSqV68OAIiIiMDmzZvx6NEjrF+/HqNGjeJ5UtWaP39+qXEvNG/ePBVMo14sLS3Rr18/bN68me9R1I6ZmRkkEgny8vIAAIaGhjJjlp6ezsd4vPH09Czz3xPDMIiMjFTRROojJSUF79+/R+3atWXe/vjxY7i6usr9YZqQQtRz2ajn8lHP5aOey0Y9l496TrhCPZePmi4b9Vw+6rl81HTZqOeEK9Rz+ajn8lHT5aOmy0Y9l4+aLkU71pXE29sbbm5uOHv2LPT09IrdJhQK0bp1a3z48EEr//ER2aysrLB06VKMHDmS71HUTlhYmEI/HP72228qmIZogiFDhiAiIgI3b96UeXvTpk3h7++Pbdu2qXgyommo56S8qOfyUc9JeVHPCVeo56S8qOfyUc9JeVHPCVeo56QiqOnyUdNJeVHTpXT5HqCqio+Px5QpU0pEHgD09PTw1VdfYfr06TxMpl5yc3MBAEZGRjxPwr/u3bvj7NmzFHkZtm/fzvcIRMOcP38eo0ePlnt7165dsWnTJhVORDQV9Vwx1PNPqOfyUc9JeVHPCVeo54qjpktRz+WjnpPyop4TrlDPFUc9/4SaLh81nZQXNV1KUPZdSEXUqVMHL1++lHv7y5cvERwcrLqB1EhMTAyGDh0KBwcHmJqawtTUFA4ODhg2bBjevn3L93i8mTNnDiIjIzFixAjcu3cPHz9+REpKSolfhMiSmZmJ9+/fIyYmpsQvbfTx40fY2trKvd3GxgaJiYkqnIhoKuq5fNRz2ajnpDKo58VRzwlXqOelo6aXRD0nlUVN/4R6TrhCPS8d9Vw2ajqpDOp5cdR0KTpjXUnWrVuHzp07w9vbGyNGjCg6Oiw3NxebNm3Cvn37cOLECZ6nVL0XL16gefPmSEtLQ2hoKPz9/Ys+v3PnThw7dgxXr16Fn58fz5OqXrVq1QAA4eHhpS6VIRaLVTWS2nn//j3Cw8ORnp4OiURS4vbBgwfzMBW/Nm7ciJUrV5a6zJU2/p1xcnJCeHi43Nvv3bsHOzs7FU5ENBX1XDbquXzU87JRz0uinstGPSdcoZ7LR02XjXpeNuq5bNT0kqjnhCvUc/mo5/JR08tGTS+Jei4bNV2KrrHOkdq1a5f4XEpKCuLi4qCrqwtnZ2cAQGxsLEQiEZycnGBjY4OHDx+qelRe9ejRA9evX8e5c+dQq1atYrc9efIEbdq0QdOmTXH48GGeJuTP/PnzFbqmybx581QwjXrJy8vDkCFDcPDgQUgkEjAMg8JvXZ8/Z9oWs02bNmHMmDFo3749WrRogVmzZmHSpEkwNDTE9u3b4eDggPHjxyMsLIzvUVVu0qRJWL9+PQ4cOIBu3boVu+3IkSPo06cPRo8ejTVr1vA0IVFX1HPFUM/lo57LRz2XjXouH/WcVBT1XHHUdNmo5/JRz+WjpstGPScVRT1XHPVcPmq6fNR02ajn8lHTpWjHOkdCQkIU+gb9XxcuXFDCNOrLysoKU6ZMwezZs2XevmjRIqxcuRKpqakqnoyos8mTJ2PdunX4/vvv0aRJE4SEhGDHjh1wcnLC6tWrERsbi507dyIwMJDvUVWqZs2acHd3x8mTJ5GcnAw7OzucPXsWrVu3Rnp6OurXr49Ro0ZhypQpfI+qcunp6WjevDmePXuGoKCgor8bT548wcOHD+Hv74+rV6/C0tKS30GJ2qGeK4Z6TiqCei4b9Vw+6jmpKOq54qjppLyo5/JR02WjnpOKop4rjnpOKoKaLhv1XD5quhQtBc+Rixcv8j2CRhAKhUXL9MhibGwMoVCowonUV25uLgCU+nxpiwMHDmDo0KGYMWMGkpOTAQAuLi5o3bo12rZti9atW2P9+vXYuHEjz5Oq1ps3bzB27FgAgJ6eHgCgoKAAAGBhYYFvv/0WGzZs0MrIW1hY4ObNm1i2bBkOHTqEAwcOAAB8fHwwZ84cTJs2DSYmJjxPSdQR9Vwx1HPFUc8/oZ7LRj2Xj3pOKop6rjhqumKo559Qz+WjpstGPScVRT1XHPVccdT0T6jpslHP5aOmSwn4HoBIJSUlwdvbGzdu3OB7FKWqU6cOtm7divT09BK3ZWRkYNu2bahbty4Pk6mHmJgYDB06FA4ODjA1NYWpqSkcHBwwbNgwvH37lu/xeJOYmIiGDRsC+PRDT3Z2dtHtvXv3xqFDh3iZjU8WFhYQiUQAAHNzcxgbG+Pdu3dFt5uZmSE+Pp6v8XhnYmKCBQsW4PHjx8jJyUFOTg4eP36M+fPna0XgCT+o59RzgHouD/VcNup56ajnhA/a0nOAml4a6rls1HP5qOnyUc8JH6jnUtrec4CaLg81XTbqeemo6XTGutoQi8WIjo4uOmKqqlqwYAE6dOiAGjVqYOjQoahevToAICIiAjt27EBycjLWr1/P85T8ePHiBZo3b460tDSEhobC39+/6PM7d+7EsWPHcPXqVfj5+fE8qeo5ODgUHTVnbGwMKysrREREoGvXrgCkPyDm5eXxOSIvAgMDi103qnHjxti4cSM6deoEiUSCzZs3F/0b02ZxcXFITEyEr6+v1sSd8Id6Tj2nnstHPZeNeq4Y6jlRJW3pOUBNl4d6Lh/1XD5qetmo50SVqOfUc4CaXhpqumzUc8VoddNZohbi4+NZhmHYc+fO8T2K0p05c4YNDg5mGYYp9qtOnTrs2bNn+R6PN927d2ft7OzYR48elbjt8ePHrL29PdujRw8eJuNfnz592C5duhR9PGTIENbe3p7dvXs3u3PnTtbOzo5t164djxPy49dff2UbNmzI5uXlsSzLslevXmUNDQ1ZgUDACgQC1sDAgD1+/DjPU/Lnr7/+Yv38/Iqej8Lvrx8/fmSDg4PZw4cP8zsgqZKo59Rz6rl81HPZqOelo54TPmhTz1mWmi4L9Vw+6rl81HT5qOeED9Rz6jnLUtNLQ02XjXpeOmo6y9KOdTWhbaFnWZaNi4tjb968yd68eZONi4vjexzeWVpasosWLZJ7+8KFC1lLS0sVTqQ+rly5wo4fP74oZjExMWz16tWLfkD09fVlX7x4wfOU6uHNmzfs6tWr2XXr1rERERF8j8Obo0ePsgKBgG3WrBm7YMGCEt9fO3fuzHbr1o3HCUlVRT2nnlPP5aOeK456LkU9J3zRxp6zLDX9c9Rz+ajn5UNNp54T/lDPqecsS00vDTVdcdRzKWq6FC0FT3jj6OgIR0dHvsdQG0KhsOhaJrIYGxtDKBSqcCL10bx5czRv3rzoYzc3Nzx//hyPHz+Gjo4OatSoAV1d+nYGAN7e3pgwYQLfY/Bu4cKFaNGiBS5cuIDk5GTMnz+/2O1NmjTB5s2b+RmOkCqGel4c9Vw+6rniqOdS1HNCVIua/gn1XD7qeflQ06nnhKga9bw4arp81HTFUc+lqOlS9K+CKNXOnTsBAIMGDQLDMEUfl2Xw4MHKHEst1alTB1u3bsW3334LCwuLYrdlZGRg27ZtqFu3Lk/TqR+BQICgoCC+xyBq6smTJ1i5cqXc2x0cHJCYmKjCiQjRbNRzxVHPy4d6TkpDPSeEe9R0xVDPy4d6TkpDPSeEe9RzxVHTy4eaTkpDTZeiHetEqcLCwsAwDL766ivo6+sjLCyszK9hGEYrI79gwQJ06NABNWrUwNChQ1G9enUAQEREBHbs2IHk5GSsX7+e5ylV4/LlyxX6uhYtWnA8iXoRCARgGKZcX8MwDEQikZImUl/GxsbIzs6We3tkZCRsbGxUOBEhmo16rjjq+SfUc9mo54qjnhPCPWq6Yqjnn1DP5aOmK4Z6Tgj3qOeKo6Z/Qk2XjXquOGq6FO1YJ0oVFRUFANDX1y/2MSmpdevWOHHiBKZNm4alS5cWuy04OBi7du1Cq1ateJpOtUJCQsoVM5ZlwTAMxGKxEqfi39y5c8sdeW3VqlUr7NixAxMnTixxW3x8PH755Rd06dJF9YMRoqGo54qjnn9CPZeNeq446jkh3KOmK4Z6/gn1XD5qumKo54Rwj3quOGr6J9R02ajniqOmSzEsy7J8D0GA1NRU9OrVCytXrkSdOnX4HofwLD4+Hm/fvgUAeHh4aN11cS5dulShr2vZsiXHkxBNFRERgcaNG8PT0xN9+vTBnDlzMHXqVOjp6WHz5s1gWRZ3796Fp6cn36OSKoZ6Tj5HPaeek8qhnhO+UM/J56jn1HNSOdRzwhfqOfkvajo1nVQONV2KdqwrUWJiIj58+IDc3FyYmprC19cXxsbGfI/FK29vb6xevRrdunWTefvx48cxfvx4REZGqngyQjTHu3fvIBAI4OLiAgDIy8vDhg0bStzP1dUVffv2VfV4auPp06eYMGECLly4gM9TFxISgvXr18Pf35/H6YgmoZ6XRD0npPKo54qhnhOuUM9lo6YTUnnU9LJRzwlXqOeyUc8JqTzquWKo6bQUPOeSk5OxdOlS7N27F7GxscVuEwgEaNy4MaZMmYIePXrwMyDPoqOjkZWVJff2rKysoqPGqrqdO3cCAAYNGgSGYYo+Los2XguHfPL48WPUqVMHq1evxnfffQcAyM7OxtSpU0vcV0dHB/7+/qhVq5aqx1QLNWvWxNmzZ5GamorXr19DIpHA29sbdnZ2fI9GNAD1vHTU80+o56QiqOeKo56TyqCel42aLkU9JxVFTVcM9ZxUBvW8bNTzT6jppCKo54qjptMZ65x6+/YtvvjiC8TGxsLf3x/GxsZ49uwZhEIhhg0bhrS0NFy6dAnx8fEYPnw4Nm3axPfIKicQCLBnzx70799f5u1TpkzB9u3bkZycrOLJVE8gEIBhGOTm5kJfXx8CgaDMr9GGa5p8bs+ePbC1tUX79u0BAJmZmRgwYECJ+3l4eGDdunWqHo8X3333HU6ePIlXr14V/Z1JTk6GnZ0d9uzZg6ZNmwIAJBIJQkJC0LVrV/z88898jkyIxqGel416/gn1vGzU85Ko54QoH/VcMdR0Kep52ajnslHTCVEu6rliqOefUNPLRk0viXpOyoPOWOfQ1KlTkZeXh/v376N27doApP/4+vXrh6ioKJw8eRISiQTLly/HzJkzUb9+fXz77bc8T618a9aswZo1awBIIzVx4kTMmjWrxP3S09ORlpaGr7/+WtUj8iIqKgoAoK+vX+xjInXixAkMHjwY//zzT9HnCgoKcPz4cTg4OMDQ0BAAwLIs/v77b3Tq1AkdO3bka1yVuXDhAnr16iXzh0IHBwd4eHgUffz111/j6NGjqhxPbZw7dw7379/HtGnTij7366+/Yv78+cjPz8fXX3+NFStWQEdHh8cpibqinstGPZeNel466rls1HPFUM9JZVDP5aOml0Q9Lx31XD5qetmo56QyqOfyUc9lo6aXjpouG/VcMdR0KdqxzqFz585h6tSpRZEHABsbG/z4449o2LAhXr9+DV9fX8yYMQP379/Hhg0btCL09vb2qFmzJgDpsjQuLi5F16koxDAMTExMUK9ePYwZM4aPMVXu82/Gsj7Wdjt37kTDhg3Rtm3bErft2bMHrVu3Lvq4YcOG2L59u1ZEPjo6GjVq1Cj2OV1dXQQFBcHMzKzY5728vLRmmaf/mj9/frF/U48fP8bIkSNRu3Zt+Pr6Yu3atXB0dMSMGTN4nJKoK+q5bNRz2ajnpaOey0Y9Vwz1nFQG9Vw+anpJ1PPSUc/lo6aXjXpOKoN6Lh/1XDZqeumo6bJRzxVDTZeiHescys/Ph7m5eYnPW1hYgGVZJCQkwNfXFwAQGhqK48ePq3pEXvTv379oGZpWrVph9uzZaNOmDc9TqR9vb2+sXr0a3bp1k3n78ePHMX78eERGRqp4Mn7cvHkT33zzjUL37datG7Zu3arkidSHRCIp9rGFhQXCw8NL3I9hGGjr1T6eP3+O3r17F328a9cumJub48qVKzA2NsaoUaOwc+fOKh95UjHUc9mo54qhnhdHPZePel426jmpDOq5fNT0slHPi6Oel46aXjrqOakM6rl81HPFUNOLo6bLRz0vGzVdquwLTBCF1alTB7t37y5xPY5ff/0VOjo6qF69etHnsrKyYGRkpOoReXfhwgUKvBzR0dHIysqSe3tWVpZWHQkVHx8PNze3Yp8zMjLChAkT4O7uXuzzLi4uSEhIUOV4vHF1dcXDhw8Vuu/Dhw/h6uqq5InUU3Z2drEXXqdOnUKHDh1gbGwMAGjQoIFW/Xsi5UM9Lxv1XD7qeXHUc9mo54qhnpPKoJ4rhpouG/W8OOq5fNT0slHPSWVQzxVDPZePml4cNV026rliqOlStGOdQ/PmzcPdu3cREBCA//3vf1iwYAHatWuHH3/8EUOHDoWdnV3RfS9fvoxatWrxOC0/9u7di7CwMLm3Dx06FPv27VPdQGqGYRi5t925cweWlpaqG4ZnhoaGJX7oMTY2xqpVq4qORC2UnZ1ddN2cqi40NBR79uxBYmJiqfdLTEzEnj17EBoaqqLJ1Iubmxvu3LkDAHj9+jWePHmCdu3aFd2ekpICAwMDvsYjao56Xjbqeemo559Qz2WjniuGek4qg3quGGq6fNTzT6jn8lHTy0Y9J5VBPVcM9bx01PRPqOmyUc8VQ03/F0s4dezYMTYgIIBlGIZlGIa1srJi//e//7EFBQXF7rd792729u3bPE3JnwYNGrAjRoyQe/vo0aPZxo0bq3Aifq1evZr18vJivby8WIFAwNrb2xd9/Pkva2trViAQsAMHDuR7ZJWpX78+27dvX4Xu27dvX7ZevXpKnkg9REVFsSYmJmxgYCB7584dmfe5c+cOW6tWLdbExISNjIxU8YTqYf78+axAIGC7du3Kent7s9bW1mxqamrR7f369dOq7zWk/KjnpaOeF0c9l496Lhv1XDHUc1JZ1POyUdM/oZ7LRz2Xj5peNuo5qSzqedmo58VR0+WjpstGPVcMNV2KrrHOsS5duqBLly5ITU1Ffn4+HBwcZB4RNWDAAB6m419ERASGDRsm9/agoCD88ccfKpyIX/b29qhZsyYA6bI0Li4ucHFxKXYfhmFgYmKCevXqYcyYMXyMyYsePXpgwYIFuHnzJho3biz3frdu3cKhQ4cwf/581Q3HI09PT+zduxf9+/dHo0aN4Ovri8DAQJiamiIrKwtPnjzB69evYWRkhN9//x1eXl58j8yLWbNmoaCgACdOnIC7uzu2b99edPRpSkoKLl68iAkTJvA7JFFr1PPSUc+Lo57LRz2XjXquGOo5qSzqedmo6Z9Qz+WjnstHTS8b9ZxUFvW8bNTz4qjp8lHTZaOeK4aa/i++9+xrs/fv3/M9gsqZmZmxP/zwg9zbf/jhB9bExESFE6mPkJAQ9uzZs3yPoTYyMjJYb29v1szMjF2+fDkbGxtb7PbY2Fh2+fLlrLm5Oevt7c1mZGTwNCk/3rx5w44YMYJ1cXEpOmKXYRjW2dmZHT58OPvq1Su+R9QoBQUF7KVLl9i0tDS+RyEaiHpeEvWcel6Iel466jm3qOekMrSx5yxLTZeHel4c9bxs1HTuUM9JZVDPZdPWnrMsNf2/qOmlo55zq6o2nWFZluV7535V0aRJE2zZsqXMa7lIJBKsXr0aCxYsQHp6uoqmUw8tW7ZEWloa7ty5U+L6HPn5+WjQoAEsLCxw5coVniYk6uT169fo2bMnnj59CoZhYGlpWXSUWFpaGliWRUBAAA4fPoxq1arxPS5vMjMzkZGRATMzM5ibm/M9jkZKSEiAs7Mzzpw5g9atW/M9zv+zd9/hUZV5G8fvSSWBBBJ6L6FLb1KlIyAivSlSRWURGzZ0BdvqWrAhTSkKuqyrooCgAtIERARFOioQERAhCIQWUp73j7wzZpiSSTIl5fu5Li7NafObMzPnfs55TkGAkeeZI8+RFeS5Z8jznCPPkRF57hkyHZ4izz1HpucMeY6MyHPPkOfICjLdM+R5zuXXTA8KdAH5yZEjR9S0aVM9/PDDunz5stNptm7dqqZNm2rSpElq3769nysMvEcffVS7d+9Wx44dtWzZMh06dEiHDh3S0qVL1aFDB+3Zs0ePPvpooMsMiMWLF2vkyJEux48aNUoffvih/wrKBapXr64ff/xR77//voYMGaIqVaooLCxMlStX1uDBg7Vo0SL9+OOPBTrgJSkqKkrly5cn4HOI88xgRZ5njjx3jTx3RJ57hjz3DvIcVuS5Z8h058hzR+S558j0nCPPYUWee4Y8d41Md0Sme4Y89458memBulQ+Pzp79qy58847TVBQkKlatapZvny5w7jg4GBTqVIls2TJksAVGmDz58830dHRJigoyPbPYrGY6OhoM3fu3ECXFzDNmzc348aNczn+7rvvNi1btvRjRUDB8ccffxiLxWLWrFkT6FKQC5DnniHPnSPPgcAhz5ERee45Mt0ReQ4EDnmOjMhzz5HnzpHpQODk10wPCXTHfn5StGhRzZo1SyNGjNC4cePUu3dv9e3bV127dtWUKVN05swZ3X///XrqqacUGRkZ6HIDZuTIkerXr59WrVqlX3/9VZIUFxenbt26KSoqKsDVBc6BAwc0evRol+MbNmyo//znP36sKPdJSkrSjh079Oeff6pNmzYqUaJEoEsCkA+R554hz50jzzNHngPwB/Lcc2S6I/I8c+Q5AH8gzz1HnjtHpmeOTAeyho51H2jVqpV++OEHTZgwQXPmzNGSJUtUp04drV69WvXq1Qt0eblCdHS0+vfvH+gychVjjM6ePety/F9//aXk5GT/FZTLvPHGG5o6darOnj0ri8Viey7H6dOnVbt2bb344otuG0kAkFXkeebIc0fkuXvkOQB/I889Q6bbI8/dI88B+Bt57hny3BGZ7h6ZDmQdz1j3gZSUFD3//PN67733FBMTo3LlyungwYNasGCBLl68GOjycoXExETt3r1bGzdu1IYNGxz+FUSNGzfWf/7zH129etVhXFJSkj744AM1btw4AJUF3vz583Xfffepe/fumjdvnt1zOUqUKKFOnTpp8eLFAawQQH5EnmeOPHdEnrtGngMIBPLcM2S6PfLcNfIcQCCQ554hzx2R6a6R6UA2Be4u9PnThg0bTJ06dYzFYjHDhg0zJ0+eNImJieaee+6xPe/ls88+C3SZAXP69GkzZMgQExoaavesl2v/vyBasWKFCQoKMq1btzZLly41v/76q/n111/NZ599Zlq2bGmCgoLsniNUkFx33XWmT58+xpj079C1z+V44YUXTLly5QJVHvKB/Pq8F2Qfee4eee4aee4aeQ5fI89xLfI8c2S6c+S5a+Q5fI08x7XI88yR566R6a6R6fC1/Jrp3Arei8aMGaMFCxaoWrVq+uqrr9SlSxfbuDfeeEO33367xo0bp759+6p3796aPn26ypcvH8CK/e+OO+7QsmXLNHHiRLVr104xMTGBLinX6NGjh+bOnat7771Xffr0sQ03xigqKkpvv/22brrppsAVGEC//PKLJk6c6HJ8bGysEhIS/FgRgPyMPM8cee4aee4aeQ7An8hzz5DpzpHnrpHnAPyJPPcMee4ame4amQ5kDx3rXrRo0SI99thj+uc//6nw8HCH8c2aNdP333+v119/XVOmTFHdunV17ty5AFQaOF999ZXuv/9+vfjii4EuJVcaOXKk+vXrp1WrVunXX3+VJMXFxalbt26KiooKcHWBU6xYMZ0+fdrl+L1796pMmTJ+rAi52aVLl3Tbbbepf//+uvXWWz2ap2jRopo/f76uu+46H1eHvIA8zxx57h557hx5jqwgz5FT5LlnyHTXyHPnyHNkBXmOnCLPPUOeu0emO0emIyvI9L/Rse5FP/74o+rUqeN2mqCgIN1///0aMGCA27OB8qvIyEhVqVIl0GXkatHR0erfv3+gy8hVevbsqTlz5mj8+PEO4/bs2aO3335bo0ePDkBlyI0iIyO1evVq9ejRw+N5ChUqpBEjRviwKuQl5HnmyPPMkeeOyHNkBXmOnCLPPUOmu0eeOyLPkRXkOXKKPPcMeZ45Mt0RmY6sINP/ZjHGmEAXkR8dP35cX3zxhfbt26fz588rKipKdevWVffu3VWuXLlAlxcwDzzwgHbt2qVVq1YFupRcKzExUfHx8frrr7/k7Od5ww03BKCqwDp+/Liuv/56GWN08803a86cObrtttuUmpqqjz/+WGXLltV3332nEiVKBLpU5BI9e/ZUmTJlNG/evECXgjyOPHeOPM8cee6IPEdWkefwFvLcNTLdPfLcEXmOrCLP4S3kuWvkeebIdEdkOrKKTE9Hx7qXXb58WZMmTdI777yjlJQUh410aGioxo4dq5dfflkREREBqjJwNm/erHvuuUclS5bUuHHjVLFiRQUHBztM16RJkwBUF1gJCQmaMGGCPv74Y6WmpkpKf9aLxWKx+3/ruILmzz//1OTJk/XJJ5/o7NmzkqSoqCj1799fL7zwgkqVKhXYApGrHDp0SDfeeKMGDx6su+66SxUqVAh0SchjyHP3yHPXyHP3yHNkBXmOnCLPM0emO0eeu0eeIyvIc+QUeZ458tw1Mt09Mh1ZQaano2Pdi1JSUtStWzetW7dOHTt21O23366GDRsqKipKiYmJ2rlzp9577z2tXbtWHTt21FdffeU04PKzoKAg2/9bwyujghxk/fr107JlyzRx4kS1a9dOMTExTqdr3769nysLrKSkJH355ZeqUqWKGjRoIEk6deqU0tLSVLJkSbvvFGAVFRWllJQUXb16VZIUEhLi8Cwui8VSIJ+7hcyR55kjz10jz50jz5Ed5Dlygjz3DJnuHHnuHHmO7CDPkRPkuWfIc9fIdOfIdGQHmZ6OZ6x70cyZM7Vu3Tq99dZbuvvuux3GN2rUSCNGjNCsWbM0fvx4zZw5UxMmTAhApYEzf/78QJeQa3311Ve6//779eKLLwa6lFwlLCxMAwcO1Ouvv24L+ZIlSwa4KuR2/fv3d7ojAXiCPM8cee4aee4ceY7sIM+RE+S5Z8h058hz58hzZAd5jpwgzz1DnrtGpjtHpiM7yPR0XLHuRS1atFD58uW1ZMmSTKft06ePjh07pm3btvmhMuQFpUqV0tSpUzV+/PhAl5Lr1KtXT0OGDNETTzwR6FIAFADkOXKCPHeNPAfgT+Q5coI8d408B+BP5Dlyikx3jUwHsof7OXjRvn371L17d4+m7d69u/bv3+/jipCX3HbbbR41EguiyZMna/r06Tpw4ECgSwFQAJDnyAny3DXyHIA/kefICfLcNfIcgD+R58gpMt01Mh3IHm4F70UWi0VpaWlZmr6gGT16dKbTWCwWzZ071w/V5C4DBgzQ+vXr1b17d40bN04VK1Z0+kygJk2aBKC6wPr2229VvHhx1atXTx06dFCVKlUUERFhN43FYtHrr78eoAqRW/3+++/64YcfdO7cOafb59tvvz0AVSG3I88zR567Rp67Rp4ju8hzZAd57hky3Tny3DXyHNlFniM7yHPPkOeukemukenIroKe6dwK3otatGihsmXL6rPPPst02j59+uj48eP67rvv/FBZ7lGlShWHBk5qaqpOnDih1NRUlSxZUoULF9ahQ4cCVGHgBAX9fQMJZ41AY4wsFotSU1P9WVaukHHduFJQ1w2cu3LlikaMGKGPP/5YaWlpslgsssZdxt8X3xk4Q55njjx3jTx3jTxHVpHnyAny3DNkunPkuWvkObKKPEdOkOeeIc9dI9NdI9ORVWR6Oq5Y96LbbrtN999/v2bOnKm7777b5XSzZs3SsmXL9Oqrr/qxutzhyJEjTocnJydr9uzZeu2117Rq1Sr/FpVLzJ8/P9Al5FpZOTMVkNJvZfTJJ5/oueeeU6tWrdShQwe9++67Klu2rF577TUdP35c7733XqDLRC5FnmeOPHeNPHeNPEdWkefICfLcM2S6c+S5a+Q5soo8R06Q554hz10j010j05FVZHo6rlj3opSUFHXp0kUbN25U586dNXz4cDVs2FBRUVFKTEzUTz/9pIULF2r16tVq27at1qxZo5AQzm3IaPz48YqPj9fnn38e6FKQC+3evVsrVqywNRarVq2qHj16qF69eoEtDLlOpUqV1L17d82ZM0cJCQkqWbKkVq9erU6dOkmSOnXqpFq1amnmzJkBrhS5EXmec+Q53CHP4SnyHDlBnnsHmQ5XyHN4ijxHTpDn3kGewx0yHZ4i0/+fgVddvHjRjBs3zoSEhJigoCC7fxaLxYSEhJg77rjDXLhwIdCl5kqzZs0yRYoUCXQZyGWuXLliRowYYfsdBQcHm+DgYGOxWExQUJAZPny4SUpKCnSZyEXCw8PN22+/bYxJ3y5bLBazdOlS2/jp06ebUqVKBao85AHkec6Q53CGPEdWkefIKfI858h0XIs8R1aR58gp8jznyHM4Q6Yjq8j0dJy+5WWRkZGaPXu2nnzySa1cuVJ79+5VYmKioqKiVKdOHfXo0UMVKlQIdJm51qpVqxQZGRnoMgJi9OjRmU5jsVg0d+5cP1STuzzyyCN67733NH78eN1zzz2Ki4uTxWLRL7/8ojfeeEMzZ85UbGysXnvttUCXilyidOnSSkhIkJS+XY6JidGBAwd08803S5LOnz+vK1euBLJE5HLkec6Q5+6R5+Q5PEOeI6fI85wrqJlOnrtGniOryHPkFHmecwU1zyUy3R0yHVlFpqfjVvB+sn//fv3vf//TiRMnVLt2bY0cOVLR0dGBLsvvnn76aafDz549qw0bNmjHjh169NFH9a9//cvPlQVelSpVZLFY7IalpqbqxIkTSk1NVcmSJVW4cGEdOnQoQBUGTokSJXTTTTfp3XffdTp++PDhWrlypU6fPu3nypBbDRo0SJcvX9ayZcskSSNHjtTKlSs1bdo0paWl6cEHH1Tjxo315ZdfBrhS5DXkeTry3DXy3DXyHFlFnsNXyPO/kenOkeeukefIKvIcvkKe/408d41Md41MR1aR6f8v0JfM5ydvvvmmqVGjhjl16pTd8GXLlpnw8HDbLTUsFouJi4tzmK4gsL7/a//Fxsaa5s2bm9mzZ5u0tLRAl5mrXL161bz55psmLi7OHDp0KNDlBER0dLSZMWOGy/EzZswwRYsW9V9ByPU2btxoJk6caK5cuWKMMea3334zNWvWtG1zqlevbvbv3x/gKpFbkeeZI8+zjjwnz5F15Dlygjz3DJmeNeQ5eY6sI8+RE+S5Z8jzrCPTyXRkHZmejivWvahbt24KDg7WypUrbcNSUlJUvnx5XbhwQTNmzFCzZs30+eef6/HHH9eECRP06quvBrBi5CXjx49XfHy8Pv/880CX4neDBg3S1atX9emnnzodf8sttyg8PFwffvihfwtDnpKWlqZdu3YpODhYtWvXVkgIT0OBc+Q5fIk8J8+RM+Q5PEWew5fIc/IcOUOew1PkOXyNTCfTkTMFMdODAl1AfrJ37161bNnSbtjatWt16tQp3X///RoxYoSuu+46Pfzwwxo0aJBWrFgRoEr9JzY2Vh999JHt76efflq7d+8OYEV5V8OGDbVhw4ZAlxEQzzzzjA4fPqx+/fppzZo1io+PV3x8vFavXq2+ffsqPj5ezzzzjM6cOWP3D8goKChIDRs2VL169QpEwCP7yHNH5Ln3kOfkOXKGPIenyHPnyHTvIM/Jc+QMeQ5PkefOkefeQ6aT6ciZgpjpBeNd+klCQoIqVqxoN2zNmjWyWCzq27ev3fA2bdrok08+8Wd5AXHhwgVdunTJ9vfUqVNVvXp11atXL4BV5U2rVq1SZGRkoMsIiDp16kiSdu3apc8++8xunPWmG3Xr1nWYLzU11ffFIVfIbgP4hhtu8HIlyA/Ic0fkufeQ5+Q5XCPP4U3kuXNkuneQ5+Q5XCPP4U3kuXPkufeQ6WQ6XCPTnaNj3YtKly6tP/74w27Yxo0bFRkZqYYNG9oNDwsLU1hYmD/LC4i4uDh99NFHateunaKjoyVJFy9ezPTMptjYWH+Ul6s8/fTTToefPXtWGzZs0I4dO/Too4/6uarc4cknn5TFYgl0GcjFOnTokKXviDFGFouFhiCcIs8dkeeeI89dI8+RGfIc3kSeO0eme4Y8d408R2bIc3gTee4cee45Mt01Mh2ZIdOd4xnrXjRgwADt2rVL33//vaKiorRnzx41atRIt9xyi92tWSRp0qRJWrlypfbs2ROgav1j4cKFGjVqlLL6NcvvPzxngoKcP5khJiZGcXFxGjt2rO644w7CDnBi/fr12Zqvffv2Xq4E+QF57og89xx5DmQfeQ5vIs+dI9M9Q54D2Ueew5vIc+fIc8+R6UD2kenO0bHuRbt27VLz5s1VrFgxXXfdddq+fbsuXbqkLVu2qGnTpnbTxsXFqVOnTnr77bcDVK3/HDhwQOvWrdPJkyc1depU9e3bVw0aNHA7z5QpU/xUHQAA9shz58hzAEBeQp67RqYDAPIK8tw18hwAAoNbwXtR/fr19fXXX+u5557ToUOH1LJlS02aNMkh5NetW6fIyEgNHDgwQJX6V61atVSrVi1J0vz58zVixAj17t07wFUFXmxsrObMmaMBAwZISr8tTb9+/XgWDgAEGHnuHHnuHHkOALkTee4ame6IPAeA3Ik8d408d45MB+BrXLGOXO306dNq0aKF3n//fbVq1SrQ5XhVWFiY3nnnHd1+++2S0m9Ls2jRIg0bNizAlQF53+jRozOdxmKxaO7cuX6oBgB5DiA7yHMg98mvmU6eA75DngO5T37Nc4lMB3yJTE/HFevI1VJTU3XkyBFdvnw50KV4XVxcnD766CO1a9dO0dHRkqSLFy/qzJkzbueLjY31R3lAnvb11187PBspNTVVJ06cUGpqqkqWLKnChQsHqDqg4CHPHZHnQObIcyD3ya+ZNjfgTwABAABJREFUTp4DvkOeA7lPfs1ziUwHfIlMT8cV68jVTp48qbJly2r16tXq1KlToMvxqoULF2rUqFHK6k8wNTXVRxUB+V9ycrJmz56t1157TatWrVLVqlUDXRJQIJDnjshzIPvIcyBw8mumk+eA/5HnQODk1zyXyHQgEApapnPFOhAgw4cPV4sWLbRu3TqdPHlSU6dOVd++fdWgQYNAlwbkW6GhoZowYYL27t2rCRMm6PPPPw90SQDyOPIc8D/yHIC3keeA/5HnAHyBTAf8r6BlOh3rQADVqlVLtWrVkiTNnz9fI0aMUO/evQNcFZD/NWzYUAsXLgx0GQDyCfIcCAzyHIA3kedAYJDnALyNTAcCo6BkelCgCwCQ7vDhw1kK+NOnT6tatWrasmWLD6sC8qdVq1YpMjIy0GUAyIfIc8B/yHMAvkKeA/5DngPwJTId8J+CkulcsQ7kUampqTpy5IguX74c6FKAXOfpp592Ovzs2bPasGGDduzYoUcffdTPVQGAI/IccI08B5BXkOeAa+Q5gLyETAdcI9PT0bEOAMh3pk6d6nR4TEyM4uLiNGvWLN1xxx3+LQoAAGQJeQ4AQN5HngMAkD+Q6enoWEeuFhYWpvbt2ysmJibQpQDIQ9LS0gJdAoAMyHMA2UGeA7kPmQ4gq8hzIPchzwFkB5mejo51+N2ff/6pY8eO6fLlyypSpIiqV6/u8rkLMTExWrt2rZ8rBAAAmSHPAQDIH8h0AADyPvIcAPyDjnX4RUJCgl544QUtXrxYx48ftxsXFBSkli1b6sEHH1SfPn0CUyCAPO23337L1nyVKlXyciVA/kaeA/Al8hzwHzIdgK+Q54D/kOcAfIlMd46OdfhcfHy82rVrp+PHj6tOnToqV66c9u7dq+TkZI0ePVpnz57V+vXr1b9/f91xxx2aNWtWoEsGkMdUqVJFFosly/Olpqb6oBogfyLPAfgaeQ74B5kOwJfIc8A/yHMAvkamO0fHOnxu0qRJunLlinbs2KEGDRpISj+bbvDgwTp8+LBWrlyptLQ0vfTSS5o8ebKaNWumsWPHBrhqAHnJvHnz7EI+LS1Nr7/+uuLj43XrrbeqVq1akqT9+/frgw8+UJUqVTRx4sRAlQvkSeQ5AF8jzwH/INMB+BJ5DvgHeQ7A18h05+hYh8+tWbNGkyZNsgW8JBUvXlz//ve/1aJFC/3yyy+qXr26HnnkEe3YsUMzZswg5D0QFham9u3bKyYmJtClAAE3cuRIu7+fe+45XblyRb/88ouKFy9uN27q1Klq27at/vjjDz9WCOR95LlvkOfA38hzwD/IdO8jz4G/keeAf5DnvkGmA38j050LCnQByP+SkpIUHR3tMLxo0aIyxujkyZO2YV27dtWBAwf8WV6u8ueff+qHH37Q5s2b9dNPP+nSpUsup42JidHatWvVuHFjP1YI5A2zZs3SuHHjHAJekkqWLKk77rhDM2fODEBlQN5FnnuOPAe8gzwHfINM9wx5DngHeQ74BnnuOTId8A4yPR0d6/C5xo0ba9GiRQ7PVZg3b56Cg4NVs2ZN27ALFy4oIiLC3yUGVEJCgh566CFVrFhRZcuWVbNmzdSuXTs1btxYRYsWVbt27fTpp58GukwgT0lISHDbSL506ZISEhL8WBGQ95Hn7pHngPeR54BvkOmukeeA95HngG+Q5+6R6YD3kenpLMYYE+gikL+tWrVKPXr0UFxcnPr27auIiAht2rRJa9as0ZgxYzRnzhzbtP369dNff/2ltWvXBrBi/4mPj1e7du10/Phx1alTR5GRkdq7d6+Sk5M1evRonT17VuvXr9cff/yhO+64Q7NmzQp0yUCe0KlTJ/3000/68ssv1bRpU7tx33//vW688UY1bNhQX3/9dYAqBPIe8tw18hzwDfIc8A0y3TnyHPAN8hzwDfLcNTId8A0yPR0d6/CL5cuX65FHHtG+ffskScWKFdOdd96pp59+WqGhobbp3n//fdWsWVPNmzcPVKl+NXDgQK1fv16rV6+2PQ8nISFBgwcPVmhoqFauXKm0tDS99NJLmjx5smbPns2zcAAP7N27Vx06dFBCQoJatmypGjVqSJJ+/vlnffvtt4qNjdW6det03XXXBbhSIG8hz50jzwHfIM8B3yHTHZHngG+Q54DvkOfOkemAb5Dp6ehYh1/99ddfSkpKUunSpWWxWAJdTsDFxsZq0qRJmjx5st3w7du3q0WLFjpw4ICqV68uSRo8eLB+/vln7dixIxClAnnOyZMn9cILL2jlypWKj4+XJFWuXFk9e/bUww8/rDJlygS4QiDvIs/tkeeA75DngG+R6X8jzwHfIc8B3yLP7ZHpgO+Q6VJIoAtAwRITE5PpNMeOHVP58uX9UE3gJSUlKTo62mF40aJFZYzRyZMnbSHftWtXLV++3N8lAnlW6dKl9eqrr+rVV18NdClAvkOe2yPPAd8hzwHfItP/Rp4DvkOeA75Fntsj0wHfIdOloEAXgPyvVatW2rVrV6bTpaWladq0aapbt64fqsodGjdurEWLFik1NdVu+Lx58xQcHKyaNWvahl24cEERERH+LhEAAEnkuTvkOQAgLyHTnSPPAQB5CXnuGpkOwJe4Yh0+d+TIETVt2lT33XefnnrqKadBtXXrVt11113auXOnevXqFYAqA2PKlCnq0aOH6tatq759+yoiIkKbNm3SmjVrNGbMGJUsWdI27YYNG1S/fv0AVgvkLfv27dP8+fN16NAh/fXXX7r2yScWi0Vr1qwJUHVA3kOeu0aeA75DngPeR6Y7R54DvkOeA95HnrtGpgO+Q6bzjHX4wblz5/TII4/o7bffVuXKlfXmm2/qpptushv3zjvvqHz58nr99dfVp0+fwBbsZ8uXL9cjjzyiffv2SZKKFSumO++8U08//bRCQ0Nt073//vuqWbOmmjdvHqhSgTxj4cKFGjVqlEJDQ1WrVi2Xt8Rau3atnysD8i7y3D3yHPA+8hzwDTLdNfIc8D7yHPAN8tw9Mh3wPjI9HR3r8JstW7Zo3Lhx2rt3r/r27auuXbtqypQpOnPmjO6991499dRTioyMDHSZAfPXX38pKSlJpUuXlsViCXQ5QJ4WFxen2NhYrVy5UiVKlAh0OUC+Qp67R54D3kOeA75FprtGngPeQ54DvkWeu0emA95DpqejYx1+lZKSogkTJmjOnDmyWCyqU6eOFi9erHr16gW6tDzh2LFjKl++fKDLAHK9iIgITZs2TXfffXegSwHyJfI8Z8hzwDPkOeB7ZHr2keeAZ8hzwPfI85wh0wHPkOnpggJdAAqOlJQUPf/883rvvfcUExOjcuXK6eDBg1qwYIEuXrwY6PIColWrVtq1a1em06WlpWnatGmqW7euH6oC8r4GDRro+PHjgS4DyJfIc0fkOeAb5DngW2S6PfIc8A3yHPAt8twRmQ74Bpmejo51+MXGjRvVoEEDTZkyRX379tW+ffu0b98+3XXXXXrttddUt25dLV26NNBl+t2RI0fUtGlTPfzww7p8+bLTabZu3aqmTZtq0qRJat++vZ8rBPKmadOmae7cudq8eXOgSwHyFfLcOfIc8A3yHPAdMt0ReQ74BnkO+A557hyZDvgGmZ6OW8HD58aMGaMFCxaoWrVqmjlzprp06WI3/vvvv9e4ceO0c+dO9e7dW9OnTy8wt145d+6cHnnkEb399tuqXLmy3nzzTd10001249555x2VL19er7/+uvr06RPYgoE8onfv3vr555918OBB1a1bV5UqVVJwcLDdNBaLRZ999lmAKgTyHvLcNfIc8A3yHPANMt058hzwDfIc8A3y3DUyHfANMj0dHevwufDwcD300EP65z//qfDwcKfTpKWl6fXXX9eUKVNksVh07tw5P1cZWFu2bNG4ceO0d+9e9e3bV127dtWUKVN05swZ3XvvvXrqqacUGRkZ6DKBPKNKlSqyWCxup7FYLDp06JCfKgLyPvI8c+Q54F3kOeAbZLp75DngXeQ54BvkeebIdMC7yPR0dKzD5/bt26c6dep4NO3Ro0c1ceJELVmyxMdV5T4pKSmaMGGC5syZI4vFojp16mjx4sWqV69eoEsDAIA89xB5DgDI7cj0zJHnAIDcjjz3DJkOwNvoWIdfHT9+XF988YX27dun8+fPKyoqSnXr1lX37t1Vrly5QJcXMCkpKXr++ef1/PPPKyIiQpGRkTp58qQmTpyop556SoULFw50iQAA2JDnzpHnAIC8hkx3RJ4DAPIa8tw5Mh2AL9CxDr+4fPmyJk2apHfeeUcpKSm69msXGhqqsWPH6uWXX1ZERESAqgyMjRs36s4779T+/fs1dOhQvfrqq4qMjNTkyZM1Y8YMlS9fXm+++aZ69+4d6FKBPCkxMVHnzp1TWlqaw7hKlSoFoCIg7yLPXSPPAd8izwHvItOdI88B3yLPAe8iz10j0wHfKsiZTsc6fC4lJUXdunXTunXr1LFjR91+++1q2LChoqKilJiYqJ07d+q9997T2rVr1bFjR3311VcKDg4OdNl+MWbMGC1YsEDVqlXTzJkz1aVLF7vx33//vcaNG6edO3eqd+/emj59usqXLx+gaoG8ZebMmZo2bZrbZ7qkpqb6sSIgbyPPXSPPAd8hzwHvI9OdI88B3yHPAe8jz10j0wHfIdMlGcDH3njjDWOxWMyMGTPcTjdz5kxjsVjMm2++6afKAi8sLMw8/vjj5sqVKy6nSU1NNdOmTTNRUVEmOjraj9UBeZd1e9K9e3fzr3/9y1gsFvPAAw+YyZMnm3LlypnGjRub+fPnB7pMIE8hz10jzwHfIM8B3yDTnSPPAd8gzwHfIM9dI9MB3yDT03HFOnyuRYsWKl++vJYsWZLptH369NGxY8e0bds2P1QWePv27VOdOnU8mvbo0aOaOHGiR+sRKOiuu+46VapUSStXrlRCQoJKliyp1atXq1OnTjp37pyaNWumu+66Sw8++GCgSwXyDPLcNfIc8A3yHPANMt058hzwDfIc8A3y3DUyHfANMj1dSKALQP63b98+jRkzxqNpu3fvroceesjHFeUeGQP++PHj+uKLL7Rv3z6dP39eUVFRqlu3rrp3765y5cqpYsWKBDzgoV9//VX/+Mc/JKU/T0qSrl69KkkqWrSoxo4dqxkzZuT7kAe8iTx3jTwHfIM8B3yDTHeOPAd8gzwHfIM8d41MB3yDTE9Hxzp8zmKxKC0tLUvTFySXL1/WpEmT9M477yglJUXX3kQiNDRUY8eO1csvv6yIiIgAVQnkLUWLFlVKSookKTo6WpGRkTp69KhtfFRUlP74449AlQfkSeS5e+Q54H3kOeAbZLpr5DngfeQ54BvkuXtkOuB9ZHq6oEAXgPyvdu3a+uKLLzya9osvvlDt2rV9XFHukZKSoptuukkzZ85U27ZtNW/ePO3YsUM///yzduzYofnz56tt27aaOXOmevXqpdTU1ECXDOQJ9erV086dO21/t2zZUjNnztSxY8d09OhRzZ49WzVr1gxghUDeQ567Rp4DvkGeA75BpjtHngO+QZ4DvkGeu0amA75Bpv+/QD7gHQXD66+/boKCgsyMGTPcTjdz5kwTFBRkXn/9dT9VFnhvvPGGsVgsHq0bi8Vi3nzzTT9VBuRt8+bNMy1atDBXrlwxxhjzzTffmEKFCpmgoCATFBRkwsPDzfLlywNcJZC3kOeukeeAb5DngG+Q6c6R54BvkOeAb5DnrpHpgG+Q6eksxlxzDwzAy1JSUtSlSxdt3LhRnTt31vDhw9WwYUNFRUUpMTFRP/30kxYuXKjVq1erbdu2WrNmjUJCCsZTClq0aKHy5ct79ByXPn366NixY9q2bZsfKgPyn0OHDmnZsmUKDg5Wt27dCsbZc4AXkeeukeeA/5DnQM6R6c6R54D/kOdAzpHnrpHpgP8UxEynYx1+cenSJd1///2aN2+ew7NfjDEKDg7WqFGj9Oqrr6pw4cIBqtL/oqKi9PLLL+vOO+/MdNpZs2bpoYceUmJioh8qAwDAEXnuHHkOAMhryHRH5DkAIK8hz50j0wH4UsE4RQkBFxkZqdmzZ+vJJ5/UypUrtXfvXiUmJioqKkp16tRRjx49VKFChUCX6XcWi8Wh0ZPZ9AAABAp57hx5DgDIa8h0R+Q5ACCvIc+dI9MB+BJXrCOg9u/fr//97386ceKEateurZEjRyo6OjrQZflNixYtVLZsWX322WeZTtunTx8dP35c3333nR8qA/KWoKCgLDeCLRaLUlJSfFQRULCQ5+Q54A3kORB4BTnTyXPAO8hzIPAKcp5LZDrgLWS6c1yxDp+bPn263njjDW3evFklSpSwDV++fLkGDBig5ORkWc/veOONN/Ttt9/aTZef3Xbbbbr//vs1c+ZM3X333S6nmzVrlpYtW6ZXX33Vj9UBeceTTz7J2aWAj5HnrpHngHeQ54B/kOnOkeeAd5DngH+Q566R6YB3kOnOccU6fK5bt24KDg7WypUrbcNSUlJUvnx5XbhwQTNmzFCzZs30+eef6/HHH9eECRMKTJilpKSoS5cu2rhxozp37qzhw4erYcOGioqKUmJion766SctXLhQq1evVtu2bbVmzRqFhHA+DADA/8hz18hzAEBeQqY7R54DAPIS8tw1Mh2AL9GxDp+rUKGC7rjjDk2ZMsU2bNWqVbrxxhs1efJkPfvss7bht956q77//nsdOHAgEKUGxKVLl3T//fdr3rx5Ds9+McYoODhYo0aN0quvvqrChQsHqEogbzh69KiCgoJUvnx5SdKVK1c0Y8YMh+kqVKigQYMG+bs8IE8jz90jzwHvIc8B3yLTXSPPAe8hzwHfIs/dI9MB7yHT7XEaDnwuISFBFStWtBu2Zs0aWSwW9e3b1254mzZt9Mknn/izvICLjIzU7Nmz9eSTT2rlypXau3evEhMTFRUVpTp16qhHjx6qUKFCoMsEcr1du3apcePGeu211zRhwgRJ0sWLFzVp0iSHaYODg1WnTh3Vr1/f32UCeRZ57h55DngHeQ74HpnuGnkOeAd5Dvgeee4emQ54B5nuiI51+Fzp0qX1xx9/2A3buHGjIiMj1bBhQ7vhYWFhCgsL82d5uUb58uU1duxY29/79+/X//73P/3rX/9S7dq1NXLkSEVHRwewQiB3mz17tipXrqzx48c7jHv//ffVunVrSVJaWpo6dOig2bNna/r06f4uE8izyHPPkOdAzpDngO+R6Zkjz4GcIc8B3yPPPUOmAzlDpjsKCnQByP+aNWumd999V4mJiZKkPXv26LvvvtONN97o8OyS/fv3F6gzxaZPn66aNWvq9OnTdsOXL1+uRo0aaerUqZo1a5buu+8+NWnSxGE6AH9bu3at+vXrp6Agx2grXbq0KleurMqVK6tq1aoaNmyY1q5dG4AqgbyLPHeNPAe8hzwHfI9Md448B7yHPAd8jzx3jUwHvIdMd0THOnxuypQpio+PV40aNdS5c2e1adNGFotFjz32mMO0S5YssZ3hUhAsXbpUcXFxKlGihG1YSkqKxowZo+DgYM2bN0+7du3SCy+8oPj4eD333HMBrBbI3Y4cOaLatWvbDQsJCVHDhg0VFRVlN7xq1aqKj4/3Z3lAnkeeu0aeA95DngO+R6Y7R54D3kOeA75HnrtGpgPeQ6Y7omMdPle/fn19/fXXatq0qY4fP66WLVtqxYoVatq0qd1069atU2RkpAYOHBigSv1v7969atmypd2wtWvX6tSpU7r//vs1YsQIXXfddXr44Yc1aNAgrVixIkCVAnlDWlqa3d9FixbVDz/8oObNm9sNt1gsMsb4szQgzyPPXSPPAe8izwHfItOdI88B7yLPAd8iz10j0wHvItPt8Yx1+EXr1q31+eefu52mQ4cO2rVrl58qyh0SEhJUsWJFu2Fr1qyRxWJR37597Ya3adNGn3zyiT/LA/KUChUqaOfOnR5Nu3PnzgJ1CyzAW8hz58hzwHvIc8A/yHRH5DngPeQ54B/kuXNkOuA9ZLojrlgHAqh06dL6448/7IZt3LhRkZGRatiwod3wsLAwhYWF+bM8IE/p2rWr3n//ff35559up/vzzz/1/vvvq2vXrn6qDEB+R54D3kOeAwgU8hzwHvIcQCCR6YD3kOmO6FgHAqhZs2Z69913lZiYKEnas2ePvvvuO914440KCbG/ocT+/fsLxNk+QHZNmjRJycnJ6ty5s77//nun03z//ffq0qWLkpOT9eCDD/q5QgD5FXkOeA95DiBQyHPAe8hzAIFEpgPeQ6Y7spiCcMN7IJfatWuXmjdvrmLFium6667T9u3bdenSJW3ZssXheThxcXHq1KmT3n777QBVC+R+y5cv19ChQ3Xp0iVVr15d9erVU5EiRXThwgXt3r1bv/zyiyIiIvTBBx+od+/egS4XQD5BngPeRZ4DCATyHPAu8hxAoJDpgHeR6fa4Yh0IoPr16+vrr79W06ZNdfz4cbVs2VIrVqxwCPh169YpMjJSAwcODFClQN7Qq1cv7dy5U2PHjtXFixe1ZMkSLVy4UEuWLNGFCxc0ZswY/fjjjwUi4AH4D3kOeBd5DiAQyHPAu8hzAIFCpgPeRabb44p1AEC+lZiYqPPnzysqKkrR0dGBLgcAAGQDeQ4AQN5HngMAkD8U9EynYx0AAAAAAAAAAAAAADe4FTwAAAAAAAAAAAAAAG7QsQ4AAAAAAAAAAAAAgBt0rAMAAAAAAAAAAAAA4AYd6wAAAAAAAAAAAAAAuEHHOgAAAAAAAAAAAAAAbtCxDgAAAAAAAAAAAACAG3SsAwAAAAAAAAAAAADgBh3rAAAAAAAAAAAAAAC4Qcc6AAAAAAAAAAAAAABu0LEOAAAAAAAAAAAAAIAbdKwDAAAAAAAAAAAAAOAGHesAAAAAAAAAAAAAALhBxzoAAAAAAAAAAAAAAG7QsQ4AAAAAAAAAAAAAgBt0rAMAAAAAAAAAAAAA4AYd6wAAAAAAAAAAAAAAuEHHOgAAAAAAAAAAAAAAbtCxDgAAAAAAAAAAAACAG3SsAwAAAAAAAAAAAADgBh3rAAAAAAAAAAAAAAC4Qcc6AAAAAAAAAAAAAABu0LEOAAAAAAAAAAAAAIAbdKwDAAAAAAAAAAAAAOAGHesAAAAAAAAAAAAAALhBxzoAAAAAAAAAAAAAAG7QsQ4AAAAAAAAAAAAAgBt0rAMAAAAAAAAAAAAA4AYd6wAAAAAAAAAAAAAAuEHHOgAAAAAAAAAAAAAAbtCxDgAAAAAAAAAAAACAG3SsAwAAAAAAAAAAAADgBh3rAAAAAAAAAAAAAAC4Qcc6AAAAAAAAAAAAAABu0LEOAAAAAAAAAAAAAIAbdKwDAAAAAAAAAAAAAOAGHesAAAAAAAAAAAAAALhBxzoAAAAAAAAAAAAAAG7QsQ4AAAAAAAAAAAAAgBt0rAMAAAAAAAAAAAAA4AYd6wAAAAAAAAAAAAAAuEHHOgAAAAAAAAAAAAAAbtCxDgAAAAAAAAAAAACAG3SsAwAAAAAAAAAAAADgBh3rAAAAAAAAAAAAAAC4Qcc6AAAAAAAAAAAAAABu0LEOAAAAAAAAAAAAAIAbdKwDAAAAAAAAAAAAAOAGHesAAAAAAAAAAAAAALhBxzoAAAAAAAAAAAAAAG7QsQ4AAAAAAAAAAAAAgBt0rAMAAAAAAAAAAAAA4AYd6wAAAAAAAAAAAAAAuEHHOgAAAAAAAAAAAAAAbtCxDgAAAAAAAAAAAACAG3SsAwAAAAAAAAAAAADgBh3rAAAAAAAAAAAAAAC4Qcc6AAAAAAAAAAAAAABu0LEOAAAAAAAAAAAAAIAbdKwDAAAAAAAAAAAAAOAGHesAAAAAAAAAAAAAALhBxzoAAAAAAAAAAAAAAG7QsQ4AAAAAAAAAAAAAgBt0rAMAAAAAAAAAAAAA4AYd6wAAAAAAAAAAAAAAuEHHOgAAAAAAAAAAAAAAbtCxDgAAAAAAAAAAAACAG3SsAwAAAAAAAAAAAADgBh3rAAAAAAAAAAAAAAC4Qcc6AAAAAAAAAAAAAABu0LEOAAAAAAAAAAAAAIAbdKwDAAAAAAAAAAAAAOAGHesAAAAAAAAAAAAAALhBxzoAAAAAAAAAAAAAAG7QsQ4AAAAAAAAAAAAAgBt0rAMAAAAAAAAAAAAA4AYd6wAAAAAAAAAAAAAAuEHHOgAAAAAAAAAAAAAAbtCxDgAAAAAAAAAAAACAG3SsAwAAAAAAAAAAAADgBh3rAAAAAAAAAAAAAAC4Qcc6AAAAAAAAAAAAAABu0LEOAAAAAAAAAAAAAIAbdKznIevWrdMdd9yhunXrKiYmRqGhoSpevLhatGihCRMmaPXq1TLGOMw3cuRIWSwWLViwwP9FI8umTp0qi8WiqVOnBroUIFNVqlSRxWLRkSNHAl0KEBDW38C1GbtgwQJZLBZZLBaFhYXpzz//dLmMpKQkFS9e3Db9s88+azd+3bp1tnEZ/0VFRalhw4Z69NFH3S4/UMgzR3v37lWfPn1UqlQpBQcH59v106FDB1ksFq1bty7QpcCJ7H4+/KYB78rp/r3FYlGjRo3cvsa2bdvs2g7ffPON3Xjr7zrjv+DgYMXGxqpdu3Z68803lZyc7M237RXkjKNly5apXbt2io6Otn2WrJ+8h/1r5Ec///yzJkyYoLp166pw4cIqVKiQKlSooObNm2vChAn6+OOPA11ipnLzb9N67GHkyJGBLsWpzZs3q1u3boqNjVVQUJDHfRS5eZ3nNUlJSZo8ebJq1Kih8PBwWSwWValSJdBlIZeiL9EzIYEuAJk7ffq0br31Vn311VeSpPLly6tNmzYqWrSozp07p927d+utt97SW2+9pcaNG2vHjh0Brjj/sFgskuT0gEZ+tWDBAo0aNUojRozw+ga0IK5PAEhOTtbChQv14IMPOh2/ZMkSnTlzxqNljRgxQlL6djQ+Pl7ffvutfvrpJy1YsEDr1q1T7dq1vVY3vOvixYu66aabdOTIETVr1kw33nijgoODM+0UAbLCl+04AN7hzf37nTt3avv27WratKnT8XPnzvWoptKlS6t79+6S0tstBw4c0DfffKNvvvlGixcv1ldffaXChQtn8Z3CX3788Uf1799faWlp6tSpk8qWLSuLxaIyZcoEujQABdwnn3yiYcOG2U4mb9OmjUqWLKm//vpLP/74o9566y0tXrxY/fv3D1iNU6dO1VNPPaUpU6ZwAqmXHT9+XDfddJPOnTuntm3bqkqVKgoKClL16tUDXVpAdOjQQevXr9fatWvVoUMHv73uP//5T7300ksqXbq0brnlFkVGRqpEiRJ+e31/WbdunTp27Kj27dvnqZML6S/Jm+hYz+XOnj2rtm3b6sCBA6pdu7ZmzJihjh07Oky3e/duvfrqq1q8eHEAqgRQUK1Zs0bJyckqX758oEsBcqUGDRpo3759mj9/vsuO9Xnz5kmSmjdvrm3btrld3rUdZQcPHlTnzp31+++/a9y4cdqwYYNX6ob3bdu2TUeOHFHr1q21adOmQJcDZNmECRM0ZMiQfHkQBvAXb+7fN2vWTN9//73mzZvntGP98uXLWrx4scqWLavg4GD9/vvvLpdVu3ZthzbGsmXL1LdvX23evFn//ve/9fTTT3v+RuFXn376qZKTkzV58mQ999xzgS4HOcD+NfKTkydPasSIEUpKStKDDz6oZ599VoUKFbKbZvv27froo48CVGH+0LdvX7Vs2VJFixYNdCkOvvrqK509e1bDhg3T+++/H+hyCqwPP/xQkrRx40bVqFEjwNUA+QO3gs/l7rnnHh04cEDVqlXT5s2bne50S1K9evU0d+5crV271s8VAijI4uLiVLt2bYWGhga6FCBXKlmypG6++Wbt2bNHW7dudRj/22+/ac2aNbr++utVt27dLC+/Zs2aeuaZZySl7ySdOHEixzXDN3777TdJYkcWeVaJEiVUu3ZtOtaBHPDm/v1NN92k0qVL6z//+Y+uXLniMP6jjz7SuXPndPvttys4ODjLtd5888267bbbJP19QBa5E22M/IP9a+Qny5cv14ULF1SuXDm9/PLLDp3qktS0aVM9//zzAagu/yhatKhq166tsmXLBroUB+RT7sDnAHgfHeu52K+//qoPPvhAkvTqq68qJiYm03latGjh8fIze15CZs9oOXjwoMaPH69atWopMjJS0dHRqlu3rsaPH6/du3c7TL9//36NGjVKlStXVnh4uGJjY9W5c2eXO+lpaWmaM2eO2rRpo2LFiik0NFSlSpVSw4YNdc899zh9xkpKSoreeecddejQQbGxsQoPD1fVqlV199136+jRo56uGtuz5qyufe7cta/95ZdfqlevXipVqpTCwsJUrlw5DR48WN9//73Hr5mZ5ORkLVq0SLfeeqtq166t6OhoRUREqFatWpo4caKOHz/ucl5jjD755BP16tVLZcqUUVhYmMqUKaO2bdvq3//+ty5fviwp/fk1o0aNkiS9++67du854y1q4uPj9e9//1udOnVSpUqVFB4ermLFiqlt27aaPXu20tLS7F4/q+vz4MGDuvPOOxUXF6dChQqpaNGiuuGGG7Ro0aJsr7+vv/5aAwcOVIUKFRQeHq6SJUuqefPmmjJlihISEhymz+pnmvE5fz/++KP69eunEiVKKDw8XHXr1tUrr7zicEuXVq1ayWKxuL0SZfr06bJYLOrbt6/DuO3bt+vWW2+1fQaxsbG68cYbtWLFCqfLyvh8os8++0ydOnVSbGys3fP3kpKS9NJLL6lp06aKioqyfVeaN2+uhx9+2OF21e6eeXTp0iW98MILatKkiaKiohQZGanrrrtOTzzxhP766y+H6Y8cOWJ7zo8xRnPmzFHTpk1VuHBhFS1aVN26ddOWLVtcrisgtxo9erSkv69Mz2j+/PlKS0uzTZMdGa9Si4+P92ieEydO6N5771XNmjVVqFAhRUZGqmLFiurcubNefvllh+k/+eQTjR07VvXq1VNMTIwKFSqkqlWravTo0Tpw4IDHtT722GOyWCy66667XE6ze/duWSwWlS5d2u65rtmt4eLFi/rnP/9pe55YuXLlNHr0aB07dizTZ0ZndTvrzLp162SxWGy38b82X6082UZLWW9PZXyPx48f19ixY1WuXDlFRETYOm4yLnvYsGEqU6aMChUqpIYNG+q///2vx+/VGU8zsX379rJYLPrPf/7jclkvvviiLBaLBg0a5PT9xcfH6/bbb1fZsmVVqFAh1axZU1OnTrW1c5zxNO/PnDmjypUry2KxaNasWQ7LuXDhgmrXri2LxaJ///vfduOy893N2E4/fPiwhg8frjJlyig8PFxxcXF64oknlJSUZDePp+24jDz9fK5d1xll3Ge4ePGiHnvsMVWvXl3h4eEqU6aMRowYoWPHjjl9fSn9FpEPPPCA6tSpo8jISEVFRal58+aaPn26UlJSXM4H5DXe3r8PCQnR8OHD9ddff2nJkiUO463tDm+0MbLyfNPt27dr8ODBqlChgsLCwhQdHa1q1aqpf//++uyzz+ymTUxM1Ntvv61+/fqpRo0aKly4sAoXLqz69evr8ccf19mzZz1+3ezuW+Wkhvj4eI0cOdKWmzVq1NCUKVN05cqVTJ8D/9FHH6l79+4qWbKkwsLCVL58ed12223au3evx+/Zul2eP3++JGnUqFEO2/2M+1ipqamaNm2aGjdurCJFiti1Q6Sc7QN/++23uummm1S8eHFFRUWpffv22rhxo23aL774Qp07d1ZMTIyKFCmirl27Zvsxhrt371b//v1VokQJRUZGqn79+nrttdeUlpbmcv/02naXu/eSUVaPfUg52691VX92jjdktGbNGvXr109ly5ZVWFiYSpUqpb59+7J/DZ86efKkpPSTzT11/vx5RUdHKyQkxO1x3J49e8pisWjGjBm2Ydn5nVgsFj311FOSpKeeesqu/ezqePjatWvVrVs3xcTEKCIiQk2aNNF7773n9n1ldZuflSx1d/x+9erVuvnmm1W6dGmFhoYqJiZGNWrU0G233Zatu90tXrxYnTt3th1zr1y5skaPHq2DBw86rWnKlCmS7NetN57t/eyzz8pisahixYrauXOnqlWrJovF4nabNn78eFksFj388MO2YdnZ55Ky3nawHhNYv369JKljx45237Vr+2b++usvTZkyRY0aNbIdT61fv76effZZXbp0yeP1ZM0U6/fe2Wtm3Mf77bffNGbMGFWsWFGhoaF236mcHONNS0vTG2+8oQYNGigyMlJly5bVXXfdZTvGnJSUpGeeeUa1a9dWRESEypUrp3vvvVcXL170+L126NDBdsLq+vXr7d5rxu9cxs989+7dGjx4sO3uTlOnTtWUKVNksVh05513unyt7777ThaLReXLl7ftr1o/4w4dOujSpUuaPHmyqlevrkKFCqlcuXIaM2aMwz6xr/ufMm4T169fr27duik2NlaRkZFq0aKFFi5cmOl69eXvwipj2yc729eAMci1XnvtNSPJxMTEmNTU1GwvZ8SIEUaSmT9/vkfDrebPn28kmREjRjiMe//99014eLiRZCpVqmT69+9v+vbtaxo2bGgsFouZMmWK3fTLly83hQoVMpJMrVq1zJAhQ0ynTp1McHCwkWRGjx7t8BqjRo0ykkyhQoVMly5dzNChQ82NN95oatSoYSSZJUuW2E1//vx506FDByPJFClSxLRv394MGDDA1KpVy0gyxYsXNzt27PBonS1ZssS2fqzrIOO/U6dO2aZ94oknjCRjsVhMmzZtzNChQ02jRo2MJBMcHGzmzp3r0WtaTZkyxUhyWIdHjx41kkzRokVNy5YtzcCBA03Pnj1NuXLljCRTsmRJ8/PPPzss7+rVq6Zfv35GkgkKCjItW7Y0Q4cONV27djXly5c3kszhw4eNMcY8+OCDpk2bNkaSiYuLs3vPzz//vG2ZzzzzjJFkqlatajp37myGDBli2rdvb8LCwowk069fP5OWlpat9fnhhx/aviu1a9c2ffv2NZ06dTKFCxc2ksyoUaOytD6NMeaee+6xvXajRo3MkCFDTI8ePUy1atWMJLN27Vq76bPzmbZv395IMo8++qgJCwszderUsa0X6/f83nvvtZtn9uzZRpK58cYbXdbepEkTI8ksXbrUbvhrr71mgoKCbO9pwIABpm3btrbP4KmnnnJYVuXKlY0kM2HCBCPJNGvWzAwdOtS0b9/ebNiwwaSmpprOnTsbSSY6Otr06NHDDB061HTp0sU27w8//OB0mdbvkFVCQoJtnUVHR5vevXub/v37mxIlSti+O9fOc/jwYSPJVK5c2YwYMcKEhoaaTp06mUGDBpmaNWsaSSY8PNx8++23LtcX4G/W38C1WWrN0M6dO5uUlBRTrlw5Ex0dbS5dumSbJi0tzVSuXNlERkaac+fO2baTzzzzjN2y1q5da9uGOfPNN9/YxnuScydOnLBlR6VKlcwtt9xiBg8ebNq1a2diY2NN0aJFHeYJDg42kZGRplmzZqZfv36md+/etm1o4cKFzaZNmxzmcZZnBw4cMJJMsWLFzOXLl53W98ADDxhJ5oEHHshxDRcuXDDNmze3tQ169eplBg4caMqWLWtKlSplRo4c6TRzjcnedtaZffv2mREjRrjMV6vMttHGZK89Zf0cRo0aZcqUKWMqVapkBg0aZDp27Gib7+WXXzZbtmwxUVFRtuW2atXK9r1avHixR+/VKjuZ+PHHHxtJpnXr1k6XmZqaaqpUqWIkmfXr1zu8v9tvv90UL17clC5d2gwcOND06tXL1nZo06aN0+9bVvP+22+/NaGhoaZQoUIOeTh06FAjydx00012bSBjsvfdtW4P7r33XhMdHW0qV65sBg0aZLp06WIiIiKMJNOnTx+7eTxtx2Xn88m4rq/9vVi3d3369DENGjQwxYoVMzfffLO55ZZbTKlSpWzZfvbsWYdlrl+/3sTExBhJpkqVKqZ3797mxhtvtA3r1q2buXr1qsN8QF7k7f37Z555xuzdu9dIMl26dLGb5pdffrFt24z5O2M2btxoN531d92+fXunr/Xss8/a2vOeWL16tQkNDTWSTMOGDc2AAQNM3759TYsWLUx4eLi55ZZb7KbfuHGjbV+2bdu2ZvDgwaZbt26mePHiRpKpXr26OX36tMPrWLdjGffjsrtvld0a9uzZY9u3KVeunBk0aJC56aabTOHChU3btm1N69atne5rJicnm0GDBtn2bVq3bm0GDhxoGjZsaCSZiIgIs3LlSo/Wt3U/Oy4uzpZ31273rftYlSpVMr179zZhYWGmc+fOZujQoaZBgwa2ZeVkH3jSpEkmJCTENG7c2AwePNg2X3h4uNm0aZOZPn26CQoKMq1bt7bbtytSpIjTYxjurFu3zpaDcXFxZsiQIaZr164mLCzMDB482OX+qbu2dMb3cu3nldVjHxnXeXb2a13Vn93sNia9fWA9HtSiRQszcOBAc/311xuLxWKCg4PNvHnzXK4XICcWLlxo24asXr3a4/msx/AmT57sdLw146Kjo01iYqJteHZ+JyNGjLBtfxs2bGjXfn777bdt01l/m//85z+NxWIxTZs2NUOGDDEtW7a0bV9effVVh1qzs83Papa6On6/YMECY7FYjMViMddff70ZPHiw6d27t2nSpIkJDg52uc1wJi0tzdx+++1GkgkJCTGdOnUyQ4YMsW3TIiMj7d7Hxo0bXa7bBx980KPXdLY9vHr1qq2/oFGjRubYsWPGGGNeeeUVI8kMGzbM6bLOnTtnihQpYoKCguyWl519Luv7y0rbwXpMoHTp0ra2SsbvWsb22Z49e0zFihWNJFO2bFnTvXt3c/PNN9vmbdSokdP9KmcefPBBl8fjra9pbQsOGzbMxMbGmjJlypj+/fubfv362T6rnB7jHTp0qImIiDDdu3c3ffr0se0jNm7c2Fy4cMG0bdvWttxevXqZokWLGkmmR48eHr1PY4x5/vnnzY033mgkmdKlS9u914zfOev6uOOOO0x4eLipUqWKGTRokLn55pvNyy+/bE6cOGHCwsJM4cKFzV9//eX0tay/hYzHhKzH7Vq1amVatmxpIiMjTc+ePW3HnySZMmXKmIMHD9rm8XX/k3WbOHHiRBMUFGTq1q1rhgwZYm644Qbbsa5rj7tlXEe+/l1YZXf7Gmh0rOdiw4cPN1L6Qfmc8HbH+vfff29CQ0ONxWIxb7zxhsNBgSNHjpjvv//e9vcff/xh2yA+++yzdjsd27Ztsx04mzNnjm14fHy8kWQqVKhgTpw44VDb3r17TXx8vN2wYcOGGUmmV69e5uTJk3bjXn31VSPJ1KhRw6SkpDh9v85ktvO1cuVKI6V3/n/11Vd249555x0jyYSGhprdu3d7/JquDlqeP3/efPbZZyYpKclu+NWrV81jjz1mJJmePXs6LM/aQVGlShXz448/2o1LS0szq1evtgtkdydUWH333Xdm165dDsOPHTtmazR9+OGHDuMzW58//fSTCQ8PN4UKFTIff/yx3bgjR46Y+vXrG0nm3XffdbmMa73xxhtGSj+x4uuvv3YYv3XrVvPbb7/Z/s7uZ2oNK0lm1qxZduPWrFlj22k9evSobfi5c+dMZGSkCQoKMr///rtDbTt37rQ1CJKTk23Dv/jiC2OxWEyJEiXsOheMSV+HFSpUMJLMunXr7MZZgyo4ONh89tlnDq+3fv16W8Pm/PnzDuO3bdvmEICudvwHDx5sJJnrr7/ebp7ExETTo0cPIzl2nlgbXdaG14EDB2zjUlJSzOjRo20H2YHcwpOOdWOMbTv93nvv2aZZtWqVkdI7BI2xP1CeUWYd65MmTbJttzJ23Lvy1FNPGUlm3LhxDgcCr1696vSgx+LFi82FCxfshqWlpZm33nrLSDLXXXedw7Jc5Zm14+8///mPw+skJyfbdrKuzZns1HD//fcbSaZu3brm+PHjtuGXL182AwYMsK3Xa2vM7nbWnczyNbNtdHbaU8b8/TlIMnfddZddnixdutRIMlFRUaZy5coOy7V2AlWvXt3j92lM9jIxJSXFtg6cnSCybNkyI8muE+Da93fLLbfY/QaOHj1qO9jz6KOP2s2X3bzP2Ka0ZuXMmTONlN5pkZCQ4FB7dr67GXewH3/8cbv2665du2wnDWzevNluPk/acdn5fIzJvGPdepDo3LlztnFnzpyx7fD/61//spvvxIkTpnjx4sZisZgZM2bY7VOcPn3adOrUyeGABZCXeXv/3tpeaNWqlQkKCrLbP3788ceNJFuHWXY61tPS0kyLFi2MJHPDDTd4VFvHjh2NJLNo0SKHcWfPnjVbtmyxG3b06FGzevVqh2MKFy9etB20HD9+vMOynHWCZnffKrs1WDvphwwZYq5cuWIb/vvvv9tO7r+2RmOMmTx5sm0/6dChQ3bj/ve//5ng4GATExPj8mCuM+6O7WTcx6pQoYLdPpZVTveBLRaLWbhwod0463GIWrVqmSJFiti1L1NSUkz//v2NJDN27FiP3+elS5dsFwc8+OCDdp/Znj17bJ0OzvZPMzsW4apjPTvHPnKyX5tZx3pWs3vOnDm2ttzOnTvtxq1fv95ERUWZsLAwuwP9gLckJibafrMWi8V06NDBPPPMM+bzzz83f/75p8v5Dh48aCwWiylVqpTd9tXKerLIPffcYzfc223cjKy/zdDQULNs2TK7cda2cNGiRR32x7Ozzc9qlrpq/1etWtVp9htjzMmTJz2++MyYv/d3SpQoYXeCcVpamm39FStWzOFz9WTdunLt9vDs2bOmS5cuRkrvcM14UsXZs2dN4cKFTVhYmPnjjz8clvXmm28aSebmm2+2G57dfS5vtl8yunTpku1kuSeeeMKuD+DixYu2k7mzetGZuwzMuD992223Of3N5fQYb1xcnDly5Iht3OnTp20XTtavX9+0aNHCbrmHDh2yHd/45ptvPH6f1mNnrk4YNcb+M3/00Uednux66623Gklm2rRpDuNOnTplwsPDTWhoqF2fVcbjdtWrV7drl1++fNnW7mnZsqXDMjNro3ijr+LaffGMJyp+8cUXLteRP34X2d2+Bhod67mYdcM0ZMgQp+N//PFHhzNZrj3LyRjvd6z36dPHaQPGFesZvk2bNnU6/uWXX7YdoLT67rvvjCTTu3dvj15j7969xmKxmHLlyjntEDTGmJ49expJDj9QdzLbsFmv7nV2do8xxvTq1ctI6WdBeSq7jY5y5cqZoKAgu/d/8uRJ25nUGU92cMeTA7LufPnll0aSGThwoMO4zNanNahffvllp+Ot3wtX36VrJScnm5IlSxpJDh31rmT3M7WGVb9+/ZzO1717dyPZd6oZ8/cBtmsDzhhj7rvvPiOlXwGQ0fXXX28kmY8++sjpa3344YdGkunfv7/dcGtQObuiMeN8EydOdDreGWc7/vHx8SYoKMhYLBaHnXdj0g82Wa+4zHiVXsZG17VX6BuTfgBeSj/Ll6vXkFt42rF+8OBBI8l06NDBNs2QIUOM9HfnbFY61tPS0kx8fLx59tlnTUhISJZ+u+PHjzeSzCeffJKdt+zAemXznj177Ia7yrO5c+e6PJj46aefGin9au2c1nDp0iVTpEgRI8l8+eWXDvP8+eefJjIy0mmN2d3OuuNpx7qrbXR22lPG/P05VKpUyelV2w0aNDCSTIsWLRw6d5OTk01sbKyR5HBCozvZzcQXX3zRSDJjxoxxmMd69vns2bOdvr+IiAinJ2NaO+Sjo6Pt3n9O2nDWOwENHjzY7Nixw7Zjfe1BLk+4+v1YtwdNmzZ1+FyMMeauu+4ykszTTz9tNzwrHetZ/Xwy61gvXLiw3QksVosXLzaSTKdOneyGP/LII0ZKv0uDM7///rsJDQ01JUuWdLoOgLzG2/v31vbC22+/bSSZqVOnGmPS7/BRoUIFU6RIEdtJPVnpWL969arZs2ePrZ2SlTZD3bp1jSRz5swZj6Z35+LFiyYkJMSULFnSYZyrA9PZ2bfKTg0bNmwwUvoV185OqFq+fLlt3WWsMSEhwURERJhChQo57fw35u922ptvvulxnZ52rF+7XbfK6T6ws33/hIQE2+s+9NBDDuO3b99upPSr3Dz13nvvGSm9s9rZ/uD06dNtr+mtjnV3XB37yMl+bWYd61nJ7tTUVNtdqlwdD7K2vTy9ghTIqv3799v2ra7916hRIzNz5kynF0BZj+Fee9LOpUuXTExMjLFYLGb//v1247zdxs3I+tt0tZ2sXbu2kWS705gx2d/mZzVLXbX/IyMjnd6NLjusnb1vvPGGw7i0tDTbPuVzzz1nN85bHevx8fGmXr16RpK58847nX5nrOvy2uMpxvz9+Vx7XCC7+1zuZKf9YmU9gaFXr15OxycmJppSpUqZkJCQLLW13GWg9TOKjY11eiW8N47xfv755w7zTZs2zUjpJ904O4HNeueKrJxknZWO9Zo1a7q8+NLa/1CjRg2H78Xzzz9vJJmhQ4c6fW1J5tNPP3VY5smTJ23Hn669Y11mbZScttMaN27sdD7rSUpdu3a1G+7v30V2tq+5Ac9Yz8OOHj2qd9991+HfL7/84rPXTE1N1apVqyRJ48aN82ge6zOqRvz/s0WvNWbMGEnSzz//bHtOeO3atRUVFaUVK1boueee0+HDh92+xooVK2SMUY8ePRQVFeV0GuszxjZv3uxR3ZlJSUnRpk2bJMnlc3es723t2rVeeU1J2rlzp6ZNm6Z77rlHo0eP1siRIzVy5EilpKQoLS3N7vNfu3atrl69qqZNm9o9g9cbkpKStGzZMj355JO66667NGrUKI0cOVKzZ8+WpCw9c1eS0tLStHLlSknS4MGDnU7TrFkzFSlSRD/88IOuXLmS6TK3b9+uU6dOqUSJEk6fUX4tb3ymN998s9PhderUkSSH56lkfBZqRsnJyXr//fcl2T8X8fTp0/ruu+8UERHh8rUy+64PGDDA6fAmTZooODhY8+bN01tvvaUTJ044nS4zGzZsUFpamho3bqwGDRo4jC9fvrxuvPFGSc7XY0hIiLp37+4wvEyZMoqJiVFSUpISEhKyVRsQKDVq1FC7du20fv16HTp0SH/99Zc+/fRTxcXF6YYbbvB4OdZnLQUFBaly5cp64oknlJKSomHDhunFF1/0aBnW57U++uij+uSTT3ThwgWP5vvll180ffp03XfffRozZowtf6zPzvN0uz9o0CAVLlxYq1ev1u+//243zvqMUFfPg81KDdu3b9eFCxdUokQJdevWzWFZJUuWVNeuXR2Ge2M7mxOuttHZaU9l1LFjRxUqVMhheI0aNSRJPXr0cHj2aEhIiO15ZM6WmZmsZuLYsWMVGRmpDz74wO45bb/88ou++uorFStWTLfddpvTZXbr1k1lypRxGN6rVy8VL15c58+ftz3LNad5P2/ePFWrVk3//e9/1bFjRyUlJemFF15Qy5YtnS7L+h6y8/vp1auX02fCulqHWZHVzyczzZo1U9myZT1e3ueffy7JdbuvfPnyqlGjhk6dOqWff/45S7UAeVF29+8HDx6swoULa8GCBTLG6Msvv9Tvv/9uy1tPZHwWZVhYmK677jotXrxYYWFheuWVVzzal5L+bmPceuut+uabb2zPnczM5s2b9e9//1v/+Mc/bPuV48ePV1hYmE6dOuX02Z3OZHXfKrs1WJ+R2r17d8XGxjos66abblKxYsUchq9du1aXL19WmzZtVL58ead1+LKN0b9/f4dh3tgH7tmzp8Ow2NhYFS9e3OV4a/sjK+0L63ofOHCgQkNDHcbfeuutHi8rK7J77MMX+7VZye4ffvhBx48fV1xcnMvjQb78vgGSVKtWLX377bfaunWrnnzySd144422Z67/+OOPuvvuu9W9e3ddvXrVbr57771XkjR9+nS74db9hC5duqhWrVpOX9PbbdzsLju72/zsZum1WrRooXPnzun222/X9u3blZaWlq3l/P777/r1118lOd8XtVgstvz15vFvqx07dqhly5bas2ePXnjhBc2aNUvBwcEO002cOFEWi0WzZ8+2W2dr1qzR/v37VatWLaf7/1L297m81X6xymz/qEiRImrWrJlSUlK0bdu2LC07M126dFHRokUdhnvjGK+z4zHWdkClSpVUr149l+OzcxzCE3369HH6PZKk5s2bq1WrVvr555/15Zdf2oanpaVp1qxZkqQJEyY4nbdYsWLq3bu3w/BSpUrZ2gTWYzue8EY77fbbb3c63Pp7/uabb5Samuow3t+/C19uu30hJNAFwLUSJUpIkk6dOuV0fK9evWSMsf3dpUsXrVmzxqc1JSQk6OLFi5LksgFzLeuXvmrVqk7HFytWTLGxsTpz5ox+//13lStXTlFRUZo/f75GjRqlJ554Qk888YTKli2rli1bqnv37ho2bJiKFCliW8ahQ4ckSXPnztXcuXPd1uNqfWZVQkKCrXPX1XuLi4uT5J0f/sWLFzV8+HAtWbLE7XTnz5+3/X98fLyk9BMVvOnbb7/V4MGD9dtvv3lUhycSEhJs81SsWNGj6V01TK2s779WrVpOg8DZMnP6mVaqVMnp8OjoaElyOCGgQ4cOqlatmg4cOKDNmzerdevWkqTly5fr1KlTuv76620BIkmHDx+WMUaXL19WeHi42/fj6rtu7SS5VlxcnF599VU99NBDmjBhgiZMmKDKlSurVatW6tWrlwYOHKiwsDC3ryll/pu3vlbGaTMqW7as0wMlUvp6/Ouvvzw6sQLIbUaPHq2NGzdq/vz5KlOmjK5cuaJRo0Z5tH2ysjZ8LRaLIiMjVbVqVXXv3t3pjogrw4cP16pVq/T++++rf//+Cg4OVt26ddW2bVsNGDBAnTp1sps+NTVVEyZM0OzZs+3aHdfydLtfpEgRDRw4UAsWLNB7772nyZMnS5L+/PNPff755ypUqJCGDh2a4xqsnfautnmuxnljO5sTrurNTnsqI1f5ZG1PuRpvPWExO9vdrGZiTEyMhg8frtmzZ2vu3LmaNGmSJGnGjBkyxmjUqFGKjIx0ukx3mVOlShUlJCTYvhM5zfuiRYtq4cKFatOmjc6dO6eePXvqgQcecLqcnP5+sroOs8Lby87q8qzt93bt2mW67FOnTqlmzZpZqgfIbXy1fx8VFaUBAwbo3Xff1ddff6158+ZJct2B7Ezp0qVtB/uCgoIUHR2tunXrqnfv3k5PWnLl+eef108//aSVK1dq5cqVioiIUJMmTdShQwfdeuutdvs1Unr29+/fX998843b5Z4/f14xMTGZvn5W962yW4MnbYzKlSvr7NmzdsOs2701a9Zk2v7zdhujVKlSTjPUl/vARYoUUUJCgtPx1vZFUlJS5sX/v8zWe7FixVS0aFGdO3fO42VmJifHPnyxX5uVrLV+33799Ve/f9+Aa7Vo0cLWYWyM0Q8//KCXXnpJixcv1urVq/X666/roYcesk3ftWtX1alTR1u3btX27dttJ4e89dZbklx3akm5p/2c3W1+VrPUlRkzZqhXr15auHChFi5cqKioKDVv3lydOnXS8OHDXb6Xa1m3/cWLF7e9z2t58/j3tQYPHqyUlBQ9++yzeuSRR1xOV6tWLXXr1k1ffvmlPv30U9sJ69bvzPjx411+Dln9zni7/WJl/c4MHz5cw4cPdzutt7fb2T0OIWV+jDckxLEL0pfHITzhrg0npZ+osWXLFk2fPt3WRl6+fLni4+PVuHFjWxvT2XJdfc+s6/DaC0zc8UY7zdV81uGXL19WQkKCSpUqZTfe378LX267fYGO9VysSZMmWrhwoXbs2KG0tDQFBfn3BgPZPZPNW/r3768uXbpo6dKl2rhxozZt2qQlS5ZoyZIlevLJJ7Vq1SrVr1/frtZGjRqpYcOGbpd7/fXX+7x2X3jssce0ZMkS1a5dWy+88IKaN2+uEiVK2Do6W7durS1btrg9aOsNly5dUp8+fXTy5EmNGjVKd999t6pXr67o6GgFBwfr4MGDqlWrVpbryPh9c3U1XkaZdXYESlZ/pxaLRSNHjtSTTz6pBQsW2ILZetWm9axPK+t6KlKkiNMrDjwRERHhctw999yjQYMGaenSpfrmm2/0zTffaPHixVq8eLGmTJmijRs3Or0azZv8va0D/GXgwIGaOHGi3n33XRUvXlxBQUEebe8yWrBgQY7rCAoK0qJFizR58mR9/vnn2rRpkzZt2qSZM2dq5syZuvnmm7VkyRLb2buvv/66Zs2apTJlymjatGlq3bq1Spcubbv6ediwYfrPf/6Tpe3+6NGjtWDBAr377ru2jvVFixYpJSVFAwYMcLjKKyc1uDuI4WycN7azOeFuG50TmW1bfbHtzc4yJ06cqNmzZ2vmzJl64IEHdOXKFc2fP18Wi0X/+Mc/clSPN9tICxcutP3/vn37dO7cOadn+Of09+PLTPT2srO6POtvbcCAAZleVWu96hHIy3y5fz969Gi9++67eumll7R27VrVqlVLbdq08Xj+2rVre6WNUaZMGX3//fdav369Vq9erU2bNmnr1q3atGmT/vWvf+n555+3Oyg+duxYffPNN2rVqpWeeuopNWzYUDExMbbOyHLlyunEiRMeb7+zum+V0xqy28aoXr16pp+Pt0+O91X7QvJ/GyOr690Tzo5/5fTYR6DbVtb3VKZMGdvVhK5YT/wB/MFisahJkyb6z3/+o0uXLmnp0qX69NNP7TrWLRaL7rnnHo0fP17Tp0/X/PnztWXLFv3www+qUqWKevXq5XL5uaX9nN1tflaz1JU6derowIED+uqrr/T1119r8+bN2rhxo77++ms9/fTTmjt3rsu7geUmI0aM0Ny5c/Xqq6+qe/fubu/Ieu+99+rLL7/UW2+9pQEDBujo0aNaunSpihQp4vJqXynr3xlvt1+srN+Z7t27q3Tp0m6nrVy5cpaWnZn8dBzCE5m93wEDBmjSpElauXKlDh8+rKpVq3p0Yo8nfN13kx3OavL37yKv9QfQsZ6L9erVSw8++KD++usvrVixwm2jITusHbKJiYlOx1uv9s2oePHiioyM1KVLl3TgwAGPrpArX7689u/fbzvr6lrnzp3TmTNnbNNmVLRoUbuztI4ePap77rlHn332mSZMmGC7FZj1Cuc2bdo43CbIV4oXL67w8HAlJSXp0KFDTm+HYn3PmV1Z7YkPP/xQkvTf//7X6Ws5u0Wm9Uyf/fv35/j1rTZs2KCTJ0+qSZMmtqshMqvDEyVKlFBERIQuX76sl19+2Ss7dtb3f/DgQRljMt3J9vdnajVixAhNnTpV//3vf/X666/r/PnztrNShwwZYjet9btusVg0b948n4RO6dKldccdd+iOO+6QlP79GT16tLZs2aJHH33U4daK17KuG1e/+YzjvLkegdyucOHCGjRokObOnaujR4+qe/fuqlChQsDqqVu3rurWrauHHnpIxhh9/fXXGjZsmJYtW6b33nvPdvDZmj+zZ892ekur7Gz327Vrp+rVq+vgwYPatGmT2rRpYzug7+wKu+zUYN2+HDlyxGUdzsb5YzubHTlpT+UldevWVZcuXbR69WqtXLlSx48f19mzZ9WjRw/bmdjOuHtskPVztv7ecpr3ixcv1qxZs1S6dGk1a9ZMn3/+uUaPHq2PP/7YYVpf/H7yi4oVK+rnn3/WI488ombNmgW6HMDnfLl/f8MNN6h69eq221U660D2F4vFog4dOthub3vlyhUtWLBA//jHPzR58mQNGDBAcXFxunjxolasWKGgoCCtWLHC4aS6ixcv6o8//sjy62dl3yq7NXjSxnB2PMXaxqhVq5ZXTmTwhkDtA2dHZuv93LlzDncJsAoNDVVycrISExOdPj7Q2eflq2Mf/mL9vhUvXjzXfN+Aa3Xr1k1Lly7V6dOnHcbdfvvtmjx5shYvXqyXX37Zdrz37rvvzjX7aO7kZJvvaZZmJiQkRD179rQ9kuP8+fOaNm2annrqKd15553q27dvpie4Wre91juNOrtq3Zc58cQTT6hu3bp68MEH1alTJ33++edq27at02m7d++umjVrat26ddqzZ48++OADpaamavjw4S6vts8qX7VfpPTvzP79+zVmzBiXj4jzt4J6jDckJER33323nnjiCc2YMUN33HGHVq1apdjYWIc7LGbkyfGnrBwH9EY7zdWxEms9hQoVyvGJ7L78XeRWuT+FCrDq1avbnqnxwAMPePV2VtLfP7Z9+/Y5jDPG2J53nVFwcLDteSRvv/22R69jbQS46oyz7qDUqFEj0w1wxYoV9dRTT0lKfxaPVY8ePSRJS5cu9eptIaxn1Dh7nk1ISIgtyF01kKzvrWPHjjmuxXqw3NkZaV9++aXTRminTp0UFham7du3254rmhnrCReunuFjrcPV7TkWLVrkctnu1mfG75b1IHRONWvWTCVKlNCpU6f06aefZjq9vz9Tq0qVKqlz5846f/68PvnkE9tVm/369XO4+q1cuXJq0KCBEhMT9cUXX3itBndq165tOxs24+/OlRtuuEFBQUH68ccftXPnTofxJ06csNXuzfUI5AVjx45V8eLFVbx4cdvJK7mBxWJR586dNWzYMEn2v3V3+bNnzx6PtgvOWA/6L1iwQNu3b9euXbtUsWJFde7c2WHa7NTQtGlTRUZG6tSpU1q9erXD+NOnT2vVqlUOwwOxnfWEN9tTuV3GZyp6elb4V199pT///NNh+IoVK5SQkKCoqCjb1Q05yfuDBw9q3LhxCgoK0vvvv68PPvhAcXFx+uSTT/TGG284LMdXvx9XMmvH5SbW9ru32n1Abufr/fu77rpLxYsXV6lSpVw+SzEQChUqpLvuuksNGjRQWlqafvrpJ0npnaCpqamKjo52+jzyRYsWZeuKnqzsW2W3hhtuuEGS9MUXXzh9TuTKlSudDu/cubPCwsK0bt06p5kVCIHaB84O63r/3//+5zTnPvjgA5fzujv+9dNPP+no0aMOw3Ny7CM3sN7lcO/evdqzZ0+gy0EB5Mk23PqYBWcdTYULF9aYMWN05coV/etf/9JHH32kQoUK2Z4n7C2+aj97c5vvKkuzKjo6WlOnTlWxYsV06dIlHTx4MNN5KlSoYOvEd5YTxhjbcF/lxAMPPKA5c+bowoULuvHGG53ux0t/3+lAkqZNm6Z33nlHUs6vMM4oJ+2XzL5ruXH/KC8d4/X2b/nOO+9UoUKFNG/ePL3yyisyxmjMmDFur3Y/e/asli1b5jD81KlTtvVkPbZj5ev+J1ftlffee0+S1LZtW6e3688KX7XrczM61nO5t956S9WrV9fPP/+s1q1b267QvtaRI0ey9HwGKf2ZbVL6rSz37t1rG56cnKxHHnlE27Ztczrf448/rpCQEE2fPt32zMuM4uPjtX37dtvfd9xxh6Kjo7Vjxw7961//spv+hx9+0LPPPitJdrf8+eGHH/Tf//5Xly9fdnh968Yp4wHKxo0bq3///jp69Kj69evn9Oygixcv6v3339fJkyddrpNrWRt2rnZCHnzwQUnSzJkzHZ5/t2DBAi1dulShoaG2A8Q5YX2Gzptvvmk3/MCBA7rrrruczlOqVCndfffdktJvQbx792678dYrFDMe1LG+54zfCWd1rFmzxmGaOXPm6L///a/L95DZ+pwyZYrCwsL00EMP6d1333V6O7bdu3frk08+cfkaGYWEhOjxxx+XJI0bN04bNmxwmGbbtm12vx1/fqYZWa/QnD9/vttbFUqy/WZGjRrlNKyNMdq6dau++uqrLNXw9ddfa8WKFUpOTnZY3vLlyyV5dquhSpUqaeDAgTLG6M4771RCQoJt3MWLFzVu3DhduXJFrVu3dvlMGiC/atmypU6fPq3Tp0+rX79+Aanhvffes8tpq8TERK1bt06S/W/dut1/66237LbLJ06c0O23357tnZYRI0YoKChIH374oa3z1DrsWtmpITIyUmPHjpUk3X///Xb5n5SUpAkTJujixYtOa/PVdjYnstOeyqt69uyp6tWr64svvtDOnTsVFxdnO8jgyuXLl3X33XfbtR2PHz9uy/W77rrLdvt1KXt5f+XKFQ0cOFCJiYn65z//qc6dOys6OloffvihwsPD9dBDDzm0n331+3Els3ZcbvLQQw+pWLFimjZtml555RVdvXrVYZrDhw/n+o4LICt8uX//4IMP6vTp0zp58qTPH93kyssvv+z0WdT79++3Xd1rbWOULl1aMTExOnv2rN3jNaT051o/9thj2a7D032r7NZwww03qGHDhkpMTNQ999xjt/3KmD3OXu+ee+7RxYsXdfPNN2vXrl0O0yQlJWnp0qVevetcZgK1D5xVAwcOVNmyZXXkyBE9/vjjdrm6f/9+Pf300y7ntR7/euqpp+ye637kyBGNGDHC6cHenBz7yA1CQ0M1ZcoUGWPUt29fp888TU1N1ddff61vv/02ABUiv5sxY4ZGjBihzZs3O4wzxuiTTz6xXYV+7R1FrCZMmKCgoCBNmzZNV69e1dChQ73+iKDMjlVmV3a3+VnJUlcuXbqkadOmOX0O98aNG3X27FkFBwd7fOXspEmTJEnPPPOMXeeqMUbPPvusfvzxRxUrVsynFw/ccccdWrRoka5evaqbb77Z5QVUI0eOVNGiRTVv3jz9+eef6tixo+rWreu1OnLSfsnsuzZu3DhVrlxZ//vf//TII484vcvwH3/84fHFjt6Ql47xWtfvzz//7HBsOztKlCihYcOG6cyZM5ozZ46CgoI0fvz4TOd78MEH7drxSUlJ+sc//qGLFy+qRYsWDo+G8HX/0/bt2/Xiiy/aDfvmm29sx+Huv//+TN9TZnzZrs+1DHK9kydPms6dOxtJRpKpUKGC6dWrl7nttttM//79TYMGDYzFYjGSTP369c2uXbvs5h8xYoSRZObPn++w7FtuucVIMhEREaZr166md+/epkKFCiY6Otrce++9RpIZMWKEw3zvvvuuCQ0NNZJM5cqVzYABA0y/fv1Mo0aNjMViMVOmTLGbftmyZaZQoUJGkqldu7YZOnSo6dy5swkJCTGSzKhRo+ymX7Jkia2uNm3amCFDhpgBAwaYWrVqGUkmLCzMrFy50m6e8+fP29ZTWFiYad68uRk0aJAZOHCgad68uQkLCzOSzL59+zxe95MmTTKSTIkSJcygQYPMmDFjzJgxY8zp06dt0zzxxBNGkrFYLKZt27Zm2LBhpkmTJkaSCQ4ONnPnzvX49YwxZsqUKUaSwzr8+OOP7T7nIUOGmE6dOpnQ0FDTqVMn07p1ayPJrF271m6+pKQk07t3byPJBAUFmVatWplhw4aZbt26mfLlyxtJ5vDhw3bTlytXzkgyjRs3NrfffrsZM2aMefHFF23TWL83YWFhplu3bmbIkCGmdu3axmKxmMcff9z2vcjO+vzwww9NZGSk7bverVs3c+utt5oePXqYChUqGElm8ODBHq/PtLQ0c9ddd9l+P40bNzZDhgwxPXv2NNWqVXO6zrLzmbZv397psqxcfa5Wly9fNjExMbY6q1SpYtLS0ly+r9dff932+6levbq56aabzLBhw0zXrl1NqVKljCTzyCOP2M1TuXJlh887o1dffdVIMtHR0aZDhw5m2LBhpm/fvrb5ihYtan744QePlnn69GnTsGFD23x9+vQxAwYMMCVLljSSTNWqVR3mOXz4sMvvjqfvAfA363fy2oydP3++kWQ6d+7s8bKsef3MM8/YDV+7dq1t2+AN1m14uXLlTM+ePc2tt95qevbsaYoWLWokmXr16pnz58/bpv/2229tGVq9enUzaNAg0717dxMREWGuu+4607dvX6frILPtnjHGdO/e3fbeLBaL+fXXX51Ol90aEhMTTdOmTY0kU6RIEdO7d28zaNAgU65cOVOiRAnbOn/uueccXjM721l3rN8JZ+0qYzzbvmW1PWVM5p+Du3aiMZnnW3bm8eS78dprr9m+G6+88orL6azLuv32201sbKwpU6aMGThwoLn55ptN4cKFjSTTqlUrc+nSJYd5s5r3Y8eONZJMp06dTGpqqt24N99805Zvf/31l214dr+7mX0urr5PnrTjsvv5uBqe2XfbXb6vX7/elChRwkgypUqVMp06dTK33nqr6dWrl4mLizOSzPXXX+90uUBe5a39+2vbC+5YM2bjxo12w62/6/bt23vjrdnaErVr1zZ9+/Y1w4YNMx06dLDl1O233243vXX/w/pbHzp0qGnTpo2xWCxm+PDhLrMxs+1YVvatslvDrl27TGxsrJFkypcvbwYNGmR69eplChcubNq0aWNatWplJJlNmzbZzZecnGyGDRtm2z9v3Lix6d+/vxk8eLBp06aNLbuuPd7hjrvM8GQfyxjf7ANn1rbJTvt2zZo1trZQ9erVzZAhQ0y3bt1MWFiYGThwoKlUqZKRZI4dO2Y336FDh0yxYsWMJFOpUiXTv39/c8MNN5iIiAjTpUsXl8dTsnPsIyf7tdn9zrtrWz300EO2dX3dddeZW265xQwZMsR06NDBtk5mzpzpslYguzJuX0uWLGm6detmhg0bZnr27GmqVKliG3fbbbc5tK0z6tOnj23a7du3u5wuu7+TP/74w7btbdOmjRk5cqQZM2aMmTdvnm2azLZnrrbD2dnmZzVLnbXF//rrL9trNmzY0AwYMMAMHTrUtGrVytbGePLJJ12uy2ulpaWZ4cOHG0kmJCTEdO7c2QwdOtR2nD4iIsKsWLHCYT5P9vtccbXOP/vsMxMeHm5CQkLMokWLnM5733332b4zH3/8scvXyO4+V3bbDsuXL7dlSq9evczo0aPNmDFj7NoKu3fvtv0+ihUrZm644QYzbNgw06dPH1O3bl1jsVhM6dKlXb4nZ9zlrSefkS+O8VqPc7lqg2a2j+lKs2bNjCRTq1Ytc+utt5oxY8bYHbfJ7DO/1o8//mhbfzfffLPL6azvp1WrVub66683kZGRplevXrbjT9b93f379zvM66v+J+s2ceLEiSYoKMhcd911ZujQoaZ9+/YmKCjISDL33nuvw3z+/l1kd/saaHSs5yGrV682o0ePNrVq1TLR0dEmJCTExMTEmCZNmpg777zTrFq1ymlDxN2X78qVK+aJJ54w1apVM6GhoaZUqVJm6NCh5pdffsl0A7Znzx4zZswYU7VqVRMeHm6KFi1q6tatayZMmGD27NnjMP3evXvNiBEjTIUKFUxoaKgpVqyY6dixo1m8eLHDtCdOnDAvvPCC6dmzp6lataqJjIw00dHRpm7duuYf//iH042QMcakpqaaDz74wPTs2dOULl3ahIaGmuLFi5t69eqZUaNGmSVLlpirV6+6X9EZXL582Tz88MOmevXqtoOizn7oK1euND179jTFixc3ISEhtoO6W7du9fi1rNwF2oYNG0znzp1NiRIlTGRkpKlXr5557rnnTFJSktsGZFpamvnggw9Mt27dTPHixU1oaKgpU6aMadeunXnppZfM5cuX7abftWuX6d27tylZsqRtQ5sx6K5evWpeeuklU79+fRMZGWliY2NNt27dzFdffeU2OD1dn4cPHzb333+/qVevnilcuLApVKiQqVy5sunQoYN54YUXzC+//JLl9bpy5Upzyy232L4XJUuWNC1atDBPPfWUSUhIcDp9Vj5Tb3QijB8/3rZOPGl07tq1y4wbN87UqFHDFCpUyERGRppq1aqZG2+80bzxxhsOBxMyC6pffvnFTJ061XTu3NlUqlTJFCpUyMTExJgGDRqYRx991Bw9etRhHnfLvHjxonn++edNo0aNTGRkpClUqJCpU6eOmTx5sjlz5ozD9HSsIy/Kix3rGzZsMPfdd59p0aKFKVOmjAkLCzNlypQxrVq1Mm+++aa5cOGCwzw//fST6d27tylbtqwpVKiQqVGjhnn44YfN+fPnXbYzPNnuffjhh7b3ltlB/ezUYEx65/rkyZNNtWrVbO91+PDhJj4+3owePdpIMrNnz3b6mlndzrrjjY51Y7LWnjIm73as79u3z0gykZGRdh3V7pZ16NAhM3ToUFO6dGkTFhZmqlevbp588klz8eJFl/N7mveLFi0ykkzp0qXNiRMnnC5rwIABRpLp27ev3fDsfHezuzNrTObtuNzUsW5MeifjP//5T9OkSRMTFRVlwsLCTIUKFUzr1q3NlClTzE8//eR0PiCvy+n+fW7sWF+0aJEZNWqUqVevnomNjTXh4eGmcuXKpkePHmbJkiVOO7c//fRT07p1a1OsWDFTpEgR06xZMzNjxgyTlpaW7U5GY7K2b5WdGoxJ374NHz7clCpVyoSFhZm4uDgzefJkc+nSJdtJ3AcOHHD6mitWrDD9+vUz5cuXt+V5nTp1zJAhQ8wHH3zgNruu5Y2OdWO8vw/si451Y4zZuXOn6du3r4mNjTWFChUydevWNS+99JJJSkoyYWFhJigoyOEYhzHpbah+/fqZmJgYEx4ebmrVqmWeffZZc/XqVZfvJTvHPnJbx7oxxmzatMnceuutpnLlyiY8PNxERUWZmjVrmj59+ph33nnH6f45kFPnz583n376qbnnnntMixYtbPsvERERJi4uzgwdOtSjk4hmzpxp67ByJye/kw0bNpguXbqYmJgYW/s5Y9s2px0/WdnmZzVLnbXFk5OTzaxZs8zQoUNN7dq1TdGiRW3rvX///mbNmjVO68zMBx98YDspJzQ01FSsWNGMHDnS5XF6X3SsG5PehipcuLAJCgpyui+/cuVKI8lUrFjRpKSkuHyNnOxzZbft8Pbbb5smTZrYLipz9vrnz583L774omnVqpVtXZctW9Y0b97cPPTQQ2bz5s0u35MzOe1YN8b7x3h91bEeHx9vhg0bZsqWLWs7GSVjDdnppC1TpoyRZL788kuX02R8PxcuXDAPPfSQqVq1qgkLCzOlS5c2I0eONL/99pvTeX3V/5Rxm7hmzRrTuXNn27agWbNmZsGCBU7n8/fvIq92rFuMMUYAAABAAZKcnKx69erp4MGD2r59u5o0aRLokpDBE088oeeee07jxo3T7NmzXU43depUPfXUU5oyZYqmTp3qvwIBAHDh8OHDql69uqKionTmzBmnj7mB923YsEHt27dX/fr1s/38YQC5T9u2bbVp0yZ98MEHGjp0aKDLQR5w22236f3339e//vWv/HkLavjN6tWr1bVrV9WqVUv79u2TxWJxOt26devUsWNHtW/f3vaIxUDr0KGD1q9fr7Vr1zo81x05R+seAAAA+db27dvtnsEpSRcuXNCECRN08OBBNWjQgE71XObEiRN66623FBQUpPvuuy/Q5QAA4ODixYtOn4UZHx+vW2+9VWlpaRoxYgSd6l526tQpHT582GH47t27bc/2HTVqlL/LAuAjK1eu1KZNm1SpUiUNGDAg0OUgD9i1a5f++9//qkiRIrrzzjsDXQ7ysNTUVE2ZMkWS9MADD7jsVEfBFBLoAgAAAABf6d+/vy5duqT69eurVKlS+vPPP/Xjjz/qzJkzio2N1YIFCwJdIv7fo48+qmPHjmn16tU6e/as7rrrLtWpUyfQZQEA4ODUqVOqV6+e4uLiVLNmTUVHR+u3337Tjh07lJSUpIYNG+qZZ54JdJn5zp49e9SxY0fVrVtX1apVU0REhA4fPqwdO3YoLS1NXbt21T333BPoMgHkQEJCgh555BH99ddfWrFihSTpxRdfVGhoaIArQ242duxYXbx4UStXrlRKSoqeeOIJxcbGBros5EHz58/Xhg0b9P3332v37t2qX7++Ro8eHeiykMvQsQ4AAIB864EHHtCSJUu0d+9ebdq0SUFBQapcubJuu+02TZo0SRUrVgx0ifh/ixcv1m+//aYyZcrovvvu0wsvvBDokgAAcKpEiRKaNGmSvv76a23btk1nz55VZGSkGjRooP79++uee+5RZGRkoMvMd2rWrKl//OMfWr9+vTZt2qTExERFRUWpdevWGjZsmO644w6FhHCoE8jLEhMTNXfuXIWEhKhatWp68MEHNXjw4ECXhVxu7ty5CgoKUsWKFTVp0iQ9/PDDgS4JedT69ev17rvvqlixYurbt69ee+012hZwwDPWAQAAAAAAAAAAAABwg4c9AQAAAAAAAAAAAADgBh3rAAAAAAAAAAAAAAC4kW8fDpCWlqbjx48rKipKFosl0OUAACBJMsYoMTFR5cqVU1AQ57dlhjwHAORWZLrnyHMAQG5FnnuOPAcA5Fb+zPN827F+/PhxVaxYMdBlAADg1NGjR1WhQoVAl5HrkecAgNyOTM8ceQ4AyO3I88yR5wCA3M4feZ5vO9ajoqIkpa/E6OjoAFcDAEC68+fPq2LFiracgnvkOQAgtyLTPUeeAwByK/Lcc+Q5ACC38mee59uOdevtaKKjowl6AECuw23TPEOeAwByOzI9c+Q5ACC3I88zR54DAHI7f+Q5D44BAAAAAAAAAAAAAMANOtYBAAAAAAAAAAAAAHCDjnUAAAAAAAAAAAAAANygYx0AAAAAAAAAAAAAADfoWAcAAAAAAAAAAAAAwA061gEAAAAAAAAAAAAAcIOOdQDIR9asWaMjR44EuoxcJzExUXPmzNHJkycDXQoA4P9t27ZNM2fO1NWrVwNdCgAAyKZDhw7pjTfe0OnTpwNdCgAAAPxo/fr1WrhwYaDL8LuQQBcAAPCO06dP67777lPFihW1YsWKQJeTqyxatEgzZszQzz//rJdeeinQ5QAAJI0ZM0bGGDVp0kTXX399oMsBAADZ8Oyzz2rbtm2SpIkTJwa4GgAAAPjLhAkTJEldu3ZVmTJlAlyN/3DFOgDkE5cvX5YkHT16NMCV5D7Hjh2TJO3bty/AlQAArIwxksQV6wAA5GHx8fGS0u8SBgAAgIKnoB3XoWMdAFBgWDtxAAC5R2pqaqBLAAAAOcS+FgAAQMFU0I7r0LEOIE+5dOmSvvjiC61YsULnzp0LdDnIYywWS6BLAABco6DtgAEAkB+xrwUAAPKjtWvXauDAgfrjjz8CXUquVdBOsKRjHUCe8sEHH+ihhx7SI488olmzZgW6HOQxBS3kASAvSElJCXQJAAAAAAAADp588knt379fK1euDHQpuVZaWlqgS/ArOtYB5CmHDx92+v+AJ7iKAgByHzrWAQDIuzh5GQAA5Gdnz56VxN323CloHeshgS4AALLi999/V1BQkMIiQnX06NFAl4M8wtqhzkEfAMh9kpOTA10CAADIJva1AABAQcAFW64VtJMO6FgHkKfEx8crunhhRUQV0rH4Y0pOTlZoaGigy0IeQQMIAHIfrlgHAAAAAAC5GScRulbQrljnVvAA8oxz584pISFBxUpFK6ZUtFJTU/Xbb78FuizkITSAACD3uXr1aqBLAAAAOcQdaAAAQH7GBVuuFbR2IB3rAPKMgwcPSpKKlyum2LLF7IaBcHfH2qHOOgKA3Keg7YABAJAfcaIcAADIz7hgy17G9VHQ7kRIxzqAPGPv3r2SpOLlYlSifIwkad++fYEsKVch3DPHOgKA3CHjTteVK1cCWAkAAPCGpKSkQJcAAADgM1ywZS/jcZ2CdsFErutYX7dunSwWi9N/3377baDLAxBAu3fvliSVqhSrkhViZbFYtGvXrgBXhbyEBhAA5A4ZD75zIB4AgLzLevIyJ8oBAID8jAu27GW8W1FB61gPCXQBrkycOFHNmze3G1a9evUAVQP4359//qldu3apU6dOdAYqPbh27NihwtERioopLIvFouLlimnXrl1KTk5WaGhooEsMOL4nmaMBBAC5w+XLl53+PwAAyFvS0tIkkecAACB/49i7vYwd6wXtgolc27Herl07DRgwINBlAAHz8MMPa/v27Xr33XfVpEmTQJcTcL///rv+/PNPVW9cyRZi5eJK6adjB7Rr1y7Wkeg09gQNIADIHehYBwAgf7hy5fL//5cr1gEAQP7FsXd7BflOhLnuVvAZJSYmFriH3gNW27dvlySdOXMmwJXkDps2bZIkVahZxjbM+v+bN28OSE3Ie2gAAUDucOnSJdv/07EOAEDeZIzR5cvpHerkOQAAyM+4YMtexrYfHeu5xKhRoxQdHa1ChQqpY8eO+v77791On5SUpPPnz9v9A/IDNtjpNmzYIEmqWKusbVj56qUVHBJkGwe4wu8IAHKXjDtgGTvZAQBA3nHlyhXbreAvXrwY4GoAAAB8hwu27BXkOxHmuo71sLAw9e/fX6+//ro+++wzPfvss9q1a5fatWunH374weV8zz//vIoWLWr7V7FiRT9WDfgOG+z0u1ds2bJFJSvEKiqmsG14aHiIKtQso3379unYsWMBrDB3oPMYAJBXZOxM50A8AAB5E3kOAAAKCo6926NjPRdp3bq1PvroI40ePVq9e/fWo48+qm+//VYWi0WPPfaYy/kee+wxnTt3zvbv6NGjfqwagC+tXr1aKSkpimvoeMJMtQbpw1auXOnvsgAAQDZduHDB9v8ciAcAIG/KmOGXLl3iwgAAAJBv0c6xR8d6Lle9enXdcsstWrt2rVJTU51OEx4erujoaLt/QH7AmVDSZ599JlmkGk2rOIyLa1hJIaHB+uyzzwg3AADyiIwH4ulYBwAgb8p4olxqamqBe74mAAAoOOinsVeQ71yUJzrWJalixYq6evVqgfuAgILeWfzLL79o+/btqlizjN1t4K3CCoWqWsOKOnLkiLZt2xaACgEAQFbZdaxfon0PAEBedO0xOo7ZAQCA/Kqg99Nc69o7FxUkeaZj/dChQypUqJCKFCkS6FIA+NH7778vSarfrpbLaazjFi5c6JeacivC3TXWDQDkLhmvcLuQeMHNlAAAILe6tiM9Y74DAADkJ1yxbo8r1nORU6dOOQzbuXOnli5dqm7duikoKNeVDPhUQd5gnzx5Up999pmKlYxSpTplXU5XulJxlalaUuvWrdPPP//sxwqR1xTk3xMA5CbWA++FCofrypUrSklJCXBFAAAgqxITEyWl57lExzoAAMi/uHDLXsbnqhe0K9ZDAl3AtQYPHqyIiAi1bt1apUqV0t69ezVnzhxFRkbqhRdeCHR5gN8V5A32/PnzlZycrCZdmmZ6Uk2zrtdp+Zx1mj17tl5++WU/VYi8piD/ngAgN7EeeC9SLFJXLibp4sWLKlq0aICrAgAAWXFtntOxDgAA8isu2LKXsTM9Yyd7QZDrLv/u06ePTp8+rWnTpmn8+PH673//q379+un7779XnTp1Al0eAD85duyYPvzwQxUtEaUaTatkOn3F2mVVunJxffnll9q3b5/vC0SeYm340AACgNzBeoVbVExhu78BAEDeYe1It+Y5HesAACC/4oItexk70+lYD7CJEydq69atSkhIUHJyso4fP66FCxeqevXqgS4NgB+99dZbSk5OVose9RUcnPmmymKxqOVNjSRJr776qo+rQ15FAwgAcgdrR3qRmEhJ0vnz5wNZDgAAyAbbiXKx6R3r5DkAAMivOK5sryB3rOfoVvBXr17V6tWrtX//fl28eFH//Oc/JUlXrlzR+fPnVaJECZ6JDuRQcnJyoEvwu3379mn58uUqWSFW1RtV9ni+8jVKq1KdstqyZYs2bdqkNm3a+LBK5CXWhg9XrDtHngPwt7NnzyqsUKgioyIkSefOnQtwRUDeR54D8LezZ89KkoqWKCKJPAe8hUwHgNwnJSUl0CXkKlevXk3/H4v5+/8LiGwn8NKlS1WpUiXdfPPNmjRpkqZOnWob99NPP6ls2bJavHixN2oECrQrV64EugS/MsbolVdekTFGrW9pLEtQ1jpCW9/cWBaLRa+88opSU1N9VCXyKs4sdESeAwiEc+fOKTwyTOGRYba/AWQfeQ4gEGwd6yWj7f4GkH1kOgDkTnSs20tKSpIkWUL+/v+CIlsd65s2bdKAAQMUHh6u119/XcOGDbMb36JFC1WvXl0ff/yxV4oECrKCdsX6pk2btHXrVlW5rrzKVy+d5fljyxZT7eur6eeff9ayZct8UCHyIp6x7hx5DiAQjDFKSEhQRJFCiowqJElKSEgIcFVA3kWeAwiUM2fOKDgkSEVLRNn+BpB9ZDoA5F50rNuz9ltZggteH1a2bgX/zDPPqFixYtq+fbtKlCjh9EBYs2bNtHXr1hwXCBR0BWmjlJaWptdefy39eem9GmV7OS2619fP24/orRlvqWfPngoLC/NekciTrFeqc8W6PfIcQCBcunRJSUlJiowqoYj/71g/ffp0gKsC8i7yHECg2E6Ui07P81OnTgW4IiBvI9MBIPfi7rj2bOsjWEpOKTh9WFI2r1jfunWrbrnlFpUoUcLlNBUrVtQff/yR7cIApEtLSwt0CX6zevVqHdh/QDWbVVFsmaLZXk7hopGq366m/jjxB2fxwg5XrNsjzwEEgnWbUrhYpIoUi5QknTx5MpAlAXkaeQ4gENLS0nTy5EkVLhap0LAQhUeGkedADpHpAJB7ccW6PWvHuiVISkstOH1YUjY71pOSkhQdHe12mrNnzyooKNuPcAfw/wpKR6AxRnPmzJElyKJm3erneHmNO9VVaFiI5s6dW6Cu+odz1ivVC9KJKp4gzwEEwvHjxyVJUcUiVTg6QpYgi20YgKwjzwEEQkJCgpKTkxUVk36SXJFikTp+4jh3CQNygEwHgNyLjnV7tvVhKXjH3LOVwtWqVdO2bdvcTrNlyxbVrl07W0UB+FtB6VjfuHGjDhw4oBpNKqtoiSI5Xl6hwuG6rk0NnTx5kmetw6ag/J48RZ4DCITffvtNklS0RJSCgoMUFVtY8b/FB7gqIO8izwEEQnx8enZbn69etESULl64yHPWgRwg0wEg96Jj3Z7tZEo61j3Tv39/bdq0SfPnz3c6/uWXX9bu3bs1ePDgHBUHQAoNDQ10CX6xYMECSVKTTnW9tsyG7WsrOCRI7777boHbuMM5rp6wR54DCITDhw9LkoqVSr8ap1jJaJ0+dVqJiYmBLAvIs8hzAIFgy/OS/5/n/5/r1uEAso5MB4Dci2es27P2t1hkClzfS0h2ZnrooYf08ccfa+zYsfrggw+UlJQkSXr44Ye1ZcsWbd68WY0aNdKECRO8WixQEIWEZOtnmqfs2bNH27ZtU6U65RRbtpjXllu4aIRqNK2i/VsPaePGjWrfvr3Xlo28iSvW7ZHnAALhwIEDCgoKUrFS6Ve4FS9bVL/tO66DBw+qadOmAa4OyHvIcwCBcODAAUlSbNmiktLz3Dq8WbNmAasLyMvIdADIvehYt5eWliZZJFnSL2YzxhSYY+/ZumK9SJEi2rhxo4YMGaJ169bpm2++kTFGL7/8sjZv3qxBgwZp9erVCg8P93a9QIETHBwc6BJ8znombuNOdby+7EYd6ti9BgomrlR3jjwH4G+pqak6cOCAYssWVXBIehunRIVYSdK+ffsCWRqQZ5HnAAJh3759CgoOUmyZ9A518hzIOV9l+p49ezRw4EBVq1ZNkZGRKlGihG644QYenQgAWUDHur3U1FRZ/r9jXSpYt4PP9qWwMTExev/99/XGG29o27ZtOnPmjKKjo9W8eXOVLl3amzUCBVpQULbOf8kz4uPjtWrVKpWsGKtycaW8vvzYMkVVuW45bd++XT/++KMaNWrk9dfILQrKGWHZcfny5UCXkGuR5wD86eeff9bly5dVtWI527DSlYpLkn766adAlQXkeeQ5AH9KSkrSvn37VKJ8MduJcsVKRCk8Iow8B3LIF5keHx+vxMREjRgxQuXKldOlS5f08ccfq3fv3po9e7bGjRvn5XcBAPkPz1i3l/GKdSm9o70gXCQqZbNjvVOnTmrTpo2eeeYZFS9eXN27d/d2XQAKiDlz5igtLU1Nu17ns47hpl2vU/ze45o1a5ZmzZrlk9dA7nbhwgVJXLl+LfIcgL/98MMPkqQy1UrahkXFFlbh6Aht3769QN06DPAW8hyAv+3du1fJyckqW/XvPLcEWVS6SnEd3ndYZ86cUWxsbAArBPImX2V6z5491bNnT7thEyZMUNOmTTVt2jQ61gHAAwXpimxPpKSk2HWsp6SkKCwsLKA1+Uu2LoXdunUrtz0A/CQ//9Z++eUXLV++XMXLFVPVehV89jplqpRUxVpltGnTJm3bts1nr4Pc6+LFi5KkS5cvBbiS3IU8B+BvW7dulSSVj/v7ahuLxaKycaX0559/Kj4+PlClAXkWeQ7A37799ltJUrk4+6tnrX9/9913fq8JyA/8menBwcGqWLGizp4965fXA4C8jivW7V29elWWIMkS9PffBUW2OtZr167NQS/AT5KTkwNdgs+88sorSktLU6ubG/n86rSWvRpJFumll17Kt2eXcTW2a9Yr1i9euBjgSnIX8hyAP6WkpGjr1q0qWjJKUbGF7cZVrFVGkrRly5ZAlAbkaeQ5AH/79ttvZQmyqFx1+8e5VaxZxjYeQNb5OtMvXryo/2PvvsOjKrMHjn/v9PSekEACCb0XlSKKXaxYV3Td1V3r/nbV3XXXVXdt4ApiB1lFQBERFUWp0jvSlE5ooRgIhCSk9zIz9/fHODGBACkzc2cm5/M8eUim3PdMSHLu3PO+583NzeXIkSO8++67LF68mGuuueacj6+qqqK4uLjehxBCtFZSWK+vqqoKFLW2ylxZWaltQB7UrML6k08+ybx589i3b5+r4xFCnMFfZ/qsXLmSH374gcRu8SR1S7jwE1oopl0kXS9OZv/+/cyePdvt4wnv4iysV1ZWyklQHZLPhRCetGvXLkpLS0nqGn/WfYm/3LZ+/XpPhyWEz5N8LoTwpKKiInbt2kmb9tGYA+q3+4xuG0FAsIX169fLxG8hmsHdOf0f//gHMTExdOrUiX/+85/ccccdTJw48ZyPHzt2LGFhYbUfiYmJbolLCCG8Vd3zGbmmXF95RTnof12x3poK683aYz0lJYUrr7ySwYMH8/jjj3PJJZcQFxfX4IrTYcOGtThIIVqzqqoqrUNwudLSUsaOHYveoOfyOy/y2LhDbu1HeupJ3n33Xa666ipiYmIu/CQfIsn93Jyt4AEqKioICQnRMBrvIflcCOFJa9euBaB9z7Mn1AWHBxLdNoItW7ZQXl5OYGCgp8MTwmdJPhdCeNKGDRuw2ey079n2rPsUnUL7Hgkc+PEo+/fvp0ePHhpEKITvcndO/9vf/sbdd99NZmYmX3/9NTab7bwLep5//nmefvrp2q+Li4uluC6EaFXqdhP2587CzVFeVo5iBEXv+Lru9Xd/16zC+pVXXomiKKiqyttvv33eFs6y15sQLeOPhfW33nqL7OxsBt7Ym/CYUI+NGxgSwJAR/Vgz60dGjx7NhAkT3N6C3pP8tbtBS6mqWm/GXFlZmRTWfyH5XAjhKaqqsmrVKoxmw1n7sTp16NmWrctS2bRp03lbUgoh6pN8LoTwpFWrVgGOvN2QDj3bcuDHo6xevVoK60I0kbtzerdu3ejWrRsADzzwANdffz233norW7ZsaXAss9mM2Wxu8jhCCOEvpLDesJqaGiorKzEEqCgGx6p+KaxfwEsvveRXxSghvJm//cH+4Ycf+Pbbb4luG0H/a3p6fPzugzpyeMdx1qxZw4IFCxgxYoTHY3AXf5yE4QrV1dXY7fbar1tTW5oLkXwuhPCUo0ePcuzYMTr2S8Jg1Df4mOTe7di6LJWVK1dKYV2IJpB8LoTwlKqqKtavX094TAgRcQ1Pkk/sGo/BqGflypX85S9/8XCEQvg2T+f0u+++m8cff5y0tDS6du3qsXGFEMJX1F3I5m91mpYoLi4GQDE4PgAKCwu1C8jDmlVYf+WVV1wchhDiXPxpFXJBQQEvvPgCeoOOa347BL1e5/EYFEXhqnsH8fWbi3ltzGtcdNFFtG3b8Ex7XyMF44ad+X2R79OvJJ8LITxlxYoVAKT0bnfOx0S3jSAkMog1a9ZQU1OD0Wj0VHhC+DTJ50IIT9m8eTPl5eV0HdzjnMU/o9lAYtd4DqUe4vjx4yQlJXk4SiF8l6dzekVFBQBFRUUeHVcIIXxF3YVsck35VwUFBQCOVvC/XLppTYV1z1e1hBBN4i/7ZquqyqhRo8jLzWPQzX2JSgjXLJaQiCAuv+siysvKef755/3me1xaWqp1CF7pzJX8srJfCCE8b8WKFegNetr3OPdkNkVRSOmTSElJCT/++KMHoxNCCCFEYyxfvhyAlL7n32M5pU9ivccLIbSVk5Nz1m01NTV89tlnBAQEyLYNQghxDnWvI8s15V/l5eUBoDOpKCbHbbm5uRpG5FnNWrHuVFZWxty5c9m5cyfFxcWEhobSr18/br/9doKCglwVoxCtjqqqtZ/7S9F39uzZrFy5knad4+g7rJvW4dB5QAeO7ctkx/YdTJkyhf/7v//TOqQWa037mDTFmSc9/tQFwlUknwsh3CkjI4MDBw7QoWdbTJbzr0JP6ZPIrjUHWLFiBUOHDvVQhEL4B8nnQgh3qqmpYc2aNQSHBxKbGHnex3bo1RadXseKFSt4+OGHPRShEP7D1Tn98ccfp7i4mGHDhtG2bVuysrKYOXMmBw4c4O233yY4ONgNr0IIIXyfFNYbdvr0aQAUk6O4Xve21qDZhfVvv/2Wxx57jMLCwnpFQEVRCA8PZ8qUKdx5550uCVKI1qZuMd0fCutHjx5l3LhxWALNXP3bISg67feAVBSFYXdfQlZ6LpM+msSQIUPo16+f1mG1SElJSe3n0kL3V9IK/vwknwsh3G3lypXAr6vXzqdN+2iCQgNYuXIlL7zwAnp9w/uxCyHqk3wuhHC3rVu3UlRURJ9hXS+4B7Q5wES7znGkpqZy6tQp4uPjPRSlEL7PHTl95MiRfPzxx3z44Yfk5eUREhLCRRddxLhx4xgxYoSrX4IQQviNugu0pLD+K2cnFJ0JdOb6t7UGzWoFv3HjRu69917Kysp45JFH+OKLL1i9ejVffvkljz76KOXl5dx7771s2rTJ1fEK0SrU1NQ0+Lkvqq6u5l//+hdVVVVcde8ggsMDtQ6pljnAxLW/uxTVrvLss8/WK0z7orqt4H39tbhSbSFd73hD6txDTEg+F0J4xooVK1B0Ch16nrsNvJOiU0ju3Y6CggJ27NjhgeiE8H2Sz4UQntCUiXJ1H7dq1Sq3xSSEv3FXTr/33ntZvnw5WVlZ1NTUkJ+fz/Lly6WoLoQQFyAr1huWlZUFgM6souhBMfx6W2vQrBXrY8aMwWw2s2HDBvr27VvvvpEjR/LnP/+ZSy+9lDFjxrBgwQKXBCpEa1J3Ra2vr6597733OHjwID2Hdia5dzutwzlLfHIMFw/vxU9L9vDqq68ybty4C86+91Z1C+tlZWVERp6/PV9r4ZxkoDODvVxa5tcl+VwI4W6nT59m165dtOschyXI3KjnJPdOJHXDIVauXMnFF1/s5giF8H2Sz4UQ7ma321m5aiUBwRbaJEc36jkderWFbxwT7O6//343RyiEf5CcLoQQ3qVuMb2mpgZVVX22duBKp06dAkBncXytmFUyMzM1jMizmrVifdOmTYwcOfKsBO/Up08f7rnnHjZu3Nii4ABee+01FEWhV69eLT6WEL6ivLy89nNfXl27YcMGZsyYQUSbMIaO6K91OOd00XU9iU+OYfHixT79xqTuz03dIntrV1xcDIAuwLFivbCwUMNovIsn87kQonVavXo1AMmNXN0GkNApFnOgiVWrVtVrfymEaJjkcyGEu+3Zs4fc07mOvdN1jbuUGBgSQHxyDNu3b5f3YEI0kuR0IYTwLmd2E/b17sKucvLkSRSDY6U6gM6iUlpaWnsd3t81q7BeXl5OXFzceR8TFxdXr8jTHCdOnGDMmDEEBQW16DhC+Bp/aOmdn5/Pf174D3qDjut+dykGU7MaZHiETqfj2t9dijnAxGtjXuPEiRNah9QsdSdhSGuaX2VnZwNgCFHrfS08l8+FEK2Xs/1rcq/Gd63R63W0755AZmYmBw8edFdoQvgNyedCCHdz5vOUJnahS+7VDrvdzpo1a9wQlRD+R3K6EEJ4Fymsn01VVU6cPIHO8utCCOfK9ZMnT2oUlWc1q7DeoUMHli9fft7HrFy5kg4dOjTn8LX++c9/MnjwYGkBKVqdoqKiBj/3FaqqMnr0aPJy8xh8cz+i20ZoHdIFhUQGMezuiykvK+ff//43NptN65CarG4x3Zc7HbhaRkYGAIZwtd7XwnP5XAjROpWUlLBlyxZiEiMJDg9s0nOd28c4V7wLIc5N8rkQwt1WrVqF0Wykbec2TXqe5HMhmkZyuhBCeJczC+nV1dUaReI98vPzqSivqFdY11ta13X3ZhXW77nnHrZt28aDDz54Vt/8U6dO8Yc//IFt27YxcuTIZge2bt06Zs+ezXvvvdfsYwjhqwoKChr83FcsXryYlStX0rZzHH2GddU6nEbrPKADnQe0Z8eOHXzxxRdah9NkdRO91WrVMBLvcuDAAVBAH+LYZ11WP/7KE/lcCNF6bdiwAavVSkrvxreBd0rqFo/eoGflypVuiEwI/yL5XAjhTj///DPp6ekkdYvHYNQ36blh0SFExoexYcMGmfwtRCNIThdCCO9yZiFdVqzD8ePHAdAF/Hqb83Pnff6uWb2Zn332WZYsWcKMGTOYNWsWnTp1Ii4ujuzsbA4fPkx1dTUDBw7k2WefbVZQNpuNJ598kkceeYTevXs36jlVVVX1Vmu2ll7+wj/l5eXVfl5UVITVasVg8N5W6nUVFBQwZsxrGM1Grrp3EIpO0TqkJrn8zos5eSib8ePHc9VVV9GuXdNa3Wmp7ip7SfIONTU17N+/H32wiqIDfYid7OxscnJyiI2N1To8zbk7nwshWrcVK1YAv65Wawqj2Ui7LnEc3HeQjIwMEhObXpwXorWQfC6EcCfnJLfm5HNwtIPftnwvGzdu5JprrnFlaEL4HcnpQgjhXZzX2BWDDtVqlxXrQHp6OgC6wDqt4H/5/Oeff9YiJI9r1or1wMBA1q1bxyuvvEK7du3Yt28fq1evZt++fbRr145Ro0axdu1aAgICLnywBkyaNIljx47x6quvNvo5Y8eOJSwsrPZDLr4JX+YsrIdGt0FVVZ9atf7+++9TVFTMoJv6EBoZrHU4TWYJMnPZHRdRVVXFG2+8oXU4TVJ3lbrdbtcwEu+xY8cOqqurMYQ6krs+zPHvpk2btAzLa7g7n5/ptddeQ1EUevXq5ZLjCSG8V2VlJevWrSM8NpSIuNBmHaNj3yQAWbUuxAVIPhdCuNPy5cvR6XW075HQrOc78/mF2lsLITyf04UQQpyfczGvzuTo2iOFdThy5AgA+sD6e6wrOjh69KhWYXlUswrrAGazmZdeeonDhw9TVFRERkYGRUVFHD58mBdffBGz2dys4+bl5fHSSy/x4osvEhMT0+jnPf/88xQVFdV+tJZe/sI/OQvrse07AZCbm6tlOI125MgRZs+eTVR8OL2GdtY6nGbr2C+Jtp3iWL16NT/99JPW4TRa3RXr0greYd26dQAYohyJ3hip1rtduC+fn+nEiROMGTOGoKAglxxPCOHdfvjhByoqKkjpk4iiNK97TYeebdHpdCxdutTF0QnhfySfCyHcISMjw1HQ69IGc4CpWceISggnNDqYNWtW1+s0KYRomKdyuhBCiAs7s7Au5zJw6NAhAPSBv96mKI5V64cPH65Xo/BXzS6s1xUSEkLbtm0JCQlp8bFeeOEFIiMjefLJJ5v0PLPZTGhoaL0PIXxVTk4OAHHJXQA4ffq0luE02tSpU1FVlUE390Wnd8mfF00oisKQW/sBMHnyZG2DaYK6xXQprDtW7S9ZugTFAIZfVqrrAhxJft26dZSXl2scofdxZT4/0z//+U8GDx7MxRdf7PJjCyG8z6JFiwDo3L99s49hCTLTrmsbUlNTZdKsEE0g+VwI4SqLFy8GWpbPFUWhU7/2lJWVs379eleFJkSr4M6cLoQQ4sIqKysB0FuM9b5urVRVZd++fegCVJQzdi7WB6tUVlZy7NgxbYLzoGZVvjZs2MDTTz9NVlZWg/efOnWKp59+ms2bNzfpuIcOHWLy5Mk89dRTZGZmkp6eTnp6OpWVldTU1JCenk5+fn5zQhbCp+Tk5GAJCiYirl3t194uLy+PxYsXE5UQ3uwWcd4kNimKxG7xbN68uba9iber24pG9liH7du3k52VjTHGjvJLtlMUMMXaqayslNbCuC+fn2ndunXMnj2b9957r0XHEUL4hqKiItauXUtkmzCiEsJbdKwuFzku5C9cuNAFkQnhnySfCyHcQVVVFixYgMGob/b+6k5dBjjy+YIFC1wRmhB+y1M5XQghROM4F2YZAhyF9bKyMi3D0dzJkycpLCxEH6yedZ/zttTUVE+H5XHNKqy/8847LFiwgDZt2jR4f3x8PAsXLuTdd99t0nFPnjyJ3W7nqaeeIjk5ufZjy5YtpKWlkZyczOjRo5sTshA+JSsri5DIWEKiYmu/9nbLly/HZrPRfVDHZrd89TbdB3UEfl115+3qzphr7bPnAObOnQs4Cul1GWMdSX7evHmeDsnruCuf12Wz2XjyySd55JFH6N279wUfX1VVRXFxcb0PIYRvWbJkCdXV1XQbmNLiYyX3TsRkMTJ37lzsdvuFnyBEKyT5XAjhDrt27SI9PZ2UPo5c3BKR8eHEJEaydu3a2q3vhBBn80ROF0II0XilpaUAGIJM9b5urXbs2AH82h22Ludtzsf4s2YV1n/66Scuu+yy8z5m2LBhTZ4916tXL+bMmXPWR8+ePUlKSmLOnDk8/PDDzQlZCJ9RWlpKcXExodFtCIt2nEifPHlS46guzNnSrWPfJI0jcZ0OPRIwGPX88MMPWofSKHVbm7f2NuelpaUsXboUXYCKPqz+ffoAMITb2bJlS6tvLeyufF7XpEmTOHbsGK+++mqjHj927FjCwsJqPxITE5s9thDC81RV5euvv0an09Hl4g4tPp7RZKBT//ZkZmaycePGlgcohB+SfC6EcIdvvvkG+HXCeUt1H9QRm83GnDlzXHI8IfyRJ3K6EEKIxnNOEDYEm+p93Vpt27YNAH3o2YV1XRAoBvjxxx89HZbHNauwnpOTQ9u2bc/7mDZt2jS5fXV0dDS33377WR/R0dGEhIRw++23N2p2vBC+zLkHRXibtoRExqA3mjh+/LjGUZ2fqqrs2bOH0KhggsICtA7HZQwmA9FtI0hLS6OqqkrrcC6opKSk9vPWPntu8eLFVFZWYmpjp6EGCqZ4R/Jv7Rd13JXPnfLy8njppZd48cUXiYmJadRznn/+eYqKimo/WvvkByF8zdatW0lLS6Njv0QCQ1xzTtDrss4AzJw50yXHE8LfSD4XQriac6u3iLgwEjrFuuSYXS7qgMli5KuvvsJqtbrkmEL4G3fndCGEEE1TUFAAChhDzIBj67vWSlVVNmzYgGIEffDZ9yuKYzHb8ePHfWKhaEs0q7AeHh5+wULfsWPHCA5u4LsrhDgv537eUQkdUHQ6IuMTOXLkCKp69iwgb1FcXExBQQFR8eFah+JyUQnhWK1Wr08GqqpSWlqKzmIAZPbcd999BwqY2jT8e2OMVlEMjnbxrfmijrvz+QsvvEBkZCRPPvlko59jNpsJDQ2t9yGE8B3Tp08HoPflXV12zOiECBI6xvLDDz9w6NAhlx1XCH8h+VwI4WpffPEFNTU19L68i8u2ejNZjHQbmEJ2djZLly51yTGF8DdyzV0IIbxLQUEBerMB/S97rOfn52sckXYOHz5MVlYWhoiGF7IBGCIc1+Kd3Y39VbMK64MHD2bOnDnnnHV+/Phx5s6dy6WXXtqi4JzWrFnTKja8FwLgwIEDAMQmOdqtxSSmUFZWxokTJ7QM67xOnz4N4Fer1Z2CwgKBX1+jtyorK8Nms2EKtQCte/bcwYMHSU1NxRBpR2dq+DGKDoxxdk6fPs2GDRs8G6AXcWc+P3ToEJMnT+app54iMzOT9PR00tPTqayspKamhvT09FZ9MiqEPzp48CBr164loWMsbTpEu/TYA67pAcDUqVNdelwh/IHkcyGEK5WUlPDFF18QGGKh2yXJLj123yu7odPpmDJlCna73aXHFsIfePqauxBCiPPLzc1FbzGitzgK63l5eRpHpJ2VK1cCYIw69wJQ533Ox/qrZhXWn376acrLyxk6dCifffYZp06dAuDUqVNMnz6doUOHUlFRwT/+8Q+XBitEa7B79250ej0x7TsB0Cale+3t3srZgtwceI4qpg9zvqa6bda9UWFhIQCGX9rSOL9ujb799lsAzPHn7/JgbuO4kDN79my3x+St3JnPT548id1u56mnniI5Obn2Y8uWLaSlpZGcnMzo0aNd/ZKEEBqaOHEiABdd19Plx07sFk9Mu0iWLFnC4cOHXX58IXyZ5HMhhCt99tlnlJaW0vfKbhhMBpceOyQiiK6XJHPkyBFZtS5EA+SauxBCK4cOHWLkyJEsW7ZM61C8Rk1NDUVFRegDDOgtBlAchfbWatmyZY7FapHnvuauM4M+ROXHH3/06wnYzTpDHjZsGO+88w7/+Mc/+OMf/wiAoii1rap1Oh3jx49n2LBhrotUiFagvLyc1NS9tEnuhtHkKJC269YHcOxZevPNN2sZ3jlVVlYCYDC69k23N9Ab9ABUVFRoHMn5OVeoGwIM6E2GVtsKvqKiggULFqAzg+E8SR4ce8HoQ1TWrVtHdnY2cXFxHorSe7gzn/fq1avBPexfeOEFSkpKGD9+PB07dmzZCxBCeI2dO3eyZs0aEjrF0q5LG5cfX1EUBt3ch4UfrWH8+PG8//77Lh9DCF8l+VwI4Sq5ublMnz6doNAAel3WxS1jXHx9L9K2/cz777/Ptddei9FodMs4QvgiueYuhNDKkiVL2LdvH+PGjeP666/XOhyv4Fydrg80oegU9BZjqy2sHzx4kEOHDmGMtqNcoARkirVTcURh6dKl3HfffZ4J0MOaXQX761//ylVXXcWkSZP46aefKCoqIjw8nIEDB/KnP/2JXr16uTJO4afsdjs2mw2dToder9c6HM1t3boVm81KUs8BtbfFtu9EQHAoGzduRFVVl+1v5ko1NTUA6I3NaoLh1Qy/vCZv34fbWVjXmQwoZn2rXbG+dOlSSktLMbc/914vdZni7VSkKXz33Xf83//9n/sD9ELuyufR0dHcfvvtZ93+3nvvATR4nxDCN9ntdsaNGwfAkFv6ue1cJbFrPG07x7FmzRq2bNnCoEGD3DKOEL5I8rkQwhUmTpxIRUUFV9wyEKOLV6s7hUQG0WtoZ3atPciXX37JAw884JZxhPBVcs1dCKEF5wQeb+/a6kk5OTkAGH7ZX10fYKy9rbVZsGABAMa48y9kAzDGqlQchfnz50thvSF9+vThgw8+cFUsopUpLi5mxIgR5OXlERISwnfffUebNq5f4eRL1q1bB0BKv8G1t+l0ejr0Gcj+jSs4cuQInTp10iq8c6otrOv9r7DunPBRVVWlcSTn51yhrjMb0Jv0rfIkSFVVvvzyS1B+bfN+IaZYlcqj8M033/DII4+02tUSks+FEC0xf/58UlNT6XxRB+Lau3Zv9boURWHobQP45u0ljB07lm+++abV/t0WoiGSz4UQLbF3716+++47ohLC6T4oxa1jXXR9bw5uTeeDDz7gpptuIjrafecPQvgiyelCCKG906dPA6APdFx3MAQaKcsvory8nMDAQC1D86iamhrmz5+PznT+NvBOOhMYIu2kpqaSlpZGly7u6YKkJf+rggmfcfjw4dp2GiUlJezbt0/jiLRlt9tZuWoVgaHhJHSuvzdppwFDAVi1apUWoV1QeXk5AEaz/13cNpod84+8vRW8s5CuM+rRmfSUlpZitzeuuOwvdu/ezb59+zBG2dFZGvccRQ+mODunT59m5cqV7g1QALBmzRpSU1O1DkMI4SJFRUW88847GE0GhtzSz+3jRbeNoMelnThy5AgzZ850+3hCiIZJPhfCv9hsNv773/+iqiqX3XEROjdPmrcEmhh0U1/Kysp466233DqWEEIIIURzOAvrtSvWfymwO29vLVauXElBQQHGWDtKI08RzfGOAvw333zjxsi006Qz5XMVaQoLC/n73/9O37596du3L0888USrbYkgGu/kyZMARLZ3zFg5deqUluFobvfu3eSePk2niy5Dp6vfFj+l3xD0BiPLly/XKLrzc66WNlr8r7Bu+iVxevsKcOfkBp1Rh+Ij+8K72vTp0wEwt23ahAJTWzsojuc72x75O8nnQghXefvttykoKGDgjX0IDvfMjO1BN/UlMMTC//73P06cOOGRMYXwRpLPhRCu8tVXX5GamkrXi5Np2ynOI2P2GNyRuPZRfP/992zcuNEjYwrhrSSnCyGE9zlrxXpA6yysz5o1CwBTQuOvuRsiVXQWR4fDsrIyd4WmmUYX1idMmIDReHZhr7KykmHDhjFhwgT27NnDnj17+OCDDxg6dKjXF6KEtpyF9Ii2yQBkZmZqGY7mnL9bXQddddZ95sAgOvQZyIEDB8jIyPB0aBfk7DwQGNLIZcI+JCA4APj1NXqr2sK6QYful33h/TFpncvx48dZuXIl+hAVfVjTnqsPAGOUoz3Ntm3b3BOgF5F8LoRwlc2bNzNnzhxi2kXQ+3LPtfayBJoYevsAKisrGT16dKuZFCVEXZLPhRCukpmZyYQJE7AEmbn0tv4eG1fRKVx5zyB0Oh2jRo2qfU8rRGsjOV0IIbxTbm4uUH+P9bq3twZpaWls3boVQ4QdfUDjn6coYGpjo7y8vHZ/dn/S6ML62rVriY2N5brrrqt3+5QpU0hNTaVHjx6sWrWKH3/8kbvvvpujR48yYcIElwcs/IezkB7RriPw6wr21khVVZYvX44lKISkHgMafEzXgVcCeOWqdeds2cCQJvx19RGBoY7JAtnZ2RpHcn7O1ek6ox5dK1yx/sknn2C32zEn2lGUpj/fnOiYcTd16lQXR+Z9JJ8LIVyhvLycl19+GZ1Ox1X3DnZ7y9gzderfnvY9Eti0aRNz58716NhCeAPJ50IIV1BVlVdeeYXy8nIuu2MAAcGenSwflRBO/2u6k5mZyfjx4z06thDeQnK6EEJ4J2cB3VlQN7TCwrpzCz5z26YvaDDFqyg6xzH8bcvaRl8B2717N1dcccVZt8+aNQtFUZg5cyZXXnklF198MV9++SVt27Zl/vz5Lg1W+BdH606FkNi26I3mVr1ife/evZw6dYpOF12G3mBo8DGdLhqKXm/wysJ6VlYWKBAc7n+FdaPJgCXI7HiNXsy5Ol0x6FBa2Yr17Oxs5s+fhz5QxRjdvFWLhlAwhNvZsGED+/btc3GE3kXyuRDCFd59910yMzPpf013ottGeHx8RVG44jcDMQeYGDdunNfnaSFcTfK5EMIVvvvuOzZt2kT7Hgl0HtBBkxguvr4XkW3C+OKLL9i6dasmMQihJcnpQgjhnXJzcx2L2IyORWytbcV6Xl4eCxYuQBeoYohs+jV3nQmMsXbS09P54Ycf3BChdhpdWD99+jQdO3asd1tNTQ1bt26lU6dO9OnTp/Z2vV7P8OHDSUtLc12kwu+cPHkSc3AoOoOBgLCIVr1H5rJlywDoOujKcz7GEhRC+94Xk5qa6nXfq1OnThEYEoDeoL/wg31QSEQgWVmntA7jvJxtwHQmAzqT4/+htLRUy5A85tNPP6Wmxtrs1epO5iTHCYK/r1qXfC6EaKmffvqJr776isj4MC6+vpdmcQSHB3Lpbf0pKytj1KhR0hJetCqSz4UQLXXq1CneePMNzAEmrrxnIEpL3ky1gN6g56r7BqPoFF566SVpCS9aHcnpQghvYLPZtA7B6+Tm5qKz/LoI0llY9/YtY13lq6++oqa6BnPb5l9zN7dzrFT/9NNPXReYF2h0Yb2yspLKysp6t+3Zs4fq6moGDx581uPj4uLkZFick81m49SpU1hCHSucLKGRlJSUUFxcrHFknqeqKkuWLMESFEKH3pec97HdBl8NwNKlSz0RWqOoqkp2drZfrlZ3CgoPpKys3Kv3sPq1sK5H/0th3ZvjdZX8/Hy++eYbdBYwxrasoGIIV9GHOrZlOHLkiIsi9D6Sz4UQLVFeXs5LL72EolO4+r4hmk+q6zYwhaTu8fzwww/MmzdP01iE8CTJ50KIllBV1bGveVk5Q+8YQFBYoKbxxCVF0f/q7mRkZPD+++9rGosQniY5XQjhDVrTlqKNoaoq+fn5GALqFtYdn7eGwnp5eTlffvklOiOY4pp/zV0fBIZIOz/99BOpqakujFBbjS6sx8fHn9Ued+PGjSiKwsCBA896fHFxMVFRUS2PUPilnJwcbDYbAWGRAAT8UmBvjfus79y5k1OnTtH5kmHoDcbzPtbRKt7I4sWLPRTdhVVUVFBVVeXxvdg8yfnaCgoKNI7k3PLy8n7ZX12H3tJ6Zs99/vnnVFVVYU60obRwe19FAUuS/++1LvlcCNES77//PidOnKD/1d2JTYzUOhwUReHKewZhshh54403OH36tNYhCeERks+FEC0xf/58NmzYQFL3BLpenKx1OABcMrw3EXGhzJw5k507d2odjhAeIzldCOENZMJOfcXFxVit1trr7AA6g6MtfH5+voaRecacOXMoKirC1NaG0sL1FJZEx/X2Tz75xAWReYdGlyGGDRvGihUrWLduHeAopk2ZMgWAG2644azH79q1i3bt2rkoTOFvnPupW0LCHf/+UlhvjfusO/dF6jH0ugs+1hIUQscBl3Lw4EEOHjzo7tAaxdllwBxo0jgS9zEHOF6bN68Azzl9unbWnLMtTU5OjpYhuV1JSQlffPEFOhOY2rim/a8hUkUXpLJo0SIyMjJcckxvI/lcCNFcu3fvZubMmUTEhXLx9b21DqdWcHggl47oT0lJCWPGjNE6HCE8QvK5EKK5cnNzeeONNzBZjJq2gD+T3qDnqnsHo6Ly0ksvUV1drXVIQniE5HQhhDcoKysDkC3WfuFcsFa3sA6gsxj8fjFbTU0N06ZNQ9GDKaHlPw/6MNCHqKxYsYKjR4+6IELtNbqw/q9//Qu9Xs+1117LgAEDSElJYc+ePdx6661n7QOTl5fHpk2buOyyy1wesPAPWVlZwK+FdXNIWL3bW4vy8nIWL15MWEw8Sd37N+o5vYbdCMC3337rztAazW53zDjS6Vu4XNiLOV+b1WrVOJKGlZeXk5ebiyHYDIAxxPHviRMntAzL7b766ivKysowtWv5anUn56p1u93OtGnTXHNQLyP5XAjRHDU1NbX7mF85chAGo7Yt4M/UfXBHEjrGsmLFClavXq11OEK4neRzIURzvfnmmxQXFzP4ln4Eh2vbAv5MbTpE0+fyrvz8889+tapJiPORnC6E8AalpaWAFNadnKvS9XVawQPoLQby8/P9+vu0YMECsrOzMcXb0Z2/wXKjOK+3q6rKxx9/3PIDeoFGlyJ69uzJggULSEpKYufOneTl5XHnnXc2eKL70UcfYbVaGT58uEuDFf7DuZLWHBwKgCXIUVhvbe07Fy5cSFlZGb2vvBlF17hfx5S+gwiJjGX+ggW1M8m05Jzdrtr9N5k4X5u3zOQ/k3NltTHUUVDXBxpR9DqOHz+uZVhuVV5ezowZn6EzgtkFM+fqMsao6AJU5s6dQ3Z2tkuP7Q0knwshmuOLL74gLS2NHkM6EZ8co3U4Z1EUhSvuGYjeoGPM2DGyP5zwe5LPhRDNsXnzZhYtWkRch2h6DumkdTgNGnhjH4LDA5k8ebLfdhEToi7J6UIIb+CsM1RVVWGz2TSORnu1hfUzVqzrLUaqq6trJyL4G6vVypSpU1B0YG5nd9lxDVEq+iCVhQsX+sX5XZPW+F133XUcPnyY7OxsSktLmT17NpGRZ++t+Le//Y2CggKuv/56lwUq/Etubi4ApqDQX/4NqXd7a2C325kxYwZ6g5G+V49o9PN0egP9r7udstJS5s6d674AGyk4OBiA6soajSNxn6oKRwu6kJAQjSNp2OHDhwEwhQcAjuKCMczM4cOHazsK+Jtvv/2WgoJCTAkt3+flTIoC5iQ7NTVWpk+f7tqDewnJ50KIpsjNzeWDDz8gIMjM4Fv6aR3OOUXEhtLvqu5kncry264jQtQl+VwI0RQ1NTW8/vrrjslod1+CovPOieMmi5Ghtw+gpqaGN998U+twhPAIyelCCK3VLRTLfuvnWbH+y9f+us/6ggULOJFxAmO8HZ3ZdcdVFDC3d3SJnTx5susOrJFmNc+NiYnBZDr3fsqBgYGEhYV57epOob2CggIATAFBjn8Dg+vd3hqsWrWK9PR0el4+nKCwiCY9t+81t2E0B/Dpp59SU6NtQTs4OBij0Uh5SaWmcbhTRanjtTX0psYbpKWlAWCKCKi9zRQRSEVFhV+2g6+urnbs82IAU1v3dEowxaroLPDNN9/49d8lyedCiMaYOHEi5WXlDLq5L5bAc//N8AYDrulJcHggH3/ycavbYki0XpLPhRCN8e2333LkyBF6XtqJ6LZNuwbhaSl9EmnbOY7Vq1ezefNmrcMRwmMkpwshtFK3mC6F9XPvse782h/3Wa+pqeGjjz5C0YEl0fWL9YzRjlXr8+fPJz093eXH9yT/3RRZeLWioiIAjBbHfl46gxGd3lB7u7+z2+1MmjQJRadj4C2/bfLzA4JD6XftbWRlZWm+al1RFBISEijJ174tvbsU55USFBREaGio1qE06MCBAwCYI37dH88cGVDvPn8yb948Tp8+7bJ9XhriaHdjo7KykhkzZrhnECGE8AFHjx5lzpw5RMaH0W1QitbhXJDRbGDQTX2prqrmww8/1DocIYQQwiuUl5fz4YcfYrIYueSG3lqHc0GKojD0tgGgwHvvvefX+5gKIYQQ3qDudmqytVrdVvBn77EO/rlAdPbs2Zw8eRJTgmtXqzspClg6OFatT5w40fUDeJAU1oUmSkpK0On16AyOqpiiKBjMFoqLizWOzDOWLVvGwYMH6XnZcCLjE5t1jIG33IfRbGHSpElUVmq7WjwxMZHy4oralun+RLWrFOeWkpSU5LUzgg8cOIAxxIzO9GtPdHNkYO19/sRqtfLxJx+j6F27z0tDTPEqOpNjX2F/3TdHCCEu5IMPPsButzP45n7odL7x1qHzRe2Jig9n7ty5HDt2TOtwhBBCCM198cUX5Ofn0++q7gQEW7QOp1Gi20bQeUAH9u7dy8qVK7UORwghhPBrVVVVtZ9XV/vfNf6mOtce64YA/1yxXlpayoeTPkQxOLZIdRdDlIo+RGXp0qXs2bPHbeO4m29cHRN+p6SkBL3JUq9QaTBbWkXxqrq6mvHjx6M3GBl61x+bfZygsEguvvEecnJyNF9R27lzZwDys/yv40Bxfhk11VY6deqkdSgNys3NJT8/H1NkYL3bnV8fOnRIi7DcZsWKFZw8cRJjnB2dm7sRKzowtbNRVlbG7Nmz3TuYEEJ4oaNHj7Js2TJik6Jo3yNB63AaTafTcfENvbHb7XzyySdahyOEEEJoqry8nOnTp2MJNNP3iq5ah9MklwzvhaIoTJkyRVatCyGEEG5ktVprP9d661lvkJeXh6JT0Jn19W53rlj3t8L61KlTKcgvwJxoc1uHWHCsWg/oaAPgzTff9NnzOymsC02UlpZiMNXvJ2EwtY7C+meffcaJEycYMPwuwmLiW3Ssgbf8lsCwCKZMmUJ2draLImw6Z2E996T/tUDJPemYndalSxeNI2nY4cOHATCF1191oDcbMAQaa/df9xeffvopKO5fre5kjldRDDBjxgw5qRRCtDqffvopqqpy0XU9vbZry7mk9GpHRFwY8+fP1/QcSQghhNDanDlzKCwspPewLhjNbrxS6gbhMaF07JfEvn372LRpk9bhCCGEEH5JVVVsNlvt13U/b63y8vLQWQxnXQvR++GK9YyMDD777DN0FjC3c3+h2xAGxhg7O3bsYPHixW4fzx2ksC40UVJSgsFUvxBoMFuoqKioNzvK32RkZPDRRx8RFB7JpXf8ocXHMwcGccW9f6KiooJx48a1PMBm6tmzJwCnM/I1i8Fdcn55Tc7X6G2OHz8OgDHs7HZ+hlALWVlZflMQTk1NZe/evRij7OgDPDOmYgBTnJ2cnBzWrl3rmUGFEMIL5Ofn8/333xMRG0qHHm21DqfJFJ1Cv6u6YbVa+frrr7UORwghhNCE3W5n5hczMRj19L7MOyeLX0j/q7sDjnb2QgghhHC9M1cN2+2eWdDkzXLzcmvbvtflLKw7W8X7OlVVee2116ipqcGSYkPxUMXYkmJH0cMbb7xBSUmJZwZ1ISmsC4+zWq2UlZVhsNRvXW0wOypl/rrPus1m46WXXqKyspJrHvgb5sAglxy31+U3kNijP8uXL2fJkiUuOWZTdejQgZCQELJ+Pq3J+O6UnZ6LXq/32sL6iRMnADAGm8+6zxhixm63c+rUKU+H5RbOduymBM+2iDEl2OuNL4QQrcHcuXOprq6m1+VdUHS+tVrdqfOADgQEmZk9e7bfTDITQgghmmLz5s1kHM+g80UdsASd/Z7RF8S0iyQ+OYZ169bVvv8VQgghhOucuUK9ta9YLy8vp7ysvLaIXpfOpEfRKZw+7R91kGXLlrFhwwYMkXaM0Z675q63gDnJRl5eHhMmTPDYuK7SrMJ6UVERu3fvpry8vMH7y8rK2L17d7MKpHv37uU3v/kNKSkpBAYGEh0dzbBhw1iwYEFzQhVeqLCwEABjQP3CujEguN79/uajjz5i69atdB14JV0HXemy4yo6HTc8+iwmSwCvjBpVu4LZk3Q6Hf3796fwdAnlxRUeH99drDU2co7n0bVrVwIDAy/8BA04k7gh6OwNxw2BjuSfk5Pj0ZjcoaamhmXLl6EzgyHcs4V1fSDoQ1U2bdrkV21+wL35XAjhu1RVZe7cuRiMerpenKx1OM1mMOrpckky+fn5rF+/XutwhHAbyedCiHOZO3cuAD2HdNI2kBbqcWknVFWVa4PC70lOF0JoQQrr9Tmvt+sDzy6sK4qCPtBIdo7vbzlXWFjIa2NeQ9FDQCc7nt4B0NxORR+kMmvWLLZv3+7ZwVuoWYX10aNHM3To0HP+gtlsNoYOHcprr73W5GMfO3aMkpISHnzwQcaPH8+LL74IwIgRI5g8eXJzwhVexvmHyRwYUu92c5CjsO4PRcAzLVmyhEmTJhEWG8/wR//l8n1KI+Lacv0j/6KstJQnn3ySoqIilx6/MQYNGgTA8YP+sToaIPNIDtYaG4MHD9Y6lHNyFnr1FsNZ9+kt/rPny6ZNmygpLsEY4/kkD2CKtWO321m5cqXnB3cjd+ZzIYTv2rt3Lz///DPJfRIxWXxrL9YzdbskBYD58+drHEnTZGZmkpWVpXUYwkdIPhdCNKS0tJSVK1cS2SaMmMRIrcNpkZTejnOSefPmndWu1psVFRVx5MgRrcMQPkRyuhDuJ23Oz3Zmh7fW3vEtO9tRNDcEnr2QzXl7TnaOz09AGDt2LAX5BZg72Dy27Wpdig4CuthQUXnppZeoqPCdBZvNKqwvWbKE6667jpCQkAbvDw0NZfjw4SxatKjJx77ppptYsmQJL7/8Mo8++ih//etfWb16NX379uWdd95pTrjCy2RmZgJgCQ2vd7slJKLe/f5iw4YN/Pvf/8YUEMSdT4/FEtTw701L9bj0Wgbdej9Hjx7lL3/5i8f3prj88ssBSE896dFx3Sl9r6PN3GWXXaZxJOdWUFDgaEGjP/vPuT7AUWz3hy4Qa9asAcAYrc3JrzHKcfFm9erVmozvLu7M50II37Vs2TIAugxor3EkLReVEE5kfDjrf1h/zpU/3ug3v/kNI0eO1DoM4SMknwshGrJ27Vqqq6vpPKCDyyf3e5rRbKBDr7acPHmS/fv3ax1Ooz399NPcfvvtPnUOIrTlrpz+008/8cQTT9CzZ0+CgoJISkrinnvuIS0tzRVhC+EzampquPHGGxk1apTWoXiVqqqq837d2pw86ahvGILPUVgPNmGz2Xy6Hfzy5ctZtGgR+lAVc1vtJi0aQsHc1s6xY8d8qiV8swrrx48fp3Pnzud9TMeOHV3Wklqv15OYmOgXxSEB6enpAASER9e7PSA8qt79/mDZsmU88cQTqIqOO/4xhpikjm4db9i9j9P7ipvYtWsXDz30kEdXKicnJ9O5c2eO7cukqqLaY+O6i81m5/CO40THRDNgwACtwzmnwsJCdOazV6sDtbcXFBR4MiSXU1WVtWvXojOCPlSbGHQW0AerbPlxi19dFPF0PhdCeD9VVVmxYgUmi5F2XdpoHY5LdOyTSHVVNT/88IPWoTRacXEx+fn5WochfITkcyFEQ5zdtlL6JmociWt07JMEwIoVKzSOpPF+/PFHAL96Dyncy105fdy4cXz77bdcc801jB8/nscee4x169YxYMAAUlNTWxKyED6lvLyczMxMZs+erXUoXqWsrAwA3S8Lt5xft1YZGRkAGEPMDd5vDDbXe5yvycvLY/To0Sh6COxm06Q7bF2WZDv6QJXPP/+89tzJ2zWrsK4oygVnrVRVVbWoFUJZWRm5ubkcOXKEd999l8WLF3PNNdecd7zi4uJ6H8I7HTx4EIDgqPoXa4Oj4gD8YrakqqpMnTqVf/7zn+iNZu557h2Suvd3+7iKonDDo8/S79rbOXDgAL/97W9rv9+ecMstt2Cz2kjb+rPHxnSXn/ecoLKsiptuvAm9Xq91OA1SVZW8vLwG28ADGPykFfy+ffvIycnBEKVNG3gnQ5RKTXUNmzZt0i4IF/NEPhdC+Jbjx4+TkZFBYrd49AbvzH9N1aFXWwCfKqwL0RSSz4UQZ6qpqWHjpo2Ex4QQEavR7GQXS+zaBr1BL/lc+DV35fSnn366djXgI488wgsvvMD69euxWq28/vrrLQlZCJ/i6x1c3MVZSwuJDKr3dWv188+O2oYx1NLg/cYwS73H+RJVVRk1ahSFhYVYkrVpAX8mRQcB3WygwH/+8x9KS0u1DumCmlVY79atG0uWLDnnvkZ2u53FixfTtWvXZgf2j3/8g5iYGDp16sQ///lP7rjjDiZOnHjOx48dO5awsLDaj8RE/5iR6492796NMSAIS2hEvdsN5gACI6LZs2ePT1/0KSsr45lnnmH8+PGERMVx38sTadetj8fGV3Q6rvvj01x+z6NkZmby+9//niVLlnhk7DvuuAOT2cTudWk+vV+NqqrsWnsARVG49957tQ7nnAoKCqipqTnnfi/6QEdhPScnx5NhuZyzJbGzHbtWjFGOn2lnPP7AE/lcCOFbNm7cCEBSt3iNI3Gd6IQIAoItbNy40af2ZRWisSSfCyHOlJqaSllpGYl+lM8NJgPxKTHs379furoIv+WunH7ppZdiMtW/dtS5c2d69uzpU9srCNFSOl2zymF+z9nt1DkZz9e7n7bU4cOH0VsMGAKMDd5vCndUow8dOuTJsFxi3rx5rF69GkO4HVOC91wfMYSAJclGVlYWb7zxhtbhXFCz/pLcd999pKWl8dBDD1FUVFTvvqKiIh566CEOHz7M7373u2YH9re//Y3ly5czffp0brzxRmw2G9XV524v/fzzz1NUVFT74attGPxdRkYGJ0+eJKJdSoMzxMLbplBcXOyzJ3WHDx/m3nvvZenSpbTr1pcH/juZ2KROHo9DURSG3P4Adzw9BhsKzzzzDK+//jo1NTVuHTciIoI7br+DotwS0ramu3Usd8o4cIrs9FyuueYar56k42z9ZQxpuLCuM+nRmw0cO3bMk2G5lNVqZf78+ShGMERqm+z1waALdLRI9peZm57I50II37J9+3YA2naK0zgS11F0CgmdYsnOzubUqVNahyOEy0k+F0Kcadu2bYB/5XOAtp0dr2fnzp3aBiKEm3gyp6uqSnZ2NtHR0ed8jHSIFf5GCusNc3Y7jUoIByA3N1fDaLRVUlLCsWPHMEWceym3KdyColPYt2+fByNruezsbMaNG4digMCu2naGbYg5SUUfojJnzhyv71DUrL8kTzzxBJdeeinTp08nOTmZ4cOH89BDDzF8+HCSk5P57LPPuPzyy3niiSeaHVi3bt249tpreeCBB1i4cCGlpaXceuut55yxZzabCQ0NrfchvM+qVasAiOrQ8MzK6A7d6j3OlyxatIj77ruP9PR0Bt16P/f+5z0Cz1iV72mdL76cB/47hZikjsycOZM//vGPZGdnu3XMxx57DJPZxE9L9mCttrp1LHew2+1sWrgTRVH4y1/+onU453X48GEAjOENJ3pFUTCGWTh27JjbJ1W4y8qVK8nNzcUYa0fR+NxXUcDUxk51dTVz5szRNhgX8UQ+F0L4lp07dxIUGlDbAs5fxCfHALBjxw6NIxHC9SSfCyHOtGvXLgDaJJ+7YOaLnPlcCuvCX3kyp8+cOZOTJ08ycuTIcz5GOsQKfyOt4BvmrBfEJkYBvt/9tCVSU1NRVRVzTPA5H6PodZgiA9l/4MB5FwN7E1VV+e9//0tpaSmWFBu6hrvca0rRQWBXG4oOXn7lZcrKyrQO6Zwa3pj3AoxGIytWrOCFF15gypQpLF++vPa+0NBQnnnmGUaPHo3R2HCrhOa4++67efzxx0lLS/NoC7vdu3czf/58j413IWFhYTz++ONnte/xBaqqOlae6vTEpPRo8DGRHbpgMFlYuHAhf/nLX7x2b+u67HY77733HtOmTcMcEMQdT4+h88WXax1Wrcj4JH43ahLLPnmbXeuXcM899/D+++/Tp4972tPHxsby4AMPMmXKFLav3MfAGz3XBt8V9m48TF5mIXfccQedOnm+20BT7N69GwBzVOA5H2OOCqQyp5T9+/e77f/cXex2Ox9++CEoYG7rHVsLmOJVqo7Dx598zG9+8xsCA8/9vfcFWuRzIYT3KigoICsriw692vrdBYfYpEgADhw4wM0336xxNEK4luRzIcSZ9u3bR0hEEIEhXrBxpgvFtIsEBZ/tcijEhXgqpx84cIC//OUvDBkyhAcffPCcj3v++ed5+umna78uLi6W4rrwaf72PtdVsrKyAIiIC8USaG7Vnd5++uknAAJiz11YB7DEBVOUm82uXbu45JJLPBFai6xatYo1a9ZgiLBjauM9LeDPpA8Cc5KNnPQcJk6cyLPPPqt1SA1qVmEdwGKx8NZbbzFu3DgOHDhAUVER4eHhdO3a1S3F0IqKCoCz2uC4k6qqvPzyy7WrQr1FQkICd911l9ZhNNnWrVtJS0sjtnMfjAENr4LSG4zEdevHyd2bWb16Nddee62Ho2ya6upqnn/+eZYtW0ZU2w7c8fQYIuO97wTTaLZw05/+TUKnHqycPp6HHnqYN94Yx9VXX+2W8R555BEWLFzAjlX76DSgPZFxYW4Zx9XKiir4cdFuQkND+dvf/qZ1OBe0ddtW9GZD7b4uDbHEBVO0P4etW7f6XGF93rx5HDlyBFMbO3ovuR6kM4C5nY2C9AKmTZvm9V0NGsPT+VwI4b0OHjwIOPYk9zdR8eHAr69RCH8j+VwI4VRYWEhOTg4derbVOhSXM5oNhEWHSD4Xfs3dOT0rK4ubb76ZsLAwZs+efd5jms1mzGZzi8cUwltIYb1hJ06cAAVCIoMIiQzk5MmTqKraKr9fmzdvRtEpWOLOX1gPiA+haG82W7Zs8frCenl5OWPGjkHRQUBn72sBfyZzokpNjsoXX3zBbbfdRrdu3bQO6SzNLqw76fV6evbs6YpYAEebidjY2Hq31dTU8NlnnxEQEECPHg2vdHaHbdu2cfjwYeyhsVg7XOyxcc9Fqa7AeGA1s2bN8rnCuqqqjpWnQNJFw8772KT+l5G5ZwuTJk3i6quv9tq9T6xWK88++ywrVqwgqccAbv/7f7EEhWgd1jkpikL/6+4gPDaBeeNf5Omn/8GECeMZNuz8/x/NERgYyAv/eYEnnniC1V9u5o6nrvPa/0cnVVVZO/tHqiqqef7ZfxMZGal1SOd17NgxMo5nENQ+/LwnOQFtQkCBH374gYceesiDEbZMfn4+b731FooBLB28Y7W6k7mdSvUp+Pjjqdx0000kJydrHZJLuDqfCyF8T3p6OuCYpe5vjGYjIRFBta9RCH8l+VwI8Ws+940J7k0VGRfGz6knKCoqIizMP1+jEOCenF5UVMSNN95IYWEh69evJyEhwaXHF8LbnWub4dbu+PHjBIcHojfoCY0K4fSJAnJzc4mJidE6NI/Kz88nNTUVc1wwOuP5JzIFxIWg6HWsW7fO67fc+uyzz8jJzsHc3nsWr52PogNLJztluxXeeustpkyZ4nWTPLyu0vX4449zzTXXMGrUKKZOncp///tf+vTpw/bt2/nvf/9LcPD5Z4q40rfffguArV1vCAjV/EMNi8Me0Y79+/ezd+9ej30fXGH58uX89NNPRKf0IKzN+Vd0B0bE0Kb7RRw8eLD2/8Abvf7666xYsYL2vS7m7mff9Oqiel3JfQdxz/PvojMY+fvTT7vtZ+mKK67g5ptvJvtYHrvWev9s8rRt6aSnnmTw4MHceeedWodzQWvWrAEgKDH8vI/TW4xYYoLZtn0bhYWFbo/LVV5//XWKi4uxdLCh87LJ0YoeAjrZqKmx8sorr2C3e1fhXwghmisjIwOAsGjfOKdpqtDoYLKysqiqqtI6FCGEEMJtavP5efYG9WWh0Y7X5XydQojGqays5NZbbyUtLY2FCxd6dPGaEN5CCutnq6io4NSpU4THOCbYh8c6rge0xknp69evR1VVghIvPHFPZ9RjaRPM/v37a1vpe6O8vDw+/uRjdCawJPrONWxjhIoh0s6WLVvYsGGD1uGcpVEr1q+++moURWH69Om0a9eu0e2jFUVh5cqVTQpo5MiRfPzxx3z44Yfk5eUREhLCRRddxLhx4xgxYkSTjtUSxcXFLFu2DDUgFDU42mPjXogttiO6ghN89913PrMSIT8/n9fGjEGnN9B52C2Nek7HoTdw+nAqb7/9Npdddhnx8fFujrJpli5dyqxZs4jt0Jk7nn4Ng9G39rxP6NyT2//2KrPfeIZ//vOffPPNN26ZtPL888+zZcsWfly0i/bd4on8pQ2rtyktLOeH77YRGBTIqFGjvG4GVEOWLl2KolMIbBd+wccGJYVTmVPKqlWrfGLSwLJly1i8eDH6UBVTgnee8BqjVYwxdrZv387nn3/OAw88oHVIjeLJfC6E8D3Z2dkABEc0vGWPrwuJCEJVVU6fPk27du20DkeIZpN8LoQ4n9p8Hu6/+Rwcr7NXr14aRyNEy3gqp9tsNkaOHMmmTZuYN28eQ4YMaW7IQvg0Kayf7ejRo8Cvnesi2ziKyocPH/b6FueutmLFCuDCC9mcgpIiqDhZzMqVK7n//vvdGFnzzZgxg8qKSgI621B8bIewgBQ7Jfk6Jk2axNChQ72qZtOowvqaNWtQFIXy8vLarxujOS/03nvv5d57723y81xtyZIlVFdXY0vqgTdtOqCGxaGaAlm0aBHPPPMMFotF65DOy2az8e9//5v8vDw6D7uFwPCoRj3PHBRClytHsG/Z1zzzzDNMmzYNo9Ho5mgbp6ioiFf/+19MlgBGPDkKkyVQ65CaJbnvIAbf9gCb5k5n4sSJPPfccy4fIywsjFGjRvGXv/yFlV9s4s6/DUev965GGaqqsvqrzVRVVDN69As+0Qbr5MmT7Nmzh8C2oegtF/4zHpwcSd7WEyxevNjrC+u5ubm8+uqrKHoI7Gbzpj+/ZwnobMdWpOO9997jsssuIyUlReuQLsiT+VwI4Xtyc3NRdAoBQV7WKsRFAkMd5825ublSWBc+TfK5EOJ8cnNzAQgK9YFen80QGOLI56dPn9Y4EiFazlM5/R//+Afz58/n1ltvJT8/n88//7ze/b/73e+adDwhfJXNZtM6BK+TlpYGQFRCOEDtwjjn7a1FSUkJGzZswBQZiDG0cTW3oKRwcjcfY9myZV5ZWC8pKeHLL79EZwZTG9+bVKIPAmO0nV27drFt2zYuvlj77bqdGlXhstvt2Gw2unTpUvt1Yz58+Q/V3LlzQVGwR7XXOpT6FB326A6UlpayevVqraO5oHfffZcNGzYQndKdxP5Dm/TcNt0H0Kb7AHbt2sWrr77qNTPKPvzwQ4oKCxl698NExp+/rb23u/TOB4lMaM+XX37J4cOH3TLGsGHDuPPOOzl9ooDty71vC4N9mw6TcTCLK664gttvv13rcBpl8eLFAAQlN24feEOQCUtsMD/++GPtRRZvpKoqr776KoWFhViSbV6/54vOCAGdbdTU1PDvf/8bq9WqdUgX1BrzuRCi8YqKirAEmlF0/ll8s/wyYaC4uFjjSIRoGcnnQojzKSoqAsAS5Fud9RrLEiz5XPgPT+X0nTt3ArBgwQJ+//vfn/UhRGsh2zme7eBBxxau0QkRAITHhqI36Dlw4ICWYXncmjVrqKmpIbhDRKOfYwgwYokLYceOHeTk5LgxuuZZsGAB5eXlmNraULxrrWOjmX9pX//VV19pHEl9PvrtdK+ff/6ZPXv2YA+LB5P3VXZsMckAzJ8/X+NIzu+zzz5j+vTpBEXF0XP4SJQm/vYqikK3a+4ktE0ic+bMYeLEiW6KtPEyMzOZNWsWEW3aMeD6u7QOp8X0BiNX/+4J7HY7//vf/9w2zjPPPEN8fDzblu8l92SB28ZpquL8UjbO30lYWCgvv/yyT6ziUVWVhQsXouh1BCc1PtEHp0Rit9tZsmSJG6NrmSVLlrBq1SoM4XavbQF/JmO0iinOzt69e/nss8+0DkcIIVqkrKwMUyM6ofgqk8XR/ai0tFTjSIQQQgj3ceY5k8U/C+sms+RzIZpqzZo1qKp6zg8hWguZaHq2ffv2odPpaleq6/U6ohLCSEtLo6amRtvgPGjp0qUATSqsOx+vqirLly93R1gtMnv2bBSdb65Wd9KHgC5IZeXKleTl5WkdTi0prDdgwYIFANijO2gbyLlYQrAHR7Fx40avbX319ddf8+abb2IJDqPfbX/EYG7eBAW9wUjfEX8gMCKayZMnM3XqVBdH2jTTpk3DarUy9K6H0Bv848Jzct9BtO3SmxUrVrht1XpwcDCjR4/Gbrez8otN2Gzazw5UVZU1s36kpqqG55//NzExMVqH1ChpaWkcOXKEwMQwdKbGb4wS3CECRafw/fffuzG65isqKmLs62NR9BDQ1e7VLeDPZOlkR2eCDz74gIyMDK3DEUKIZqusrERv9LFNt5rA8Mtrq6ys1DgSIYQQwn2qqqoA0Bv985Kf5HMhhBDNJYX1+qxWK/v27SMqIaw2vwLEJEZRXV3ttlqBtykuLna0gY9qfBt4p6D2EaD8Wpj3FkeOHOHQoUMYIu3ovGOH5WZRFDDH27FaraxcuVLrcGo1qjK4bt26Zg8wbNiwZj9XC3a7nfkLFoDBiD2irdbhnJM9Ohl7eh6LFi3iwQcf1Dqcer7++mteffVVTIEh9LvzUSyhTZvlcyZTYDD973yU7d9MYvz48QA88sgjrgi1SYqKipgzdy7hsQl0G3K1x8d3F0VRGHzb7/j2zWeZMWMGo0aNcss4gwcP5je/+Q3ffPMNO1ft56LrerplnMY68ONRTqRlceWVV3LTTTdpGktTLFy4EICQjlFNep7eYiQgIZTU1FR+/vlnkpOT3RFes02YMIGC/AIsHW3om3b+ojmdASydbJTvq+L11193a/eHlmpN+VwI0XQ2mw2Tzn8L687ONHIxRfg6yedCiPNx5jmdzj8L684ta6Sdr/AHktOF8Cx5L1jf4cOHqaqqIiax/na3cUlR7N1wiD179tC9e3eNovOc1atXY7VaiewQ1+TnGgKMBLRxtIPPzs4mLq7px3CHFStWAGCM8d3V6k7GaJWKw7B8+XLuuecercMBGllYv/LKK5vdItnX/lj9+OOPZGdlYYvtCF58YdEelQTHdzBv3jweeOABr2lhPXPmTF5//XVMgSEMuPtRgiJdswrYEhJO/7sfZ8fsjxg/fjzV1dX83//9n0df9/Lly6mqrGTInbej8+KfjeZI6TuYsJh4li5dyvPPP4/F4p7K5tNPP83atWvZuiyVlL6JRMSGumWcCykvrmDjvB0EBwfz4osves3vz4VYrVYWLlyI3mwgMKHp37uQjlGUnyhiwYIFPPXUU26IsHnS0tKYPXs2+kAVc1vfTPbGaBVDhJ1169axYcMGhg4dqnVIDWpN+VwI0XSKokgrSCF8gORzIcT5OP8+qKrqM+91hWitJKcL4VlWq1XrELzK7t27AYhrX38BV+wvX+/evdtrCpnu5CxCB7dv3gLRoPYRVJwqYeXKlfz2t791ZWjNtnHjRlDAGOn713h0ZtCHqGzbto3Kykq31a6aolGF9ZdeeqnVnIzPmzcPcKwI92oGE7bwBA4dOsT+/fvp0aOH1hHxySef8O6772IODmPAXY8SGOHa1toBoREM+M2f2PHtZD788EOqq6v561//6rGfzY0bNwLQbbD/rFZ3UnQ6ug6+ih8XfMHu3bsZOHCgW8YJDg7mhRde4KmnnmLtNz9y25+v0eRvyw9zt1NVUc2zLz1HbGysx8dvrk2bNpGbm0tY91gUfdNXHwQmhqMz6Zm/YAFPPPGE16xgGD9+PHa7naCOvtUCvi5FgYCOdkq26XjnnXcYMmSI13x/62pN+VwI0XRGoxGbzX/3UHNuRWMy+eees6L1kHwuhDgfwy/b1tntKnq9//2tcOZzo9GH+5oK8QvJ6UJ4lnQ7qW/Pnj0AxLWPrnd7REwopgBjbeHdn5WVlbFh40ZMEQFNbgPvFJQUQe7m46xYscIrCusVFRXs3r0bfbCK4h+7GWMIV6nKqGHXrl0MGjRI63AaV1h/5ZVX3ByGdygpKWH58uWolhDU4Ka1WdaCPSYZfX4Gc+bM0bywPnXqVMaPH48lNIIBdz1KQJh7vn+WkHAG3P0ndnw3hY8//hi73c7f//53j5yEHjlyBEtwKCFRvlOIbYq49p0Bx+t0V2Ed4KqrruLaa69lxYoVHPzpZ7oNTHHbWA05fiCTwzuO0b9/f+666y6Pjt1S3377LQAhnZr3+6Uz6AhOjiT7YBYbN27ksssuc2V4zZKamsq6deswhNt9fgadPghMsXbS0tJYtWoV1157rdYhnaW15HMhRPOYzWaKyyu0DsNtrNWOVT1ms1njSIRoGcnnQojzca7isVZb0Qf432QyZz73htVKQrSU5HQhPEsK6/Xt3r0bU4DxrK6yik4hLimKnw/+TFFREWFhYRpF6H4//PADNdXVRPRIaPYxDIFGLLHBbNu2jYKCAiIiWrY1ckvt378fq9WKOcy3r7XXZQhVqcIxGcQbCuvet5xOQ4sXL6aqqgpbTAq+sGxSDWsDpgAWfv89lZWVmsXx2WefMX78eMeK8rsec1tR3ckcHEr/ux4jKDKWadOmMXHiRLeO52Q0GkG1++1MUmfrV0/M+n7uuecIDAxk04IdVJZVuX08J2u1lXWzt6LX63nppZe8ckXxuWRnZ7N69WrMUYGYo4KafZzQLo5OErNmzXJVaC3yySefAGBp7x8ntub2dlAck42EEMLXhISEUF3lvyvWayodry0kJETjSIQQQgj3CQ4OBqC60j9zuvN1OV+nEEIIIZquuLiYo0ePEpsYhaI7u97hXMW+d+9eT4fmUStXrgQgqH14i44T1D4cu93OmjVrWh5UC+3fvx9wtE/3F87Xsm/fPo0jcWhxI4ANGzawc+dOiouLCQ0NpV+/fl67t+yFzJ49GxQd9hgvbwPvpOiwxaRQenIvy5cv59Zbb/V4CAsWLODNN9/EEhxG/7seIyAs0iPjmoNC6H/XY2z/ZhKTJ08mMjKS+++/361jJiUlcfDgQXKOHyY2qZNbx9JCxoFdgON1ultcXBxPPvkk48aNY9PCnVw10jOzjLat2EtxXikPP/wwnTr51v/hl19+id1uJ7RbyzommKMCMccEsXbtWo4dO0b79u1dFGHTZWVlsXLlSvQhKoZwzcJwKX0AGKPs7N27l9TUVHr16qV1SI3mT/lcCNE8oaGhVFVUY7fbfWryWWNVllcDjtcphL+SfC6EcOa5yrIqQiKaPynbW1WVOybnSz4X/k5yuhDCnZxt4Nt0iG7w/rhfbt+1axeXXnqpx+LypJqaGtatW4cxxIwpPKBFxwpKiiDvpxOsWrWKO+64w0URNs/hw4cB0Af5T2FdMYFi/PW1aa3ZhfWNGzfyxz/+sfaFqKpau5K3c+fOTJs2jSFDhrgmSg/Yu3cv+/fvxx7RDoy+007KFpOC/uRevvnmG48X1rdt28aLL76I0RJIvzsf8VhR3ckcFEL/Ox9h69cfMG7cONq1a8cVV1zhtvFuv/12li9fzsbvPuW2v77qVyvXC3My2ffDUtq1a8dFF13kkTHvvfde5s2bx/7NB+g2MIX45Bi3jleQU8yOVftJSEjg8ccfd+tYrlZaWsqsWbPQBxgJSWn571l4rzZkrz7Cp59+yssvv+yCCJtnwYIF2O12AhL8Y7W6kylBpSYX5syZ4xOFdX/L50KI5ouIiAAVKsuqCQzxnfPhxqoodXR4ioz07DmrcK3t27eTkpJCeHi41qF4FcnnQggnZ56rKPVcdzhPcr4uyee+7eDBgwQGBpKYmKh1KF5HcroQwhN27XIssjtnYT0pqt7j/NGPP/5IWVkZYT3jWlzrMYaYMUUEsHHjRsrLywkMDHRRlE2Xnp4OCuhaNlfAqygK6AJUMjIysFqtGAzabh7frKUoe/fu5frrr+fQoUNce+21vPbaa0ybNo0xY8Zw3XXXkZaWxvDhw71mWX5jONsi2+J8axUr5iDs4fHs2LGDQ4cOeWzYnJwc/v7006gq9Ln1AYIitdl33BIaQd8Rf0RnMPKvfz3L8ePH3TbWZZddxoABA0j7cS27Vi1w2zieVlNdxYL3X6GmqpInn3wSvV7vkXENBgMvvvgiiqKwbvZP2G3uK66qqlo7xr///W8CAnwrq3z++eeUlpYS1iMWRd/yFYRBieEYQy3MnTuXzMxMF0TYPCtWrEDRgSnaf2bPARjCVXQmWLFyBTabTetwzssf87kQovmiohxvnMtL/HOfdefrkgvxvis9PZ0HH3yQ5557TutQvIrkcyFEXa0lnztfp/BNd999Nw888IDWYXgdyelCCE9xFsxjkxrOp5YgM+Gxoezevdtv96Zfvnw5AEFJ4S45XlD7CKqrq/nhhx9ccrzmSk9PR2dWUfysEaE+UMVqtXLq1CmtQ2leYX306NFUV1ezaNEili5dynPPPceDDz7Is88+y5IlS1i0aBGVlZWMHj3a1fG6RVFREYsWLUK1BKOGxmkdTpPZYh2TATy1Z7Ld7ihOFuTn0/mKWwlvq23r/JDYBLpdexfl5WU888wz1NS4Zx8xnU7HuHHjCAsPZ/m0t9m3cYVbxvEka3UVc95+nlNH9nP77bdz0003eXT8Pn36cPfdd5OXWcju9QfdNs6h7cc4eSibq6++2q1dDdyhsLCQTz/9FEOAkbAWtoF3UnQKkf0TsFqtfPDBBy45ZlPl5+ezb98+9GF2FG0nmLmcooAhyk5+Xj4HDhzQOpzz8rd8LoRomZgYR/eY8mL/vBBfVlRBSEiIz02wE786ffo04GiNKn4l+VwIUdev+bxS40jco6zIcZ4SG6vNAg/hOrm5uVqH4HUkpwvhXv7UgbYl7HY7u3fvJjw2FEuQ+ZyPa9MhmpKSEscKaD9jtVpZtWoVhkAjlthglxwzqH0EAMuWLXPJ8ZqjoqKC3NxcdAH+tZANQGdxvKaMjAyNI2lmYX3NmjXcfffd3HDDDQ3ef8MNN3D33XezevXqFgXnKfPnz6eqqspRoPbBP65qeDyqOYj58xdQWlrq9vG++uortmzZQkzHnrTtM9jt4zVGm679SOh5Cfv27WPq1KnuG6dNGyZ/9BHBQUF8/8Gr7Fg+x21juVtlaQnfvPEM6Xt+4uqrr+all17SJI6//vWvRERE8NOSVEoLy11+/KqKajbO247FYvHJ1U0TJ06krKyM8D7x6Iyu6yYQ1CECc2Qg8+fPZ+/evS47bmOlpqYCYAjzvyQPYAh1vC4tvrdN4W/5XAjRMnFxjgmmpYX+W1iXi/DCH0k+F0LU5cx1ZW54f+0NnIV15wQCIfyJ5HQhhCccPnyY0tJS2iQ33AbeydkmfseOHZ4Iy6N+/PFHCgoKCGof4bIJF6ZwC8YwC2vWrKG8XJvzsJMnTwL+1QbeyfmafLawXlRURHLy+VcpJycnU1RU1KygPMlutztWeuv02GO0XXndbIoOW2xHKirKWbhwoVuHyszM5L333sMUEES3a+70qllena+4FUtIOJMnT67dh8gdevTowdSpU4mMiGD5tHdY9flE7Dar28Zzh4KsE3z+yv+RsW8Hw4cP56233sJoNGoSS1hYGP/4xz+oqaphw9ztLj/+j4t3U15SyZ/+9Cfi4+Ndfnx3OnDgAN988w2m8ABCu57/RKepFEUhamAiqqoyduxYj7f0cW5doXfNhECvow92FNbT0tI0juT8/CmfCyFazllYLyvyvwvx1hoblWVVta9RCH8i+VwIUVftRDk/zOcApYXlREZGanYNQwh3kpwuhHupqn8u8GkqZ6E8IeX8E8/bpDgmsW3f7vpr9lr7/vvvAQhOdt1WcYqiEJwSSVVVFStXrnTZcZvCWXR2ru72J87XdOLECY0jaWZhPSEhgc2bN5/3MVu2bCEhIaFZQXnSli1bOHbsGLaoJDCcu+2Ft7PHpICiY9asWW5LEKqqMmbMGCoqKuh8xa2YAr2rImYwmel69R1YrVZGjx7t1kJhjx49+OKLL+jYsSNbF83im3HPUFHiGye1R3duZsaLj5KfeYyHH36YN954Q/M3pCNGjKB///4c2XWcjINZLjtu7skCUn84RHJyss/t3WWz2Xhl1CvY7XaiBiWi6Fy/KUpAmxCCkyPZtWsXs2fPdvnxz6egoAAAxeR/SR5A+SWdFBYWahrHhfhTPhdCtFxtYd0PV7hJ21jhzySfCyHqCg0NxWKx+OVEOXBsWSMT5YS/kpwuhPCErVu3AhCfcv7uLxG/tIp3Pt5flJWVsWzZMowhZswxQS49dkiKY8/6+fPnu/S4jeUsOsuKdfdqVqVmxIgRrFmzhhdffJHKyvp7NlVWVvLyyy+zevVqbrvtNpcE6U7ffPMNAPbYzhpH0kJGC7bIRA4fPszOnTvdMsSKFStYu3YtkUmdievazy1jtFR0cjdiu/Rhx44dzJnj3jbtCQkJfP7551x99dUcS93KZy8+Sna6965OVe12Ns6Zzrdv/gu7tYaxY8fyt7/9DZ0bCrZNpSgK//nPf9DpdPwwZys2W8snRaiqyvrvtqKqKv/+9781nzzQVDNnzmRv6l5COkYRGB/qtnGiLklEbzbwzjvvkJXlukkNF1JWVgaA4rru9l7F+bo8sT1HS/hTPhdCtJyzpWqZH+6x7tw3XtrGCn8k+VwIUZeiKMTExPjlHus1VTVUV9ZIPhd+S3K6EMLd7HY7P/30E8HhgYRGnX/hpKIotO0US2ZmplcUM11l6dKlVFZWEtIpyuUdmY0hZixxwWzZsqW2LbsnOf+f9H64Yl0xOD684WexWRW1F198keTkZMaMGUNSUhK33HILDz/8MLfccgvt27fn1VdfJTk5mRdffNHV8bpUbm4uq1atwh4YgRrsupYPWrHHdgRwy8rT4uJixowZg85gpOvVd3hVC/gzdbniVgxmC2+//TanT59261jBwcG8++67PPHEExSfzmLmy/9H6rolbh2zOarKS5nz7n/44ZupxMfH8/mMGdxyyy1ah1VP165dGTlyJAXZxezdcKjFxzuyK4NTR09z3XXXMXjwYBdE6DlHjx5l/Pjx6AOMRF2S6NaxDIFGIi9uR1lZGS+//LLHWiIFBgYCoNo8MpzHOV9XUJBrZz26mr/kcyGEawQGBhIYGEh5if9diHcW1qOjXbu1ihDeQPK5EOJMzsK6v7W8dU4WkHwu/JXkdCGEux06dIi8vDzadWnTqBpPuy5tANi0aZO7Q/OYr7/+GhSFkE7uOZ8I7RKDqqp8++23bjn++dTusW7x+NBupyiOdvAnTpzQ/By3WYX1qKgoNm/ezIMPPkhpaSmLFi1i2rRpLFq0iJKSEv74xz+yefNmIiO9u1j9/fffY7PZsMemaB2KS6ghMaiWEJYtW1a7GtRV3njjDXJzc0kZfC2B4VEuPbarmYNC6XTZTZSUlDB69Gi3/5LpdDoef/xxPvjgAwIDLCya9BorPn0Xm9U79l3PPfEzM158jMPbfuDSSy9l1qxZdO/eXeuwGvTnP/+ZkJAQflq6h6qK6mYfx2a1sWnBDoxGI08//bQLI3Q/q9XKf/7zH6qrq4kZkoTeYnD7mCGdoghsF8bGjRtru3i4W0REBABqtfdO0mkJ9Zcf3/DwcE3juBB/yedCCNeJiIigsrRK6zBcrqLM8Zqiorz7PFacnzdP7tWS5HMhxJkiIiKw2+1UV9ZoHYpLVZQ5CuuSz4W/kpwuhHtpXYzzBuvXrwcgsVt8ox7vfNwPP/zgtpg8KTU1lb179xKUFIYhyOSWMYLaR6C3GPj222+prm5+jaM5Tp06Vbuy2x8pZpXy8nJKSko0jaPZPaCjo6P55JNPKCoqYteuXaxfv55du3ZRVFTExx9/7BOzRxcuXAiKDntUe61DcQ1FwRbdgcrKSlauXOmyw65evZp58+YRGteOxAGXu+y47pTQayCRSZ1Ys2YN8+bN88iYl112GV999RVdu3Zl+7Lv+Hrs3ykvLvTI2OdyeNsGPn/pcfJPZfDoo4/ywQcfeHWhLzw8nMcff5yq8mp2rNrX7OPs3XiYkvwy7r//ftq1a+fCCN3vo48+IjU1lZCOUQQlRXhkTEVRiLm0A3qLgTfffJOff/7Z7WN27doVAJt3d0pvNluJ48K/83V6M3/I50II14mIiKCyzP8K687X5M3nQUK0hORzIURdznznb5PlKsscF6fDwsI0jkQI95GcLoT7SGEd1qxZg6JTSOzauMJ6aGQwkW3C2LRpExUVvr9t3IwZMwAI7RbrtjF0Bh0hnaPJz89n8eLFbhunITk5OShm//0515kd/+bk5GgbR0sPYDQa6d27N0OHDqV3794+s4/x8ePHOXDgAPaweDC4Z2aKFpyTBJYtW+aS450+fZqXXnoJvcFIj+Ej0el8Y0NkRVHoft1vMJgtjBk71mP7LiQmJjJjxgxuuOEGMvbvZMaLj5F7wv1FyjOpqsqWBTOZ887z6FB55513eOqpp9Drvf//795776VNmzbsXnuwWa1oa6qtbFuxl+DgYB555BE3ROg+O3bsYPLkyRhDzEQPSvLo2IZAI9FD2lNZWclzzz1HTY17Vzb07NkTRVGoKWhxGvJKNQWOwnqfPn00jqTxfDWfCyFcKzg4mJpqK3abXetQXMrZCSckJETjSIRwL8nnQgj4Nd9V+dmKdWc+Dw0N1TgSIdxPcroQwtVycnLYvXs3bTvGYglsfE0suXc7Kisr2bhxoxujc7+srCyWLl2KKSKAgDbuvTYQ1i0WRafw2WefeWxCh91up6SkBJ3BfwvrzpX4xcXFmsbRooYAVVVVLFq0iB07dlBUVERYWBj9+/fnpptuwmw2uypGt1i9ejUA9kjfWs16QZZg7IHhbPxlBlFAQECzD2W323nhhRcoLCyk61W3ExTpvlk87mAJCafb1XeSuvgLnn32WaZPn+6Rk9CAgADeeOMNOnfuzPvvv8/MV/7M7X97lfa9Lnb72AA2q5Xl095h9+oFtImPZ+L77/vEqlkns9nMo48+yquvvsquNQcYcmu/Jj1/36bDVJRU8uc//9mnZrGXlJTw7LPPoqoqsZcnozN5fhJEcPsIyrtEs2/fPiZOnMjf//53t40VGRnJwIED2bJlC7ZK0PvRvi+qFax5Otq3b+8zv3u+nM+FEK4VHBwMQHWVtUlvtL1dTZVji56goCCNIxHCfSSfCyGcnPmuxs8K6858HhgYqHEkQriX5HQh3MNu968J5E21fPlyVFUlpW/TFnSl9Elk2/K9LFu2jGuuucZN0bnfjBkzsNlsRPWMc/s2Y4YgE0EdIkhLS2PDhg1cdtllbh0PoLKyErvdjs5P28ADKL9MGigt1bYNbrO/xfPnz+exxx7j9OnT9WZcKIpCbGwskydP5tZbb3VJkO6wadMmAOzhjWt54UvU8HhqMvezfft2hg4d2uzjfP7552zcuJHolO607TPYhRF6TlzXvuSlH2TPnm1MmjSJJ5980iPjKorCY489RlJSEv/5z3+Y/cYz3PznF+k2+Gq3jltTXcX8CS9zZPsGevbsycSJE32yRdTtt9/ORx99xN6Nh7joup6YLI2bEGGz2dm15gCBQYH89re/dXOUrqOqKq+++iqnTp0ion8ClthgzWKJviSRyuxSpk2bxpAhQxg82H2/+7fddhtbtmyhKkNHYGf/ObGtylRQbY7X5wt7wfp6PhdCuJbF4pjpZKuxAv5TWLdWOy7Et2TSqRDeTPK5EKIuZ76z1lg1jsS1JJ+L1kByuhDuY7PZtA5BU0uWLEFRFFL6NG2xaXTbCMJiQli9enWLF3NqpaioiG9mf4MhyERwSqRHxgzv3YbSo/l8/PHHHims116H9t8F67WvTevOzM3qwbty5UruuusuCgsLeeihh/j0009ZvHgxn376KX/84x8pKCjgzjvvZNWqVa6O1yXsdjs7d+5EtYSC0Y+WSf7CHuJYWb5jx45mH+Po0aO8N3485qAQul97t08Uh86ly1W3ERAWydSpU9mzZ49Hx77hhhuYMmUKAZYAFrz/CrvXfO+2saorK5j9xjMc2e6YAfXJJ5/4ZFEdwGQycf/991NdWcP+LUca/byjuzMoLSznrjvv8qnV6gsXLmTx4sVYYoOJ6K3tZB+dUU/csGRQ4N///jdFRUVuG+vGG28kObkD1ad02MrcNoxH2auhKkNPREQ49913n9bhXJCv53MhhOs5V8BYq/3rgoPN6ng9ssJH+CPJ50KIM/lrPrdKPhd+TnK6EO7VmvdYP3HiBDt37qRt5zgCQ5pWGFcUhc7921NRUcHatWvdFKF7ffHFF1SUVxDWMw5F55mtSc0RgQS2C2Pr1q3s3LnT7eMZjUZHHc9/1q+dRbU76pQmk7YLQZr1E/Tyyy8TEBDA1q1bmTJlCg888ADDhw/ngQceYOrUqfz4449YLBZefvllV8frEidPnqSsrAx7sGdmpnia+svrOnjwYPOer6qMGjWKmupqul5zJ6ZA7VbPuoLBZKbH9fdgV1VeeeUVrFbPztgeMGAAn346jfDwcJZOGUfqusUuH6OmqpLv3nqOjH07GD58OBMmTPD51mh33XUXJrOJfRsPN/qkZ+/GQwA+UdB0OnnyJK+99ho6k57YYckoOu0nsZijgojon8Dp06cZNWqU2046DQYD//jHP0GF8oN6VB9P+qoK5Qd1qFb4y1+eqG2n7M18PZ8L4Qtyc3OZNGkShYWFWofSKM5tc2x+1iLP9sue8bI3pfBHks+FcL+amhqmTJlCenq61qE0ijPf+VvLW7tV8rnwb5LThXCv1rxi/fvvHQv+ulzcoVnP73KR43kLFy50UUSeU1ZWxozPP0dvMRLaxbMLESP6OBbRTZ482e1jGQwGoqKisFdpX19wF7XK8W9cXJymcTSrsL5jxw5GjhxJr169Gry/T58+3HPPPWzfvr3Jx/7pp5944okn6NmzJ0FBQSQlJXHPPfeQlpbWnFAblJGRAYBqCXHZMb2KwQwGc+3rbKqVK1eyfft2Yjv3Jialh4uD00Z422QSel5CWloa8+bN8/j4Xbt25eOPPyY0LIzFk1/n0Nb1Lju23WZl3oSXOL5vO8OHD+f111/3izeZYWFhXH/d9RTkFJN9LPeCjy/KLSXzcA6DBw8mMTHRAxG2nN1u54UXXqCsrIzoQUkYg71n1n14zzZY2oSwfPlyt54wXXHFFdx5553YShQqjnhmtqC7VGUoWPN1DB06lN/85jdah9Mo7sznQgiHiRMn8r///Y+pU6dqHUqjGAyOnaKcF679hfP1OF+fEP5E8rkQ7rdq1SomTJjAc889p3UojeLMdzY/y+fOiXKSz4W/kpwuhHu11sK6qqrMnz8fg8lASu/mXTcPjw0lNimKH35YT27uha/Ve5Mvv/ySkuJiwnrEojN4toW4JTaYgPgQ1q9fz969e90+XmJiIvZKxecXsJ2LrVxBp9PRpk0bTeNoVhUjMDCQmJiY8z4mNja2WStmx40bx7fffss111zD+PHjeeyxx1i3bh0DBgwgNTW1OeGeJS8vz/GJH7aBd7IbLb++ziaaMmUKiqKj49AbXRyVtlKGXI/eaGLq1KmazNru3Lkzkz/6iACLhQXvv8Kpw/tcctwVn77H0R2buPzyyxk7dqxfvcG87bbbADi0/dgFH3t4h+MxI0aMcGtMrvTFF1+wdetWgtpHeGxvl8ZSdAqxl3VAZ9QzZswYsrOz3TbW888/T+cunanO1FGZ4Zsz6qqzFSp/1hMbF8vYsWPReailUEu5M597YqKcEL7g0CFHN5Vjxy6cy7zBryvc/KtFnl0uxAs/JvlcCPdzbpHliQuirlA7UU7yuVdoza2HRdO4M6cLIfB4J1tvsWvXLo4fP05Kn3aYLM1fkNftkmRsNnvt6ndfUF5ezvTp09GbDYR1j9Ukhoi+CQBMmjTJ7WP17t0bVLCVuH0oj1PtYC9V6NGjh2+2gr/22mtZsWLFeR+zYsUKrrvuuiYf++mnn+bYsWNMmDCBRx55hBdeeIH169djtVp5/fXXmxPuWaqrqx2f6LTd4N6tdHqqnK+zCdLS0ti3bx/RHXsQGB7lhsC0Yw4KIa5rP06cOMG2bds0iaFHjx6888472G1W5r73AmVF+S063q5V89m5ch7du3fnrbfe8ouV6nVdfPHFhIeHc3R3BuoFLggc2XUck8nEVVdd5aHoWiYjI4P33nsPfYCRmCFJjv1PvIwx2EzUwERKS0sZPXq02y4GWCwWPvjfB7Rp04bKo3qqTnnf9+J8avIUyg/qCQkJYdKHk4iIiNA6pEZzZz73xEQ5IYTr1V6I97OZ/Ha7ik6n85mJT0I0heRzIcSZaifK2fxruZKvFtZb6wpJ0XTuzOmidTlx4kSzt4n1Z621sP7dd98B0H1gxxYdp9OADugNer6b853PTBr76quvKCwsdKxWN2pTDwxoE4IlLpg1a9awf/9+t441YMAAAGoK/O/ah7XIsRK/f//+WofSvML6W2+9RU5ODg888MBZ7cYzMjL4/e9/T25uLm+99VaTj33ppZeeNdugc+fO9OzZ02U/dLXH97O9puqx2zA1o8i6efNmAGI79XZ1RF4htpOjlZLzdWph6NChPP3005Tkn+b7D/6L2syfw5zjh1nx6XtERET4xZ7qDTEYDFx55ZWUFVWQm1lwzseVFZWTe7KAgQMH+sS+1qqq8vLLL1NVVUX0oCT0LZgp6G4hnaIISAhl3bp1LF682G3jtGnThqlTpxIREUFFmp6qTN8ortfkKpTv1WMxW/jwww/p3Lmz1iE1iTvzuScmygkhXM95nux3rWOtNs1nNAvhLpLPhRBn+jWf+1dB13l+YjZ7zzZqjdFaCzmi6dyZ00XrcvPNN3P33XeTlZWldShepbrOQkRfKQy3VElJCYsXLyYsOoSEji1bsW0JNJHSN5GjR46yc+dO1wToRuXl5UybNu2X1era7skd2c+xav2DDz5w6zhDhgzBYrFQc1rB337Ea0476gXXXHONxpFAo6Z4Xn311WfdFhERwcyZM/nqq69ISkoiLi6O7Oxsjh8/js1mo0+fPvz+979n5cqVLQ5SVVWys7Pp2bPnOR9TVVVFVVVV7dfFxcXnfGxU1C8rsWsqWhybt9LVVBKd0K7Jz3O2KQ2JjXd1SF4hOMbxByw9PV3TOB544AG2bdvG6tWr2bZ0NhffeE+Tnm+trmLB+6OwWWt4/fXXNd9Twp0uu+wy5s6dy/EDp4hp13C79IyDWbWP9QVz587lp59+IigpnOAO3r26WVEUYi5tz4m5+xg7dixDhw4lLCzMLWO1b9+eadOm8fDDD5N3KA/sNsztvPcMoPq0Qvl+PQGWAD788EP69u2rdUgX5Ml8fumll551m6snygkhXM95odpa418X4q01UlgX/kPyuRDiQpw5z9/yuXOigK/ldFmxLs5F62vuwn85t0EtLi726+vGTVVZWVn7udVq9bvurw2ZP38+lZWV9L+2G4qu5QuZeg7pxKFt6cyaNcsrVg6fzxdffEFhYSGR/RPQmbTtXm2ps2p979695611tkRgYCBXXnklS5YswVYKhhC3DONxqg1qTuuIjY31ip+7RhXW16xZc877rFYrR48e5ejRo/Vu37Vrl8taG8+cOZOTJ08yevTocz5m7NixjBo1qlHH69ChAwBKRZErwvM+NZVgrap9nU1RUeGYbKA3+tbs38YymByvq24S1YKiKIwaNYrdu3ezbtZkOg4YSkRc20Y/f9PcGeSdTOf+++9v8GKXPxk4cCAAmYezuejahhPOyUOO/b8HDx7ssbiaq6CggLfffhudUU/04CStw2kUY7CZiP7x5P10gnfffZdXXnnFbWN17NiRTz/9lIcefojTR06j2m1YkryvuF6d7Wj/HhwUxKRJH/lEUR20z+eunignhHA9Zwecmir/WllVU2UlKMhP3lG2Yq1lVcmFSD4XQlyIP+dzwOc69tn9uWOmaBGtc7rwf3L+XJ+z9gGO1czuWjzkLWw2GzO/mInBqKf74Ja1gXeKT4khKiGcpUuX8ve//524OG1Xgp9LaWkpn376qWO1eg/tY1QUhcj+bclccpD//e9/bl25fuedd7JkyRKqM3UYuvrHOUjNaQXV6nht3rDFX6MisNvtzfpwxYzMAwcO8Je//IUhQ4bw4IMPnvNxzz//PEVFRbUfZ7bLqSs+Pp6oqGh0Jafxu34IgK44B4DevZvezt3ZRrum0j9X89dUOV5XUFCQxpE4ZqA+//zzWKurWPnpe41+Xl7mcX5cOJOEhASeeuop9wXoJSIiIujYsSNZP+eec3+4Uz+fJjwinJSUFA9H13Tjx4+nqKiIyAEJGAJ9Z5Z9WPc4TJGBfPvtt+zatcutY3Xo0IHpn04nPj6eyp/1VB7zrjeMVVkK5Qf0hIaEMnXqxz5TVAdt8zn8OlFu5MiR53zM2LFjCQsLq/1ITEx0ydhCiMZxXqiurqzROBLXqq6s8YrzPyFcQfK5EOJCnDmvxg/zOXjHNZ2mkMKWOBetc7rwfzKxp766hfW6n/urNWvWkHE8gy4XJ2MJcs1CSkVR6HtFN6xWKzNnznTJMd3h888/p6ioiLCecZrtrX6mgDYhBMSHsH79ere20h80aBDt27enJkeHvfrCj/d2qgpVJ3XodDruuusurcMBmrnHuqdkZWVx8803ExYWxuzZs9Hrz/0LYDabCQ0NrfdxLoqiMHTopSjVFShl+e4IXVNKwQmgeW2x27dvD0BZnn/uv1KW63hdycnJGkficP3113PppZdydNdmjuzY1KjnrJ7xPjarleeff97nZmk3V//+/amptpKfdXaXifLiCorzSunXt5/Xz9jds2cP3333HabIQEK7tmxPG09TdAoxv6ywHzNmjNvfxCUmJjJ9+nTatWtHZbqeygzv+L+tzlGoOKgnPCKcTz75xG1te/yROybKCeFLfOWCakSEY4uSyrKqCzzSd6h2laryasLDw7UOpVF85WdFtE6Sz4XwDc58XuFH+Rwc5yfBwcEYDI1qwOk1pLAlhPCkun9z5O9PfWVlZQ1+7o9UVWXy5MkoikK/q7q59NidB7QnKCyQWbNmUVTkfV2hi4qKmD59OvoAI2HdvesafGR/R9fiiRMnum0MnU7H73//e1Q7VGV6dQm4UayFCrZSheHDh3vN1hZe+10tKirixhtvpLCwkCVLlpCQkODS4990000A6E4fvcAjfUxNFfqCkySnpNClS5cmP33AgAEA5GccdnVkXsH5uvr166dtIL9QFIVnn30WnU7HulkfoV7gZOf4/h0c3bWZIUOGcMUVV3goSu05VwRnpeeedV/Wsdx6j/FWdrudsWPHoqoq0YMSXbKnjadZYoMJ6RjFvn37mDNnjtvHi4+P55NPPqFNmzZUHtVTlant96wm17FSPSQkhKlTptK1a1dN4/El7pooJ4QvcBZJfaVYGhkZCUBFqbbb5rhSZXkVqqrWFhm8nVz8Et5K8rlozXwljzuFh4ejKIpf5XNwnJ84z1V8ieR2IYQn1dTUNPi5gJKSkgY/90dr1qxh3759dOqfRHiMa8/F9QY9/a/uTnl5OdOnT3fpsV3h008/pbS0lPBebbxmtbqTJTaYwHZhbNmyhR9//NFt49x2221ERERQnalD9fGdgaqOO2oCDz30kMaR/KpFUzxPnDjB6tWryczMrLd/mpOiKLz44otNPm5lZSW33noraWlprFixgh49erQkzAYNHjyYxMREMk4cxRbXBUwWl4+hBX3mPrDbGHnPPc1avdu1a1diY2PJO7ofu82G7jwXS3yNqqqcPpxKYFAQF110kdbh1EpJSeG2225jzpw5pG1dR9eBV57zsRu//RSAv/71r16/OtuV+vTpA0DO8TwY2rnefdnH8gDvL6wvWrSIPXv2EJwSSUCc7+7xGnlRO8qOFzLh/fe54YYbarePcJf4+HimTp3KAw88QP7hfHQWG8ZIz1/UspZC+QE9FouFSZMm+V1R3V35HOpPlFu/fr3LJ8oJ4e18rbDu3B+ttKBc40hcp+SX1+ItM5svpO7FL5vNdt7ipRB1ST4Xwn28cTXW+RgMBqKioygt9J98brPaKCuuoGdX38jndUlhSzSVO3O68H91f2YqK/1rglVLFRcXN/i5v7Hb7UycOBFFUbh4eNO3C26MHkM6sWPVfj7//HN++9vfEh0d7ZZxmio3N5eZM2diCDIR2jVG63AaFNk/gfITRUyYMIEZM2a4pc5jsVh44IEHGD9+PFWZCpYk37gmdSZrEVgLdVx++eV06+bazgst0ezC+jPPPMP48ePrtQNWVbX2h8D5eVOTvM1mY+TIkWzatIl58+YxZMiQ5oZ4Xnq9nocffphXXnkF057FbhlDKxGRkdxxxx3Neq5Op2P48OHMmDGD/ONpRCd3d3F02ik6dYyKonxGjBiByeRde1s//PDDzJ07l58WfnXOwnp2ehrH921n6NChra79dIcOHQgODib72Nkr1nOO5aHT6bz6e1JZWcl7772HzqAj6qJ2WofTIoZAI+G925C//SSffPIJTz31lNvHbN++PRMnTuQPf/gD5furCe5vRe/BXRDs1VCeagC7wptvvFk70cNfuCufg2cmygnh7Zy/W75yQSMyMhKz2UxxfqnWobhMyS+vxVcKgXUvhFVVVbWarX9Ey0g+F8K9Tp06pXUITZYQn8DefXux2+3odF7bsLLRSgvLQXVMvvY1DRVGhTgXd+Z00TqUl/86qao17CPeFAUFBQ1+7m++//570tLS6DYohYhY93SOMhj1XDK8F2u+/pFJkybxwgsvuGWcppoyZQoVFRXEXNoencE7z3/MUUEEtY9g165drF27liuvvNIt49x3331MmzaN0hPFmBOsKL61kw4AlemO/8M//elPGkdSX7O+lVOmTOHtt9/muuuu409/+hN33XUXf/jDHxg+fDjr1q1j6tSp3H777fz5z39u8rH/8Y9/MH/+fG699Vby8/P5/PPP693/u9/9rjkhN+i2227jyJEjZGX5z37iiqJwxx13tOgC3IgRI5gxYwaZe7f6VWH91L6tgOP1eZv27dtzxRVXsGbNGk5nHCUmMeWsx+xatQCA3//+954OT3M6nY7evXuzadMmqiqqMQc4JkbY7XZyMvLo2LGjV190/uqrr8jOzia8TxsMQd41qaM5wnrEUXzwNJ999hn33XcfMTHun/3Xu3dvXn31VZ599lnK9+kJHmBD8cC5kapC+QEd9ir429/+6rYTHa24M597aqKcEN4uOzsbgJMnT2ocSeMoikJycjJHfj5c7wKeLyvIdqxESE5O1jiSxqk7CaOiosKrz3GEd5B8LoT7ZWZm1n5eUlJCSIj3dyFLTk5m9+7dlOSXERbt/fFeiK/l87p8ZYKl0J47c7poPeoW1ktL/WfCtCvk5uY2+Lk/qaqqYsL7E34pfLtntbpTt4Ep7FpzgNmzZ3P//fdrnqNPnjzJ119/jTHUQkgn71hBfy6R/RMoO17AhAkTGDZsmFsmQQYFBfHwww/z7rvvUnlCR0AH39qapqZAwVqo44orrvC6hW7NKqxPnjyZDh06sHjx4tr/8A4dOjBy5EhGjhzJPffcw3XXXcdvfvObJh97586dACxYsIAFCxacdb8rC+sGg4F//etfLjuev+jWrRvdu3fnwMF9VJWVYA7y/Tdg1uoqstN207ZtWy655BKtw2nQ7bffzpo1a9i/YTkx9z5e7z67zcrBzauIi4tj8ODBGkWorT59+rBp0yZyjueT+Evrt4KsYmqqrF73h7Wu8vJypkydgt5sILyX77Wsa4jOoCOiXwKnN6Tz8ccf89xzz3lk3Jtuuont27cza9YsKo7oCOzs/pOBqgwFa4GOK6+80qv2cXEVd+ZzT06UE8JbVVRUkJ+fDzgK7L7S1jslJYUDBw5QUlBGaKR7t/zwhPysQgA6duyobSCNdGZhXYgLkXwuhPvVnSCXmZnpE1tDOfNe3qlCvyis558qBHwnn9cl+Vw0ljtzumg96u4dXlZWpmEk3icnJ6f2c+ckeH8zY8YMsk5lMeDanoREBLl1LJ1ex5Bb+7Ho43W8++67TJgwwa3jXcgHH3yA1Wolrn8Sis67FwmYwgMI6RTNoUOH+P7777n11lvdMs59993HjBkzyDuRiznBjs5H1vypKlQedeTBJ598UuNoztasaRAHDhzghhtuqDeLwmq11n5+xRVXcPPNN/PWW281+dhr1qxBVdVzfgjPuPvuu1Ht9tpV3r4u68AObNVV3HXXXV7bAm3o0KFYLBYOb99w1n0n0vZQUVrMtdde6xMX5N3BWTzPPna69rbs47n17vNGCxYsoLiomNDusehNPthv5RxCOkZhCDbx3XffeXRPomeeeYYuXbtQnamjJs+9J0jWUqhK1xMbG8Orr77qF6s2z+TOfF53otzvf//7sz6EaA3qvmm32Wzk5eVpGE3jOds85xzP1zgS18g5nk9ERETt/vHerm5hvbq6WsNIhK+QfC6E+9W9+O4rF+K7d3d0IPSXfJ593HEe5SvbUdS9himt4EVjuTOni9ajbjFdCuu/stvtZGdnE9W2PeA7+bwpcnNzmTJlCgEhFgZc45l82b5nW9p2imP16tVs2bLFI2M2JC0tjQULFmCKCiSoQ4RmcTRFZL8EFL2OiRMnUlNT45YxAgICeOKJJ1BtUPmzd9bFGlKTrWArVRgxYoRXTmht9ncyPDy89vOgoKCzLhR27dqVvXv3Njswoa2bb76ZwMBATu7Zgmr3rRYRZ1JVlZO7N2MwGJq997wnWCwWBg4cSO6Jnykrqv/G91jqNgAuvfRSLULzCr17O1rXZB/79W+N83NvLqzPmjULRa8jtKv726V7kqJTCOseS0VFRYPdRdzFbDbz+tjXMRoNVKTpsbvnnAPVDhUH9Kgq/Pe/r9XLef7GXflcJsoJcfaebYWFhdoE0kS1OTf99AUe6f3KSyooziulT58+PjNBqu4barkQLxpL8rkQ7lNZWVlv0lNRUZGG0TRer169UBSF7HTfb3WrqirZx/KIi4sjNjZW63AapW4xVPK5aAq55i5aqm4reCms/yo/P5+amhqiEztiNAf41fbAThMnTqS8vJxBN/bBZDF6ZExFURh6+wBQ4I033sBms3lk3DNNmDABVVWJGtDWZ977G4JMhHaLITMzk6+//tpt49x+++2OhWpZOqwlF3681lQrVP6sx2w289RTT2kdToOaVVhv27YtJ06cqP26Y8eOZ81GSU1NJSjIva0mhPsEBQUxYsQIKosLOH10n9bhtEjhiaOU5p7iuuuuIzrau/fWuOiiiwA4eXBPvdtPHtyDoigMGDBAi7C8QkREBO3atSPneH7tRbyc43kEBgZqvn/LueTm5nLo0CEC4kMwBHjmZMaTgpMjAdi8ebNHx+3cuTNPPvkU9mqoOOyemXaVx3TYyhR++9vf+vVeopLPhXCvM9t+1r3A4c169eqFxWLhxCHfn8F/8pfX4K1bATWk7oV4rS5KCN8i+VwI9/LVfB4cHEz37t3JSs/FWm298BO8WEFOMeXFFT6Vz+vmcMnnorEkpwtXqJu3ZCuKXzk7yoVERBMcGc3p074/kbyugwcP8t133xGVEE63QSkeHTu6bQTdB3YkLS2NefPmeXRsgB07drB27VoC4kMISAj1+PgtEdE7Hp1Jz0cffeS2c0y9Xs/zzz0PQMUhx0Iyb1aZrsNeDY899pjXdh5sVkVi6NCh9Qopt912Gzt27ODxxx/n+++/5/nnn2fx4sUMGzbMZYEKz/vd736Hoiik/7Tap1cjpP+0GoAHHnhA40guzLnyOvPIr5MZVLudrJ8P0LFjR4KDfX+f05bo2bMnFaWVlBVVYK22kp9VRPfu3b22Pf6uXbsAsMT55/+bIdCEMdTM9h3bPT72Aw88QJ8+fajJ0VGT69pZiNYSqMrQ0a5dO/7617+69NjeRvK5EO5l/6Xrj8Foqve1tzOZTFx00UXkZRZSVuTbF2IyDjpWIQwePFjjSJrHl8/B3cFXVh54muRzIdzLWRR15nNfKpIOGTIEm9VG5pGcCz/Yi2UcOAX4Vj6vm8Mln4vGkpwuXKFulwx3tZf2Rc4OcoFhEQSFRZKfn+83f59VVeWtt95CVVWG3jZAk61wB93UB6PZyIT3J3h0EqKqqowfPx6ASB9are6ktxgI7xlHQUEBn3/+udvGufjiixkxYgS2EoXqTO/9HllLoCpTR4cO7fnDH/6gdTjn1KzfsN///vd07NiRY8eOAY49b/v168eUKVMYMWIE48aNo3379rz55psuDVZ4Vvv27bnhhhsoyT5BzuE9F36CF8rPOEz+8UMMHTqUXr16aR3OBXXv3t3Rqu3ntNrbCrJPUF1R7jP7iLlTt27dAMg9kU/eqUJUu1q7b5w3cp686vxob/Uz6Yx6bFbPX1jS6/W8+uqrGI1GKg7rUV20AENVoSJNDyqMHj2awMBA1xzYS0k+F8K9nG/onBfgtXhz21xXXnklAD+nnjj/A72YzWYnPfUkbdq0oUuXLlqH0yy+dlHA3fzlwperST4Xwr2c+duX8/nRPb6bzwF+3nMCnU7HZZddpnUozSL5XDSW5HThCnU7YElh/VelpaUAmAOCMAUEYrPZ/Garjg0bNrB582ba90igXZc2msQQGBpA/6u7k5ebx7Rp0zw27qZNm9i2bRtBSeFYYnxzcVtYjzj0FiPTpk2juLjYbeM8/fTThIaGUvmzHnvlhR/vaar91+vyL730MiaTSeuQzqlZ7wauvPJKFi9eTPv27QFHe6nNmzfz9ddfM2bMGL744gv27NlTe7/wXU8++SRGo5HD677HWuWFv23nYbPWkLZ6Hoqi8Le//U3rcBolKCiIxMRETh8/XHvb6eNHgF+Lyq2Z86J43qki8rOK6t3mjZxFWXuVb7fdOxdVVbFVWQkICNBk/JSUFB5//HHsVVCR7pqLW1UnFGylCnfddZdPtRlsLsnnQriX0ejYBkS1/7LSzeA7E62uvvpqAI7sOq5xJM2XeTibyvIqrr32Wp+6oF23YONLxRuhHcnnQriXM3/7Yj7v06cP0dHR/LznBHabb3TOOVN5cQWZR3MYMGAAUVFRWofTaHU763lrlz3hfdyZ00tLS3n55Ze54YYbiIyMRFEUPv30Uxe/AuEN6nZWqVtkb+2c3wudwYBeb6h3my+z2Wy88847KIrCkFv7axpL3yu7ERQWwPTp08nNzXX7eKqqMmnSJAAi+iW4fTx30Rn1hPeOo7S0lJkzZ7ptnKioKJ577jlUG5Sn6byuJXxVhuO6/N133+311+VddqXGaDRy99138+yzz3LvvffKXi9+IjExkccee4zKkkIOrp7rU6tEDv+wiLL8HO6//36fKkp36tSJ8uJCyosLAcg9kV57e2vXuXNnAPKzfy2sd+zYUcuQzqtXr17o9XpK0wu0DsUtqvPKsZZW07+/didtDz30EMkpyVRn6rCVtexY9mqoOq4nIiKcv//9764J0AdJPhfCdZyFdSdfuhAfGxvLRRddxMnD2ZQUtPAPrEYO/vQzADfccIPGkTSNXIgXriD5XAjX8eV8rtPpGD58OBWllWQcPKV1OM2Sti0dVLjxxhu1DqVJJJ8LV3FVTs/NzWX06NHs37+fvn37ujhK4U3qbkHmK9uReYLzb7HdasVms9a7zZctXryYQ4cO0W1gCpFtwjSNxWgycMkNvamoqGDy5MluH2/r1q3s2LGDoKRwzJG+3XU0tGsM+gAjM2bMoKzMfddgbrnlFi6//HKsBTqqs71nAYKtzHFdPjY2hqefflrrcC6oWYX1lJQUJkyYcN7H/O9//yMlJaVZQQnv8vDDD9O/f3+yDuwgY8d6rcNplMy9WzmxcyOdO3fmqaee0jqcJnHOOi3MPglAQVYGAB06dNAqJK8RFxeH2Wyi6HQJRbklAF698iYyMpKrr76a6vxyyk+5r42LVgr3ZgNw9913axaD0WjkmX8+AypUHG3ZXLHKYzpUKzzxxJOEhWl7Iuopks+FcK8zVxv72urj2267DdRfC9S+pLqyhqN7TtC+fXv69OmjdThNUrdg4w8XeoT7ST4Xwr3O/Fvsa3+bR4wYAcD+LUc1jqTpVFXlwE9HMZlMDB8+XOtwmkQ60IjmcGdOj4+P59SpUxw7dkxayfu5usX0uqvXWztnZ9HqynJqKivQ6XRYLBaNo2oZq9XKBx98gN6g4+Lh3rENbrdLUgiLCeGbb74hMzPTrWN99tlnAIT3iXfrOJ6gM+gJ6x5LSUkJ8+bNc9s4iqLw0ksvERQUROURPXYv2A1BVaH8oB7VDq+8MoqQkBCtQ7qgZp3ZpaenU1hYeN7HFBYW1u4HI3yb0WjkrbfeIi4ujkPrF5F1YIfWIZ1X7tH9HFj5LWFhYbz33nuatalurnbt2gFQeNoxm7zodBYGg4HY2Fgtw/IKOp2OhIS2lOSXUZxXRmhoqNcXQB9++GF0Oh2nN6Rjq/b99kJOpen5lP6cT+/evRk8eLCmsVx22WUMGjQIa74Oa0nzjmGvgupTOpKTk7nzzjtdG6AXk3wuhHud2X7c1y6qXn/99QQGBbJ/8xGfW+mQtvVnrNVW7rzzTp9qA38mX/uZEdqQfC6Ee52ZR3wtr3Tv3p1u3bqRnnqS8uIKrcNpkqz0XPJPFXHNNdd4/Xv/M9X9OZF8LhrLnTndbDbTpo02ey8Lz6pbTPe193HuFBoaCkBlWQmVZSWEhIT4XE4/04IFC8jIyKDHkE6ERHhHh/4zDXcAAQAASURBVCqdXsclw3tjtVqZMmWK28Y5ceIEa9euxRIbjCXaO157S4V2iUHR6/jiiy/c2jm6TZs2/Otf/0K1Qvkh7VvCV51QsJUojBgxgssvv1zbYBrJbWd2RUVFmM1mdx1eeFhsbCwffPABoaGh7Fs6y2uL67lH97Pn+8+xmM1MnDiRpKQkrUNqsri4OABKC3Jr/42Ojva5WfHu0qZNGypKKynOLfGJNwQ9e/bk//7v/7CWVpOzPh3V7jvbKZxLdWEFuRuPYwkI4PXXX9f8IoGiKDz++OMAVB1vXixVGTpQ4dFHH/Wp1o6eIPlciObz9QvxQUFB3HrLrZQUlHF8v++0j1VVldSNhzAajdx+++1ah9Nkdd9Ay4Ww+nztd8ibSD4XwnV87W+Roijcc8892O129m0+onU4TbJ3wyEARo4cqXEkTSf5/Nx8aZtJb+TJnF5VVUVxcXG9D+H96u4b7g97iLuKc4JWZVkplaUlPjdh60w1NTVMnjwZg1HPgGt6ah1OPZ36JxERG8rcuXM5dco91xIWLlyIqqqEdo1xy/G1oLcYCOoQwbFjx9izZ49bx7rjjjsYPHgw1jwdNae1O7e1VUBVup7IyEj+9a9/aRZHUzW6erBu3bp6X6enp591GzhmRGVkZDBz5ky6dOnS8giF1+jSpQuTP/qIRx97jL1LZ2GtqqRd3yFah1Ur68AO9i37GovZzAcffEC/fv20DqlZoqKiACgvLqj9N7Gz7K/uFBPjSJbWGhvR0dEaR9M4jzzyCNu3b2fTpk2c3nSMmEvb+9zFGKea0ipOLTuErdrKmHGvec3klYsvvphevXqRujcVe5UdXRPeY6o2qM7WERcX53P78DaH5HMhPMcfLhrec889zJo1iz0/pNGhZ1utw2mUzCM55J8q4uabbyYyMlLrcJqs7goTad1Ynz/8TrmK5HMhRFPcfPPNvP322+zdeJj+1/RAr/f+FdTlxRUc3nmcjh07MmDAAK3DaTLJ5+cmhb76vDmnjx07llGjRnlkLOE61dXVDX7e2gUHBwNQXVFGdUUZwW1847ryuXz//fecOHGC3pd3ISjMuzr26nQ6Lrq+Fys+38jUqVN58cUXXT7G4sWL0Rl0BCWFu/zYWgpJiaT0SB6LFi1y67Z2iqLwyiuvcNttt1F5pApDhBWd0W3DNUhVoSJNh2qH//znPz412aXRhfUrr7yythCkKArTp09n+vTpDT5WVVUUReH11193TZTCa/Ts2ZPpn37Ko48+xsHVc6kqLSLl0uGaFglVVeX49nUcXr+IkNBQPvzgA/r27atZPC0VEREBQEVxITVVldRUVdbeJqj3vfCV74vBYOC9997jkUceYc+ePegMOqIGJvpccd1aVs2pZYewllfz7LPPctNNN2kdUi1FUfjNb35Damoq1VkKlvaNv/Bek6ugWh0z9YxGD59BaEDyuRCec+bqJF+8qNqlSxcuueQSfvrpJwpyiomIDdU6pAvave4gAPfff7/GkTRPTU1N7edy4Vmci+RzITznzHzui6uPAwMDueOOO/j888/5f/buOzyKem3j+HdLeg8Q0kNC7713EBABQcSGiu1Vjx574SjnYEU5CvZesKDiQcEKonSQ3nsJEEJCSO91s2XeP9YEIgmk7O7sJs/nurgIW2aeDMneM/Nrpw+epU0P5+ggfSmHt57EYrZw8803u9y1M0ieX8qFx0Y4d6Y//fTTPPbYY5X/LigoICoqyiH7FvVXVlZW+bXB4AQLKDuJipkeTOUGTMZyl57NqWK0uk6vo+eoTmqXU602PaPZ9cdBfvjhB+666y7Cw8Nttu20tDQSEhLwiQ5E69a4Zvn1CvNH665jy5Ytdt9XREQE//znP3n99dcpO63Fu51jz3GNGRpMeVqGDx/OmDFjHLrvhqp1w/ozzzyDRqNBURReeOEFhg8fzogRIy56nU5nHbY/cuRIOnbsaMtahZNo27Yt33zzNf/4xz9I3LmOssI8Oo6Zhlbn+OmTFYuF+A2/cnb/FkJDQ/noo4+Ii4tzeB22VDGyqjg/p3LUuqs0IDtCxXo4f//a2Xl7e/P+++9z1113EX80HsWi0HxAtMvcIDAWGkhdGY+x0MD999/PLbfconZJFxk3bhwvv/wy5ZlleMbUvvGqPMP6fzB58mR7leZUJM+FcJy/jw5w1dEC06dPZ+fOnRzaFM/QqX3ULueSCnOLSTyUQpcuXejatava5dTLhTeb5cZzVa7YmGUvkudCOM7f89tVGyluuukmvvnmGw5tinf6hnWz2cKRLSfx8/NjwoQJapdTLxc2pkueVyXHoypnznQPDw+XbnxsqkpKSiq/Li4uVrES51I5+5VGA3/9zrmqirXVuw5th2+gt9rlVEur1dJnXFdWf72Fjz76yKazX+zcuROwNkI3NhqtBs+Wvpw+fZrMzMzK2Xvt5ZZbbuHXX3/lxIkTuIdZ0PvZdXeVFBOUJejw8PBg1qxZLtNGUqHWLaHPPfdc5dcbNmzgjjvuYMaMGfaoSbiAiIgIvvrqKx566CH27t2LobiAbhNvRe/huGlHzMZyDq34lqyEI7Rv34H333+PkJAQh+3fXry8vPD19aMoN4vCnEzg/Lrr4vy0PX//2hUEBgayYMEC7rnnHo4ePYpittBiUCs0WucOjvKCMlL/iMdUXM5DDz3E3XffrXZJ1fLx8WHo0KGsXr0acwnoanFeaTGBKVdLly5diIyMtH+RTkDyXAjHKSoqAsDLN4DSovzKf7uaESNGEBoayvGdp+l/VXfcPZ13do/DW06gKArTp09Xu5R6kxvxNXPFWR/sRfJcCMf5e567aiNFdHQ0Q4YM4c8//yTrXC7Nw523A//pA8kUF5QyY8YMvL2ds8HgcmSN45rJ8ahKMl3YWn5+PgBanZ6CggKVq3EepaWlALh5eOLm7lGlA4IrKS0t5d1330XvpqP3Fc61tvrftekZzZ41h/npp5+YMWMGrVu3tsl2T5w4AYBHM9c8R7gcj2Y+lCTnc/LkSbs3rLu5uTFr1izuuOMOSk/o8O1pxhFt3GVJWizlcPcDd9t0NgNHqdeiSuvWrZOAFwQGBvLJJ58wZswYcpNPsXvJRxiKCx2yb2NZCXt/XEBWwhEGDRrEl19+0Sga1SuEh4eRn5lGfmYaAKGhoSpX5Dy8vM533nDFC+zAwEA+/fRTunXrRuHJbDI2JqA48egrQ24pqSuOYyou54knnnDaRvUKo0ePBqzTu9eGKUcDyvn3NTWS50LYV2amtYNci+jWVf7tavR6Pddffz3lZUZO7Dmjdjk1MpvMHN2eQGBgIOPGjVO7nHqTNVlrJiPWqyd5LoR9NZY8B7jxxhsBOLzlpMqVXNqhLdYb5jfccIPKldSf5HnNXHmUqL1JpgtbyMnJAY0G76AWZOfkqF2O08j561h4+QXg5R9Ibm6uyhXVzxdffEFmZiY9RnbE29+51lb/O61Wy8CJPbBYLMybN89mn/9nzljvS7j5e9pke86m4vuq+D7trU+fPowfPx5zoQZjpv1b1c2lUJ6iJSIigttvv93u+7OHejWsV9i7dy8zZ87k6quv5oorrqh8/MyZM3z33XeVH1ai8fLw8GD+/PncdNNNFGWmsuf7DyktsG8oGYoL2bPkY/LPJTJp0iTeffddfHx87LpPR4uOjsZQUsS5E4cAiImJUbki53Fhw7qnp2uGp7+/Px9//DF9+/alKDGXtHUJKGbnu1FsyC4h9ffjmMtMzJ49m9tuu03tki5r6NChaLVajDm1izdjtvVkYfjw4fYsy+lJngthH0lJSQC06tqnyr9d0TXXXINOp+PwXze6ndHpQymUFpZxzTXX4O7urnY59XbhzXdpSK5KjselSZ4LYR8VNzUr8txRNzntYfDgwYSFhRK/KxGjwTlnRcnLKODcyQwGDhxIdLRzT1l/KReOypaG9aokzy9PMl00RHp6Oh4+/nj6B1JSXExhoWMGwjm7tDTrADa/4BD8glqQlZXlcjOEnT17lk8XfIpvoDc9R7nGMk/RHcOJ7hDG5s2bWbt2rU22mZ+fDxrQejSu9dUr6LysE41XzD7hCA8//DBubm6Undah2Dmmy05rUSzw6KOPuuxyI/VuWJ85cyZ9+vRh/vz5LFu2jHXr1lU+VzH94ldffWWTIoVz02q1PP3009x///2U5GWxZ8lHdmtcLy8pYu/SjynKSuWWW25hzpw5uLk573Sk9dWqVSsATuzeVOXfgioftq76wQvWacvff/99Bg8eTElyHmnrTmExOc/FpSGrmNQ/4lGMFubMmcP111+vdkm1EhAQQPfu3TEXaLBcZnY5RbFOAx8aGkqbNm0cU6ATkjwXwn6OHz+ORqOhwwDrrBjHjh1TuaL6a968OSNGjCArJZesc87Zs//YjlMATJ06VeVKGubCm81yI17UluS5EPYTHx8PQGSH7vgGNef48eMqV1R/Op2Oa66ZitFg5NSBZLXLqdaxnQkAXHvttSpX0jAXjsqThmRRF/bM9HfffZc5c+bw2WefAdZ1mufMmcOcOXMc2oAj7MdkMnHu3Dk8/YPw9A8GrI2x4vxxCGwRRkBIGIqikJKSonJVtacoCi+++CLlhnIGTe6Fm4drtIloNBqGXNMbnV7Hyy+/bJMl8srKytDqdS63LndtafXWZtuysjKH7TMiIoJbbrkFSxkYztnvuJoKwZippXv37owdO9Zu+7G3ejWsf/7558yfP5+JEydy4MABnn766SrPt2rVin79+vHLL7/YpEjh/DQaDffddx+PPPIIZQW57F36ic2nhTeWlbL3h08pzsngjjvuYObMmWi1DZp0wWnFxsYCUJSTibe3d6Oa5r6hGkvDOlhH3L/99tsMGzaMkrP5pK076RQj1w3ZxaSuPIFisjB37lyuvvpqtUuqkyFDhoACptxLnwSYC0ExWl/fWE/ELkfyXAj7sVgsHDp0mODwaAJbhuPfvCWHDh1y6akvJ0+eDMDxnadVruRiJQWlJB9Lo3v37i7fIfHCm+9yI17UhuS5EPZ14MABtFodITFtCY3rQGpqKllZWWqXVW8V13fOmOeKRSF+VyJ+fn6MGDFC7XIaRKaCr5krnw/bm70zff78+cyePZsPPvgAgB9++IHZs2cze/Zsl50WW1SVnJyM2WzGO6g5PkHWtZlPn3a+z3s1JCYmAhAUFkVwaBTgWrPQLFu2jC1bthDTKZzW3aPULqdOAkP86T2mMxkZGbz55psN3p67uzuK2dJo80QxW78vR8/Ed9ddd+Hr50t5kg7lMgPW6qvstLU979FHH3Xp+/H1apV8//336dixI0uXLqVLly7V/gd36NCBEyecd6pIYR933XUX999/P6X52ez/+XPMxnKbbNdiNnHg14UUZaVy8803u/wv3uVcOPV7TExMo/5e6+rCzxtXb1gH6/fzxhtvMGLECEpTCkjfoO6a64bc0spG9VdeeYWrrrpKtVrqa/DgwcDlG9Yrnq94fVMkeS6E/Zw+fZqiokLC23QGIKxNZ7Kysjh37pzKldXfkCFDCAgI4NS+JBSLc13AntqfhKIoTJw4Ue1SGuzCqWMv/FrI8aiJ5LkQ9mM0Gjl85AjNo+Nw9/QivE0nAPbv369yZfUXERFB7969OXcyg5KCUrXLqSItMZOivBLGjh3r8tf70rBeswuPR2NtFKkve2d6YmIiiqJU+8fVO6cKq5MnTwLg26wlPs1aVnmsqUtISECr1RHUMoLgiJjKx1xBdnY2r7zyX9w93Rg2ra9LthX0HNWRZmGBLF68mD179jRoW76+vigWxSkGqNmDxWC97vX19XXofgMCArjrzruwGMGQYvufMVOedfbYIUOG0Lt3b5tv35Hq1bB+5MgRxowZg16vr/E1LVu2JCMjo96FCdf1j3/8g+uvv57CjBSOrPzOJifJx9f9TF5KAldeeSUzZ850yfCoi8jIyMqvo6JcqweavV14ce3K66deyN3dnfnz5zNw4ECKk/LI2JSoysWlsaCMtJXxWMrNzJkzhyuvvNLhNdhCx44dCQwMxJSj5VKH0ZijRavV0r9/f8cV52Qkz4Wwn4MHDwLWBnWg8kZ8xeOuyM3NjdGjR1OUV0LaGecaqXdyXxJarbbKGpSu6sJ1/qQhuaqSkhK1S3BKkudC2M+pU6cwlJVV6SgHcOjQITXLarCxY8eiKAoJTjYd/Ml9SQAuey16Icnzml2Y5waDQcVKnI9kumioiuVKfJuH49s8tMpjTZmiKBw/fpygsEj07h60iIoDXOfYzJs3j/z8AgZM6I5fkI/a5dSLTq9jxI39QQPPP/98g9a3DwsLA8BUZJtBnc7G+Nf3FR4e7vB9T58+naCgIAxndZddZrWuShN1ADzwwAO23bAK6tWwrtfrKS+/9A/tuXPnHN6jQjgHjUbDU089Rb9+/cg4cZDkvZsatL3UI7s5d2gHnTt35sUXX2y0079fKDg4uPLrli1bqliJ82lMU8FfyMPDg7feeouePXtSlJBD9i7Hrn9kKjWSuuoEplIj//nPf5g0aZJD929LWq2WgQMHYjGApYYBGIoJzIUaunXrhp+fn2MLdCKS58JWzp07x80338zChQvVLsVpVFygt2zV9q+/2wHn12l1VWPGjAEg8ZDzrNNXWlRG2uksevXqRfPmzdUup8EuvMFwuc/opkbW/qye5LmwlfLycu69916ef/55tUtxGseOHQMuzHPr365yI74mFR3RTjtRniuKQuKhFAICA+jTp4/a5TTYhZ/LkudVXZjneXl56hXihCTTRUMdPXoUAL+QcNw8vfH0D+LIkaMqV6W+5ORkCgsLK6/LA1qE4enj5xId5fbs2cPy5ctpGdOMzoPaql1Og7SMbkbXIe1ISEhg0aJF9d5OxWy/5XmOW4PckYz51u8rOjra4fv29vbmrrvuQjFBuQ1HrZvywJyvYcSIEXTu3Nlm21VLvVoou3btytq1a2ucyqikpITVq1e7/HB+UX9ubm68+uqrNG/enFObVlCYUb9pT0vysjm+7if8/P15/fXX8fT0tHGlzunCEfnNmjVTsRLnc+HPQGP7efDy8uKdd96hdevW5B9OJ+9wukP2azGaSVt9AmOhgfvvv5/rr7/eIfu1p0GDBgFgyqn+BMCYpwHl/OuaKslzYSv79u3jwIEDzJs3T+1SnEbFlHLNIloB0Dwytsrjrqpfv354e3tz+lCK2qVUOnPkHIqiMHLkSLVLsYmysrJqvxZVb8TL1LHnSZ4LWzl79ixbtmxhyZIlapfiNCpyuyLHPX388Atu4fJ5HhISQpcuXUg5mUF5Wf1HjNlS9rk8CnOLGT5s+CVH67qKC0diS55XdWFjunSaq0oyXTSEoigcOnQIL/9g3Dy9AfALiSArK5O0tDSVq1PX7t27AYho1xWw3nsPb9eFM2fOkJXlXLOxXUhRFF5//XUAhkztg0br+rP49r2yG54+Hnz00UcUFhbWaxsdO3YEwJBVbMvSnEZZVhGenp6qLdFx3XXXERAQQHmKDsVGq9mUJVmbou+55x7bbFBl9WpYv/POO4mPj+cf//jHRVP2FBQUcPvtt5OWlsbdd99tkyKFa2rWrBkvvfQSFouZI6u+x1LHNaUUxcLRVUswG8t5ZvZsVaa+cAYBAQFql+BUGuuI9QoBAQF8+OGHhISEkL0zmeLkPLvuT1EUMv48jSG7hGnTpvGPf/zDrvtzlIEDBwJgrGGd9Yr11Zt6w7rkubAVGYVzsaSkJHyDmuPu6QWAl18AHt6+JCc715SrdeXu7s7AgQPJyyggP6tI7XIAa8M6wPDhw1WuxDZKS0ur/VpUnTpWjs15kufCVi7Mc1kT2qoit4NCzy/XFtgygtTU1AZNYeoMhg0bhsVs4Wy8czS2VOT5sGHDVK7ENiTPa3ZhnssyL1VJpouGOHPmDDk5OfiHnR/lGhBmHdm7d+9etcpyClu3bgUgulOvyscqvt62bZsqNdXGrl272L9/P3HdomgZ3TgG33l6u9NzVEcKCwv57rvv6rWNLl26oNPpKE2vX8O8M7OUmynPLaVr1664ubmpUoO3tze33HILFiOUpzW8M4epyLq2+oABA+jatasNKlRfvRvWb7zxRhYsWECLFi1YsGABYB3BEhERwZIlS7jtttuYNm2aTYsVrmfQoEFMmzaNosxznN2/uU7vTTu6l7yUBMaMGdMo1teqq9tuuw1/f3+6dOmidilOpTGPWK8QGhrKe++9h6eXFxkbT2PIsd+FZs6eFIqT8hg4cCCzZs2qMluCK2vZsiWtW7fGnK9FsVz8vClXg5+fX5P//ZI8F7YiN8SqMplMpKamERhyvlOgRqMhICSM5ORklx9pO2TIEACSj9VvRiJbqmgQiIqKqpwOztUVFZ3vsFBc3Dh74NfXhWvUynq150meC1uRxq6LpaSk4O7phZdfYOVjgSHhWCwWUlNT1SvMBoYOHQpA0lH18xysdeh0uspO0q7uwjy/8GsheX4pkumiITZvtt57D45qXflYcFQbADZtathSra7MZDKxefNmAlqEERx+vtNBXPf+AGzcuFGt0i5r6dKlAPQY2VHlSmyr86C2uLnrWfrD0nrdH/Hy8qJ79+4YsooxGxpXjpScKwDF+rmvphtvvBFPT08MZ3U09BaWIdnaDH3HHXfYoDLnUO/FqhctWsRHH31EbGwsKSkpKIrCrl27iI6O5oMPPuCzzz6zZZ3ChT3yyCMEBgZyevsayktqdzFhKjdwctMKvLy9eeqpp+xcoXN64okn2Lx5Mx06dFC7FKfS2EesV+jQoQOvvvIKislC2tqTmO0wPV9hQjZ5B9No1aoV8+fPV60XnL0MHDgQxQzmgqqPW8rAUqqhf//+6HQ6dYpzIpLnwhZkCseqzp07h9lsIrBlRJXHg0KjKC0tJTMzU6XKbKOiYT3puPoNCulJ2RhKyxk8eLDapdhMQUFBtV+LqjffZTRtVZLnwhYuzHPJduvsXmfOnCGwZWSVDsgVo9cTExNVqsw2OnXqRGBgIMnH01Tv9GcoLSftTBbdunXD399f1VpsRfK8ZtKwfmmS6aK+fv/9dzQaDc1izzfC+rYIw9MvkHXr1l00C0JTsXv3bgoKCmjTa3DV5VcjWhEYEs6ff25yyllojEYj69evI6C5Hy1jGsdo9Qrunm7EdoskOSmZ+Pj4em1j2LBhoEDJ2cZ1zlry1+y1as/gExgYyOTJk7GUgSm7/gPxLOVgzNTStm3bRtN5EhrQsA5w9913s3//foqKijh79iwFBQUcPnyYe++9t97bLCoq4tlnn+XKK68kODgYjUbDF1980ZAyhcoCAgJ44IEHMBnKSNy5rlbvSd77J+Ulhdx1552EhITYuULhSi5sTG+sI9YrjBw5kgcffBBTUTlp6xNQLNUMva4nQ3YJmZvP4OvryzvvvNNobl5cqH9/a89TY17VqKuYHr7ieWGfPBdNS05OTuXXMi08nDhxAji/HmuF5n+tt37y5ElHl2RToaGhxMXFkXIiHbNJ3cbN5GPWxv3G1LCem5tb+fWFv1tCGikuR/JcNNSFnzkXfhY1VSkpKZSUlNA8slWVx5s1kjzXarUMHDiQwtxi8jLU/UxNOZGOYlEa1VJdF/4Oye9TVReuqSt5Xj3JdFFXR44cYd++fQS3ao+Hj1/l4xqNhtCOvSgsLGTZsmUqVqieNWvWANCmz9Aqj2s0Gtr2HUZRUSHbt29Xo7RLOnbsGMXFJUR1CG00M4xeKKp9GGCd7r4+Ro8eDUBRYuPJWMVsoTg5j7CwsMp15NU0ffp0AAwp9f/5Kz+nAcW6rcb0c9yghvUKXl5ehIeH4+vr2+BtZWVl8cILL3D06FG6d+9ug+qEM5g6dSqRkZGkHNyOofjSa1+YDGUk79lEUFAQt956q4MqFK7iwg9gd3d3FStxjP/7v/9j7NixlKUVkr07xSbbNJeZSF93CiwKr776Kq1atbLJdp1N79690Wq1mPOqPm7Kt/4MqT2ljjOyZZ6LpuXCG/HSEAj79+8HIDSu6qwzoXHtqzzvygYNGoTRYCItMUvVOpKPp6LX6+nbt6+qddhSdnZ2tV8LSEpKqvZrUZXkuaivCzNcPn/gwIEDQHV53qHK866somNa0jF1Z6FJaoQd5S78HcrMUvd8ydlcmOHJyckqVuL8JNNFbSiKwltvvQVAdK+LR7lGdh+EVu/Ghx9+RFlZmaPLU5XFYmH16jV4+QUQ1aHbRc+36zscgFWrVjm6tMuq6MDXIjJY5Urso+L7OnXqVL3e36pVKzp06EBpSn6jmQ6+JCUfS7mZ8ePHO0UjdFxcHP3798eUp8VcWvf3KwqUp+nw9fVlwoQJti9QRTZpWLelsLAwUlNTOXPmDPPmzVO7HGEjbm5u/N///R8Wk5HkfZdeaz3l0A6MhlJuv/12vL29HVShcEWNeSr4ChqNhhdffJHWrVuTfzidwoSGNVgpFoX0jQkYiww8+OCDlevqNUZ+fn60b98ec2HVddbN+VqCgoKIjY2t+c1CiDqRETlVbdu2DZ2bO2FtOlV5PKJdVzRaLdu2bVOpMtupnA7+qHo34suKDWQk5dCrVy98fHxUq8PWsrKy0Hu7ofPUkyU34isZDIYq0wQeOnRIxWqEaJxkxHpVFXkd1bFHlcf9glsQ2DKCHTt2uPw01hUjxNVsWFcUheTjqQQEBtCpU6fLv8FFVCz94x7sRUF+vlNOM6yWgwcPVvu1EKJ+/vjjD7Zs2UKzVu2rrK9ewcPHj+heQ0lLS+X9999XoUL17Nu3j8zMDNr2GYZWp7/o+fA2nfALDmHNmjVO9zmdlpYGgF9w47nWvVDF91XxfdbHxIkTUSwKRacbxwCPwpPWTnnO1Ag9bdo0AMpT696UbMrRYDHApEmT8PLysnVpqrr406QGcXFxdd64RqOpc48TDw8PQkND67wv4fwmTZrE22+/zblD24ntPxqd/uL1nBWLhbP7t+Dt7c11112nQpXClTS2NcFr4u3tzVtvvcUNN9xA1pYzeAR74R5YvzDK3X+O0nMFjBw5krvuusvGlTqfXr16cfToUcxFoPcHi8G6xnqPgT2couefGhyV56JpuXAd1ry8PPUKcQKpqakcPXqU2O79cXOv2gHM08ePiHZd2bdvH9nZ2TRr5rrrpPXu3RsPD3eSjp1j4KQeqtSQfDwVRWlc08aCtWFd6+kGFoXMrEy1y3EaBw4cwGg04h5qoTxNW+8pAxsLyXNhDxdOydzU11g3mUxs2LAB38BmhES3uej5uB4D2PPHUnbv3u3SS0y1aNGCDh06cPLUCYzlJtzca32b0GZyMwoozClm/Pjx6HQ6h+/fXrKzs0GjwT3Qi/KcUrKzs+V+J9bPlhMnTqAPtGAp07Br9y4sFgtardON/XIYyXTREKmpqbz44ovo3NxpP3Jyja9r1XckGfEH+OKLLxg6dGijmvHrUlasWAFAx4Gjqn1eo9XSvv8Idq34ju3bt1d2IHcGRUVFAHh4Nc4ZW93c9ej02irLg9TVhAkTeP2N1yk8kUVAB9deTthUaqTkbD6dOnWiXbt2apdTafTo0QQEBFCYno9nrIW63E4vT7O++Nprr7VTdeqp9RlzYmIiOp0Ovd7xJ9m1YTAYMBgMlf+WNXqcj7u7O1OnTuXTTz8l89RhQtv3uOg1OcknKSvI5brrrsPPz+/ijQhxgaZ04RUTE8NLL73EI488Qvr6BCImdEDrVrebDiUp+eTuTyUyKpI5c+Y0iePXpUsXAMyFGvT+CuZCa6B363bx9E9NhbPnuXBNFzamN/Ub8b/99hsA7fuNqPb59v1HcPbYfv7444/K9apckaenJ/369efPP/+kKK8E30DHzzJ05sg5AIYNu3i6Q1dVXl5OcXExXgH+oCgUpBZgNpsbVUNDfe3btw8At2YKpkKFAwcOYDKZmmyeSZ4Le7gwz5t6R7nt27eTk5NDr7FT0VRz3dS+/0j2/LGU3377zaUb1gGGDh3KsWPHSDmRTqvOEQ7ff9Jfed7YZlPLyclB56lH52UdEJCbmysN61iXRFIUBX2AgtkDCtILSExMrFfjcmMhmS7qq7y8nCeeeIKCggI6jpmGV0DNHbd1bu50vvJGdn/3AU88+SRLvv+eFi1aOLBaxzMajfzxxx/4BAYT1alnja/rOHA0u1Z8x/Lly52qYd1isU6/2Zjv32o0msrvsz6aN2/OiOEjWLt2LYbsEjyaue7sx4WnslEsCtdcc43apVTh5ubGxIkT+eabbzDlaHBrptTqfRYjGLO1dOjQgfbt29u5Sser82/liBEjWLhwIQUFBZSWll72j6PMnTuXgICAyj9RUVEO27eovSlTpgCQfnxftc+nH9tX5XVCiPNGjx7NjBkzKM8rJXtn3dYhM5UaydiUiJu7G6+/9jr+/v52qtK5dO7cGQBzkbVB3fTX341pisH6ctY8F67HYrHImqx/URSFn376Cb2bO+37j6j2NR0Hjkar0/Hjjz+iKLW7IHFWFQ3aFQ3cjmQxW0g6lkpYWCht2lw8ktBVVTRk6Tz1aD2sN1ebemeVCseOHQNA56eg91MoKyuTddaRPBe2dWGGN+U8B/jxxx8B6Dz0ymqfj2zXlYCQMH7//XdKSkocWZrNVeb5UcfnOUDikRS0Wm2jWl8drJmu9dCh+yvPZXkFq+PHjwOg8wO9n1LlsaZOMl3U1auvvsqBAwcI69SbsE59Lvt6/9Ao2gybQE52No8//rjTTX1ua1u3biU3N5eOA69Aq625o3Jo644EhUayevVqp8r0iplazSazypXYh6IomM2WBs9IWzEauuCE6872pigKhfFZeHh4ONU08BUmT7bOhlGeXvvh6sYMDSjn39vY1Lph/ciRIzz88MPs27ePG2+8kfDwcB599FGnWQvn6aefJj8/v/JPcnLdGp2EY8TExNChQwdyzsRjKjdUec5iMZOZcISwsHC6du2qUoVCOLdHHnmEDh06UBCfRfGZ2l2YK4pC5uZEzKVGHn/scTp27GjnKp1HdHQ07u7ulQ3rlmLr4840pY6jOXueC9eTlZWFyWSq7B1/7pw6N2Wdwfbt20lMTKTDwFF4ePtW+xpv/yDa9raODHP137vhw4cDcPrQWYfvOy0xi7JiA8OHj2hUS3tUNqx76CtvxDf1UaMVkpOT0ehA4w5aL+uN+LNnHf+z5ywkz4U9nDt3Dk//YEBDaqp6a26rLTMzk9Wr1xAS04bQuA7Vvkaj1dJtxERKSkpYtmyZgyu0ra5duxIYGEjioRSHd/orKyknNSGT7t27Exwc7NB925PFYiG/IL9KnktHOauK+7VabwWtV9XHmirJdFEfv/32G4sXL8a3RTjtR11T62uiyO6DaNm+B3v37uWdd96xc5Xq+uWXXwDoPHTcJV+n0WjoPPRKysrKWL16tSNKq5WK2XwNpeUqV2IfpnITikXB17f6eye1NXjwYFqGhlKUkIPFRTshlKUXYSwoY9y4cU45i3OHDh2Ii4vDlK1FMdXuPeUZWrRaLVdeWX0nVVdX64b1Dh06MH/+fM6ePcvSpUsZOHAg7733Hj169KBPnz588MEHqp4kenh44O/vX+WPcE4jRozAYjaTe7bqWkAFacmYDKWMGDG8Ud0gFcKW3NzceOWVV3B3dydrWxJmw+XTrCghh5Kz+QwePNilpx2uD51OR+vWrbGUalAUMJdo8A/wp3nz5mqXphpnz3Phek6ePAlA8zhrp52mvNbfN998A0DPsZdeP6rn2KkAfP3113avyZ7CwsLo0KEDKSfSMRocO9qhojF/xIgRDt2vvVWMZtN66tF5SsP6hc6dO4fGQ0GjAa2n9bGm3PAneS5sLTs7m9zcXHybh+IVEMTJkyddfmaV+lq8eDFms4meY6de8t5Et5ET0end+Oabbxo0janadDodw4cPpzi/hMyzjh1VfeZICopFqeys11gUFhZiMVusM9B4yoj1C1V0wtV6gNbT+hnTlPMcJNNF3aWmpvLCCy+gd/eg64Rb0OlrP+JXo9HQ8Ypr8QkO4fPPP2f79u12rFQ9hYWFrFu3juZRcYTEtL3s6zsPGQvAr7/+au/Saq3i3mVRnvOMoreliu+roUsS6HQ6rp06FUu5maLTrpm1BfHW0fbXXXedypVUT6PRMHHiRBQLGLMv325nLgNzgYaBAwc22nvwdZ4KXqfTMWXKFH755ReSk5N5+eWXKS4u5p///Cfh4eHccsstMiWfuKQBAwYAkHc2ocrjucmnqjwvhKheXFwc9913H6ZSIzl7Ui75WrPBRPbOs3h6efHss882yU4r0dHRKGZQysFSpqFVTCu1S3IKkufCVvbv3w9AcFQbvIOac/DgQZe+uVxfCQkJbNiwgYh2XQmrYXRbhaiOPQiJacvKlStJSbn057izGzVqFGaTmaRjjrshqigKiYdS8PHxoV+/fg7bryNU3HTXeZxvWL9wqYWmqri4mPz8/Mob8BV/u/rvjy1IngtbOXDgAAD+LSPxC4kkOzu7Sc5CU1payv8WL8bbL4BOg8de8rU+AcF0HHQFCQkJbNq0yUEV2seoUaMAOH3QsSOHE//qKFex/8aiIrt1Hm6S53+TkpKC1gM0WmvjesVjQjJd1F7Fz0a7EZPxDqx5XfWaVKy3rtFqef755zEYDJd/k4tZu3Yt5eXldBo8plb3QgNahBHZvhs7duwgKyvLARVeXmxsLAC56QUqV2IfOX99X61atWrwtq65xjprQ+FJ5/i/qwuzwUTxmTzi4uLo3r272uXUqGLkeXnG5X+fjJmaKu9pjOrcsH6hli1b8q9//YujR4+yatUqgoOD+fbbb9m3b5+NyhONUZcuXdDr9eSnnqnyeH6a9eSwZ8+eapQlhEu57bbbaN26NQXxmZTnl9X4uryDaZjLjPzz/vsJCwtzYIXOIzIyEgBTgQYs5/8tzpM8Fw2xc+dONBoNAeGtCIyIo6CgoEmuk/j555+jKAr9Jl1+ZhCNRkO/SdMxm818+eWXDqjOfs7fiHfclNy5afnkZxUydOjQBq/H5mwq1jTWebmh87R+b3Ij/vxMGLq/poytmDq2YsYMYSV5Lhpix44dAARGxhEUGVflsabkhx9+ID8vj57jrsXN3eOyr+838SYAPv30U3uXZlcDBw7Ew8PDocu7mIxmko6m0qpVq8rGg8aismHdSy95foHS0lLOnTuH1svaCVejs3aWkzy/mGS6qMnWrVtZv349QVFtCO3Yq97b8QuJIKrnUJKTk1m0aJENK3QOFVO6t+8/stbvaT9gJBaLhbVr19qrrDpp164dOp2OtNOuu3b4pVR8X506dWrwtkJDQxk0aBBl6UWXvE/ujIoSclDMFqZOvfRMSWqLioqiY8eOmPMuPx28MVOLTqdj5Mja//65mgY1rIP1Zup9993HtGnTSElJITw8XBotxCV5eHjQtm1bijJTUS4Y0VaUcY7w8HCCgoJUrE4I1+Dm5sZDDz0ECuTur34kianUSMGxDFq2bMlNN93k4AqdR0WHAlO+9eQkPDxczXKcluS5qA+DwcC+ffvwDYnAzdOLoMjWQNO7EZ+WlsayZctoHhlLm56DavWeDv1HEBgSzg8//ODSN1rbtWtHeHg4Z46cw2x2zEwFCX+Nphs9erRD9udIBQXWXvs6Dz1aWZO10qFDhwDQ+f01Yt3NeiP+4MGDTXaq6ppInov62rFjBzo3dwJCowiKapoN60ajkS++/BI3Dy96XWZZlwrNI2Np03sIe/fuZe/evXau0H68vLwYPHgwOan55GcWOmSfZ4+nYiw3Nco8r8hurYcenYeuymNN2dGjR7FYLOguWE5X56uQmZlJenq6eoU5Kcl08XeKovDee+8B0HbYhAY3wsX2G4WblzefLlhASUnjmW7cYDCwdds2WkTFEdQyotbva9tnKAAbN260V2l14u3tTY8ePUhPyqakoFTtcmyqYhY6Ly8vm43SnjJlCoDLjVovPJmFTqdj4sSJapdyWaNGjbJOB59b82ePxQDmQg39+vUjICDAgdU5Vr0a1rOysnjjjTfo1q0bAwYM4LPPPmP06NEsX76cM2fO0KtX/XtLAbz77rvMmTOHzz77DLCubTFnzhzmzJkjJ6KNRLt27TCbjJTmW28kG0uLMRQX0K5dO5UrE8J1jBw5ko4dO1J0OgdTSflFzxfGZ2IxWbj77rvx8Lj8aIvGKiQkBLCGOjR87Z7GxN55Lhq/Q4cOYTQaCYqwjjKquBG/e/duNctyuEWLFmEymeg38SY02tqdXmt1evpOuBGDwcDixYvtXKH9aDQaRo8ejaG0nHMnHXNDNOHAWdzc3BgyZIhD9udIFQ3rWncdWnfrjfjCQsc0cDizis8Unf/5RnRdgEJeXh6nT59WqyynIXkuGqqgoIATJ07gHxqNVqfHOygEd29fdu/eo3ZpDrVq1SrSUlPpNmoiXr7+tX5f/79mq3H1WWgqGrgTHDQdfMJfs900xob1iuyWPK+qMs8DLsjzv7J9z56m9XlTE8l0cSm7d+9m//79tGjTBb8WDR80ovfwJKrnUAry81m6dKkNKnQOe/bswVBWRmyPui0369+sJc0jY9m5cydGo9FO1dXNVVddhWJROLo94fIvdiHnTmaQn1XI2LFjcXd3t8k2R44ciZ+fH0WnclAsrtH52pBbiiG7hGHDhtGsWd2XdXC0ihHol1pn3ZhjfW748OEOqUkttW5Yt1gsLFu2jKlTpxIREcHjjz8OwGuvvUZKSgpLlixh/PjxaGt5M/FS5s+fz+zZs/nggw8A61Rcs2fPZvbs2ZXrDgrX1qZNGwCKczIAKPrr74rHhRCXp9FouP7660GBwpPZVZ5TFIXCk9l4eXsxadIklSp0Ds2bNwfAXFz1302VI/NcNH4VN8cCw60N6x4+/nj5B7N3794ms866wWDghx9+qFxntS46D70STx8/vvv+e6e5cK+P8zfi7T99bEFOEVkpuQwYMABfX9/Lv8HFlJZaRyJo9Vq0euvncGMaPVIfiqKwa9dOtB6g9Tz/uP6vm/K7du1SqTJ1SZ4LW9q3bx+KohAY3gqwXmcEhMWQmnqOtLQ0dYtzoG+//RaNRkOfK6+r0/si2nUlrE0n1q1bR2pqqp2qs7/hw4ej0+kckucWs4XEwymEhITQuXNnu+/P0SqyW6vXodFq0ei0TT7PwToCG85nOIA+UKnyXFMkmS5qq6IDV0xv2zVYRXYbgM7Nna+++gqT6TLzO7uIihl3Yjr3rvN7Yzr3pqSkpHLGLLVdddVV+Pv7s3/DMcrLXPeewYUURWHnHwcBbDrDqoeHB1dddRWmknJKU11jXfqK0fWTJ09WuZLaadeuHcHNgq3TwdfQd8H012j2wYMHO7Ayx6t1IkdGRjJ58mTWr1/PXXfdxY4dOzhw4ACPPPKIzRspEhMTURSl2j+tWrWy6b6EOirWzyrJtTaol/zVsN7Y1tUSwt7Gjx+Pu7s7RYlVpxE2ZJdgLDQwdsxYvL29VarOOQQHB1u/MGuq/ruJcmSei8bvzz//RKPREBh5Pr+DoluTl5fnNBei9rZ69Wry8/PpNnIiOn3d1vt29/Si87ArycrM5M8//7RThfbXo0cPgoKCSDx41u49w0834tFtcP5GvMZNh9ZNV+WxpurEiRPk5OSiC7Rw4WyXFTfit23bplJl6pI8F7ZUMeVpUFTryseCottUea6xO3nyJPv27SO2xwACWoTV+f09r5iCxWLhp59+sn1xDhIQEECfPn1IT8yiON++U86mns6krNjAqFGjGmVjYWXDupu28u+mnufl5eXs2bMHrY+C9oJTZp0vaPRNN89BMl3UTmJiIhs2bCAgvBUBYdE2266bpzdhnfuSmprKmjVrbLZdNW3duhWtTk9k+251fm90l96V23AGvr6+3HnnnZQVG9ix4oDa5djEyb1nOHcqg5EjR9q8c9356eCzL/1CJ6BYFIoScggMDGTYsGFql1MrGo2GgQMGYjGApZpTRUUBU76W0NCWxMTEOL5AB9LX9oVpaWm4ubnRvXt3EhMTeeaZZy77Ho1Gw/LlyxtUoGicKjpIFOdkAlCSm1nlcSFE7fj4+NC3b182b96MqcSI3tt6hVqaYl02Y8SIESpW5xyCgoKq/DswMFCdQpyE5LmwlYSEBPbt20dwdFvcPM934Alp05Vzh3by448/0q1b3S9kXc2PP/4IQNfhE+r1/m4jJrJ7xff89NNPjBo1ypalOYxOp2PkyJH88MMPZCRn0zLGfjcATx88i1arbbT5Vjl1rJuOii7gTX3q2IpGPbfgqp02dF6g9VLYunUr5eXlNptC0FVIngtbKS0tZcWKFXj4+FWOWAdo0bozJ9b/wo8//sh1113X4HVcnV1Fg3i3EfVb37J9/5Gs/vJNfvrpJ+69916XbSweNWoU27dvJ/HwWToPamu3/TT2jnJFRUUAlZ3kNG66yseaqu3bt2MwGPAIqZrnGg3oAy0kJydz+vTpJjngRjJd1MY333yDoihE9xpq821H9RjM2f1b+Oqrrxg3bpzNt+9IGRkZHD58mJgufXDz8Lz8G/4mulMvtDo969ev5/7777dDhXV366238tNPP3Hwz3had48iLC5E7ZLqraSglE0/7sHT05OZM2fafPudO3emdevWJCSexmwwofOodfOnw5Wk5GMuNTLx2om4udVtkIaaevXqxfLlyzEVaNB5V810Sxko5dCzZ69Gf+1Qp58so9HIhg0bav36xn7wRP1FRESg1+vJT0si9egeclOsayM2xRNoIRpq0KBBbN68mdK0QvzirKOxS9MK0Wg09OvXT+Xq1Ofl5YVer6+c0qqpN6yD5LmwjQULFgAQ2X1glceDY9riHdicn3/5hXvvvZfQ0FA1ynOIpKQktm/fTlSnngS2rN8ady2i4giN68jGjRtJS0tz2eM1evRofvjhBxIOnLVbw3ppURmpCZn06NHDJdYfq4/8/Hw0f00DrygKGq2G/Px8tctSjaIo/Prrr2i0FzesA7g1VyhKLmLDhg2MGTNGhQrVJXkubOH777+noKCA2AFXoLmgMdjTN4DmrTtz6NAhtm/fzoABdVun1JUYDAZ+/uUXvP0Dad1z4OXfUA03D086DBjNgXW/smXLFoYMGWLjKh1j1KhRzJ07l4SD9mtYVxSF0wfP4u/vT+/edZ+m1xXk5eUBoP3rhr7OXVf5WFO1bNkyANyaX7xclFsLBWOW9TUPPvigo0tzCpLp4lIKCwv5+eef8fQPokVcJ5tv3zuwGc1jO7J//34OHTpEly5dbL4PR/n1118Ba4e3+vDw8iauxwCO7t7EiRMnaNvWfp3Masvd3Z05c+Zw220zWPXVFq5/YjyePh5ql1VnFouF1d9spbSojFmzZhEZGWnzfWg0Gq655hrmz59PUUIOAR2dtxNCQbx1GvhrrrlG5Urqpnv37gCYCzQQWvUa3VxgzaYePXo4uiyHq3XD+unTp+1Zh2hi9Ho9bdu25ejRoxz5YzEAYWFh+Pn5qVyZEK6nYkSoIbsYv7hgFEXBkFVCXFwc/v7+KlenPo1Gg5+fH7m5uQBN/nNG8lzYwqlTp1i2bBm+zcNoHtexynMajZZW/UZxZOV3fPjhhzz33HPqFOkAFZ0Leo2d2qDt9Bo7ld8+fIkvvviCp556yhalOVz//v3x9vbm9KGzDJzUwy77OHPkHIqiNNrRbQBZ2VnoPK2XaBqNBq2nnuxs55/Gzl527txJQkICbiEWNNVcubqHWjAka/n222+bXMO65LmwheLiYj799FP0Hp5E9bh4HcTYfqPJPHmIt956i/79+zfahpylS5eSl5vLwCm31XlZlwv1GjuVA+t+5dNPP2Xw4MEuebxCQ0Pp3LkzR48dxVBajoeX7WcDyT6XR2FuMZMmTXKpEVp1UZHdOq+/Gta93CjJzqekpKRJLtWWlZXFqlUr0Xor6Kq5HHdrpqBxgyVLl3DPPffg4eF6DUYNIZkuLuenn36itLSUNr1HVukEZ0tRPQeTlXCERYsW8fLLL9tlH/ZmMBhYtGgR7l7edBxY/9nguo+axMndm/jiiy946aWXbFhh/XXv3p0HHniQt956i1VfbWbCPSNcbnacnb8f5Gx8GiNHjuTGG2+0236uvvpq3nrrLQqOZ+LfoYVTno+ZisspOZtH165dadeundrl1ElcXBxu7m6Yi8oves5cZD3WnTrZvgOQs6l1w3pjnxNfON68efPYs2dP5b9duTecEGpq3749Wq0WQ7Z1zTZToQGL0WzzdWpcWUXDupu7W5O7SP87yXNhC2+88QYWi4XWg69Eo7n4Yi60Q0/O7N7Ajz/9xIwZM4iLi1OhSvs6duwYP/30E82j4mjXp2HrYXUcdAVbf/qSxYsXc/3117vk8fLw8GDIkCGsXLmSnLR8gkMDbL6PhIPJAC47Zf7lWCwWMjMy0Qefn7JQ7+1Genq6dfS6E94QsLfPP/8cAI+Ii0e3Aei8QR9sYefOnS4/uqauJM+FLXz22Wfk5ubSetCVVZZ1qeAXEk7Ldt05dGg/K1eudPnpYauTn5/PBx9+iIeXD72vnNagbYXEtKFN7yHs3r2JNWvWcMUVV9ioSscaPXo0hw8f5syRc7Tr3crm2084kFy5n8YqIyMDrV5bORW87q8l29LT05vkTI3ffPMNRqMJr1gL1Z3OaHTWznI5yTn8+uuvTJvWsN9FVyOZLi7FbDbz7bffotO7Ed6lr932ExTZGp9mLfn999957LHHaN7cfst72ctXX31FRkYGAybfioe3b723E9d9ACExbfn111+55ZZb6Nix4+Xf5AB33nkn+/fvZ/369Wxfvp+Bk3qqXVKtndqfxO5Vh4mKjmLOnDl2vbYNCgpi7NixLF++nLK0QrzCnG/QWf7xDFDghhtuULuUOnNzc6NN6zYcO34URaFKrpuLrX+7WmeB+nCtbi2iUYmJieGaa66p/OMMU6sI4Yq8vLyIjIzEmF8GQHme9e82bdqoWZZT8fHxsf7t7aNyJUK4vk2bNrFhwwaCotrQrFX7al+j0WppM+QqLGYzr7zyCopy8RTOrsxoNPLMM89gsVgYPeOhBo8a0On1jLzlAUwmE88++yxms9lGlTpWRQNCRQO4LRkNRpKPpdG+fXuioqJsvn1nkJ2djclkQu9zfnSg3scDg8HQJKePPXjwIJs2bUIfaEF/iXshnlHWRvcPPvjAQZUJ0TicPXuWL774Ek/fAKJ6XjxavULrwePQ6nS89tprlJaWOrBCx3jllVfIy81l0LV34O0f2ODtjbz5n+jd3Hnp5ZdddimPigbv03bIc7CeJ3h4eDBo0CC7bN8ZpKamovNxr2w4cPsr29PS0tQsSxW5ubksWrQIrTu4t6z5msAj0oJGC59++ilGo9GBFQrh3NatW0dycjKhHXtV2wnOVjQaDVE9BmM0Glm0aJHd9mMviYmJfPDhh/gEBtN/0s0N2pZGq2XUrQ+iKArPPvus03wmabVa5s6dS1xcHHvXHiV+d6LaJdVK1rlc1i7ahre3N2+/9bZDZle9+Wbrz0De4XS776uuLEYzhcezCA4O5sorr1S7nHpp06YNisW6pvqFLCVawsLCmsTsPNKwLoQQjUBcXBzmUiNmg4ny/NLKx4RVRaA3hWAXwp6Kiop44YUX0Gi1tBs+6ZK9jJu1ak/z2I5s2bKFn3/+2YFV2t8nn3zC0aNH6TZyEjGdbbM2aJteg+k4aAz79u3jyy+/tMk2HW3o0KG4ubmRsN/2N+LPHDmH2WRu1KPbzp49C4De94KG9b++rniuqTCbzZVTUHq2qn60egV9IOgDLWzcuJGNGzc6oDohXJ/FYuG5556jvNxAm2ET0LnVPN23V0AzonsPJzU1lbffftuBVdrfmjVr+PXXXwlv07nBo9UrBIVGMnjaXWRlZjrN9LF1FRcXR1xcHGeOpmIsN9l023kZBeSk5jN48GC8vLxsum1nUVJSQk5ODm5V8tw6a1pTy3OAt956i5KSEjyizVQz0VUlrTu4h1tISUnhiy++cFh9Qjgzi8XChx9+iEajIbpXw2ZJq43Qjr3w8PFj0aJFLtWx12g08tRTT1FuMDD2zifwsMGgmuhOPek2ciJHjx51qg68vr6+vP322/j5+bF+8XYykpx72bDSojJ+X7ARk9HMf//7X4cNAuvatSu9e/em5Gw+htwSh+yztgriMzEbTEyfPt1lZ1WtaHOwlJy/J6iYwWJoOu0R0rAuhBCNQMXUYcZCA8YCQ5XHBJU3bfT6Wq+AIoT4G0VReP7550lNTaVVv1H4Ng+95Os1Gg3tR01B7+HJy3PnkpCQ4KBK7evEiRN8/PEnBLQIY+QtD9h021fc/gi+Qc147733OHPmjE237Qi+vr4MHjyYrJRc8rOKbLrtU3811o8dO9am23UmSUlJALj5nb+4rvjaFX8eGuKdd97h0KFDuLW0oK/FqgJebayj3P4z+z9NcjSgEHX12WefsX37dlq07kxI226XfX2rfqPwCW7J119/zYYNGxxQof0VFBTw4osvonf34Kr7ZqHV6my27b4TbiCiXRdWrFjB+vXrbbZdRxozZgymchNJx1Jtut2mkOfJydbvUe93fmmXijyvyPqmYsWKFSxduhSdr4J7+OVnsPKMsaD1gPfee6/K0pFCNFU//PADx48fJ7Rjb7yD7D81u07vRkzfURQXF/POO+/YfX+28s4773D48GG6jphA2z5Dbbbdkbc8SGDLCD799FN27Nhhs+02VExMDPPmzcNiVvj98z8pKXTOGYXMZgsrv9xMQU4xDzzwACNHjnTo/u+55x4Acveec+h+L8ViNJN3KB1fX1+7rjNvb9HR0QBYLvjRM//1dVNpj5CGdSGEaAQqpsU1FhgwFRnQaDRERESoXJXzqGhYb4rr0wphK1988QW///47gRGxtOpXuzWuPf0C6TjmOkpLSnj44YcpKCiwc5X298orr2A2mxh71xN4eNl2FgwvX3+uuP0xysvLmT9/vk237ShjxowB4NQ+2zUEGw0mzhw9R2xsLK1bt7bZdp1NReO5W8AFN+L/+ropNaz/8MMPLFiwAK03eLe59Gj1Cjof8GxtJjcnl/vuu4/CwkI7VymE69q0aRPvvPOONaOvuLZW58c6vRtdrpqOTu/Gv/71r0bRWe7jjz8mOzubwdfeQXBYtE23rdXquPKep9Hp9bz66qtOM4VsXdgjz8G6xqqbmxvDhw+36XadSWJiIgDuARd0lPsrzyueawr27t3Lv//9bzR68O5ornZt9b/T6MG7gwmzxcxDDz3UpI6XEH+XkpLCa6+9ht7Dk9aDxzlsvxFd++PbPJTvvvuOLVu2OGy/9bVp0yY+//xzgsOiGT3jYZtu28PLm0kPPodWp+NfTz1FdrbzjA4fPHgwjz76KEV5JfzxxSbMJudbTm7rL3tJOZnOmDFjuPvuux2+/4EDB9KrVy+Kk/Ioy7Rtx//6yj+SjrnUyO23305AQC16kDupisZzc+n5cLf89XVFo3tjJw3rQgjRCISHhwNgKjZgKiqnRYsWuLm5qVyV86iYWqexrfMshKNs2LCBN954A0+/QLpcdXOdRnWFtOlCTN+RJCYm8uSTT2Iy2XZKUUfav38/27dvJ67HAGK79bPLPtr2GUpUp56sX7+e48eP22Uf9jRy5Ejc3Nw4uc92I7ISj6RgKjcxfvx4m23TGZ0+fRoAN//zDevuf92Ir3iusdu+fTvPP/88Wnfw6WJCU4eJZjzCFTwiLJw8eZInnngCi6V2jfJCNCUJCQk88cQTaLQ6uk6agZtX7adK9W0eSscx11FcbB115KrrhwPk5+fzv8WL8W8eSp/x19tlH83Co+lxxTUkJyfz+++/22Uf9tSuXTtiY2NJPHwOo8E252656flkpeQydOhQfH19bbJNZ1Rdnus89Og89U0mz5OTk3nggQcwmYx4dzKjq0NfVH0geLUzk5+fz/33398oOuYKUVfFxcU89PDDFBUV0W7EZDx87L8mdQWtTkensdej1emZOXOmU8+0kZ6ezqxZs9C7uXP1Q8/j7mn7JUbC4jow7MZ/kJWZyaxZs5zqGuO2227jqquuIjUhk80/O9csH8d3nebAxuO0adOGOXPmqDLQSaPR8OijjwKQtT1Z9XvCpuJy8g6m0axZM2655RZVa2moigF+F66xXjF6vak0rMucuEIIl9O2bVun6iXoDMLCwgAwFZVjKi4nLC5M5Yqci1Zr7UcmI9aFqLvExERmzvwXWr0b3SbdhoePX5230XrQWIqz09myZQtvvfUWjz/+uB0qtb8lS5YA0G/idLvtQ6PR0G/CjSQf2cvSpUuZNWuW3fZlD35+fgwdOpS1a9eSk5ZPcGjDe2Gf2JMIwLhxjhupoYYTJ05U3nivoPNyQ+uu4+TJkypW5hglJSU8PetpFCz4dDahq8d9Mc/WFsxlsGXLFr799ltuvvlm2xcqhIsqKiriwQcfpLi4mC5XTcc/pO6zW7Vs352inHQSt69h5syZfPDBB5Xn2a5k2bJlGMrKGHzddej09uuM3PeqG9jzxxK+//57Jk2aZLf92INGo2H8+PG8//77JB4+S9terRq8zRN7rKPfG3tHuYrMdg+sGmRuAZ4kn03GYDC47JqqtaEoCs8++ywFBQV4tTfjFlT3hgyPUAVLqYXkpGTmz5/PCy+8YIdKhT0kJCTw4EMPkZmZqXYpdtOvb1/efPNNuy01WFxczD//+U/ijx8nsvsgwjr2sst+LsUvJIL2o6ZwdNUS/u///o/PPvuMyMhIh9dxKQaDgccee4zc3FzG3Pk4ITH2W7u7z/jrSTqyly1bNvPee+/x4IMP2m1fdaHRaHjuuec4lXCKQ5uO0yIymI791Z/hLSM5hw3f7cDPz4+3334bb2/bzvRXFz169GDChAksX76cwhNZ+LdroVotWTuTsZgsPPbYY/j41L5zqzPy9vamWbNgckvOt89UjFivaHRv7FzvCkgI0eR9++23rFixQu0ynEqLFtYTg/K8UhSLQkhIiMoVOZeKBnW1eycK4WrMZjNPPf00JSXFdBwzDb+Q8HptR6PR0vnKG/EJDuGLL75wqvXJaqu8vJzVq9cQ0CKMqI497Lqv2G798AkIZuXKlZjNzjel2+VMmDABgPjdDR+VVVpURtKRVDp37kxcXFyDt+esioqKSEpKwj3Yq0onMI1Gg3uQF6dPn6akpETFCu1v1apVZGZk4h5pQV/PQTkaDXi3t6DRwddff2XbAoVwcfPmzSMpKYlWfUfSsl33em8nbsAVNI/rxJYtW1i0aJENK3ScFStWoNXq6DR4jF3349+8JdGderF3717S0tLsui97uOqqqwDrqLOGUhSF+N2J+Ph4M2LEiAZvz5kdOXIEnacenXfVThsewd5YzBaXnJGoLuLj49m5cyf6YAseofW//vZsZUHro/DLL7+Ql5dnuwKFXX388ccknTlDiVlLscaz0f0pKTOwYcMG1q1bZ5fjl5qaym233c7u3btp2b4H7Yar1ykrvHNf2gwZT2pqKrfcciv79+9XrZa/M5lMPP300xw4cIAuw66kx+jJdt2fRqNhwj/+TVBoJB9//HFlh3tn4OXlxVtvvkVAYAAbl+wk9bS6nVpKCkr5/bONmE0W5s2b5xSNrI899hi+vr5k7zqLqaRclRqKk3IpTsylZ8+eTJw4UZUabC0yMgqLQUPFrXZLGU1qaVoZsS6EcDmNuXd3ffn7++Pu7o4h23rTvVmzZipX5FykQV2I+lm7di2HDx0itGOvBt2EB9C7e9D5yhvZ8e07vPPOO3z1lWs1em3YsIGiokL6jZxk99kvtDo97fqNYO+qH9i2bRuDBw+26/5sbfjw4fj5+RG/K5F+47s1aDTjyb1nsFgsLjfSr6527dqFoih4hlw8Na5niC9l6UXs2bOHIUOGqFCdYxw7dgwAt+YNm15R6wa6AAtnz6ZQVFTUqKcbFqK2zpw5w48//ohfi3BiBzasMVmj0dJpzDS2LpzPBx9+yLRp0/D09Lz8G51EUlIS+/fvp1XXvnj7B9l9fx0GjebM4d0sX76cu+66y+77s6WYmBi6devGwUMHKc4vxSeg/lPspp3OpCC7iClTprjUz0tdpaamkpycjHdU4EXni54hvuQfzWDHjh1069ZNpQrt73yeN+waXKMBt2YKhiQzJ0+epE+fPrYoT9jZiRMnQOeGsfsE639iI6PJT8ft2Dri4+MZM8a2nbPWrFnDM88+S0F+PpHdB9Fu+CQ0Ks8KE9NnBDp3T+LX/cztt9/Oww8/zK233opOV/ul4WytpKSEWbNmsWbNGqI79WLsXU86ZHZKT18/rn3yFRY9/0+ef/55SktLueWWW5xiZsyIiAhef+117rnnHn7//E+ufWQs/sGOvwYylZtY8dlGivJKeOKJJ5zmPkZISAhPPPEEzz33HJlbzxA6qo1D/9/MZSaytiXh5u5mXfbMBWd7qk5UVBT79+9HMYDG0zpivWXLlk1madrG8b8ohBBNnEajISgoCMVkvRkdHBysckXOSRrYhaib9evXAxDTe7hNtucXEkFwVBv27dvnUiNPzGYzCxYsAKDr8AkO2We3kdb9fPzxx061jltteHh4cNVVV1GUV8LZ+PQGbevo9gT0en3lqLnGqmL9Xe/Ii6fOr3jMFdforYvycuvoAY0N7tNVbMNoNDZ8Y0I0Ahs3bkRRFKJ6DUWrbfgvmZuXD+Gd+lKQn+9UI9hq45NPPgGg20jHdNjq0H8U7p5efP3NNxQXFztkn7Y0ZcoUFItCfANHrR/dngDA5Mn2HVWotj/++AMAn2ry3CvcH41Ww+9//N6or0sNBgNgqzy3HifJc9cREREBZiOa0ny1S7ELTaF1NLAtR2RmZWUxc+ZMHnnkEYqLS+g4ZhrtR05WvVG9QmS3AfSc+n/oPLx57bXXuOOOOzh16pQqtSQkJDBjxgzWrFlDTJc+TH3iv+jd3B22/+CwaG78z9v4BjXj1Vdf5T//+Q9FRUUO2/+l9OvXj3//+9+UFpax/OP1lDl4ZLbFYmHNoq2kn8lmypQpzJgxw6H7v5ypU6cyaNAgSpLzKTyR5bD9KopC5tYzmEqMPPjAg8TGxjps3/ZW8TloKQPFAhaDxumWjLAn5/iEFkII0WBBQedHXAQGBqpXiBOq6InYWHoFCuEoWVnWCw7vINutQ1WxrYptu4J33nmHw4cP03nolTQLj3bIPlu2ake7fsPZs2cPH3/8sUP2aUvXXHMNAEe31f+mS2ZyDlkpuYwYMaJKxjU2586d448//sA90BOP5hevteYZ4oubvye//fabS04lXFtlZWXWL2wxeEDzt20K0cRVrHXrY9M8b15l265gxYoV/PTTT4TEtKVdv2EO2aeHtw/9Jt1MVmYmzzzzjMst8XLllVfi4eHB0e2n6t0YXF5m5NS+JKKioujdu7eNK3Qe5eXlfP3N12j1WnxaXXzeovPQ4x0dyPFjx9m2bZsKFTpGZfba4tJb8tzl3HTTTQDoT20Dc+PqEKEpykafepTg4GaMHTu2wdszm83873//4+qrr2bFihUEhMXQ7+aHCe/c1wbV2lZQVGv63/IILdv3YO/evVx77bW88cYbDluqymg08tlnn3Hddddx/Phxeo27lmkz5+HuWf+ZVOqreWQst774CWFtOvHLL79wzdSpbNq0yeF1VOe6667jjjvuIDe9gN8+3YCx3OSQ/SqKwqYfdnNqfzL9+vXjmWeecYqR/BfSaDS8+OKL+Pv7k70jmfK8Uofst/BkNsVncunbt6/TdTZoqIqGdXOZBou1Tx3h4fVbPtIVyVTwQgjRSPj7n1+QNCDg4h7yQkasN3WKopCcnFw5KlItnp6eREREON2FRnUqb/7asNSK79sVbiwbjUbmz5/PokWLCA6LYvSMhxy6/7F3Pk766Xjee+89SkpKeOihh9DrXeP0vVOnTnTo0IH4Q/GUFJbh7Vf3aV8Pbz0JwLRp02xdntNQFIUXX3wRk8lESLfoaj8XNBoNQd3DyPjzNC+99BJvv/22S3x+1IXJZGLXrl1o9KC1wQzBOh8FI7Bt27bKTh6icUlLS1N9hJBWq6VVq1Yu0XGzcuYTW352aLRVt+3EFEXhf//7H6+88goe3r5M/Odsm4zcr63+k27mzOHdrFy5EpPJxEsvveQyy1T4+flx5ZVX8vPPP3PuVAYRbVrWeRvxuxMxlpu49tprG11+Xej9998nPS2dwC6h6DyqP18L6hZGcWIuL730Et9//z1eXo5vFLK3HTt2AKDzbvi1t+6v/obbt29n5MiRDd6esL+BAwdy00038e2336I/vhFTu2Ggd/1pgTXFObgd34gGhZdffglvb+8Gbe/w4cO88MILHDlyBDcPL9qPuoaIrv3QaJz3nMLNy4cu428itENP4tf/zGeffcby335j1tNPM2rUKLvtd/PmzbzyyiucPn0a36BmTHzoSdr0UneKcb/gFkx/5j22/fIVW3/8kvvuu4+RI0fy5JNPqr6m+COPPEJmZibLli3jjy/+ZPydw9Dp7XvOs/23AxzafIIOHTrw1ltvOe1U4CEhIbzwwgs88sgjpG9IIGJCR7R6+/3OleeVkr09CT9/P1566SVVl1Cwh4pGdMVg/QO2nc3D2bnGnTkhhBCX5efnV/n1hY3s4rzGfCNHXFpBQQH/+c9/WLdundqlADBhwgSeeeaZBl+Q21N6ejqHDx/G0z/IpjefvQKbAbBu3Trat29vs+3a2oEDB3j++eeJj4+nRVQc02bOw9PH7/JvtCFv/yBu+PebLHl1Jp9//jk7duzgmWeeoVOnTg6toz40Gg3XXnstL730Esd2JNBrdN1qLi8zcmLPGcLDwxk4cKCdqlSXoii88cYbbNq0Ce8If3xjax6V7xsXTOGpbNavX88777zDQw85tpOHvX3xxRecO3cO93CLTdr93FsqGM7Au++9y/Dhw2WJnEbEaDTy5ptvsnDhQrVLAaBnz568+uqrhIaGql1KjcrKytiyZQsajQYvG64pXpHn69evZ+LEiU7bwSA5OZmXX36ZTZs24RMQzNTH59I80rHTcOr0eqY+/l9+euPfrF27lmumTuU///43w4YNc4nrk2nTpvHzzz9zeMvJOjesK4rCka0n0el0jXoa+GXLlrFgwQLc/D0I6h5W4+s8gr0J6NySM4fP8K9//YvXX3/dZTpN1sbGjRvZsGEDOn8FnQ0uc/SBClpP+O6775gyZQodOnRo+EaF3c2cOZO8vDxWrFiB29E1GNsPA3fnve69HE1eKm4nt6BRzLw0Z06D1o02mUx88MEHfPrpp1gsFsI69aHNkPG4e7tGZyuA5rEdCIpqzZmd6zmzaz0PP/ww48ePZ/bs2VXuSzbUyZMnmT9/Pps3b0ar1dF73DQGT7vT4dfkNdHp9Qyeegft+41g9Zdvsm7dOv7880+mT5/OPffco9qAJ61WywsvvEBRURHr169n5cLNjJ0x2G6N67tWHmTP6sPExMTwwQcfOH3HwdGjR3PDDTewePFisnedpcUA+8xIaDFZSN+QgMVk4cUXXiQsrOZzA1dVcf1jKdNg8az6WFPQeM7ehBCiibvw5MXZT2TUIiPWm6YTJ07w0EMPc/ZsMhbfZig+zVSsRkFTmMXy5cs5duwYb7/9NtHRjplavLZMJhMrVqzg9ddfp6ioiPajbDvaM7RDT87s2sB7771Hamoq9913n1OdfCcmJvL++++zYsUKAHpcMYUR0+9XZZo5gMCQcG594SPWfv0OB9cv58Ybb2TSpEn84x//UL03/OVMnDiR119/nSNbT9JzZEc02to3HsTvTsRoMHLdddc5bWNNQxiNRl5++WWWLFmCe4AnIUPjLtm4otFoaDkslpTfjvPJJ59QWFjIzJkznXY0QG2ZTCYWLFjAu+++i9YDPFvZZuSr1gM8YsxknM7g9jtu5+233qZVq1Y22bZQT1ZWFk888QS7d+9G8fTFEhCGTadUqSNNWQF79+7l+utv4LXX5tO3r3NN3aooCtu3b+fVV1/lxIkTRHQdgJvXxctN1FdgeAwB4a1YtWoV9913H4899phTdZjLzs7ms88+49tvv8VoNBLXfQDj7p6JX7DtpsOvCw8vb6771zy2/vwV235ayAMPPMCAAQN46KGH6Nq1qyo11Vb37t1p27Ytpw6cqvMsNOlnsslKyWXcuHE0b97cjlWqQ1EUFi5cyOuvv47OQ0/oyDZo3S7deNGsVwTluaWsW7eOf/7zn7z66qsuP+Ocoij8/vvvzJ49G40WvNvaZmYqjRa82pgpPgR33/1/vP76G073WSsuptfrmTt3LgEBAfzvf//D/fBqjO2Govi43tJO2vQT6M/swcPdnVdemcfo0aPrva2ysjIeeeQRNm/ejFdAMB3HXEdQZJwNq3Ucnd6NuIFjCO3QgyOrlrBixQqOHTvGxx9/3OBr+/z8fN577z2+++47zGYzsd36MeLmf9IiyjmPVfPIWG6Y9SbxOzew4dsPWbhwIT//8gsPPfgg1157rSqjlN3c3Jg/fz4PPfQQW7ZssVvj+q6VB9mx4iCRkZF8+umnLpPzTz75JHv37iX+WDxeYX74xtj+syl7ZzLluaXcdNNNDfrccGYtWljPqS3lVE4F37Jl3Wc2clUapZG2MhQUFBAQEEB+fr6M3BRCNAlz585l0aJFACxdupR27dqpXJHz+M9//sPPP/9MTEwMy5YtU7UWyae6acjxUhSFX3/9lRdefBFDWRnmiM6YI7rYdhrU+rBY0CXvR5d2HB9fX1584QXGjBmjakmKonDy5El+//13fv7lF9LT0tDq3Wg9aBxRPYfYfDRVSW4WB3/7hqLMc+h0OkaOHMnEiRMZPHgwnp42mAe6Hk6fPs0nn3zC8uXLraMHWndk1K0PEtHOeW52Jx3dy7qv3iU9MR6dTs/VV0/i7rvvduoG9ueff54lS5Yw4Z4RxHSs3XpbiqLw3fwV5GcUsWrVKpe5QK+tXbt28fLLL3PixAk8mnkTOqoNeh/3Wr3XVFxO6pqTlOeU0K59e/49axa9evWyc8W2Zzab2bhxI++88w4nTpxA6wk+XUyVU77agqJA2WkthmQter2em2++mVtvvdWmF/uS6bXX0GO1a9cuZs6cSWZmJuZm0Zhj+4FO5XECioI24xT6M3vQauDhhx/m9ttvV70zUFpaGn/88Qe//PIL8fHxAER2H0TbYRPQ2viYGctKOPLHd2SdPgpA3759mTRpEqNGjVKtoTArK4uFCxfyv//9j9LSUgJCwhhx03206zfCaUaHZ6ecYd2i90jYuxWAYcOGce+999KtWzeVK6vZ4sWLmTNnDv0ndKf3FZ1r/b4132zl+K7TfPbZZ42uQfT06dP897//ZcuWLei93Qkd3RqPZrULMovRTPrG05Qk5xESEsKTTz7JuHHjnOZntLYURWH//v188MEH1pkx9ODd0YxbsG1vNRvSNJTG60CB8ePHc88999CmTRubbV/yvPbqcqwUReHrr79m3rx5KFodxjaDUAJdZP1dRUGXtA9d2nGCg5vx3nvv0qVLlwZtcs6cOSxevJjmcR3pPO5G9B7qXPfamsVi5tTm30navZFu3brx1Vdf1ftcaM2aNTz//PPk5ubSLCKGkbc8SFz3/jau2H7MJiN7/ljKlh++wFBaTNeuXXnppZeIjXXsTDkVysrKePjhh9myZQutOkcw7vYhNmlcVxSFnb8fZNfKQ0RGRvL555871WCJ2khISOCGG27AaDERcXVH3Hw9bLbtojO5pK87Rfv27fnmm2/w8LDdtp3NgAEDKNMUoQtQKE/RsmTJElU72joyz6VhXQghGok333yTBQsWAPDbb785dUOLo1U0rEdHR7N8+XJVa5F8qpv6Hq+MjAz++9//smrVKtC7Y4zrjxLkXGv9aLPOoE/cCWYTkyZN4sknnyQoyHG9+EtLS9m9ezebNm1i48aNJCcnA6B39yS0Uy9ieg3D04ZTxv6dxWIm/fh+kvduojAjBQAPDw8GDhzIkCFDGDx4MJGRkXbbf4XTp0/zwQcf8Pvvv6MoCiExbRh87Z206W37DgW2oFgsxO/cwOaln5N19jQ6nY4JEyY47Qj2Y8eOcd1119GqcwRX/d/wWr0n9XQmP769ivHjx/Pqq6/auULHsFgsbNq0iUWLFrF582YA/Nu3oFnfqDqv62YxmsnamUxhfBYAQ4YMYfr06QwePFj1Br3LSUpKYsWKFSxdupTU1FQA3MMseMZa0Npp8L0xS0PpKR2WMuvUiKNGjeLqq69m8ODBuLvXrkNDTSTTa6++x6qkpIT33nuPr776CgUNpqhuWELbq99J7gKawizcTm6B8hL69OnDs88+69AZEkwmEwcPHmTTpk38+eefHD1qbeTWaHWEtOlCTJ8R+IXYryFDURRykk6QtHsjOUknANDqdPTq2ZOhQ4cyePBg2rVrZ/dMzc7OZsGCBXz3/fcYysrwCw5hwJRb6TZiAjonXd83+dh+Ni/5jKQjewAYPHgw9913H927d1e5sosVFxczatQodB4abv7PpFrlTVmxgS+f+4mY6Bh++uknpzyvqo8DBw7w7bff8vvvv2MymfCODKDFoBj03nXLFEVRyDuYRu7+VBSzhc6dO3PzzTczbty4BueTvWVnZ7Ny5UqWLl3K8ePHAdAHW/BqbbHJFPDVMRVA6Ukd5kLrz1GfPn2YMmUKo0aNavD005LntVefY7V+/XqeePJJDAYDprj+WJq3sm+RDaVY0J3aji77DK1bt+aDDz5o8DTOJpOJ/v37o/PyZcCMx23e0c0Z7P/lS7ISjtSrYc1isTBv3jy+/vpr9O4eDL3u/+g1bho6F10qozg/h/XfvM/hTX/g4eHBK6+8otqo5bKyMh599FE2bdpEdMcwrrxjGPrLzKxyKYqisP23/exZfYTo6GgWLFjgco3qFX766Sdmz56NZ0tfwse1r9MMezUxFZdz9pcjuGn0fPfdd6p1qnCUiRMnkpx6Bl2ABWOmlnXr1qk6MEIa1m1AToqEEE3NRx99xLvvvgugepA5Gxmx7rrqerwMBgNff/01H330MaWlJVj8QjC17g8eNhwGaUulBehPbUdbnI2fnz///Of9XH/99XaZ3llRFOLj49m0aRNbt25lz549GI1GAPQenjSLaU+LNl1oHtsBnZtjb+YVZqaSceIAmaeOUJydVvl4VHQ0gwYOZNCgQfTv3x8fH9v9P+bl5fHWW2/x448/YjabaRnbnkHX3Oa0Dep/V9HAvuXHL8lMOoVOp+f666/jgQcecLrPlltvvZV9+/dx638m4xd8+f/DVV9v4cTuRD7//HP69OnjgArtw2KxcODAAdauXcvKlStJSbF2IPEM9aN5n0g8mjfs57kss4jsXWcpSy8CICoqijFjxjBq1Ci6du3qNI3sSUlJrF69mpUrV3L48GEANDpwa2nBI9xi01HqNVEsUJ6hofyctvKGvK+vL6NHj2bMmDEMHDiwXo0Ykum1V9djZbFYWL58OW+8+SaZGRkoXv6Y4vqj+Kq5nMslGA3oE3ehzUlG7+bGrbfcwt13323TtUYvdPbsWTZv3syWLVvYvn07xcXFAGh1eoKiWtOidWdC2nSx6dTvtVFWkEv6X3mef+4MYL3d1Kx5cwYOGMCgQYMYNGgQzZrZ7v/RYDDw2Wef8fnnn1tHqLcIo//Vt9B1+HinbVD/u7PHDrDlxy9IPLgTsK7/+eSTTxIR4VydQitGW171f8Np1fnyte1dd5Stv+zlqaee4uabb3ZAhfahKAqnTp1i3bp1rFq1qrLzinugF8G9I/CJCmzQ9o2FBrJ3n6X4TC4oEBQUVJnn/fr1c5plX7Kysli3bh0rV65kx44dWCwW0IBbc2ue6wPtX4OigClHgyFFgynXep7j5u7G0CFDGTNmDMOHD6/X567kee3V91gdPHiQf/zjHxQUFGBqPcB5G9cvaFTv2bMn7777rk1+JkwmE0OGDMGoaOh/y6MutaZ6bVjMZvYs+Yj81DP88ssvdW5MnD9/Pl9++SUtolsz+eEXCA5zruXy6uvEzo0s/2AOZmM57733HoMHD1aljvLych5//HHWr19PVPtQxt85DL173TstKIrC1l/3sW/dUWJjY/n0008JCQmxQ8WOoSgKM2fO5Pfffye4ZzhB3RvWEVVRFM79EU9ZWiEvvPAC11xj22UVnVHFvR59gAVTnpY9e/aoet4iDes2ICdFQoim5osvvuC1114DYMuWLXa7keeKXn/9dT7//HOGDBnCBx98oGotTSmfDAYDzzzzDF999RW5ubl069aNOXPm1Gna89oeL4vFwm+//cZbb71FWloauHliiuyGpUWsU41qq5ZiQZt+En3KITCVExUVxWOPPcbo0aMb3MCrKAp79uxhxYoVrFu3joyMjL+e0eAXEk5wTDuaxbQjICwGrQprf1WnrCCX7DPxZJ+JJzf5FCZDKWBdq69fv36MGTOGMWPGNGiK2W3btvHkzJnk5ebSPCqOYdffQ+teg1yiQf3vFIuF+F0b+XPxJ+SkJtG8eXNef/11evbsqXZplX799VdmzZpF7zGd6X/VpUfhlRaVsfD5n2gVE8uPP/7ocv8nqamp7Ny5kx07dvDnpk3kZGcDoHXT4RMbREDHEDyCbDuUy5BTQv7RDIpP52AxWdcob9a8GcOGDqNv37707dvX4aMIEhISWLlyJatWraqcihoN6IMsuIcouDVT0Kg0AMVUBMYMLcZMLZYy62Pe3t4MHz6csWPHMmTIkFovSdFUMt2ReQ6wc+dO5s2bZ2280uowhXXEEt4RtM6RU5eiyTmLPmkfGkMRAQEB3HfffTbrMHfy5EmWL1/O2rVrSUhIqHzcO6h5ZZ4HRbZ2eOe4mhhLi8k+c4KcpHhyzpzAUFxQ+VzXrl254ooruOqqqxr0+ZSYmMhDDz3E6dOn8Q1sxqBr76Tr8KtcdoRbSvxBNvzvI84e24+npyfPPPMMkyZNUrusSvHx8Vx77bXEdApnwt0jLvlaxaLwzdxfKS8ysXbtWpe7Ns3NzWX37t3s3LmTP//8s3JmJ41Wg3dkAAEdQ/AM9bPpeYqxyEDBsUwKT2ZjLrN2fvXx8WHw4MH069ePvn37Ehsb69Bzo4yMDFatWsWqVavYs2cPFbeQdf4K7iEW3FooaFX6yDGXgjFTQ3mGFkux9Zjo9XoGDBjA2LFj67QkheS5ffL8744fP84dd9xJYVERxvbDUAKcb5Sr7swedGnx9O7dm/fffx9vb9udt3/yySe8/fbbePoH0WH0VJrFNI7lG4uyUjm6+gcK0pIYM2YMr732Wp0+p9LS0rjyyvEEhIRzy/Mf4unrWnlxOedOHmbRc/+kQ4f2LF68WLU6jEYjTz75JGvWrCG6QxhX3lm3keuKorBt+X72rjlCXFwcCxYsaBQDugoKCph67VQyMjKImNARj2b1/53PO5xG9s6z9fo9cFUPPPAAGzZsQOul4KX1Zdu2barWIw3rNtBUToqEEKLCokWLmDt3LgC7d+92+unjHCkrK4vPPvuM66+/3qFTc1anKeXTTTfdxJIlS3jkkUdo27YtX3zxBTt37mTdunUMGTKkVtuozfE6efIkzz33HPv37wetDnPLdpjDO4LexX4HjAZ05w6jSz8JioV+/frx3HPP1Xt67y1btjBv3jxOnjwJgJuXD81bdaBZq/YERbfB3cGj2OrDYjFTmH7W2tB++hgF6WcB65TxN9xwA//85z/rfLPj0KFD3HrrraDRMuzGe+k17lq0LtBYczlmk4ldKxaz6ftP0Wm1/O9//6Nt27ZqlwVYb+KNvmI0RrOBW5+dgk5X80jqvWuPsPXXfTz99NNMnz7dgVXWT0pKCrt27WLXrl3s3LmzclQ6gM7LDZ+oQHyiA/EK80Nzie/bFiwmC6VpBRQn5VGSnI+51Fj5XGRkJH379qVPnz706dOH8HDbTwudlZXFsmXL+OWXXzhxwjodtEYLuiAL7s0V9M0Uu033Xh+KAuZCMGZpMWZpsJRab3x4eXkyevQVTJ48mX79+l1y5H9TyXRH5XlWVhZz585l5cqVAJibt8Ic2dV5Z52picWMNi0efepRMJUTGxvH888/V+8OTydOnGDu3Lns3Gkdzaxzcyc4ui3NWrWnWUw7uy7bYiuKolCck0HOmXiyE4+RezYBxWJBq9Uybtw4nnzySVq0aFGnbebk5DBlyhRyc3PpPf46hl73f7h72mkOagdSFIVj29ay+vPXKS0q4LXXXmPs2LFql1VpxowZ7N2397Kz0CQfT+XXD9cxdepUnn/+eQdWWD85OTns3r27Ms8rcgxA667DOyLAmucR/ujqMcqvLhSLQllmkTXPk/IwFhoqnwsODq7M8759+xIXF2fzG/fFxcWsXLmSn3/++Xxjugb0/taGdLdmClonWxraXGJd+sWYdX5mGp1Ox6BBg5g8eTIjRoy45Bq3kue2zfNL2bdvH3feeRdGBco7jwVP5xm5rc1MQJ+wg7Zt2/Lll1/avEOQoih88sknvPfee1gsFoKiWhPdaxjNWrVDo3GOmabqoiD9LEl7/iQjfj+KojBp0iRmz56Nl5dXnbazZ88ebrvtNnqNncoVtz9qp2rVo1gsfPL4dIzF+WzbulXVWoxGI48//jjr1q0jtmsk424bgraW16i7Vh1ix28HiI2N5bPPPmsUjeoVtm3bxt133417oBeRkzrW67q9PK+Us78eJSgwkJ9+/Mmhyzyq6emnn7bODKtRCAsNr7yOU4s0rNtAUzkpEkKICkuWLKm8aXHgwIEm0TPOFTWVfNqxYwf9+/dn3rx5PPHEE4B1bacuXboQEhLCli1barWdyx2vDRs28Nhjj1FeXo65WTTmqO6udwP+78oK0SftQ5ubgre3N++++y59+/at0yYOHTpU2SjZskNPwjr1ISgiFo2TTA1dX2WFeaTH7yflwDZK83O46qqreOWVV+q0jYqlISY//ALt+4+0U6XqObh+OSs+/i833ngj//73v9Uup9K8efNYuHAh4+4YSutu1XcWURSFRXOXUVZQzrp165xydFtxcTFr165l27Zt7Ny5s3KdcACdhx7PUD+8Qv3wDPXDPdBTtSxWFIXyvFLK0gopTSukLK0Is8FU+Xx4eDh9+vRhwIABjB49ukGjcc6ePcs777zD77//jsViQaO1rrNacfNd4wL9VhQFLMVQnqnFmHm+kT08PJz77ruPSZMmoatmVo+mkOmOyvMTJ05w9913k52djcWvOeaY3ig+Ln5DymhAl3IIXfpJtFoNs2fPZtq0aXXaRF5eHuPHj6eoqIhmMe0I79qfZq3au8wU5zUxlpWSefIgZw9sozAjhbi4uDqvwf3111/zyiuv0P/qmxl+4z/sWK06slMSWfDkrfTs2ZOFCxeqXU6lillo+oztQr/x3Wp83R9f/Mmp/cl8++23dOnSxYEV1o7RaGTz5s1s2rSJnbt2knDq/CwQWr0WjxBfvP7KdI/m3qqdQyuKgqmonNLKPC/EVFxe+XxQUBB9+vShX79+XHHFFQ1q6MjPz+eTTz5h8eLFlJVZp3TRB/6V583VG5leV5YyKM/UYMw838ju5+/HHbffwS233FJto5/kue3yvDYqPkcUnyCMna5wihlpNCV5uB1ehb+vD999951dl+KIj4/nzTff5M8//wTAKyCYsM59CevQ0+k7yxnLSkiPP0Dq4Z2Vnd47dOjAo48+yqBBg+q1TYPBwOjRoyksLGL0bQ/TY/Rkl79vUcFQUszKz17j6JZVjB07tnKWUTUZjUYeeOABtmzZQscBrRlxfb/Lnn8d2XaS9Yt3EBERwcKFC116+veazJ07l0WLFhHUPYzgnnX7/VcsCikrjmHILOadd95hxIgR9inSCb388st8++23ALRv354lS5aoWo8j89w158cSQghxkQtHqEujulDbkiVL0Ol03HPPPZWPeXp6ctdddzFr1iySk5PrPRK7QmFhITNnzqTcrGBsNxQlyLnWoaw3Tz9M7YaizT5DScIOHnv8cdasXl2nWSiSk5NRFAW9pzfNYzsSEBbdKC5OPf0CaRbTntK8HFIObiMpKanO2wgLCwPgwPrlhMS0JSg00tZlqiY75QyHN1l7CDt66u/Lufbaa1m4cCFHt56ssWH93KkM8jMLmTx5stM1qu/atYslS5awevVqDAbryDGdpx6fmKC/GtJ9cQ/0cpr81Wg0eAR54xHkTUDHltaG9txSytILKU0rIj09g19++YVffvkFT09PxowZw7Rp0+jVq1ed9nPy5Emuv/46jEYTOl8FrzALbiEKWhe7ytRoQOcLXr4WPFtZR7KXp2lJTT/H7Nmz2bFjBy+//LLaZarCEXkO8K+nniI7OxtTdE8soe2cfxmX2nDzwNyqN5ZmMbid3MyLL75Iv379iI6u/bqhOTk5FBUVAdC8dWeCIuNcvlEdwM3Ti+CYdhhKiijMSCElJQWTyVSnKfMr8vzU3q207T2U8Lad7VWuwxXn57Bj+f8A58vzMWPGMHfuXI5uP0WfcV2qndWjtKiM04dS6NChA507O9f/y6lTp/juu+/47bffyMvLA6zLtXhF+Fsb0luq25D+dxqNBjc/D9z8PPBv27xKQ3tZWiGF6YWVU7XP/e9chg4ZyuTJk7niiivqdE5SWFjIlGumkJWZhdYTPGPNuLdU0NY8yNtpaT3BM0rBM8qMuQTK07UUpxXy9ttv8/vvv/Pdd99V21musXNUntfGpEmT2L17N0uXLkWXvB9zTN3OP23ObEJ/cgtYzMydO9eujeoA7dq14/333yc+Pp5FixaxYsUKErb8QcKWPwiMiKVlu+60aNMFDx/nuB4ylRvISjhKevx+shOPo1jM6HQ6Ro0axfTp0+nX7/INs5fi4eHBhx9+yP3338+qz19nz8ofGHjNbbTvN9xlz3lKC/PZu+pHdq34jrLiQnr16sUzzzyjdlkAuLm58cYbb3DXXXdxaNshgkL86TGyY42vTzmRzsbvdxEUFMQnn3zSKBvVAR5++GE2bNjAuYPn8IkJwiO49h3P849lYMgsZsKECU2qUR3A1/f8rCM+Pi4+yKmOnPKWhy3WfBFCiKbGFms3CmEre/fupV27dhf1EOzXrx9gnQKuugt3g8FQ2WgF1t6GNUlPT6ekpASLfwiKpx+UFdqoeudg8Q7C4hVAXm4Oubm5tGzZstbvveKKK5g6dSo//vgjh377Bp2bO4HhrQiKak1AWAzuPq4zEsNiNlGYkUJeymlyk09Smp8DQEREBP/5z3/qvL077riDAwcOsGXLFhY8cQtxPQfSceBoQuM6uGZDjqJw7uQRjm5dzen921EsFkaMGMEtt9yidmVVxMXF0aNHD/bv3096Ujae3hd3FDm8xbpswdSpUx1d3iWlpKRwxx13WP+h0eDXtjk+0YG4B1SdB9V0wVStzkir1+IdEYB3hHW90fK8MoqTcik8lc2vv/7Kr7/+yqpVq+rUiLN3716MRutIeI9ICzp/BcUIZuNl3ujkNG7gHmZBMVvXY9++fbvaJanGEXlusVg4nZCA4u6NEtASDEU2qt45KG4emP1CIPsMiYmJdWpYj4uL45FHHuH999/n+NofiV/3M/5h0QRFtSYwLAavwGaAi2SXYqEoO538c4nkJJ2kKMs644e/vz/PPvtsna9lRo4cyfTp01m0aBFfP/sPIjt0p+PA0UR37o3WRRvNcs6d4eiW1cTv3Iip3EC7du2YOXOm2mVV4enpyVVXXcXixYs5secMoa0uHiEdvzsRi9nCNddc4zQdzircfPPNFBcXA+ATHYhPqyA8mvlUOQU0FZXX8G7n4dXSF6+WvgQRhrG4nJLkfApPZbNhwwY2bNjAe++9x7Bhw2q9vcTERLIyswBwDzfj1kxBsVjXMXdpGnAPsYAChmQt8fHxFBQUNJkpei/kiDyvi3/961/s27ePU6fiUTz9sKi43ro+aT+a0gJmzJhRp9+bhmrXrh3PPfccM2fOZOXKlSxfvpwdO3aQl3Ka+PU/ExARS0jbrgRHtUbj8F6rCgVpyWScOEj2mXgsJuvJfceOHZkwYQITJkyw6VTgXbp04bvvvuPDDz/kp59+Ztm7z7PG1592/UfScdBo/IJdoDFXUUhLOMaRzas4fWA7FrOZoKAg7r/3MaZPn37J5Sgczdvbm3feeYebbrqRrb/uI7CFH0GhARe9zmgwsfKrzeh0Ot566y2Hdb5Rg7e3N88++yz33HMPmVvO0HJYbK3eZy43k7v3HIGBgfzrX/+yc5XO58LG9Asb2ZsCp2xYv/322y9a8+Wqq66q05ovQgjR1Hh6OtlCZ6JJS01NrRxJdKGKx86dO1ft++bOnVvrdRjj4uLo27cvO3fuxP3Ab/Uv1smNHDmyTo3qYO1o8/zzz3P77bezbNky1q9fT3x8PNln4u1UpWP4+/sz/MorGTt2LCNGjKhXhyJvb28++OAD1qxZw6effsqR3Zs4uXuTHap1vG7dunH33XczfPhwp7uRDTBlyhT27dvH0jf+qPE1UdFR9V6H2F5KSy+4q6woFJ7IovBElnoF2VHF1K+1NWjQIHr16sWePXsoOeaaDVmXExAQwIwZM9QuQzWOyHOtVssNN9zAN998g9vB3+tfrJNrFRtb56VdAO666y4mT57M8uXLWbNmDfsPHCD/XKLtC3QgDw8PBg8ezNixYxk3bly9RrhotVqefvpprrzySj766CO2bNnC2WP77VCt40XHxHDH7bczefJkp+w8fc0117B48WLWfFPzWrFubm5cddVVDqyqdi7MueKkPIqT8tQrxo5KSkrq9PrWrVszZswY1q5dS1kClCVc/j2uxsPDg6uvvprAwEC1S1GFI/K8Lry8vHjttde48aabKEvcbfPt11W3bt145JFHVNm3t7c3U6ZMYcqUKWRlZbFq1SpWrlzJ7t27yTur/i9ju3btGDt2LGPHjiU2tnaNjfURGhrKc889x913383//vc/fvvtN/av+Zn9a3622z7tpXPnzkyaNIlrrrmmQUtu2VPz5s2ZP/81brvtNn5bsPGSr501a5bTXaPbw8CBA5k0aRK//vorST8cqtN7Z86c2SQ7bV0406CzzTpob063xrozrfkihBCupKSkhA8//JCOHTsyfvx4tcsRNWgq+dS6dWvat2/Pb79VbfBOSEigdevWvPHGG9VeuFbXIz4qKqrG41VSUsKXX35JRkaGzb8HZxAREcGMGTPqNA18TbKysti3bx8HDhygsNB1RvdrtVpat25Nz549adeunc2nbkxISGDlypWkp6fbdLuOFB4ezrhx4+o0ClINBoOB999/v8aRLhqNhvHjx9er4cmeLBYLH330UaP9nKnQsmVL7r333np1ykhOTmb58uUu/Xv0dzqdjoEDBzJs2LAaG7aaQqY7Ks8tFgvfffcdx48ft/n34Az8/f25/fbbbXLDraioiP3797Nv3z6yslyrk09kZCQ9evSga9euNjm3uVBmZiZ//PEHp06dsul2HSkgIIAxY8bQqVMnp+wgV0FRFBYuXEhiYmKNr+nduzcTJ050XFG19MMPP3Dw4EG1y7ArX19f7rnnnnrd4M7Ozub333/n5MmTdqhMHRqNhq5duzJmzJgaR9NJntsuz+tq7969/Prrr6jZPOHr68ttt91m0xHYtpCVlcWaNWs4duyYKvuPiIjgiiuuoFWrVqrs32w2s3v3btatW1fnzr9qCQkJ4corr7RrBwRb27hxI+vWravx+Xbt2nHjjTc69XmJLRUUFPDFF1+Qm5tb6/e0b9+eG264ockcowvl5OTw6aefYjAYmDZtGh071rysgCM4Ms+drmF95syZvP766+Tk5FT55ufOncusWbNISkqq1bQTTeGkSAghhOtpKvnUpUsXWrZsyZo1a6o8fuTIETp37syHH37Ivffee9ntNJXjJYQQwvU0hYySPBdCCNHYNYWMkjwXQgjR2Dkyo7R23Xo91GbNl+oYDAYKCgqq/BFCCCGEOsLCwkhNTb3o8YrHwsPDHV2SEEIIIepI8lwIIYRwfZLnQgghhO04XcN6Q9Z8CQgIqPxTm1HtQgghhLCPHj16EB8ff1FHt+3bt1c+L4QQQgjnJnkuhBBCuD7JcyGEEMJ2nK5hvbS0FA8Pj4se9/T0rHy+Ok8//TT5+fmVf5KTk+1apxBCCCFqNm3aNMxmMx9//HHlYwaDgc8//5z+/ftLBzghhBDCBUieCyGEEK5P8lwIIYSwHb3aBfydl5cXBoPhosfLysoqn6+Oh4dHtQ3yQgghhHC8/v37c9111/H000+TkZFBmzZt+PLLL0lMTGTBggVqlyeEEEKIWpA8F0IIIVyf5LkQQghhO07XsB4WFkZKSspFj8uaL0IIIYRrWbhwIbNnz+arr74iNzeXbt26sWzZMoYNG6Z2aUIIIYSoJclzIYQQwvVJngshhBC24XQN6z169GDdunUUFBTg7+9f+bis+SKEEEK4Fk9PT+bNm8e8efPULkUIIYQQ9SR5LoQQQrg+yXMhhBDCNpyuYX3atGnMnz+fjz/+mCeeeAKo35oviqIAUFBQYLdahRBCiLqqyKWKnBKXJnkuhBDCWUmm157kuRBCCGcleV57kudCCCGclSPz3Oka1m215kthYSFArRvihRBCCEcqLCwkICBA7TKcnuS5EEIIZyeZfnmS50IIIZyd5PnlSZ4LIYRwdo7Ic43ihN3xysrKmD17Nl9//XXlmi8vvvgi48aNq/U2LBYL586dw8/PD41GY8dqL6+goICoqCiSk5OrTG8v5NhcihybmsmxqZ4cl5o507FRFIXCwkLCw8PRarWq1uIKJM9dgxybmsmxqZkcm+rJcamZsx0byfTac6Y8B+f7WXIWclxqJsemZnJsaibHpnrOdlwkz2tP8tx1yLGpnhyXmsmxqZkcm5o507FxZJ473Yh1sM2aL1qtlsjISBtW1XD+/v6q/3A5Kzk2NZNjUzM5NtWT41IzZzk20gu+9iTPXYscm5rJsamZHJvqyXGpmTMdG8n02nHGPAfn+llyJnJcaibHpmZybGomx6Z6znRcJM9rR/Lc9cixqZ4cl5rJsamZHJuaOcuxcVSeSzc8IYQQQgghhBBCCCGEEEIIIYQQ4hKkYV0IIYQQQgghhBBCCCGEEEIIIYS4BGlYdwAPDw+effZZPDw81C7F6cixqZkcm5rJsameHJeaybERtiA/RzWTY1MzOTY1k2NTPTkuNZNjI2xFfpaqJ8elZnJsaibHpmZybKonx0XYivws1UyOTfXkuNRMjk3N5NjUrKkeG42iKIraRQghhBBCCCGEEEIIIYQQQgghhBDOSkasCyGEEEIIIYQQQgghhBBCCCGEEJcgDetCCCGEEEIIIYQQQgghhBBCCCHEJUjDuhBCCCGEEEIIIYQQQgghhBBCCHEJ0rAuhBBCCCGEEEIIIYQQQgghhBBCXII0rAshhBBCCCGEEEIIIYQQQgghhBCXIA3rQgghhBBCCCGEEEIIIYQQQgghxCVIw7oQQrigkpISpk6dyjfffKN2KU5p/PjxLFq0iNLSUrVLEUIIIWokeX5pkudCCCFchWR6zSTPhRBCuArJ80uTTLeShnUHUhSFtWvXsmLFCgoLC9UuRwiXUFJSQrNmzZg3b57apTgVb29vVq9eTUlJidqlOKWEhARuueUWWrZsyW233cbq1atRFEXtskQjIXkuRN1JnldP8vzSJM+FPUmeC1F3kuc1k0yvmeS5sCfJcyHqRzK9epLnlyaZbqVXu4DG6t///jdbtmxh3bp1gDXkx44dy9q1a1EUhejoaNasWUPr1q1VrtT+9uzZU+f39OrVyw6VOJ+cnJw6vyc4ONgOlTgvb29v9Ho9Pj4+apfidIYMGcLWrVu5++671S7F6Rw/fpydO3fy9ddf89133/H1118TGhrK9OnTufnmm+nRo4faJQoXIXl+nuR5zSTPL0/yvGaS5zWTPBe2InlelWR69STPL0/y/NIk06sneS5sRfK8KsnzmkmmX55kes0kz2smmW6lUZpidwIH6NChA5MnT+aVV14B4Pvvv+eGG27gpZdeonv37tx7772MGDGCr776SuVK7U+r1aLRaGr1WkVR0Gg0mM1mO1flHOpybCo0lWNzofvvv59jx46xZs2aOh+vxiwhIYFx48Zxww038I9//IPIyEi1S3JKFouFVatW8fXXX/Pzzz9TXFxMx44dmTFjBtOnT5fjJi5J8vw8yfOaSZ7XjuR59STPa0fyXDSE5HlVkunVkzyvHcnzmkmmX57kuWgIyfOqJM9rJpleO5Lp1ZM8r52mnOnSsG4nfn5+vP7665W9Wm666SYOHjzIoUOHAHj55Zf54IMPSE5OVrNMh/jyyy/r/J7bbrvNDpU4n+eee67OofXss8/aqRrntXHjRu6//36aN2/O3XffTatWrfDy8rrodU2l12UFPz8/TCYT5eXlAOj1ejw8PKq8RqPRkJ+fr0Z5TikvL497772X77//HrCeaI8YMYJHH32UCRMmqFydcEaS5+dJntdM8rx2JM+rJ3led5Lnoq4kz6uSTK+e5HntSJ7XTDK9biTPRV1JnlcleV4zyfTakUyvnuR53TW1TJep4O1Er9djMBgAa4+wNWvWMGPGjMrnW7ZsSVZWllrlOVRDAttisXD27FlCQ0Nxd3e3YVXO4bnnnlO7BJcwYsSIyq///PPPi55var0uK1x77bXSm7CWNm3axNdff82SJUvIycmhS5cuzJgxAzc3Nz777DOuvvpq/v3vf/PCCy+oXapwMpLn50me10zyvHYkz6sneV57kueiviTPq5JMr57kee1IntdMMr12JM9FfUmeVyV5XjPJ9NqRTK+e5HntNdVMlxHrdjJ06FCMRiMrVqzgxx9/5O6772b16tWMHDkSgGeeeYbPP/+8yfSgq6/09HTCw8NZtWoVo0aNUrscoZLa9sBsKr0uRe0cOXKEr7/+mm+//ZakpCRCQkKYPn06t95660Xrvdxzzz0sXbqU7OxsdYoVTkvy3DYkzwVInov6kTwXtiB5bjuS6ULyXNSH5LmwBclz25E8FyCZLupHMl1GrNvNM888w6RJk2jevDkAgwcPrgx5gOXLl9O3b1+1ynMpjbnvx549e+r8nqY29QpIeIu669GjBwcPHsTDw4PJkyfz/vvvM27cOLRabbWvHzlyJJ9++qmDqxSuQPLcdiTPq5I8F+LyJM+FrUie21ZjzXTJ89qRPBd1JXkubEXy3LYaa56DZHptSaaLupJMt5KGdTsZM2YMe/bsYdWqVQQGBnLDDTdUPpebm8uwYcOYPHmyihUKZ9CnT59aTyvSVKdeEZd39uxZ9u7dS35+PhaL5aLnL5wWq6kIDAzk448/5rrrrsPf3/+yr588eTKnT592QGXC1Uiei9qQPBe2IHl+MclzYSuS56I2JM+FrUimVyV5LmxF8lzUlmS6sAXJ84tJpltJw7odderUiU6dOl30eFBQEG+88YYKFQln8/nnn6tdgssoKytj6dKl7Nmzp9ow02g0LFiwQKXq1FFWVsZtt93G0qVLsVgsaDSayt6mF548NrWQLysrY+rUqbRt27ZWAQ/g7e1NTEyMnSsTrkryXFyO5HntSZ5fTPK8epLnwtYkz8XlSJ7XnuR59STTLyZ5LmxN8lzUhmR67UmmX0zyvHqS6edJw7oQKmrIdCsWi4WzZ88SGhqKu7u7DatyPmfOnGHkyJEkJiYSGBhIfn4+wcHB5OXlYTabad68Ob6+vmqX6XCzZs3ihx9+4KWXXmLgwIGMGDGCL7/8krCwMN58803OnTvHwoUL1S7T4Tw9PXnqqad46623GDZsmNrlCCGaAMnz2pE8r57kefUkz4UQjiZ5XjuS5zWTTL+Y5LkQQg2S6bUjmV49yfPqSaafV/3E96LOtFoter2e8vLyyn/rdLpL/tHrpV+DqL/MzExiY2PZtGmT2qXY3ZNPPkl+fj7btm0jPj4eRVFYvHgxRUVFvPLKK3h5efHHH3+oXabDLVmyhDvuuIN//etfdO7cGYCIiAiuuOIKli1bRmBgIO+9957KVaqjc+fOJCYmql2GcEGS58LRJM8lzyXPayZ5LupL8lw4muS55DlIptdE8lzUl+S5UINkumS65HnNJNOtJGls5JlnnkGj0VSGd8W/hbCniilIGru1a9dy//33069fP3JycgDr9+7h4cGTTz7J0aNHeeSRR1i+fLnKlTpWRkYG/fr1A8DLywuA4uLiyuevvfZaXnjhBT744ANV6lPTSy+9xPTp0xk5ciRXXHGF2uUIFyJ5LtQgeS55LnlePclzUV+S50INkudNO89BMr0mkueiviTPhVok05t2pkue10wy3Uoa1m3kueeeu+S/hRD1V1JSQqtWrQDw9/dHo9GQn59f+fzAgQN54oknVKpOPS1btiQ7OxuwrlcSFBTE8ePHmTRpEgAFBQWUlZWpWaJq3n33XYKDgxk3bhyxsbHExsZWnghV0Gg0/PzzzypVKJyV5LkQ9iN5Xj3J85pJnov6kjwXwn4kz2smmV49yXNRX5LnQtiXZHr1JM9rJpluJQ3rwqn5+vry7LPPEhcXp3YpQkXR0dGcPXsWAL1eT0REBNu2bWPq1KkAHDlyBE9PTzVLVEX//v3ZtGkT//rXvwCYNGkS8+bNIywsDIvFwhtvvMGAAQNUrlIdBw4cQKPREB0djdls5uTJkxe9Rno5C+E4kucCJM9rInleM8lzIZyPZLqQPK+ZZHr1JM+FcD6S5wIk02sieV4zyXQraVi3kYULF9brfTNmzLBxJc4tKSmJpKQkhgwZUvnY/v37ee211zAYDNx0001MmTKl8jkfHx+effZZFSoVzmTUqFH8/PPPlT8Lt99+O3PnziU3NxeLxcJXX33V5H6XAB566CG+//57DAYDHh4evPjii2zdupVbb70VgNatW/P222+rXKU6ZK0XUV+S57UjeS7qQ/K8epLnNZM8F/UleV57kumiriTPayaZXj3Jc1Ffkue1J3ku6kMyvXqS5zWTTLfSKE1lwQg702q1dX6PRqPBbDbboRrnNWXKFIqKili9ejUA6enpdOzYkfLycvz8/MjIyOD777+v7BUlapaenk5YWBirV69m1KhRapdjV0lJSezcuZOJEyfi4eFBWVkZDzzwAEuXLkWn0zFx4kTefvtt/P391S5VdRaLhYMHD6LT6ejQoUPlOlRCiNqRPK8dyXPbkTyXPK+O5LkQDSN5XnuS6bYheS55XhPJdCHqT/K89iTPbUcyXTK9OpLn4kLSsG4jZ86cqdf7YmJibFyJcwsPD+fhhx+unEZj3rx5PPPMMxw6dIjY2FiuvPJKioqK2LJli8qVOr+mFPJC1NeGDRtYvnx55Wd0TEwMEyZMYPjw4SpXJpyV5HntSJ7bjuS5EJcneS7qSvK89iTTbUPyXIjLkzwXdSV5XnuS57YjmS7E5TX1TJduFTbSFAO7PnJycggJCan897Jlyxg+fDitW7cGYOrUqcyaNUut8oRwahs3bqzX+4YNG2bjSpxfeXk5N910Ez/99BOKohAYGAhAXl4er732Gtdccw3ffvstbm5u6hYqnI7kee1IngtRf5LntSd5LupL8rz2JNOFqD/J9NqRPBf1JXlee5LnQtSf5HntSaZbScO6cKgWLVpU9mLJy8tj27Zt/Pe//6183mQyYTKZ1CrPpfj6+vLss88SFxendik298ILL9T5PRqNhtmzZ9uhGucxYsQINBpNrV+vKEqTnQLr+eef58cff+SJJ57g8ccfp2XLlgBkZGTw2muvMW/ePF544QVefPFFlSsVwjVJntuO5HlVkucXkzyXPBfCniTTbUPyvKqmkOcgmV5bkudC2J/kue1IplfVFDJd8rz2JNOtZCp4O0pLS2PBggXs2bOH/Px8LBZLlec1Gg1r1qxRqTp13HHHHfz888/85z//Yf369axYsYL4+HhiY2MBuP/++9mwYQOHDx9WuVLHS0pKIikpiSFDhlQ+tn//fl577TUMBgM33XQTU6ZMUa9AB5I1lKq3YcOGer2vqUzBcqHY2FhGjBjB559/Xu3zt99+O+vXrycxMdGxhQmXJHl+Mcnzmkmenyd5Xj3J89qTPBe2JHlePcn06kmenyd5XjPJ9NqRPBe2JHlePcnzmkmmnyeZXj3J89qTTLeSEet2cuDAAUaMGEFpaSnt27fn4MGDdOrUiby8PFJSUmjdujVRUVFql+lw//3vf4mPj+eJJ57A3d2d+fPnVwa8wWDgu+++Y/r06SpXqY6HHnqIoqIiVq9eDVjXcxk5ciTl5eX4+fmxZMkSvv/+e6ZOnapypfb395NiYdUUw7q+UlNT6d+/f43P9+/fn//9738OrEi4Ksnz6kme10zy/DzJ8+pJntee5LmwFcnzmkmmV0/y/DzJ85pJpteO5LmwFcnzmkme10wy/TzJ9OpJnteeZLpV3buoiFp56qmn8PX15fjx46xevRpFUXjrrbdITk5m8eLF5ObmVpmOpalo2bIlmzdvJjc3l4KCAh5++OHK5ywWC2vWrOG5555Tr0AV7dixgzFjxlT+e+HChZSWlrJ//35SUlIYPXo08+fPV7FCIVxHZGQk69evr/H5DRs2EBkZ6biChMuSPK+e5HnNJM+FsB3Jc2Erkuc1k0yvnuS5ELYjeS5sRfK8ZpLnNZNMF8J2JNOtZMS6nWzevJmZM2cSHR1NTk4OcL5H0HXXXcemTZt48skn6z3NhKsLCAi46DEvLy+6d++uQjXOIScnh5CQkMp/L1u2jOHDh9O6dWsApk6dyqxZs9QqTzihO++887Kv0Wg0LFiwwAHVOJfbbruNZ599lsDAQB599FHatGmDRqPhxIkTvPnmm3z//fc8//zzapcpXIDk+aVJnl9M8lzUleR5zSTPha1Inl+eZHpVkueiPiTTqyd5LmxF8vzyJM8vJpku6kryvGaS6VbSsG4nFouFli1bAhAYGIhOp6sMfICuXbs2iV+8hQsXAnDrrbei0Wgq/305M2bMsGdZTqlFixacOXMGgLy8PLZt21all6XJZMJkMqlVnqpiY2PRaDSXfI1Go+HUqVMOqsg5rF279qLjYjabSU1NxWw206JFC3x8fFSqTl2zZs3i1KlTfPzxx3zyySeVawhZLBYUReG2226Tk2ZRK5LnVpLntSd5XjPJ8+pJntdM8lzYiuT5eZLptSN5XjPJ85pJpldP8lzYiuT5eZLntSeZXjPJ9OpJntdMMt1KGtbtJDY2ltOnTwOg1WqJjY1l9erVXH/99QBs2bKFwMBAFSt0jNtvvx2NRsONN96Iu7s7t99++2Xfo9FommTIX3HFFbz99tv4+/uzfv16LBYLU6ZMqXz+yJEjTXadoOHDh1cbZmfOnGHz5s106dKFnj17qlSdehITE6t93Gg08tFHH/Hmm2+yatUqxxblJHQ6HV988QWPPfYYv/32W+UJdExMDFdddRXdunVTuULhKiTPrSTPa0/yvGaS59WTPK+Z5LmwFcnz8yTTa0fyvGaS5zWTTK+e5LmwFcnz8yTPa08yvWaS6dWTPK+ZZLqVRlEURe0iGqMnnniCX375hfj4eADeeOMNHn/8cUaNGoWiKKxfv57HH3+cV199VeVK7evCX6wL/305Fa9vStLT05k6dSpbt27F3d2dV/6fvTuPiqr+/zj+usOO7CqKKKKQuyKKhomJS4pp7mlpJu77VmpmFlpZpuWuiPv+tbTctVTE3XLHfUkUF1xR3BeWz+8PfoxOcy/MwMxckNfjHM6Je+/MvGcYePr93rn3/vST9no4L168gLe3Nzp06ICpU6eqPGnuEhsbi8aNG2Pp0qVo2LCh2uPkKn379kV8fDw2btyo9igWd+XKFRQuXBgODg6y6589e4Y7d+7Ax8fHwpNRXsOep2PPDceeZw97row9Z88p59jzV9h0w7Dn2cOeZy6/Np09J1Nhz19hzw3HpmcPm64sv/YcYNMzcMe6mdy/fx9xcXGoUqUKbGxsIITA2LFj8fvvv8PKygrNmjXDyJEjYWtrq/aolMs8ePAADg4OOu+NZ8+e4fz58yhRogQ8PDxUnC53ioiIwIYNG3D48GG1R8lVoqKiMHToUDx69EjtUSzOysoKS5YsQYcOHWTX//rrr+jQoQNSU1MtPBnlNew5ZRd7bjz2XB57zp5TzrHnlF3sufHYc2X5tensOZkKe045waYbj02Xl197DrDpGXgqeDNxd3dH9erVtd9LkoRRo0Zh1KhRKk5FeYGrq6veMgcHBwQEBKgwTd5QpEgRnD59Wu0xcp2tW7fC0dFR7TFUkdVnxpKTk7XXgCHKDHtO2cWeG489l8eeK2PPyVDsOWUXe2489lxZfm06e06mwp5TTrDpxmPT5eXXngNsegbuWLewuLg4vHjxAuXLl1d7FIuoX7++0beRJAnR0dFmmCZ3Wbx4MQCgU6dOkCRJ+31W8uO1cDKTmJiIefPmoXjx4mqPYnHffvut7PKkpCTs2rULR44cwYgRIyw8lXoePnyIpKQk7feJiYm4cuWK3nZJSUlYsWIFvLy8LDgdvWnY86yx55ljz3Wx5/rY83TsOZlTfus5wKYrYc9NIz/3HGDTX8eekyWx54bJDz0H2HRTyc9NZ891sen6eCp4M5k6dSr27duHFStWaJd16dJF+4c8MDAQmzZtgqenp1ojWkRoaCgkSTL6djExMWaYJnfRaDSQJAnPnj2Dra2tQZ/kkSTpjT+NhhylfywmJSXh7NmzePnyJZYsWYKPP/7YwpOpS+k94+7uDj8/P3Tv3h09evTI1u9gXjRmzBjFf/j8lxAC33//PUaOHGnmqSivY8/TsefK2HPDsefy2HNd7DmZA3v+Cpsujz03HHuujE1/hT0nc2DPX2HPlbHphmPT5bHnuth0fTxi3Uzmzp2LevXqab//66+/sGjRIvTq1QuVK1fGqFGjMGbMGMyYMUPFKc1vx44dao+Qa126dAkAtNd1yfie9KWlpemFSpIklCpVCg0bNkTXrl1Rrlw5laZTT1pamtoj5CqNGjWCk5MThBAYPnw4Pv74Y1SrVk1nG0mSUKBAAVSvXh1BQUEqTUp5CXuejj1Xxp4bjj2Xx57rYs/JHNjzV9h0eey54dhzZWz6K+w5mQN7/gp7roxNNxybLo8918Wm6+OOdTOJj4/XOf3Mb7/9hlKlSiEyMhIAcPPmTSxZskSt8SgXKFmyZKbf0yv8xyIZolatWqhVqxYA4MmTJ2jTpg0qVaqk8lSU17HnlBX23HDsORmCPSdzYM8pK+y54dhzMgR7TubAnpMh2HTDselkCDZdH3esm8l/z7C/ZcsWtGjRQvu9r68vbt68aemxcpVHjx7hwYMHsp8A8vHxUWEiotxN7tolhsiPv08RERE63z948ABOTk6wsrJSaSLKq9jzrLHnRMZhzw3HnpOpsOeGYdOJjMOmG4Y9J1Nhzw3DnhMZhz03HJuejjvWzaRMmTJYvXo1evfujb/++gsJCQlo0qSJdv21a9fg5uam3oAqioyMxMSJExEXF6e4TX64ponSNUwyI0kSoqOjzTBN7pJxbSRjffrppyaeJHfx9fXN1rVb8sPvk5xDhw5h1KhR2LVrF16+fIktW7agfv36uHv3Lrp164YhQ4YgNDRU7TEpl2PPlbHn6dhzZey5PPbcOOw5mQJ7njk2nT3PDHuujE03HHtOpsCeZ449T8emK2PT5bHnxmHTuWPdbIYOHYoOHTrA3d0dT548Qfny5dG4cWPt+u3bt6Nq1arqDaiSWbNmoV+/fmjcuDG6du2Kr776CkOGDIG9vT0WLlyIIkWKYODAgWqPaRFy1zDJyn8/mfmmCg8P11uW8Vr99zV4/TV80yM/f/58neeblpaGKVOmID4+Hh07dkTZsmUBAGfPnsXy5cvh6+ubb36f/mvfvn2oX78+vL298cknn2Du3LnadYUKFcKDBw8QFRX1xkeeco49l8eev8KeK2PP5bHnhmPPyVTYc2Vsejr2XBl7roxNNwx7TqbCnitjz19h05Wx6fLYc8Ox6em4Y91MPvroIxQsWBCbNm2Cm5sb+vbtC2vr9Jf73r178PDwQKdOnVSe0vKmTZuGxo0bY/PmzUhMTMRXX32Fpk2bon79+hg+fDiCgoKQmJio9pgWwWuYKLt06ZLO90lJSejcuTNcXV0xYMAAnZhNmzYNjx49wqJFi9QY1aL++4+fsWPH4vnz5/j3339RsGBBnXWjR49GSEhIvj0F1siRI1G+fHn8/fffePTokU7kAaBevXr54j1DOceey2PPX2HPlbHn8thzw7HnZCrsuTI2PR17row9V8amG4Y9J1Nhz5Wx56+w6crYdHnsueHY9P8niCzIzs5OzJgxQwghxIMHD4QkSWLz5s3a9ePGjROlS5dWazzKpcLDw0XDhg1FWlqa3rrU1FTRoEEDER4ersJk6ipevLj45ZdfFNdPmDBBlChRwoIT5R6Ojo5iypQpQggh7t69KyRJEtHR0dr1c+bMEQ4ODmqNR5TnseeUHey5PPZcGXtOZH5sOhmLPVfGpstjz4nMjz2n7GDT5bHnytj0dDxinSzK1dUVKSkpAAAXFxc4Ojri6tWr2vXOzs759tM+r3v06BEePHiAtLQ0vXU+Pj4qTKSuNWvWYOzYsbKn8dFoNGjdujVGjRqlwmTqSkxMxNOnTxXXP336NN98GvW/bGxsZH9/Mly/fh1OTk4WnIjozcKeG4Y918Wey2PPlbHnRObHpmeNPdfFnitj0+Wx50Tmx54bhk3XxabLY8+VsenpuGPdjI4fP45p06bhyJEjsn+wJUnCxYsXVZpOHZUqVUJsbKz2++DgYERGRuL9999HWloaoqKiUKZMGRUnVFdkZCQmTpyIuLg4xW1SU1MtOFHuIITA2bNnFdefPn0631wL53XBwcGYPHkymjRpgurVq+usO3ToEKZMmYK3335bpenUFRwcjFWrVmHw4MF66548eYIFCxagbt26lh+M8iT2XB97njn2XB57Lo89V8aekymx5/LYdGXsuTz2XBmbLo89J1Niz+Wx55lj0+Wx6fLYc2Vs+v9T72D5N1tMTIyws7MTRYsWFc2aNROSJIkGDRqI2rVrC41GIypXrpwvT6Mxf/58UbNmTfH8+XMhhBB79uwR9vb2QqPRCI1GI+zs7MSGDRtUnlIdkZGRQpIkERYWJn744QchSZL47LPPxMiRI0WxYsVEYGCgWLBggdpjqqJz587C2tpa/PLLL+LJkyfa5U+ePBE///yzsLa2Fp07d1ZvQJWcOnVKFC5cWGg0GvHOO++Izp07i86dO4t33nlHaDQaUahQIXHy5Em1x1TF33//Lezs7MT7778vlixZIiRJEhMnThRz5swRZcuWFY6OjiI2NlbtMSkPYM/lsefK2HNl7Lk89lwZe06mwp4rY9PlsefK2HNlbLo89pxMhT1Xxp4rY9OVseny2HNlbHo67lg3kzp16ojy5cuLBw8eiDt37uhca+Dvv/8W7u7uYtOmTSpPmTtcvHhRTJ48WUybNk2cO3dO7XFUU6FCBREWFiaE0L8+RVJSkvD39xc///yzmiOqJikpSbz77rtCkiRha2srSpYsKUqWLClsbW2FJEkiJCRE3L9/X+0xVXHz5k0xePBgUbZsWWFvby/s7e1F2bJlxZAhQ8SNGzfUHk9V0dHRokyZMkKSJJ0vf39/sWPHDrXHozyCPTcce56OPVfGnitjz5Wx52QK7Llx2HT2PDPseebYdHnsOZkCe24c9jwdm66MTVfGnitj04WQhMiH53KwACcnJ4wZMwaff/457t+/j4IFC+Kvv/7Ce++9BwD48ssvsXXrVhw6dEjlSSm3sLe3x8SJE9G3b188fPgQbm5u2LRpE8LCwgAAP/30E2bPnp0vT2eUYe3atdi8eTPi4+MBACVLlsT777+PDz74QPZaMJQ/CSHw6NEj2Nrawt7eHseOHcOFCxeQlpYGPz8/VK9ene8XMhh7TsZiz7PGnpMh2HMyJfacjMWeZ409J0Ow52RK7DllB5ueNTadDMGmv8JrrJuJtbU1nJ2dAQBubm6wsbHB7du3tetLly6N06dPqzVervD48WPcv39f9jodPj4+KkykLldXV6SkpAAAXFxc4OjoiKtXr2rXOzs74+bNm2qNlyu0aNECLVq0UHsMyuVevnwJDw8P/PDDDxg+fDiqVq2KqlWrqj0W5VHsedbYc13sedbYczIEe06mxJ4bhk1/hT3PGntOhmDPyZTYc8Ow57rY9Kyx6WQINv0V7lg3E39/f1y4cAEAIEkSypUrh9WrV6Njx44AgI0bN6Jo0aJqjqiK58+fY8yYMZg3bx4SExMVt0tNTbXgVLlDpUqVEBsbq/0+ODgYkZGReP/995GWloaoqCiUKVNGxQkpNzpz5gwWLFiAuLg42X80S5KE6OholaZTh52dHYoWLQo7Ozu1R6E3AHsujz1Xxp5TdrDn+thzMiX2XBmbLo89p+xi03Wx52RK7Lky9lwZm07ZwZ7rY9Nf4Y51M3n//fcxf/58/Pjjj7C2tsZnn32GLl264K233gIAXLx4ET/++KPKU1pe3759sWjRIrRs2RJ16tSBu7u72iPlGp988glmzZqFFy9ewM7ODmPGjEHDhg21nyS0sbHB77//rvKU6hBCYPbs2Zg3b542Zv8lSZL204f5xZIlS9ClSxfY2NigbNmysr9P+fVqH+Hh4Vi8eDH69OkDW1tbtcehPIw9l8eeK2PPlbHn8thzZew5mQp7roxNl8eeK2PPlbHp8thzMhX2XBl7roxNV8amy2PPlbHp6XiNdTNJTk7Gw4cP4eHhob2uwNKlS/H777/DysoKzZo1Q3h4uLpDqsDNzQ3t27dHVFSU2qPkCXFxcVi/fj2srKzQqFGjfPvpuWHDhmHixImoWrVqpv84jIiIsPBk6vLz84OHhwc2b96MQoUKqT1OrvLrr7/iu+++w4sXLxAeHg5fX184ODjobde6dWsVpqO8hD2Xx54bhz1Px57LY8+VsedkKuy5MjbdcOx5OvZcGZsujz0nU2HPlbHnxmHT07Hp8thzZWx6Ou5YJ4tyd3fHuHHj0KtXL7VHoTzE09MToaGh+O2339QeJVdxcHDAxIkT0adPH7VHyXU0Gk2W20iSlC9PgUVkCuw5ZQd7Lo89V8aeE5kfm07GYs+Vseny2HMi82PPKTvYdHnsuTI2PR1PBW9mqampOHz4MC5fvgwAKFWqFKpVqwYrKyt1B1NJixYtsG3bNkY+C48fP5a9dgcA7Wlq8pNnz56hYcOGao+R61SpUgUJCQlqj5ErxcTEqD0CvWHYc13suWHYc13suTz2XBl7TqbGnutj07PGnutiz5Wx6fLYczI19lwfe24YNl0Xmy6PPVfGpqfjEetmtHDhQnz55Ze4ffu29o+1JEkoXLgwfvjhB3Tt2lXlCS3v4sWLaNeuHapXr45evXrBx8dH9h89Hh4eKkynrufPn2PMmDGYN28eEhMTFbd70z/tI6dly5YoXLgw5syZo/YoucrevXvx4YcfYtWqVXjnnXfUHofojcWe62PPlbHnythzeew5kWWw5/LYdHnsuTL2XBmbTmR+7Lk89lwZm66MTZfHnlNWuGPdTKKiotCnTx9UrVoVvXr10l6n49y5c4iKisLx48cxY8YM9O7dW+VJLev1U0VkXAtHTn4MWdeuXbFo0SK0bNky02uadO7c2cKTqS8hIQGNGzfGxx9/jF69eqFgwYJqj5QrNG/eHBcuXMD58+dRoUIF2X80S5KEtWvXqjSheu7du4dr166hSpUqsutPnDiB4sWLK/6eEWVgz+Wx58rYc2XsuTz2XBl7TqbCnitj0+Wx58rYc2Vsujz2nEyFPVfGnitj05Wx6fLYc2VsejruWDeT0qVLo0SJEti2bRtsbGx01iUnJ6N+/fq4fv064uLiVJpQHaNHj8407hkiIiIsME3u4ubmhvbt2yMqKkrtUXIdZ2dnpKWl4fnz5wAAe3t72Zg9ePBAjfFU4+vrm+XvkyRJ+e7vDJD+j+Fz587h77//ll3/zjvvoHz58pg3b56FJ6O8hj2Xx54rY8+Vsefy2HNl7DmZCnuujE2Xx54rY8+Vseny2HMyFfZcGXuujE1XxqbLY8+VsenpeI11M7l58yY+//xzvcgDgI2NDT766CMMHz5chcnUNXr0aLVHyLUkSUK1atXUHiNXatOmjUH/OMxvMq4lRfq2b9+OPn36KK7/4IMPMGvWLAtORHkVey6PPVfGnitjz+Wx58rYczIV9lwZmy6PPVfGnitj0+Wx52Qq7Lky9lwZm66MTZfHnitj09Nxx7qZBAYG4vz584rrz58/j6pVq1puoFzq2bNnAAAHBweVJ1FfixYtsG3bNvTq1UvtUXKdhQsXqj0C5TF37txBoUKFFNcXLFgQt2/ftuBElFex54Zhz19hz5Wx52Qs9pxMhT03HJuejj1Xxp6TsdhzMhX23HDs+StsujI2nYzFpqfTZL0JZce0adPw22+/YcqUKdqQAelRmzRpEn777TdMnz5dxQnVc+XKFXTp0gVFihSBk5MTnJycUKRIEXTt2hXx8fFqj6ear7/+GnFxcejZsycOHz6MO3fu4N69e3pfRHIePXqEa9eu4cqVK3pf+ZGXlxeOHj2quP7w4cMoXLiwBSeivIo9V8aey2PPKSfYc13sOZkKe545Nl0fe045xaa/wp6TqbDnmWPP5bHplBPsuS42PR2vsW4iVapU0Vt279493LhxA9bW1ihWrBgAICEhASkpKfDy8kLBggURGxtr6VFVdfbsWYSEhCApKQnvvfceypcvr12+ZcsWuLu7Y8+ePShbtqzKk1qeRvPqcy6ZnYIlNTXVEuPkSteuXcPRo0fx4MEDpKWl6a3/9NNPVZhKXZGRkZg4cWKm13TJj++ZIUOGYMaMGVi1ahWaN2+us27t2rX48MMP0adPH0yZMkWlCSm3Ys8Nw54rY8+zxp7rY8/lseeUXey54dh0eex51thzeWy6Pvacsos9Nxx7roxNzxqbro89l8emp+OOdRMJDQ3N1vUoYmJizDBN7tWyZUvs27cP0dHRqFy5ss66kydPokGDBnjnnXewevVqlSZUz+jRow16D0VERFhgmtzl+fPn6Ny5M37//XekpaVBkiRk/Ol6/TXLbzGbNWsW+vbti8aNG+Pdd9/FV199hSFDhsDe3h4LFy5EkSJFMHDgQISHh6s9qsU9ePAAISEhOH36NAICAlCpUiUA6X9nYmNjUb58eezZswdubm7qDkq5DntuGPZcGXuujD2Xx54rY88pu9hzw7Hp8thzZey5MjZdHntO2cWeG449V8amK2PT5bHnytj0/yeILMjNzU189913iuu//fZb4ebmZsGJKC8YMmSIsLa2FuPGjRM7d+4UkiSJxYsXi61bt4qmTZuKwMBAceLECbXHtLgKFSqIsLAwIYQQd+/eFZIkiejoaCGEEElJScLf31/8/PPPao6oqsePH4tvvvlGVKpUSTg4OAgHBwdRqVIlERERIR4/fqz2eER5GntO2cGey2PPM8eeE5kXm07GYs+VsenK2HMi82LPKTvYdHnseebYdCF4jfVc4u7duyhdujT279+v9ihmlZycDAcHB8X1jo6OSE5OtuBEudezZ890rheUn61atQpdunTBF198gYoVKwIAvL290bBhQ2zYsAFubm6YMWOGylNa3sWLF/HBBx8AAGxsbAAAL1++BAC4urqie/fumDlzpmrzqa1AgQIYM2YMTpw4gadPn+Lp06c4ceIERo8ejQIFCqg9Hr2h2PN07Pkr7Pkr7Lk89jxz7DmpIb/0HGDTDcWev8KeK2PTlbHnpAb2/BX2/BU2/RU2XR57njk2HeCO9VwiNTUVly9ffuP/qAcGBmLu3Ll48OCB3rqHDx9i3rx5qFatmgqT5Q5XrlxBly5dUKRIETg5OcHJyQlFihRB165dER8fr/Z4qrl9+zZq1qwJANp/JD558kS7vk2bNvjjjz9UmU1Nrq6uSElJAQC4uLjA0dERV69e1a53dnbGzZs31Rov17hx4wZiY2N13jNE5sKes+cAe66EPZfHnhuGPSdLyi89B9j0zLDn8thzZWx61thzsiT2PF1+7znApith0+Wx54bJz03njnWyqDFjxuDixYsoV64cRo4ciYULF2LhwoX48ssvUa5cOVy8eBFjxoxRe0xVnD17FtWqVcOSJUtQrVo1DBo0CIMGDUL16tWxePFiBAUF4dy5c2qPqYoiRYogMTERQPonLN3d3XVei4cPH+L58+dqjaeaSpUqITY2Vvt9cHAwIiMjcf36dVy9ehVRUVEoU6aMihOqa+3atShXrhyKFy+OatWq4Z9//gGQ/onlwMBArFmzRt0BifIw9lwZe66MPZfHnmeOPScyLzZdHnuujD1XxqYrY8+JzIs9V8amK2PT5bHnmWPTwWus5xY3b97UuVbDm2zr1q2iatWqQpIkna/AwECxbds2tcdTTYsWLUThwoXF8ePH9dadOHFCeHp6ipYtW6owmfo+/PBD0axZM+33nTt3Fp6enmLp0qVi8eLFonDhwqJRo0YqTqiO+fPni5o1a4rnz58LIYTYs2ePsLe3FxqNRmg0GmFnZyc2bNig8pTqWLdundBoNKJ27dpizJgxen9fmzZtKpo3b67ihPSmYs/Zc/ZcGXsujz1Xxp6TWvJTz4Vg0+Ww58rYc2Vsujz2nNTCnrPnQrDpmWHT5bHnytj0dJIQQqi9c5+AW7duwcvLC9u2bUP9+vXVHscibt68qT3VSsmSJVG0aFGVJ1KXu7s7Pv/8c4waNUp2/XfffYeJEyfi/v37Fp5MfXv27MHKlSsxfvx42NnZ4erVq2jYsCEuXLgAAPDz88OGDRtQtmxZlSdVX1xcHNavXw8rKys0atQo3356rkaNGnByckJMTAwSExNRuHBhnb+vY8eORVRUFK5cuaLypPSmYc/Zc/ZcGXtuOPY8HXtOasmPPQfY9Nex58rYc+Ow6ew5qYc9Z88BNj0zbLrh2PN0bHo6a7UHoPyraNGi+T7sr0tOTtZey0SOo6MjkpOTLThR7hESEoKQkBDt9yVKlMCZM2dw4sQJWFlZoVy5crC25p8zAChdujQGDRqk9hiqO3nyJCZOnKi4vkiRIrh9+7YFJyJ6c7HnuthzZey54djzdOw5kWWx6a+w58rYc+Ow6ew5kaWx57rYdGVsuuHY83Rsejr+VpBZLV68GADQqVMnSJKk/T4rn376qTnHypUCAwMxd+5cdO/eHa6urjrrHj58iHnz5qFatWoqTZf7aDQaBAQEqD0G5VKOjo548uSJ4vq4uDgULFjQghMR5W3sueHYc+Ow55QZ9pzI9Nh0w7DnxmHPKTPsOZHpseeGY9ONw6ZTZtj0dDwVfC7xpp6aRqPRQJIkPHv2DLa2ttBoNFneRpIkpKamWmC63GX79u0ICwtDwYIF0aVLF+3pRM6dO4dFixYhMTERf/75J+rVq6fypOa3a9eubN3u3XffNfEkuUvG75MxJElCSkqKmSbKvdq2bYtz587h6NGjePDggc5paW7evInKlSujWbNmWLBggdqj0huGPX+FPWfP2XN57Lnh2HNSy5vac4BNNxR7/gp7roxNNwx7Tmphz3Xlx54DbPrr2HR57Lnh2PR0PGI9l7C1tUXdunXh7u6u9igmdenSJQDpz+/170lf/fr1sWnTJgwbNgzjxo3TWVe1alUsWbIkXwQeAEJDQ42KmRAiX/zj8JtvvjE68vnV2LFjERwcjBo1auDDDz+EJEn466+/sH37dkRFRUEIgYiICLXHpDcQe07s+SvsuTz23HDsOanlTe05wKYbij1/hT1XxqYbhj0ntbDnBLDpr2PT5bHnhmPT0/GIdTO6ffs2rl+/jmfPnsHJyQn+/v5wdHRUeyzKA27evIn4+HgAQMmSJfPddXF27tyZrdvVrVvXxJNQXnbq1CkMGjQIMTExeD11oaGhmDFjBsqXL6/idJSXsOeUXew5e045x56TqbDnlF3sOXtOOceek6mw55QTbDqbTjnHpnPHusklJiZi3LhxWLFiBRISEnTWaTQaBAcH4/PPP0fLli3VGVBlpUuXxuTJk9G8eXPZ9Rs2bMDAgQMRFxdn4cmI8o6rV69Co9HA29sbAPD8+XPMnDlTb7vixYujXbt2lh4v17l//z7+/fdfpKWloXTp0ihcuLDaI1EewJ5njj0nyjn23DjsOWUHe541Np0o59h0w7HnlB3sedbYc6KcY8+Nk5+bzlPBm1B8fDzq1KmDhIQElC9fHsWKFcPp06eRnJyMrl27IikpCTt37kSbNm3Qo0cPzJo1S+2RLe7y5ct4/Pix4vrHjx9rPzX2plu8eDEAoFOnTpAkSft9Vj799FNzjkW53IkTJxAYGIjJkyejf//+AIAnT55g6NChettaWVmhfPnyqFy5sqXHzFXc3d1Ro0YNtcegPIQ9zxp7/gp7TtnBnhuPPSdjseeGYdPTseeUXWy6cdhzMhZ7bhj2/BU2nbKDPTdevm66IJNp27atKFy4sIiNjdUuu3v3rmjQoIEICwsTQgiRmpoqxo0bJzQajZgzZ45ao6pGkiSxfPlyxfWfffaZ8PDwsOBE6pEkSWg0GvHixQvt91l9aTQalae2rKVLl4o///xT+/3Dhw/FBx98oPfVv39/Fae0rH79+onSpUuL1NRU7bK7d+9qf7cuX74sLl++LOLi4oSPj4/o16+fitOqZ9u2bWL8+PE6y+bNmydKlCghPD09xeDBg0VKSopK01Fux55njT1/hT3PGnuujz03DHtOOcGeG4ZNT8eeZ409l8emZ409p5xgzw3Dnr/CpmeNTdfHnhuGTU/HHesm5O7uLsaOHau3/NChQ0Kj0YgLFy5ol7Vr104EBgZacjzVTJ48WZQqVUqUKlVKaDQa4enpqf3+9S8PDw+h0WjEJ598ovbIFpHxx/i/32f1lV9s3LhRaDQasXXrVu2yjJgVLVpU+Pr6Cl9fX1GyZEmh0WjEpk2bVJzWcipUqCCGDh2qsyzjdYmOjtZZPmLECFGhQgVLjpdrhISEiI4dO2q/P378uLC2thbVqlUT7dq1ExqNRowbN07FCSk3Y8/lsefy2PPMsefy2HPDsOeUE+y5MjZdH3ueOfZcGZueNfaccoI9V8aey2PTM8emy2PPDcOmp+Op4E3oxYsXcHFx0Vvu6uoKIQRu3boFf39/AMB7772HDRs2WHpEVXh6eqJixYoA0k9L4+3trb1ORQZJklCgQAFUr14dffv2VWNMiytZsmSm3+d3ixcvRs2aNdGwYUO9dcuWLUP9+vW139esWRMLFy5EkyZNLDmiKi5fvoxy5crpLLO2tkZAQACcnZ11lpcqVSrfnObpv86cOYM2bdpov1+yZAlcXFywe/duODo6onfv3li8eDG++OILFaek3Io9l8eey2PPM8eey2PPDcOeU06w58rYdH3seebYc2VsetbYc8oJ9lwZey6PTc8cmy6PPTcMm56OO9ZNKDAwEEuXLkWfPn1gZWWlXT5//nxYWVmhTJky2mWPHz+Gg4ODGmNa3Mcff4yPP/4YAFCvXj2MGjUKDRo0UHmq3Kd06dKYPHkymjdvLrt+w4YNGDhwIOLi4iw8mTr+/vtvdOvWzaBtmzdvjrlz55p5otwjLS1N53tXV1ccPXpUbztJkiCEsNRYucqTJ090/ofXn3/+ibCwMDg6OgIAatSogaVLl6o1HuVy7Lk89tww7Lku9lwZe5419pxygj1XxqZnjT3XxZ5njk3PHHtOOcGeK2PPDcOm62LTlbHnWWPT03HHuglFRESgSZMmqFChAlq1agUHBwfs3bsX0dHR6NatGwoXLqzddteuXahcubKK06ojJiZG7RFyrcuXL+Px48eK6x8/fpyvPgl18+ZNlChRQmeZg4MDBg0aBB8fH53l3t7euHXrliXHU03x4sURGxtr0LaxsbEoXry4mSfKnUqUKIGDBw+ia9eu+Pfff3Hy5El8/vnn2vX37t2DnZ2dihNSbsaeZ409V8ae62LP5bHnhmHPKSfYc8Ow6fLYc13suTI2PWvsOeUEe24Y9lwZm66LTZfHnhuGTU+nUXuAN8l7772HNWvWwNraGuPHj8eYMWNw6NAhDB8+HDNmzNDZtk2bNhg/frxKk6pnxYoVCA8PV1zfpUsX/Pbbb5YbKJeRJElx3cGDB+Hm5ma5YVRmb2+v948eR0dHTJo0SXuKpwxPnjyBra2tJcdTzXvvvYdly5bh9u3bmW53+/ZtLFu2DO+9956FJstdOnbsiNmzZ6N58+Zo3Lgx3N3d0aJFC+36w4cP63yqmeh17HnW2PPMseevsOfy2HPDsOeUE+y5Ydh0Zez5K+y5MjY9a+w55QR7bhj2PHNs+itsujz23DBs+v9T7/Lub7Z79+6JGzduiLS0NLVHyVVq1Kghevbsqbi+T58+Ijg42IITqWvy5MmiVKlSolSpUkKj0QhPT0/t969/eXh4CI1GIz755BO1R7aYoKAg0a5dO4O2bdeunahevbqZJ8odLl26JAoUKCAqVaokDh48KLvNwYMHReXKlUWBAgVEXFychSfMHZKTk8XIkSNF1apVRWhoqNi1a5d2XWJioihSpIj44YcfVJyQ8gr2XB57ros9V8aey2PPDcOek6mw58rY9FfYc2XsuTI2PWvsOZkKe66MPdfFpitj0+Wx54Zh09PxVPBm4u7unuU2169fh7e3twWmyT3OnTuHrl27Kq4PCAjA//73PwtOpC5PT09UrFgRQPppaby9vfXeE5IkoUCBAqhevTr69u2rxpiqaNmyJcaMGYO///4bwcHBitv9888/+OOPPzB69GjLDaciX19frFixAh9//DHefvtt+Pv7o1KlSnBycsLjx49x8uRJ/Pvvv3BwcMDy5ctRqlQptUdWhbW1NcaOHYuxY8fqrfPw8MDNmzd1liUnJ2P//v0ICAiAq6urpcakPIA9l8ee62LPlbHn8thzw7DnZCrsuTI2/RX2XBl7roxNzxp7TqbCnitjz3Wx6crYdHnsuWHY9P+n9p79N0lwcLA4fvx4ltulpqaKX375Rbi4uFhgqtzF2dlZ/Pjjj4rrf/zxR1GgQAELTpR7hIaGim3btqk9Rq7x8OFDUbp0aeHs7CwmTJggEhISdNYnJCSICRMmCBcXF1G6dGnx8OFDlSZVx8WLF0XPnj2Ft7e3kCRJ+1WsWDHRo0cPceHCBbVHzFNu3rwpNBqNiI6OVnsUygXY86yx58rYc13seebYc9Niz+l17Llh2HR57Lku9jxrbLrpsOf0OvbcMOy5MjZdF5ueOfbctN7UpktCCKH2zv03hZeXFxITEzF48GCMGTMGDg4Oetv8888/6N27N2JjY9GsWTOsW7dOhUnVU7duXSQlJeHgwYN61+d48eIFatSoAVdXV+zevVulCSk3+ffff9GqVSucOnUKkiTBzc1N+ymxpKQkCCFQoUIFrF69Gm+99Zba46rm0aNHePjwIZydneHi4qL2OHnSrVu34OXlhW3btqF+/fpqj0MqY8+zxp6TMdhzw7DnOcee0+vYc8Ow6WQo9txwbHrOsOf0OvbcMOw5GYNNNwx7nnNvatM1ag/wJjl79iy6du2KX375BRUrVsTGjRu16x48eIDevXujdu3auHfvHv744498GfkRI0bg5MmTqFevHtavX4+4uDjExcVh3bp1CA0NxalTpzBixAi1x1TFihUrEB4erri+S5cu+O233yw3UC7g7++PY8eOYdmyZfjoo4/g6+sLW1tblCxZEu3bt8fSpUtx7NixfB14AHB2doa3tzcDT2Qi7HnW2HNl7Lk+9tww7DmRabHnhmHT5bHn+thzw7HpRKbDnhuGPVfGputj0w3DnpMiNQ+Xf1Pt27dPVKpUSWg0GtGmTRsxa9YsUaRIEWFjYyOGDh0qnjx5ovaIqlqwYIFwcXERGo1G+yVJknBxcRHz5s1TezzV1KhRQ/Ts2VNxfZ8+fURwcLAFJyLKP27evCkkSXrjTktDOcOeZ449l8eeE6mHPSc57HnW2HR97DmRethzksOeZ409l8emE6nnTW26tdo79t9EtWrVwtGjR9G/f3/Mnj0bq1evRvny5bFt2zZUqlRJ7fFUFx4ejtatW2Pr1q24ePEiAMDPzw+NGjWCs7OzytOp59y5c+jatavi+oCAAPzvf/+z4ES5z4sXL3DkyBHcvn0btWvXRqFChdQeiYjeYOx55thzeex51thzIrIk9jxrbLo+9jxr7DkRWRJ7njX2XB6bnjU2ncg43LFuBikpKfjxxx+xePFiuLu7w9HREefPn8fChQsxZswYFChQQO0RVefi4oI2bdqoPUauIoRAUlKS4vr79+8jOTnZcgPlMlOnTsXo0aORlJQESZKwdetW1K9fH3fv3kW5cuUwfvz4TP+RRERkLPY8a+y5PvY8c+w5EVkae24YNl0Xe5459pyILI09Nwx7ro9NzxybTmQ8XmPdxHbv3o0qVaogIiICrVq1wpkzZ3DmzBn07t0bkydPRoUKFfLttV5e9+jRI5w8eRK7d+/Grl279L7yo8DAQPzvf//Dy5cv9da9ePECy5cvR2BgoAqTqW/BggUYPHgwwsLCMH/+fAghtOsKFSqE+vXrY8WKFSpOSERvGvbcMOy5PvZcGXtORJbGnhuOTdfFnitjz4nI0thzw7Hn+th0ZWw6UTapdxb6N0/Xrl2FRqMR/v7+YuvWrXrrDx48KAIDA4VGoxEtW7YU165dU2FKdd29e1d89NFHwsbGRudaL//97/xo06ZNQqPRiHfeeUesW7dOXLx4UVy8eFGsXbtWBAcHC41GIzZs2KD2mKqoWLGiaNmypRAi/T303+tyjBs3ThQrVkyt8egN8KZe74Wyhz3PGnuujD1Xxp6TubHn9Dr23DBsujz2XBl7TubGntPr2HPDsOfK2HRlbDqZ25vadJ4K3oSWLl2KL7/8El9//TXs7Oz01gcFBeHQoUOYMmUKIiIiUKFCBTx48ECFSdXTo0cPrF+/HgMHDkSdOnXg7u6u9ki5RpMmTTBv3jwMGjQILVu21C4XQsDZ2Rlz5sxB06ZN1RtQRf/++y8GDhyouN7DwwOJiYkWnIhys6dPn+KTTz5BmzZt0LFjR4Nu4+rqigULFqBixYpmno7yAvY8a+y5MvZcGXtOxmDPKafYc8Ow6fLYc2XsORmDPaecYs8Nw54rY9OVselkDDb9Fe5YN6Fjx46hfPnymW6j0WgwZMgQtG3bNtM/Wm+qLVu2YMiQIRg/frzao+RK4eHhaN26NbZu3YqLFy8CAPz8/NCoUSM4OzurPJ163NzccPfuXcX1p0+fRtGiRS04EeVmjo6O2LZtG5o0aWLwbezt7dG5c2czTkV5CXueNfY8c+y5PPacjMGeU06x54Zh05Wx5/LYczIGe045xZ4bhj3PHJsuj00nY7Dpr3DHugm9HvmEhAT8+eefOHPmDB4+fAhnZ2dUqFABYWFhKFasGEqUKIHVq1erOK06HB0d4evrq/YYuZqLiwvatGmj9hi5yvvvv4/Zs2ejb9++eutOnTqFOXPmoGvXripMRrlVSEgI9u/fjx49eqg9CuVB7HnW2POssef62HMyFntOOcGeG4ZNzxx7ro89J2Ox55QT7Llh2POssen62HQyFpueThJCCLWHeJM8e/YMQ4cOxdy5c5GSkoL/vrw2Njbo3r07fv75Zzg4OKg0pXo+++wznDhxAlu3blV7lFzr0aNHiI+Px/379/XePwDw7rvvqjCVuhISEvD2229DCIEPPvgAs2fPxieffILU1FT8/vvv8PLywoEDB1CoUCG1R6VcIi4uDo0bN0b79u3Ru3dvFC9eXO2RKI9hzzPHnmeNPdfHnpOx2HPKKfY8a2x65thzfew5GYs9p5xiz7PGnmeNTdfHppOx2PR03LFuQikpKWjUqBF27NiBevXq4dNPP0VAQACcnZ3x6NEjxMbGYvHixYiJiUG9evWwZcsWWFlZqT22Re3btw8DBgxA4cKF0bNnT5QoUUL2NahWrZoK06krMTER/fv3x++//47U1FQA6dd6kSRJ578z1uU3t2/fxsiRI/HHH38gKSkJAODs7Iw2bdpg3Lhx8PT0VHdAylWcnZ2RkpKCly9fAgCsra31rsUlSVK+vO4WZY09zxp7row9zxx7TsZgzykn2HPDsOny2PPMsedkDPaccoI9Nwx7roxNzxybTsZg09Nxx7oJTZs2DYMGDcKMGTPQp08fxe1mzZqFvn37YurUqejfv78FJ1SfRqPR/ndGvF6Xn0PWunVrrF+/HgMHDkSdOnXg7u4uu13dunUtPJm6Xrx4gb/++gu+vr6oUqUKAODOnTtIS0tD4cKFdd5TRBnCw8Nl/8b814IFCywwDeU17HnW2HNl7Lk89pyygz2nnGDPDcOmy2PP5bHnlB3sOeUEe24Y9lwZmy6PTafsYNPTcce6CdWsWRPe3t4GXculZcuWuH79Og4ePGiByXKPRYsWGbRd586dzTxJ7uPk5IS+ffti/Pjxao+SqwghYG9vjylTpqB3795qj0NE+QB7njX2XBl7Lo89JyJLY88Nw6bLY8/lsedEZGnsuWHYc2Vsujw2nSj7rNUe4E1y5swZdOvWzaBtw8LCMGzYMDNPlPvkx3gbytHREb6+vmqPketIkoS33noLd+/eVXsUIson2POssefK2HN57DkRWRp7bhg2XR57Lo89JyJLY88Nw54rY9PlselE2ccd6yYkSRLS0tKM2p4owyeffILVq1ejb9++ao+S64wcORKfffYZPvzwQ5QtW1btcSgPuXbtGo4ePYoHDx7I/n3+9NNPVZiKcjv2nHKCPVfGnlN2seeUHew55QR7row9p+xizyk72HPKKTZdGZtO2ZXfm85TwZtQzZo14eXlhbVr12a5bcuWLZGQkIADBw5YYLLco2vXrlluI0kS5s2bZ4Fpcpd9+/ZhwIABKFy4MHr27IkSJUrAyspKb7tq1aqpMJ26Bg4ciOjoaJw/fx6hoaHw9fWFg4ODzjaSJGHKlCkqTUi5zfPnz9G5c2f8/vvvSEtLgyRJyMjd6/8jKz9eW4qyxp5njT1Xxp4rY8/JWOw55QR7bhg2XR57row9J2Ox55QT7Llh2HNlbLoyNp2Mxaan4451E5o6dSqGDBmC6dOno0+fPorbzZo1C/369cOkSZMwcOBAC06oPl9fX71PDqampuLGjRtITU1F4cKFUaBAAcTFxak0oXo0Go32v+U+XSmEgCRJb/wfJTmvvzZK8utrQ/I+++wzTJs2Dd9//z1q1aqF0NBQLFq0CF5eXpg8eTISEhKwePFiVKpUSe1RKRdiz7PGnitjz5Wx52Qs9pxygj03DJsujz1Xxp6Tsdhzygn23DDsuTI2XRmbTsZi09Nxx7oJpaSkoGHDhti9ezcaNGiATp06ISAgAM7Oznj06BGOHz+OJUuWYNu2bQgJCUF0dDSsrXk2fgBITk5GVFQUJk+ejK1bt6JUqVJqj2RxixYtMmg7XjOHKGs+Pj4ICwvD7NmzkZiYiMKFC2Pbtm2oX78+AKB+/fooW7YsIiMjVZ6UciP2PPvYc/acyJTYc8oJ9jxn8nvT2XMi02HPKSfY85zJ7z0H2HQiU2LT03HHuok9ffoUQ4YMwfz58/WuLSCEgJWVFbp06YJJkyahQIECKk2Ze/Xt2xfx8fHYuHGj2qNQLnTy5Els2rQJly9fBgCUKlUKTZo0eeM/AUXGs7e3x/Tp09G9e3c8ffoUTk5OWLt2LT744AMAwIwZM/Dtt9/i1q1bKk9KuRV7njPsOWWGPSdDseeUU+x5zrHppIQ9J0Ox55RT7HnOseeUGTadDMWmp+PHt0zM0dERUVFR+Oabb7B582acPn0ajx49grOzM8qXL48mTZqgePHiao+ZawUEBGDJkiVqj0G5zIsXL9CrVy8sWbIEQgjtaWrS0tIwYsQIdOzYEXPnzoWtra3Kk1JuUaRIESQmJgJI/7vs7u6Oc+fOaSP/8OFDPH/+XM0RKZdjz3OGPSc57DkZiz2nnGLPc45Np/9iz8lY7DnlFHuec+w5yWHTyVhsejruWDcTb29vdO/eXfv92bNnsXLlSvzwww8oV64cwsPD4eLiouKEudPWrVvh6Oio9hiq6Nq1a5bbSJKEefPmWWCa3OWLL77A4sWL0bdvXwwYMAB+fn6QJAn//vsvpk6disjISHh4eGDy5Mlqj0q5xNtvv409e/bgiy++AAB88MEHmDBhAry8vJCWloZJkyYhODhY5SkpL2DPs4c9zxx7zp6TYdhzMhX2PPvya9PZc2XsORmLPSdTYc+zL7/2HGDTM8Omk7HY9HQ8FbwJTZ8+HVOnTsW+fftQqFAh7fINGzagbdu2SE5ORsbLXbp0afz999862+UH3377rezypKQk7Nq1C0eOHMGIESPwww8/WHgy9fn6+kKSJJ1lqampuHHjBlJTU1G4cGEUKFAAcXFxKk2onkKFCqFp06aK18Tp1KkTNm/ejLt371p4Msqt9uzZg5UrV2L8+PGws7PD1atX0bBhQ1y4cAEA4Ofnhw0bNqBs2bIqT0q5EXueNfZcGXuujD0nY7HnlBPsuWHYdHnsuTL2nIzFnlNOsOeGYc+VsenK2HQyFpv+/wSZzHvvvSfCwsJ0liUnJwtPT0/h6OgoFi5cKE6ePCl++uknYW1tLQYPHqzSpOqRJEn2y8PDQ9SoUUNERUWJtLQ0tcfMVV6+fCmmTZsm/Pz8RFxcnNrjqMLFxUXMnDlTcf3MmTOFq6ur5QaiPCk1NVUcO3ZMnDhxQiQnJ6s9DuVi7HnW2HPjsefsOZkGe06GYs8Nw6Ybhz1nz8k02HMyFHtuGPbceGw6m06mkR+bziPWTah48eLo0aMHIiIitMu2bt2Kxo0bY+TIkfj++++1yzt27IhDhw7h3LlzaoxKeVDfvn0RHx+PjRs3qj2KxbVr1w4vX77EmjVrZNe3aNECdnZ2+O233yw7GBG9kdhzMif2nD0nIstgz8mc2HP2nIgsgz0nc2PT2XQiY/Ea6yaUmJiIEiVK6CyLjo6GJElo1aqVzvLatWvjjz/+sOR4qvDw8MDs2bPRtm1bAOmnpWndujUqVaqk8mR5T0BAAJYsWaL2GKr47rvv0K5dO7Ru3Rr9+vWDv78/AODChQuYMWMG4uPj8euvv+LevXs6t/Pw8FBjXFLBrl27snW7d99918ST0JuAPdfHnpsOe86ekzL2nEyJPZfHppsGe86ekzL2nEyJPZfHnpsOm86mkzI2XR53rJtQkSJFcPPmTZ1lu3fvhqOjIwICAnSW29rawtbW1pLjqeLx48d4+vSp9vvRo0fD39+fkc+GrVu3wtHRUe0xVFG+fHkAwIkTJ7B27VqddRkn3ahQoYLe7VJTU80/HOUKoaGhetdLyowQApIk8T1Csthzfey56bDn7DkpY8/JlNhzeWy6abDn7DkpY8/JlNhzeey56bDpbDopY9Plcce6CQUFBWHRokUYMGAAnJ2dcerUKRw4cAAtWrSAtbXuS3327FkUL15cpUktx8/PD6tWrUKdOnXg4uICAHjy5Inep5z+Kz9+6unbb7+VXZ6UlIRdu3bhyJEjGDFihIWnyh2++eYbo/6AU/4TExOj9gj0BmHP9bHnhmPPlbHnlBX2nEyJPZfHphuGPVfGnlNW2HMyJfZcHntuODZdGZtOWWHT5fEa6yZ04sQJ1KhRA25ubqhYsSIOHz6Mp0+fYv/+/ahevbrOtn5+fqhfvz7mzJmj0rSWsWTJEnTp0gXGvs3e9E+0yNFoNLLL3d3d4efnh+7du6NHjx6MHRGRmbHn+thzw7HnRES5A3suj003DHtORJQ7sOfy2HPDselEZGo8Yt2EKleujO3bt2Ps2LGIi4tDcHAwhg4dqhf5HTt2wNHRER9++KFKk1pOp06dULNmTezYsQO3bt3C6NGj0apVK1SpUkXt0XKdtLQ0tUcgIiKw53LYc8Ox50REuQN7Lo9NNwx7TkSUO7Dn8thzw7HpRGRqPGKdLKpUqVKYMmUKmjdvrvYoqvPw8MDs2bPRtm1bAOmnpWndujWvhUNkAl27ds1yG0mSMG/ePAtMQ/TmYc9fYc+JzIc9JzI/Nj0de05kPuw5kfmx56+w6UTmw6an4xHrZFGXLl0yavu7d++iZs2aWLZsGWrVqmWmqdTx+PFjPH36VPv96NGj4e/vz8gTmcD27dv1TuGUmpqKGzduIDU1FYULF0aBAgVUmo4o72PPX2HPicyHPScyPzY9HXtOZD7sOZH5seevsOlE5sOmp+OOdcrVUlNTcfnyZTx79kztUUzOz88Pq1atQp06deDi4gIAePLkCe7du5fp7Tw8PCwxHlGedvnyZdnlycnJiIqKwuTJk7F161bLDkWUj7Hn+thzoqyx50S5z5vadPacyHzYc6Lc503tOcCmE5kTm56Op4KnXO3WrVvw8vLCtm3bUL9+fbXHMaklS5agS5cuMPZXMDU11UwTEeUfffv2RXx8PDZu3Kj2KET5Anuujz0nyjn2nMjy3tSms+dE6mHPiSzvTe05wKYTqSm/NJ1HrBOppFOnTqhZsyZ27NiBW7duYfTo0WjVqhWqVKmi9mhEb7yAgAAsWbJE7TGI6A3AnhOphz0nIlNhz4nUw54TkSmx6UTqyS9N5451IhWVLVsWZcuWBQAsWLAAnTt3RvPmzVWeiujNt3XrVjg6Oqo9BhG9IdhzInWw50RkSuw5kTrYcyIyNTadSB35pencsU6US1y6dMmo7e/evYuaNWti2bJlqFWrlpmmIsqbvv32W9nlSUlJ2LVrF44cOYIRI0ZYeCoiyg/YcyLTYc+JSC3sOZHpsOdEpCY2nch02PR03LFOlEelpqbi8uXLePbsmdqjEOU6o0ePll3u7u4OPz8/zJo1Cz169LDsUEREMthzImXsORHlFew5kTL2nIjyEjadSBmbno471omI6I2Tlpam9ghERESUQ+w5ERFR3seeExERvRnY9HQatQcgyoytrS3q1q0Ld3d3tUchIiKibGLPiYiI3gxsOhERUd7HnhMRZR+PWCeLu337Nq5fv45nz57ByckJ/v7+cHR0lN3W3d0dMTExFp6QiPKaK1euZOt2Pj4+Jp6EKP9gz4nI1NhzInWw6URkSuw5kTrYcyIyNTZdHnesk0UkJiZi3LhxWLFiBRISEnTWaTQaBAcH4/PPP0fLli3VGZCI8jRfX19IkmT07VJTU80wDdGbiz0nInNiz4ksh00nInNhz4kshz0nInNi0+VxxzqZXXx8POrUqYOEhASUL18exYoVw+nTp5GcnIyuXbsiKSkJO3fuRJs2bdCjRw/MmjVL7ZGJKI+ZP3++TuTT0tIwZcoUxMfHo2PHjihbtiwA4OzZs1i+fDl8fX0xcOBAtcYlypPYcyIyN/acyDLYdCIyJ/acyDLYcyIyNzZdHnesk9kNHToUz58/x5EjR1ClShUA6Z+ma9++PS5duoTNmzcjLS0NEyZMwMiRIxEUFITu3burPDUR5SXh4eE6348dOxbPnz/Hv//+i4IFC+qsGz16NEJCQnDz5k0LTkiU97HnRGRu7DmRZbDpRGRO7DmRZbDnRGRubLo8jdoD0JsvOjoagwcP1gYeAAoWLIiffvoJW7Zswb///guNRoMvvvgCbdu2xcyZM1WcNu+wtbVF3bp14e7urvYoRLnOrFmz0LNnT73AA0DhwoXRo0cPREZGqjAZUd7FnpsHe06kjD0nMg823fTYcyJl7DmRebDn5sGmEylj09PxiHUyuxcvXsDFxUVvuaurK4QQuHXrFvz9/QEA7733HjZs2GDpEXON27dv4/r163j27BmcnJzg7+8PR0dH2W3d3d0RExNj4QmJ8obExEQ8ffpUcf3Tp0+RmJhowYmI8j723HDsOZFpsOdE5sGmG4Y9JzIN9pzIPNhzw7HpRKbBpqfjEetkdoGBgVi6dClSU1N1ls+fPx9WVlYoU6aMdtnjx4/h4OBg6RFVlZiYiGHDhqFEiRLw8vJCUFAQ6tSpg8DAQLi6uqJOnTpYs2aN2mMS5SnBwcGYPHkyDh8+rLfu0KFDmDJlCt5++20VJiPKu9jzzLHnRKbHnhOZB5uujD0nMj32nMg82PPMselEpsemp5OEEELtIejNtnXrVjRp0gR+fn5o1aoVHBwcsHfvXkRHR6Nbt26YPXu2dtvWrVvj/v37+eZTYfHx8ahTpw4SEhJQvnx5ODo64vTp00hOTkbXrl2RlJSEnTt34ubNm+jRowdmzZql9shEecLp06cRGhqKxMREBAcH46233gIAXLhwAX///Tc8PDywY8cOVKxYUeVJifIO9lwZe05kHuw5kXmw6fLYcyLzYM+JzIM9V8amE5kHm/7/BJEFrF+/XlSoUEFIkiQkSRLu7u5ixIgR4uXLlzrbLV26VBw4cEClKS2vbdu2onDhwiI2Nla77O7du6JBgwYiLCxMCCFEamqqGDdunNBoNGLOnDlqjUqU59y8eVMMHjxYlC1bVtjb2wt7e3tRtmxZMWTIEHHjxg21xyPKk9hzeew5kfmw50TmwabrY8+JzIc9JzIP9lwem05kPmy6EDxinSzq/v37ePHiBYoUKQJJktQeR3UeHh4YOnQoRo4cqbP88OHDqFmzJs6dO6e9Fk779u1x4cIFHDlyRI1RiYiItNhzXew5ERHlVWz6K+w5ERHlVey5LjadiMyJ11gni3J3d0fRokUzDfz169ctOJG6Xrx4ARcXF73lrq6uEELg1q1b2mXvvfcezp07Z8nxiIiIZLHnuthzIiLKq9j0V9hzIiLKq9hzXWw6EZmTtdoD0JuvVq1amD17NipXrpzpdmlpaZg8eTLGjBmDBw8eWGg6dQUGBmLp0qXo06cPrKystMvnz58PKysrlClTRrvs8ePHcHBwUGNMojzpzJkzWLBgAeLi4nD//n389wQtkiQhOjpapemI8h72XBl7TmQ+7DmR6bHp8thzIvNhz4lMjz1XxqYTmQ+bzh3rZAGXL19G9erVMXjwYIwZM0Y2VP/88w969+6N2NhYNGvWTIUp1REREYEmTZqgQoUKaNWqFRwcHLB3715ER0ejW7duKFy4sHbbXbt2ZfkPJSJKt2TJEnTp0gU2NjYoW7Ys3N3d9bbhlVCIjMOeK2PPicyDPScyDzZdHntOZB7sOZF5sOfK2HQi82DT/58aF3an/CUpKUn06tVLaDQaUapUKbFhwwa9dVZWVsLHx0esXr1avUFVsn79elGhQgUhSZKQJEm4u7uLESNGiJcvX+pst3TpUnHgwAGVpiTKW0qXLi2CgoLEnTt31B6F6I3BnmeOPScyPfacyDzYdGXsOZHpsedE5sGeZ45NJzI9Nj2dJER++PgA5Qb79+9Hz549cfr0abRq1QrvvfceIiIicO/ePQwaNAhjxoyBo6Oj2mOq5v79+3jx4gWKFCmS6fVwiChrDg4OmDhxIvr06aP2KERvHPY8c+w5kemw50TmxaYrY8+JTIc9JzIv9jxzbDqR6bDp6bhjnSwqJSUF/fv3x+zZsyFJEsqXL48VK1agUqVKao+WJ1y/fh3e3t5qj0GU67399tto1KgRvvvuO7VHIXojsec5w54TGYY9JzI/Nj372HMiw7DnRObHnucMm05kGDY9nUbtASj/SElJwY8//ojFixfD3d0dxYoVw/nz57Fw4UI8efJE7fFUUatWLZw4cSLL7dLS0jBx4kRUqFDBAlMR5X0TJ07EvHnzsG/fPrVHIXrjsOf62HMi82DPicyLTdfFnhOZB3tOZF7suT42ncg82PR0PGKdLGL37t3o1asXzp49i48//hiTJk2Co6MjRo4ciZkzZ8Lb2xvTpk1D8+bN1R7Vory8vJCYmIjBgwdjzJgxcHBw0Nvmn3/+Qe/evREbG4tmzZph3bp1KkxKlLc0b94cFy5cwPnz51GhQgX4+PjAyspKZxtJkrB27VqVJiTKm9hzeew5kXmw50Tmw6brY8+JzIM9JzIf9lwem05kHmx6Ou5YJ7Pr1q0bFi5ciNKlSyMyMhINGzbUWX/o0CH07NkTsbGxaN68OaZPn55vTr3y4MEDfPHFF5gzZw5KliyJadOmoWnTpjrr5s6dC29vb0yZMgUtW7ZUd2CiPMLX1zfL6yZJkoS4uDgLTUSU97HnythzIvNgz4nMg02Xx54TmQd7TmQe7LkyNp3IPNj0dNyxTmZnZ2eHYcOG4euvv4adnZ3sNmlpaZgyZQoiIiIgSRIePHhg4SnVtX//fvTs2ROnT59Gq1at8N577yEiIgL37t3DoEGDMGbMGDg6Oqo9JhER5WPsedbYcyIiygvY9Myx50RElBew51lj04nIHLhjnczuzJkzKF++vEHbXr16FQMHDsTq1avNPFXuk5KSgv79+2P27NmQJAnly5fHihUrUKlSJbVHIyIiYs8NxJ4TEVFux6ZnjT0nIqLcjj03DJtORKbGHetkUQkJCfjzzz9x5swZPHz4EM7OzqhQoQLCwsJQrFgxtcdTTUpKCn788Uf8+OOPcHBwgKOjI27duoWBAwdizJgxKFCggNojEuVZjx49woMHD5CWlqa3zsfHR4WJiPI+9lwee05kPuw5kXmw6frYcyLzYc+JzIM9l8emE5lPvm66ILKAp0+fir59+wpbW1uh0WiEJEk6X7a2tqJv377i6dOnao9qcbt27RLly5cXkiSJDh06iFu3bolHjx6JAQMGCCsrK+Hj4yPWrl2r9phEec7MmTOFv7+/0Gg0il9EZBz2XBl7TmQe7DmRebDp8thzIvNgz4nMgz1XxqYTmQebLoRG7R379OZLSUlB06ZNERkZiZCQEMyfPx9HjhzBhQsXcOTIESxYsAAhISGIjIxEs2bNkJqaqvbIFtOtWzeEhoYiOTkZW7ZswbJly+Dp6QknJydMnToVf//9NwoWLIhWrVqhVatWuH79utojE+UJs2bNQr9+/eDv74/vv/8eQggMHjwYI0aMQNGiRREQEIB58+apPSZRnsKeK2PPicyDPScyDzZdHntOZB7sOZF5sOfK2HQi82DT/5+6+/UpP5g6daqQJEnMnDkz0+0iIyOFJEli2rRpFppMfba2tuKrr74Sz58/V9wmNTVVTJw4UTg7OwsXFxcLTkeUd1WoUEGEhYUJIYS4e/eukCRJREdHCyGESEpKEv7+/uLnn39Wc0SiPIc9V8aeE5kHe05kHmy6PPacyDzYcyLzYM+VselE5sGmp+M11snsatasCW9vb6xevTrLbVu2bInr16/j4MGDFphMfWfOnEH58uUN2vbq1asYOHCgQa8jUX5nb2+PiRMnom/fvnj48CHc3NywadMmhIWFAQB++uknzJ49GxcvXlR5UqK8gz1Xxp4TmQd7TmQebLo89pzIPNhzIvNgz5Wx6UTmwaans1Z7AHrznTlzBt26dTNo27CwMAwbNszME+Uerwc+ISEBf/75J86cOYOHDx/C2dkZFSpUQFhYGIoVK4YSJUow8EQGcnV1RUpKCgDAxcUFjo6OuHr1qna9s7Mzbt68qdZ4RHkSe66MPScyD/acyDzYdHnsOZF5sOdE5sGeK2PTicyDTU/HHetkdpIkIS0tzajt85Nnz55h6NChmDt3LlJSUvDfk0jY2Nige/fu+Pnnn+Hg4KDSlER5S6VKlRAbG6v9Pjg4GJGRkXj//feRlpaGqKgolClTRsUJifIe9jxz7DmR6bHnRObBpitjz4lMjz0nMg/2PHNsOpHpsenpNGoPQG++cuXK4c8//zRo2z///BPlypUz80S5R0pKCpo2bYrIyEiEhIRg/vz5OHLkCC5cuIAjR45gwYIFCAkJQWRkJJo1a4bU1FS1RybKEz755BOcPHkSL168AACMGTMGZ86cgY+PD3x9fXHu3Dl8//33Kk9JlLew58rYcyLzYM+JzINNl8eeE5kHe05kHuy5MjadyDzY9P+n5gXeKX+YMmWK0Gg0YubMmZluFxkZKTQajZgyZYqFJlPf1KlThSRJBr02kiSJadOmWWgyojfPxYsXxeTJk8W0adPEuXPn1B6HKM9hz5Wx50SWw54T5RybLo89J7Ic9pwo59hzZWw6keXkx6ZLQvznHBhEJpaSkoKGDRti9+7daNCgATp16oSAgAA4Ozvj0aNHOH78OJYsWYJt27YhJCQE0dHRsLbOH1cpqFmzJry9vQ26jkvLli1x/fp1HDx40AKTERER6WLPlbHnRESUl7Dp8thzIiLKS9hzZWw6EZlT/vhLSqqytrbGpk2bMGTIEMyfPx/R0dE664UQsLKyQrdu3TBp0qR8E3gAOHPmDLp162bQtmFhYRg2bJiZJyIiIpLHnitjz4mIKC9h0+Wx50RElJew58rYdCIyp/zz15RU5ejoiKioKHzzzTfYvHkzTp8+jUePHsHZ2Rnly5dHkyZNULx4cbXHtDhJkpCWlmbU9kSkT6PRGP37IUkSUlJSzDQR0ZuJPZfHnhOZBntOZDlsuj72nMg02HMiy2HP5bHpRKbBpsvjqeBJVWfPnsXKlStx48YNlCtXDuHh4XBxcVF7LIupWbMmvLy8sHbt2iy3bdmyJRISEnDgwAELTEaUt4wePTpb/wiOiIgwwzRE+Q97zp4TmQJ7TqS+/Nx09pzINNhzIvXl554DbDqRqbDpCtS7vDvlF9OmTRNvvfWWuHPnjs7y9evXCzs7O6HRaIQkSUKSJOHn56e33ZtsypQpQqPRiJkzZ2a6XWRkpNBoNGLKlCkWmoyIiEgXe66MPScioryETZfHnhMRUV7Cnitj04nInHjEOpldo0aNYGVlhc2bN2uXpaSkwNvbG48fP8bMmTMRFBSEjRs34quvvkL//v0xadIkFSe2nJSUFDRs2BC7d+9GgwYN0KlTJwQEBMDZ2RmPHj3C8ePHsWTJEmzbtg0hISGIjo7OV9fDITLW1atXodFo4O3tDQB4/vw5Zs6cqbdd8eLF0a5dO0uPR5SnsefK2HMi02LPicyLTZfHnhOZFntOZF7suTI2nci02PT/UHvPPr35vL29xejRo3WWbdmyRUiSJL766iud5R06dBBlypSx5Hiqe/LkiejZs6ewtrYWGo1G50uSJGFtbS169OghHj9+rPaoRLna8ePHhZWVlZg2bZp22d27d7Wfzn39y9raWhw/flzFaYnyHvY8c+w5kWmw50Tmx6YrY8+JTIM9JzI/9jxzbDqRabDp+vgxHDK7xMRElChRQmdZdHQ0JElCq1atdJbXrl0bf/zxhyXHU52joyOioqLwzTffYPPmzTh9+jQePXoEZ2dnlC9fHk2aNEHx4sXVHpMo14uKikLJkiXRt29fvXXLli3DO++8AwBIS0tDaGgooqKiMH36dEuPSZRnseeZY8+JTIM9JzI/Nl0Ze05kGuw5kfmx55lj04lMg03Xxx3rZHZFihTBzZs3dZbt3r0bjo6OCAgI0Flua2sLW1tbS46Xa3h7e6N79+7a78+ePYuVK1fihx9+QLly5RAeHg4XFxcVJyTK3WJiYtC6dWtoNBq9dUWKFEHJkiW133fo0AHr1q2z5HhEeR57bhj2nChn2HMi82PTs8aeE+UMe05kfuy5Ydh0opxh0/XpvxJEJhYUFIRFixbh0aNHAIBTp07hwIEDaNy4sd61S86ePZuvPik2ffp0lClTBnfv3tVZvmHDBlStWhWjR4/GrFmzMHjwYFSrVk1vOyJ65fLlyyhXrpzOMmtra+01lF5XqlQpxMfHW3I8ojyPPVfGnhOZDntOZH5sujz2nMh02HMi82PPlbHpRKbDpuvjjnUyu4iICMTHx+Ott95CgwYNULt2bUiShC+//FJv29WrV2tPHZEfrFu3Dn5+fihUqJB2WUpKCrp16wYrKyvMnz8fJ06cwLhx4xAfH4+xY8eqOC1R7peWlqbzvaurK44ePYoaNWroLJckCUIIS45GlOex58rYcyLTYs+JzItNl8eeE5kWe05kXuy5MjadyLTYdF3csU5mV7lyZWzfvh3Vq1dHQkICgoODsWnTJlSvXl1nux07dsDR0REffvihSpNa3unTpxEcHKyzLCYmBnfu3MGQIUPQuXNnVKxYEcOHD0e7du2wadMmlSYlyv2KFy+O2NhYg7aNjY3NV5/UJTIF9lwZe05kOuw5kfmx6fLYcyLTYc+JzI89V8amE5kOm66P11gni3jnnXewcePGTLcJDQ3FiRMnLDRR7pCYmIgSJUroLIuOjoYkSWjVqpXO8tq1a+OPP/6w5HhEecp7772HZcuW4ZtvvoGnp6fidrdv38ayZcvQsWNHC05H9GZgz+Wx50Smw54TWQabro89JzId9pzIMthzeWw6kemw6fp4xDqRiooUKYKbN2/qLNu9ezccHR0REBCgs9zW1ha2traWHI8oTxk6dCiSk5PRoEEDHDp0SHabQ4cOoWHDhkhOTsbnn39u4QmJ6E3FnhOZDntORGphz4lMhz0nIjWx6USmw6br4xHrRCoKCgrCokWLMGDAADg7O+PUqVM4cOAAWrRoAWtr3V/Ps2fP5ovTaBBll6+vL1asWIGPP/4Yb7/9Nvz9/VGpUiU4OTnh8ePHOHnyJP799184ODhg+fLlKFWqlNojE9Ebgj0nMh32nIjUwp4TmQ57TkRqYtOJTIdN1yeJ/HAleaJc6sSJE6hRowbc3NxQsWJFHD58GE+fPsX+/fv1rofj5+eH+vXrY86cOSpNS5Q3xMXF4aeffsLGjRuRkJCgXe7l5YWmTZti+PDh8Pf3V3FCInrTsOdEpseeE5GlsedEpseeE5Ea2HQi02PTX+ER60Qqqly5MrZv346xY8ciLi4OwcHBGDp0qF7gd+zYAUdHR3z44YcqTUqUd5QuXRpRUVEAgEePHuHhw4dwdnaGi4uLypMR0ZuKPScyPfaciCyNPScyPfaciNTAphOZHpv+Co9YJyIiIiIiIiIiIiIiIiIiyoRG7QGIiIiIiIiIiIiIiIiIiIhyM+5YJyIiIiIiIiIiIiIiIiIiygR3rBMREREREREREREREREREWWCO9aJiIiIiIiIiIiIiIiIiIgywR3rREREREREREREREREREREmeCOdSIiIiIiIiIiIiIiIiIiokxwxzoREREREREREREREREREVEmuGOdiIiIiIiIiIiIiIiIiIgoE9yxTkRERERERERERERERERElAnuWCciIiIiIiIiIiIiIiIiIsoEd6wTERERERERERERERERERFlgjvWiYiIiIiIiIiIiIiIiIiIMsEd60RERERERERERERERERERJngjnUiIiIiM/D19YUkSXpfTk5OCAgIwJdffonExETZ24aHh0OSJCxcuNCyQ+dCO3bsgCRJCA0NVXsUWaGhoZAkCTt27FB7FD1CCEyYMAGVKlWCg4OD9j34pktOTkZ0dDSGDRuGGjVqwM3NDTY2NihatCiaN2+OjRs35uj+nzx5ghIlSiAgIABpaWna5ZcvX4YkSfD19c3hM8j9Mt73r38VKFAAXl5eqF27NgYMGIDt27dDCKH2qEREREREREREJmOt9gBEREREb7LatWvD398fAJCWloaEhATs27cP48aNw+LFi7F7926ULl1a5SnV4+vri/j4eFy6dClf7JC0pMjISAwfPhyurq5o0qQJXFxc1B7JInbu3In33nsPAFC0aFGEhISgQIECOH36NNavX4/169ejZ8+emDVrVrY+aPDDDz/g2rVrmDVrFjQa9T6nvGPHDtSrVw9169ZV7YMdAQEBqFq1KgDg5cuXSExMRGxsLPbt24fp06ejSpUqWLhwIQIDA1WZj4iIiIiIiIjIlLhjnYiIiMiMunfvjvDwcJ1lN2/eRN26dXH+/HkMHz4cq1at0ln/448/YsSIEfDy8rLgpJQdixcvxtOnT+Hj46P2KHp+++03AMDKlSu1O5rzA41GgzZt2mDQoEGoU6eOzrpff/0VHTt2xOzZs1G7dm18+umnRt339evX8csvv6BGjRpo2rSpKcfOk1q2bInRo0frLd+9ezeGDh2KAwcOICQkBDt37kRQUJDlByQiIiIiIiIiMiGeCp6IiIjIwooWLYphw4YBAKKjo/XWe3l5oVy5cnB1dbX0aGQkHx8flCtXDo6OjmqPoufKlSsAgLfeekvlSSyrfv36WLVqld5OdQBo37699oMuixcvNvq+Z86ciRcvXqBbt245HfONVqdOHezevRshISF4+vQpOnTogNTUVLXHIiIiIiIiIiLKEe5YJyIiIlJB0aJFAQApKSl667K6xvqKFSvQoEEDeHh4wM7ODiVLlkTXrl1x/vx52e1v3LiBQYMGoUyZMrC3t4ejoyNKlCiBBg0a4Oeff9Zut2DBAkiShMaNGyvOnZCQABsbGzg4OGivEf/6taWFEJg9ezaqV6+OAgUKwNXVFY0aNcL+/ft17mfhwoWQJAnx8fEAgFKlSulcr1nu1NbJycn46aefULFiRTg4OKBgwYJo3bo1zpw5ozjv/fv3ERERgapVq8LZ2RmOjo6oXLkyvv/+ezx9+lRv+7S0NO3RzBnX5vb09ERAQAAGDBiAy5cv62yvdI31Fy9eYMKECahevTqcnZ1ha2uLokWLokaNGhg+fDju3bunOLOce/fuYeTIkahYsSIcHR3h7OyM6tWrY/z48Xj27JnsTJcuXQKg+9rKHV2s5Pr16xg2bBgqV64MZ2dnFChQAGXKlEF4eDj27dunt/21a9cwYMAAvPXWW7C3t4erqytq166NqKgo2Z2qGe+B8PBw3Lt3D4MHD4afnx/s7OwQGhqqs210dDRat24NLy8v2NrawtPTE61atdJ7Xxki47TkV69eNep2L1++xJw5c2BnZ4ePPvoo021TUlIwfvx47Xu1UKFCaNeuHc6ePat4m2fPnuGXX35BcHAw3NzcYG9vj7Jly2L48OHa37UMoaGhqFevHoD0U9+//rvz+iUV7ty5g6lTp+L9999HqVKl4ODgABcXFwQFBeGnn37C8+fPjXoNjGFra4tZs2YBAC5cuIA1a9aY7bGIiIiIiIiIiCyBp4InIiIiUsGBAwcAABUrVjT4NkIIhIeHY/HixbC2tsa7774LT09PHDlyBAsWLMCvv/6K33//HWFhYdrb3Lx5E0FBQUhISICPjw/CwsJgb2+PhIQEHDt2DIcPH8bQoUMBAB06dMAXX3yBrVu34vz58yhTpozeDFFRUUhJSUGnTp1QsGBBvfVdunTB8uXLUadOHTRr1gzHjh3D1q1bsWvXLuzcuRNvv/02AMDf3x+dO3fGqlWr8OTJE7Rp0wZOTk7a+8n44EGG5ORkvP/++9i3bx/effddlC9fHgcOHMDq1asRExODo0eP6l2j/fTp0wgLC8PVq1fh5eWFkJAQ2NjY4MCBA/j666/x+++/Y8eOHTpnBujevTsWLFgAe3t7hISEoHDhwrh37x7i4uIwffp0NGjQIMtrwaelpaFp06aIjo6Gi4sL6tSpAzc3N9y5cwcXLlzAhAkT0KFDB3h4eGR6Pxni4uJQv359xMfHo3Dhwnj//feRnJyMmJgYfPHFF/j111+xbds2uLu7AwDCwsLg6+sr+9pmXA87K9HR0Wjbti2SkpLg6emJBg0awNbWFpcvX8by5csBAO+88452+4MHDyIsLAz37t2Dj48PWrZsiQcPHmDHjh3Yt28fVq9ejXXr1sHW1lbvse7evYugoCAkJSWhTp06qF69us52Q4cOxS+//AKNRoOgoCDUqVMHV65cwdq1a7F+/XrMmTMHXbp0Meh5Aek7eQEYfamFvXv34s6dOwgJCcnybBLt27fH+vXrUbduXVSpUgUHDhzAypUrsXnzZmzZsgW1atXS2T4hIQFhYWE4ceIEPDw8UKNGDTg7O+PIkSOYMGECVq5ciR07dqBkyZIAoP09/uuvv1CkSBGd3/lChQpp//uvv/7CoEGD4O3tDX9/fwQHB+POnTv4559/MGLECKxduxYxMTGws7Mz6rUwVMWKFREYGIijR49i69ataNOmjVkeh4iIiIiIiIjIIgQRERERmVzJkiUFALFgwQLtstTUVHHt2jUxbdo0YWdnJ6ysrMT69ev1btu5c2e92wohRGRkpAAgChUqJI4ePapdnpaWJiIiIgQA4ebmJm7fvq1dN2bMGAFA9OzZU6Slpenc38uXL8W2bdt0ln311VcCgBg4cKDeXC9fvhRFixYVAMThw4e1yy9duiQACACiZMmS4ty5c9p1KSkpomvXrgKAaNSokeLrdOnSJb11QggRExOjve/AwEBx48YN7bpnz56Jxo0ba5/f654+fSr8/PwEADFq1Cjx4sUL7bonT56Ijz/+WAAQXbp00S6Pj48XAETx4sV1HifD6dOnRXx8vM6yunXrCgAiJiZGu2znzp3aeR8+fKh3PwcPHhR3796Vfb5y3n77bQFANG/eXDx+/Fi7/Pbt26JatWoCgOjQoYPe7bJ6bZVcuXJFuLq6CgBixIgROq+dEELcunVL7N69W/v98+fPtY/Vu3dv8fLlS+26ixcvCl9fXwFAjBw5Uud+FixYoP3ZNmjQQDx48EBvltmzZwsAwt/fX8TGxuqs27lzp3B2dha2trbi/PnzBj23GzduaJ/b1KlTDbpNhlGjRgkAYtiwYbLrX/89KFSokM68KSkpYsCAAdrfkefPn2vXpaWlidq1awsAolu3bjrvmeTkZPH5558LAKJevXo6j5fxu1G3bl3FmU+fPi3279+vt/zevXuiUaNGAoAYP368oS+BVsb7PiIiIsttu3fvLgCIkJAQox+HiIiIiIiIiCg34angiYiIiMyoS5cu2lM0W1lZoXjx4hgwYACqVKmCnTt3olmzZgbfV8Zp27/55hudI48lSUJERASqVKmCpKQkzJkzR7vu1q1bANKPcJUkSef+bGxs0KBBA51lffv2hY2NDRYtWoQnT57orPv9999x8+ZN1KpVC9WqVZOdcdq0aTpHultZWWHs2LEA0k9ZnZycbPDzfZ0kSViwYIHOkez29vYYM2YMAGDbtm062y9atAgXL15Es2bN8N133+kcAe3o6IjZs2fD09MTS5Yswf379wG8eq2qVaumd8Q8AJQvXx4+Pj5ZzppxP3Xq1IGzs7Pe+qCgINmj/eXs2bMH//zzj3bmAgUKaNcVLlwYs2fPBpB+eYBr164ZdJ9ZmThxIh48eIAPPvgAP/74o95R5p6enggJCdF+v3LlSsTHx6NYsWKYPHkybGxstOtKly6tfd9OmzZN9tTjNjY2mD17NlxcXHSWp6WlaU9dv2LFClSpUkVn/bvvvouvv/4aL1++RFRUVJbPKyUlBZ988gkePHiAypUro1evXlne5nVHjx4FkP4+yMqoUaN05rWyssKECRPg7e2N+Ph4/P7779p1f/31F/bu3YuqVati1qxZOu8Za2trjB8/HpUqVUJMTAxOnjxp1Mzly5dHcHCw3nJ3d3dMmzYNQPrPz5wyjqD/7+nsiYiIiIiIiIjyGu5YJyIiIjKj2rVro3Pnztqvpk2bokSJEjh48CCGDBmiPS11Vq5du4aLFy8CADp37qy3XpIk7emwY2JitMtr1qwJABgxYgT++OMPPH78ONPHKVasGNq2bYsHDx5gyZIlOutmzJgBAOjfv7/sba2trXVOSZ2haNGicHd3x4sXL7K9c83HxwcBAQF6yzN2cl6/fl1n+caNGwGkn5JbjpOTE4KCgpCSkoKDBw8CAMqVKwdnZ2ds2rQJY8eO1V6j3FjVqlWDlZUV5s+fjxkzZuDGjRvZuh8A2mu3h4WFoUiRInrrq1evjoCAAKSlpWHnzp3ZfpzX/fnnnwCAnj17GjXjRx99JHtK8datW8Pd3R2PHj3C4cOH9dYHBgaidOnSesuPHj2KhIQE+Pn5oXr16rKPnXEtdrlrvv9X7969ER0djYIFC2LVqlWyp6XPTMYHJgz5UITc76idnZ32/ZjxmgGv3qtt2rSBtbX+lbo0Gg3effddAIY9z/9KTU1FdHQ0vvvuO/Tt2xddunRBeHi49gMv586dM/o+jZGWlgYAeh/sISIiIiIiIiLKa3iNdSIiIiIz6t69O8LDw3WWpaSk4JtvvsGPP/6IunXr4ty5c7JHNr8uY8dxwYIF9Y7szeDn56ezLQB06tQJW7duxbJly9CmTRtYWVmhQoUKCAkJQdu2bVG/fn29+xk4cCD+97//YcaMGejduzcA4Pjx49izZw+KFCmCtm3byj6+l5eXztHKr3NxccH9+/dlj1g2hNKR4hmvxYsXL3SWx8XFAUh//p06dcr0vu/cuQMAcHZ2xoIFC9ClSxeMGjUKo0aNgpeXF4KDgxEWFoYOHTroXAdeiZ+fHyZNmoRhw4ahf//+6N+/P0qWLIlatWqhWbNm+PDDDw3eqZvxsyxVqlSmjxcbG6v34YLsio+PB5D+QQNDZDWjJEkoVaoU7t+/Lzuj0jXrM36GFy9ezHKnbMbPUMmgQYMwb948uLu7Y+vWrTpnVTDUgwcPAEDx9y+Dm5sb3NzcZNdlvEavn10g43l+/fXX+PrrrzO976ye539duHABrVq1wqlTpxS3efjwoc73Q4cOxd27d/W2W7hwoVGPnSHjvjw8PLTL5s6diz179uhtO2LECIPfd0RERERERERElsYd60REREQWZm1tje+//x5z5szBjRs3sHjxYvTr188sj6XRaLB06VKMHDkSGzduxN69e7F3715ERkYiMjISH3zwAVavXg0rKyvtbYKDg1GzZk0cOHAAO3fuRN26dbVHq/fs2VNxp7BGY76TIRl73xlHySod6f26kiVLav+7TZs2aNiwIdatW4fdu3dj7969WL16NVavXo1vvvkGW7duReXKlbN8/AEDBqBdu3ZYt24d9uzZgz179mDFihVYsWIFIiIisHv3bnh5eRn1nN5UDg4OssszfoZFixZF48aNM72PjNONy/n8888xdepUuLm5YcuWLQgMDMzWnBk7y/+7Izo7hBDa/854niEhIdoPxyipWLGiUY/Ttm1bnDp1Cs2aNcPw4cNRoUIFuLi4wMbGBi9fvpQ9w8CqVau0H654XXZ3rB85cgQAdH5v9uzZg0WLFultGx4ezh3rRERERERERJRrccc6ERERkQo0Gg18fX1x9+5dnDlzJsvtvb29AaRfp/jhw4eyR81mHPmase3rKlSogAoVKmDYsGEQQmD79u3o0KED1q9fj8WLF2tPI59h4MCB+OSTTzB9+nQEBARg2bJlsLa21h7BntuVKFECZ8+eRbdu3RSPsFfi6uqqc6T71atXMWDAAKxduxb9+/c3+JTrRYoUQY8ePdCjRw8AwNmzZ9G1a1fs378fI0aMkN2x+F8ZP8uMn62czH7u2eHj44Nz587h7Nmz8Pf3N8mMGafVN2bGEiVKAEg/S0N2d+oOHz4cEydOhKurK7Zs2YKgoKBs3Q+Qfm15IOtrhSclJSEpKUn2qPXLly8DAIoXL65dlvE8W7RogaFDh2Z7vv86e/Ysjh8/Dk9PT6xevVrvNPNKl6HImNEUTp06hWPHjgEAGjVqpF2+cOHCbP9MiYiIiIiIiIjUwmusExEREakgLS1NuwPLkNOLFy9eXHs0q9wOKSGEdnm9evUyvS9JktCgQQN06NABALQ7vl7Xrl07eHl5Yc2aNRg7diyePHmCVq1aoVixYlnOaoyMo99TUlJMer9NmjQBAPz22285vq8SJUpgzJgxAORfK0OVK1cOX3zxhVH3k3EN8T///FN7je/XHT16FMeOHdO5DndOhYWFAQDmzJlj1Iy//vqr7Kn+V69ejfv378PZ2VnxWulyatSogUKFCuH06dOZnspcyYgRIzBhwgS4urpi69atqFGjhtH38bpq1aoBAE6fPp3ltkuWLNFb9vLlS/z6668AXr1mwKv36sqVK3WOZM9KVr879+7dAwAUK1ZM9trtS5cuNfixsuPly5faD+KUK1cOzZs3N+vjERERERERERGZG3esExEREVlYSkoKRo0apb32sKE7nDKOZv3uu+8QGxurXS6EwPfff49jx47Bzc1Ne4Q0ACxevBiHDx/Wu69Hjx5hx44dAHRPhZ7BxsYGffr0QUpKCn7++WcAQP/+/Q17gkbIOHI3OztOM9OzZ0+ULFkSK1euxBdffIFHjx7pbXPz5k2dncdHjx7Fr7/+imfPnultu379egDyr9V/bd++HZs2bUJycrLOciEENmzYYPD9AOmnB3/77bfx7Nkz9OrVC0+fPtWuu3v3Lnr16gUA+Oijj7RHPufUZ599BmdnZ6xbtw6jRo3Sex63b9/WuT72hx9+CB8fHyQkJOCzzz7T2dF76dIlfP755wDST49vb29v8Bw2NjaIiIiAEAKtWrWSvSZ3amoqtm/fjr///ltn+ahRo/DTTz/Bzc3NJDvVgVcfWNm/f3+W23733Xc4efKk9vu0tDR88cUXuHbtGkqUKIE2bdpo17Vo0QI1atTAgQMH0KVLF9nrqN+/fx+zZs3SeW0zfncuXLig9zMCgDJlysDKygonTpzQ/q5nWL9+PSZNmpTl88iuvXv3ok6dOtizZw+cnJywbNkys14qgoiIiIiIiIjIEngqeCIiIiIzmjt3rs5OrcTERMTGxuLq1asAgK+++grvvPOOQffVq1cv7Nu3D0uWLEFQUBDq1q0LT09PHDlyBOfOnYODgwOWL1+OwoULa2/zxx9/oHPnzihWrBiqVq0Kd3d33L9/H3v37sWDBw9QqVIlnR3x/328sWPH4sWLF6hSpYrJjoh+XZs2bRATE4NPPvkEjRo1gru7OwBg2LBhKFu2bLbvt0CBAti4cSOaNWuG8ePHY/bs2ahSpQqKFy+Op0+f4vz58zhz5gw8PT21zz8+Ph4fffQRHBwcUK1aNZQoUQIpKSk4ceIEzp07B1tbW4wfPz7Lxz5+/DiGDBkCFxcXVKtWDcWKFcOzZ89w5MgRxMfHw9XVFd9++63Bz2X58uWoX78+1q5di1KlSuHdd99FcnIyYmJi8PDhQ1SrVg3Tp0/P9mv1Xz4+Pli1ahXatm2LsWPHYu7cuahVqxZsbGwQHx+Po0ePokOHDggJCQEA2NnZYdWqVQgLC0NkZCQ2bdqE4OBgPHr0CNu3b8fz58/RuHFjREREGD1L//79ceXKFUyYMAF16tRBxYoV4e/vDwcHB9y8eRPHjh1DUlISIiMjERwcDABYt24dxo4dCwDw9/fHjBkzZO+7UKFC2g+NGKJ27dooXLgwDh06pHiqdyD99atevTqqVauG0NBQFCxYEAcPHsTFixdRoEABLF++XOcDBhqNBmvWrEHTpk2xaNEirFq1CgEBAfDx8cHLly8RFxeHEydOIDU1FeHh4dqjz318fBAUFIRDhw6hcuXKCAoKgr29PQoVKoRx48ahUKFC6N+/P6ZMmYIGDRqgTp06KFasGM6dO4cjR45g1KhR+P777w1+/nLWrFmjPfNGcnIy7t27h2PHjuHmzZsAgICAACwIgm3EAAEAAElEQVRcuBBVq1bN0eMQEREREREREeUKgoiIiIhMrmTJkgKA3petra0oWbKkaN++vYiJiZG9befOnQUAsWDBAtn1y5cvF6GhocLNzU3Y2NiIEiVKiPDwcHH27Fm9bXft2iUGDx4satasKYoWLSpsbW1F0aJFRa1atcS0adPE48ePM30eb7/9tgAgoqKiFLe5dOmSACBKliypuE3G63Hp0iWd5ampqeLHH38UFStWFPb29trXKeO1iYmJEQBE3bp1Fe874zZyHj58KMaPHy9q1aqlfb28vLxEjRo1xLBhw8S+ffu02964cUOMGzdOvP/++6JUqVLC0dFRuLi4iAoVKoh+/frJvr5169bVmVcIIf79918xevRo0aBBA+Hj4yPs7e2Fu7u7qFKlihgxYoS4evWq4nNRkpiYKL788ktRvnx5YW9vLxwdHUVgYKAYN26cePr0qextlF5zQ8XHx4tBgwaJsmXLCnt7e+Hk5CTKlCkjunbtKvbv36+3/ZUrV0S/fv1E6dKlha2trXB2dha1atUSkZGRIjk5WW/7BQsWCACic+fOWc6yd+9e0bFjR1GyZElhZ2cnnJ2dRZkyZUTLli3F3Llzxb179/TuN6uvzN6vSkaOHCkAiJkzZ+qte/33IDk5WYwdO1aUK1dO2NnZCQ8PD9GmTRtx6tQpxft+/vy5mDVrlqhXr54oWLCgsLa2Fp6enqJq1aqiX79+4q+//tK7TXx8vOjQoYPw8vIS1tbWes8rLS1NzJs3T1SvXl04OTkJV1dXERISIlasWCGEyPx3JzMZ7/vXvxwcHLR/W/r37y+io6NFWlqa0fdNRERERERERJRbSUIYcSE/IiIiIso3zp8/j3LlysHV1RXXr1+Ho6Oj2iMRqer69evw8/NDpUqVcOjQIbXHISIiIiIiIiIiC+KF7oiIiIhI1jfffAMhBPr06cOd6kQAvL298fnnn+Pw4cPYsGGD2uMQEREREREREZEF8Yh1IiIiItJat24d1q5di1OnTuGff/5B0aJFcebMGcXrSRPlN0+ePEG5cuXg4eGBo0ePQqPhZ5WJiIiIiIiIiPIDa7UHICIiIqLc48iRI5g/fz6cnZ3RsGFDTJw4kTvViV5ToEABXL16Ve0xiIiIiIiIiIjIwnjEOhERERERERERERERERERUSZ43kIiIiIiIiIiIiIiIiIiIqJMcMc6ERERERERERERERERERFRJrhjnYiIiIiIiIiIiIiIiIiIKBPcsU5ERERkpB07dqBHjx6oUKEC3N3dYWNjg4IFC6JmzZro378/tm3bBiGESR5LkiRIkmSS+8pvQkNDIUkSduzYYdTtUlNTsWrVKnz55Zdo1KgRChYsCEmSYG1tne1ZHj58iK+//hpNmzaFn58fXF1dYWtri2LFiqFFixbYuHFjtu87w9q1ayFJEn755Red5QsXLtS+j6pUqYK0tDTZ2+/ZsweSJMHX1zfHs+QHGa9reHi4Ube7fPlyvnmdM34HX/8qUKAAvLy8ULt2bQwYMADbt2832d9LIiIiIiIiIiJzyv7/O0hERESUz9y9excdO3bEli1bAADe3t6oXbs2XF1d8eDBA5w8eRIzZszAjBkzEBgYiCNHjqg8cc75+voiPj4ely5dyhc7AgHg0aNH+PDDD016n7dv38b3338PJycnVKpUCQEBAdBoNPj333+xbt06rFu3Dn379sWMGTOydf8vXrzAZ599hhIlSqBfv36K2504cQJLly7Fp59+mt2nkufs2LED9erVQ926dY3+kMWbIDc8/4CAAFStWhUA8PLlSyQmJiI2Nhb79u3D9OnTUaVKFSxcuBCBgYGqzEdEREREREREZAjuWCciIiIyQFJSEkJCQnDu3DmUK1cOM2fORL169fS2O3nyJCZNmoQVK1aoMCWZgo2NDTp27IjAwEBUq1YNHh4e2p2C2VW0aFHs378fQUFBeke+x8TEoFmzZpg5cyaaN2+Oxo0bG33/06ZNQ1xcHGbMmAF7e3vZbRwdHfH06VN88803aN++Pezs7LL1XIiM1bJlS4wePVpv+e7duzF06FAcOHAAISEh2LlzJ4KCgiw/IBERERERERGRAXgqeCIiIiIDDBgwAOfOnUPp0qWxb98+2Z3qAFCpUiXMmzcPMTExFp6QTKVAgQJYunQpPv/8c9SrVw+urq45vk8nJycEBwfLnk6+Xr16+OijjwBAezYEY6SmpmLatGmwt7dHx44dFberVasWatasifj4eMycOdPoxyEytTp16mD37t0ICQnB06dP0aFDB6Smpqo9FhERERERERGRLO5YJyIiIsrCxYsXsXz5cgDApEmT4O7unuVtatasqfN9fHw8fvrpJ9SvXx8+Pj6ws7ODm5sbQkJCEBUVpXjdayWnT59GREQEateuDW9vb9ja2qJgwYJo2LAhfvvtt0xve/36dQwbNgyVK1eGs7MzChQogDJlyiA8PBz79u0D8Or60fHx8QCAUqVK6Vwn+b+nlD5w4ADatWuHYsWKwdbWFp6envjggw+wdetW2RlevHiBCRMmoHr16nB2doatrS2KFi2KGjVqYPjw4bh37x6A9NfeysoK7u7uePr0qeJzqlixIiRJwqZNmwx9CXOVjB3u2TmKfN26dbhy5QpatmyZ5YcAfvrpJwDA2LFj8fDhQ+MHVWDu9+Prnj17hl9++QXBwcFwc3ODvb09ypYti+HDhyMxMVFn29DQUO2HYHbu3KnzHs64tEHdunUhSRL+97//Kc44fvx4SJKEdu3aGfnKZC4lJQXjx49HxYoV4eDggEKFCqFdu3Y4e/as4m1M/fwB4M6dO5g6dSref/99lCpVCg4ODnBxcUFQUBB++uknPH/+3KTP+3W2traYNWsWAODChQtYs2aN2R6LiIiIiIiIiCgneCp4IiIioixs2LABaWlpcHd3R7NmzbJ1H0uWLMHXX3+NUqVKoUyZMqhduzZu3LiB/fv3Y+/evdiyZQtWrVoFSZIMur+JEydi3rx5KFeuHCpXrgw3NzdcuXIFMTExiI6Oxt9//42JEyfq3S46Ohpt27ZFUlISPD090aBBA9ja2uLy5cvaDw+888478Pf3R+fOnbFq1So8efIEbdq0gZOTk/Z+ihYtqv3vOXPmoHfv3khLS0NgYCBCQ0MRHx+PDRs2YMOGDRg9ejQiIiK026elpaFp06aIjo6Gi4sL6tSpAzc3N9y5cwcXLlzAhAkT0KFDB3h4eMDPzw9NmzbF+vXrsWzZMvTo0UPvOcXExOD06dPw8/NDkyZNDP6Z5BYHDx7Er7/+CkmS8MEHHxh9+4wdkQ0bNsxy29DQUDRp0gSbN2/GTz/9hLFjxxr9eHLM/X7MkJCQgLCwMJw4cQIeHh6oUaMGnJ2dceTIEUyYMAErV67Ejh07ULJkSQBAWFgY7O3t8ddff6FIkSIICwvT3lehQoUAAIMGDcKuXbswffp0fPzxx3ozpqWlITIyEgDQv39/k7xeGdq3b4/169ejbt26qFKlCg4cOICVK1di8+bN2LJlC2rVqqWzvTmePwD89ddfGDRoELy9veHv74/g4GDcuXMH//zzD0aMGIG1a9ciJibGbJcPqFixIgIDA3H06FFs3boVbdq0McvjEBERERERERHliCAiIiKiTHXq1EkAEA0aNMj2fRw4cECcOHFCb/n169dFQECAACB+++03vfUAhNw/2Xbs2CEuXryot/zs2bOiePHiAoD4559/dNZduXJFuLq6CgBixIgR4sWLFzrrb926JXbv3q2zrGTJkgKAuHTpkuzzOn78uLC2thaSJInFixfrrNu0aZOwtbUVAMSWLVu0y3fu3CkAiMDAQPHw4UO9+zx48KC4e/eu9vutW7cKACIgIEB2hjZt2ggA4pdfftFZXrduXQFAxMTEyN7OUJcuXRIAhJWVVY7uJ8PXX38tOnfuLNq1aydq1KghAAhbW1sxbdq0bN1fiRIlBABx6tQp2fULFizQef8eO3ZMaDQa4ejoKBISErTb7d69WwAQJUuWNHoGS7wf09LSRO3atQUA0a1bN533TnJysvj8888FAFGvXj2d+4mJiREARN26dWVnT0lJ0b7Pjxw5ord+/fr1AoCoUqWKzvKM17Vz586y96sk4/0EQBQqVEjExsbqzDJgwADtz+H58+dmf/5CCHH69Gmxf/9+veX37t0TjRo1EgDE+PHjjXqeQrz6HYyIiMhy2+7duwsAIiQkxOjHISIiIiIiIiKyBJ4KnoiIiCgLd+/eBQAULlxYdn1sbCzCw8P1vvbs2aPdpkaNGqhUqZLebYsVK4bx48cDAFauXGnwTHXr1kXp0qX1lpctWxZff/01AGDVqlU66yZOnIgHDx7ggw8+wI8//ghbW1ud9Z6enggJCTF4BgCYMmUKUlJS0KpVK3Tq1ElnXZMmTdCzZ08AwIQJE7TLb926BSD9+srOzs569xkUFISCBQtqv2/YsCEqVqyI2NhYndcUAK5du4a1a9fC0dERXbt2NWp2taxbtw6LFi3Cb7/9hoMHD8LJyQnTp09Hnz59jL6vu3fv4urVq9BoNChbtqxBtwkICECHDh3w9OlTjBkzxujHlGOJ9+Nff/2FvXv3omrVqpg1a5bOe8fa2hrjx49HpUqVEBMTg5MnTxo8u5WVFfr16wcAmDFjht766dOnA4B2G1MaNWoUqlSpojPLhAkT4O3tjfj4ePz+++/adeZ6/gBQvnx5BAcH6y13d3fHtGnTABj39yk7Mo6g/+/p7ImIiIiIiIiIcgueCp6IiIgoh65evYpFixbpLQ8NDdXZMfjixQts2bIFBw8exO3bt/HixQsIIfDo0SMAwLlz54x63MePH2Pz5s04evQo7t69i5cvXwIAbty4IXt/f/75JwBod3abQsa11sPDw2XXd+vWDdOnT8fu3buRmpoKKysrVKtWDVZWVpg/fz7KlCmD1q1bw8vLK9PHGThwIHr16oXp06frvKZRUVFISUlBly5d4ObmZqJnZV7Hjh0DADx8+BDnzp3D5MmT0bNnT6xYsQJr1qyR/bCBkowPKbi6usLKysrg23333Xf47bffMG/ePHz22WcoU6aMUc9Bjrnfjxs3bgQAtGnTRntN+tdpNBq8++67OHnyJPbt2yf7QRYl3bt3x+jRo7F8+XJMmDAB7u7uAIB///0XW7ZsgZubGz755BOD789QnTt31ltmZ2eH9u3bY+LEidixYwc6dOgAwLzPHwBSU1OxY8cO7Nu3Dzdu3MCzZ88ghIAQAoDxf5+MlZaWBgAGXw6DiIiIiIiIiMjSuGOdiIiIKAsZR1LeuXNHdn2zZs20O5+A9COso6Ojdbb5+++/0b59e1y5ckXxcR4+fGjwTOvXr0eXLl0yPbrzv/cXHx8PAChXrpzBj5OV69evAwBKlSolu97Pzw8A8Pz5cyQmJsLT0xN+fn6YNGkShg0bhv79+6N///4oWbIkatWqhWbNmuHDDz/UO3r5k08+wYgRI/DHH3/gxo0b8PLywsuXLzFnzhwAhl/7es2aNdprkr+ue/fuRh+tn1MuLi6oUaMGli1bBjc3N8ycORNjxozBzz//DCD9aPShQ4fq3a5cuXIYMWIEAODBgwfa+zKGr68v+vbti8mTJ2PkyJF6R5O/bujQodqzNrxu4cKF2v+2xPsxLi4OAPD1119rj4JXovS7qsTd3R2dOnVCVFQU5s2bp33dZ86cCSEEunTpAkdHxyzvx5CfWQY3NzfFD4Nk/D5du3ZNu8ycz//ChQto1aoVTp06pbjNf39+hrwvjJFxXx4eHtplc+fO1TtLBQCMGDHCpH/HiIiIiIiIiIgMwR3rRERERFmoVq0alixZgiNHjiAtLQ0ajXFX03n69ClatmyJW7duoUuXLujTpw/8/f3h4uICKysrnD9/HmXLltXZOZ+Z69evo3379nj27BmGDx+Ojh07wtfXF05OTtBoNNiyZQsaN25s8P2pYcCAAWjXrh3WrVuHPXv2YM+ePVixYgVWrFiBiIgI7N69W+codkdHR/To0QPjx4/H7NmzERERgd9//x23bt1CnTp1dE6nnZljx44ZdHYBS+vSpQtmzpyJ1atXa3esP378WHbWunXranfSZuyYNeZDGRm++uorzJ8/H7///jsOHDiguN2qVau0O8Ffl7ED1VLvx4wjmkNCQrQf2FBSsWJFo+9/4MCBiIqKQmRkJD777DM8f/4cCxYsgCRJBp8G3pCfmTFef83M+fzbtm2LU6dOoVmzZhg+fDgqVKgAFxcX2NjY4OXLl7Czs9O7TVbvC2MdOXIEAFC5cmXtsj179si+nuHh4dyxTkREREREREQWxx3rRERERFlo1qwZPv/8c9y/fx+bNm1Cs2bNjLr9rl27cOvWLVSrVg3z58/XW3/hwgWj7m/9+vV49uwZWrVqhZ9++sng+/Px8cG5c+dw9uxZ+Pv7G/WYSry9vXHx4kXExcXJnno64yhbe3t7nSNRAaBIkSLo0aMHevToAQA4e/Ysunbtiv3792PEiBF6O9T69euHX375BbNnz8bIkSO117429Gh1ABg9ejRGjx5tzFO0iAIFCgAAbt++rV3m6+ub5c5oT09PAEBSUpL2VPuGKlSoEIYNG4avv/4aI0aMwLfffiu73eXLlzO9H0u9H0uUKAEAaNGihexR4TlVoUIFNGzYENu2bcPmzZuRkJCApKQkNGnSJMsd2RkM+ZllSEpKQlJSkuxR6xmvefHixbXLzPX8z549i+PHj8PT0xOrV6/WO8280s8vq/eFMU6dOqW9REKjRo20yxcuXJjtHfVERERERERERKZm3OFWRERERPmQv78/2rdvDwD47LPPtKffNtS9e/cApO9IlLN06dJs3V/JkiX11gkhsHz5ctnbhYWFAYD29OmGyDgle0pKiuz60NBQAMpHqWZ8kKBOnTqy14V+Xbly5fDFF18AeHUd8tf5+PigZcuWSEhIwDfffIN9+/ahWLFiaN26tQHPJHfLuHSAsdc6L1SoEEqUKAEhBM6ePWv04w4ZMgRFixZFTEwMNm/ebPTtAcu9H5s0aQIAWLlypVFHv2f1Hn7doEGDAADTp0/HjBkzABj3wQ1jLVmyRG/Zy5cv8euvvwJ49fsFmO/5Z/z8ihUrJvs7auzfJ2O9fPkSvXv3BpD+N6B58+ZmfTwiIiIiIiIiouzijnUiIiIiA8yYMQP+/v64cOEC3nnnHezcuVN2u8uXL+tcFxkAypcvDyB95+np06d11s2ePVu7E81QGfe3atUq3LhxQ7s8NTVVu8NZzmeffQZnZ2esW7cOo0aNQnJyss7627dv613POOOIWaVrLw8aNAjW1tZYs2aN3g64LVu2ICoqCgB0jrDdvn07Nm3apPf4Qghs2LABgPxO2ozHA4Bx48YBAHr16pXlDntLCg0NhSRJekfFL1++HIcPH9bbXgiBP/74A6NGjQIA9OzZ0+jHrFevHgBg//79Rt+2QIEC+OabbwAAkydPNvr2gOXejy1atECNGjVw4MABdOnSRfY64vfv38esWbN0diJnvIcvXLig9xj/9f7778Pf3x9//vknYmNj4efnp92hbQ7fffcdTp48qf0+LS0NX3zxBa5du4YSJUqgTZs22nXmev5lypSBlZUVTpw4gR07duisW79+PSZNmpTTp6lo7969qFOnDvbs2QMnJycsW7bM6EttEBERERERERFZSu75fyGJiIiIcjF3d3fs3bsXHTp0QHR0NEJDQ1G8eHFUrVoVbm5uePbsGS5cuIATJ05ACIHKlSsjKCgIABAYGIgWLVpg7dq1CAwMRGhoKDw8PHDs2DGcO3cOI0eOxNixYw2e5YMPPkD16tVx+PBhlClTBnXr1kWBAgXwzz//ICEhAV988YXsKbl9fHywatUqtG3bFmPHjsXcuXNRq1Yt2NjYID4+HkePHkWHDh10rjXepk0bxMTE4JNPPkGjRo3g7u4OABg2bBjKli2LypUrY8aMGejTpw86deqESZMmoVy5coiPj8e+ffsghMDo0aN1Tu98/PhxDBkyBC4uLqhWrRqKFSuGZ8+e4ciRI4iPj4erq6viacnr1KmDwMBAHD16FDY2NtnaEW2Ivn37aq/5/OLFCwDpO4qDg4O12zRt2hRff/21zu0yroNtY2Ojs3zLli3o2LEjihcvjipVqsDNzQ2JiYk4e/as9jrV/fr1y9bzadmyJRYvXoytW7eie/fuRt++R48emDRpktGXJMhgqfejRqPBmjVr0LRpUyxatAirVq1CQEAAfHx88PLlS8TFxeHEiRNITU1FeHi49gMXPj4+CAoKwqFDh7S/l/b29ihUqJD2AxoZNBoN+vfvj8GDBwNIfx9IkpSt1yUrPj4+qF69OqpVq4bQ0FAULFgQBw8exMWLF1GgQAEsX74c9vb2OrOZ4/kXKlQI/fv3x5QpU9CgQQPUqVMHxYoVw7lz53DkyBGMGjUK33//fY6e65o1a7Snjk9OTsa9e/dw7Ngx3Lx5EwAQEBCAhQsXomrVqjl6HCIiIiIiIiIisxJEREREZJRt27aJrl27irJlywoXFxdhbW0t3N3dRbVq1USvXr3E1q1bRWpqqs5tXr58KSZMmCAqV64sHB0dhYeHh2jUqJHYsmWLuHTpkgAgSpYsqfdYAITcP9kePXokRo4cKcqWLSvs7e2Fp6enaNmypTh06JCIiYkRAETdunVl54+PjxeDBg3S3tbJyUmUKVNGdO3aVezfv19n29TUVPHjjz+KihUrCnt7e+08MTExOtv9/fffom3btqJo0aLC2tpaFCxYUDRt2lRs2bJF7/H//fdfMXr0aNGgQQPh4+Mj7O3thbu7u6hSpYoYMWKEuHr1aqav/xdffCEAiI8//jjT7erWrSs7qyEybpvZV+fOnXVuk5ycLFxdXYWdnZ24fPmyzro9e/aIgQMHiqCgIFG0aFFhY2MjHB0dRZkyZUTnzp3F7t27jZ4xQ0pKivZ1vHfvnt76BQsWCACiQYMGivfx22+/aZ+X3PswK5Z6PwohxPPnz8WsWbNEvXr1RMGCBYW1tbXw9PQUVatWFf369RN//fWX7GN06NBBeHl5CWtr60yf55kzZwQA4ejoKO7fv6/4nDNe1/++D7Ly+u97cnKyGDt2rChXrpyws7MTHh4eok2bNuLUqVOKtzfH809LSxPz5s0T1atXF05OTsLV1VWEhISIFStWCCGU/w5lRe73yMHBQRQtWlTUqlVL9O/fX0RHR4u0tDSj75uIiIiIiIiIyNIkIYy4QB8RERERkYpSU1Ph5+enPSK+Vq1aao+ktXfvXoSEhGDIkCGYOHGiRR/7559/xrBhwzB16lQMGDDAoo/9phk1ahTGjh2Lnj17ai9lQERERERERERExB3rRERERJRnREZGom/fvqhVq5bitbvVEhERgcmTJyMuLg4FCxa06GO/ePECFSpUwMuXL3HhwgWdU4iT4W7cuIEKFSrg4cOH/8fefYdHUbVtAL83vfcQAgkhFAm9ho6EjkgCIQFCDb0HVJoiVUUQEAERkV6UF6mCKCogHUQ60nvoLYEE0svz/cG3Q5bdTSPJJuT+XVcuZcqZZ2Zn5pwz58wcnD17Vhk/noiIiIiIiIiIiA3rRERERJSvXbp0CTNnzsSDBw/wxx9/QESwf/9+1K9f39Ch5StbtmxB+/btMXPmTIwaNcrQ4RQoH3/8Me7evYudO3fiwYMHGDRoEL7//ntDh0VERERERERERPkIG9aJiIiIKF/bs2cPmjRpAjMzM/j4+GDy5MkIDAw0dFj0FilZsiRu3bqFokWLonPnzpg+fTrMzc0NHRYREREREREREeUjbFgnIiIiIiIiIiIiIiIiIiJKh5GhAyAiIiIiIiIiIiIiIiIiIsrP2LBORERERERERERERERERESUDjasExEREeUzfn5+UKlU2LNnj6FDKTR69eoFlUqFFStWGCyGmzdvQqVSoWTJklle9/bt2/jhhx8wYMAA1KxZE+bm5lCpVOjXr1+OxLZlyxYEBASgaNGiMDMzQ5EiRVC/fn189tlnWsv269cPJiYm+O+//7K9PfU1oFKpMGvWLL3L9evXDyqVCpMnT872tgqT7N5bVqxYofwe6j8zMzO4uLigQoUK6Nq1KxYtWoTo6OjcCZwKnCtXrmDYsGGoUKECrK2tYWFhAQ8PD/j6+mLYsGHYuHGjoUPMUMmSJaFSqXDz5k1Dh6JFfU326tXL0KHodOjQIbRs2RJOTk4wMjLKUv76JutS7ti3bx9CQkLg4eEBc3NzuLi4oGbNmvjwww+RlJSUJzHs2bMHKpUKfn5+ebK9vFJYyvzPnj3D0KFD4eXlBTMzs7fytyQiIqLCgw3rREREREQF3MaNGzFo0CAsXrwYJ06cQGJiYo6km5iYiE6dOqF9+/bYuXMnKlasiODgYFSqVAnXrl3DvHnztNaZPHkyTE1NMXz48ByJYdq0aXj27FmOpFVQ5IeOHrpYW1sjNDQUoaGhCAkJQYMGDWBsbIyff/4ZAwcORLFixTBv3jyIiKFDJQPatGkTKleujO+++w6PHj1CgwYNEBQUhCpVquDu3bv47rvvMHDgQIPGOHnyZHbKySX37t3D+++/j507d6JSpUro3r07QkNDUaZMmVxdN6+9rQ29aYkIPvjgAzRu3BibNm2Ct7c3goKCUKNGDTx69Ahz5sxBQkKCocOkLDBUh6EBAwZgwYIFMDIyQocOHRAaGorWrVvnaQxEREREOcXE0AEQERERERnatGnT8PHHH8Pd3d3QoWSLt7c3wsLCUKNGDdSoUQPr1q3D1KlT3zjd/v37Y/369Wjfvj0WL14MFxcXZV5qair+/fdfrXU8PDzQr18/zJ8/H1u3bkVAQEC2t29lZYXIyEhMnz4d06dPz3Y6lDNcXFx0Nvbfv38fM2bMwNy5czFixAjcuXMHM2bMyPsAyeAePnyI0NBQJCQkYOTIkfjiiy9gYWGhsczx48exYcMGA0X4dggMDETdunVhb29v6FC0/PXXX3j27Bm6du2Kn376Kc/WpZw3efJkzJ07F/Xr18eaNWvg5eWlMf/o0aNa1zfR65KSkrB582ZYWFjg9OnTsLOzM3RIRERERG+Eb6wTERERUaHn7u4OHx+ffNlIkRnt2rXDvHnz0KtXL1SpUgUmJm/ef3bXrl1YtWoVKlWqhHXr1mk0qgOAkZER6tatq3Pdvn37AgDmzJnzRjGEhYXByMgI8+bNw717994oLco97u7u+OabbzB//nwAwMyZM7F//34DR0WGsG3bNrx48QLFihXDrFmzdDa61axZE9OmTTNAdG8Pe3t7+Pj45MvOYLdu3QIAlC1bNk/XpZx16dIlfPnll3Bzc8Nvv/2m1agOAL6+vjlS3qC32/3795GcnAw3Nzc2qhMREdFbgQ3rRERERLns7NmzCAoKgouLC6ysrFC5cmXMmTMHqampWfokY0bjMGb0advjx48jNDQU3t7esLCwgJOTE6pWrYrRo0cjPDxca/l///0XnTp1QrFixZRxtf39/bFjxw6d6SckJGDmzJmoWbMmbG1tYWZmhqJFi8LX1xdjxoxBZGSkVvpjxoxB7dq1lbG73dzc4O/vj507d+rcRtpxZaOiovDRRx+hZMmSsLCwQNmyZfHVV18hNTUVAHD37l0MHDgQnp6eMDc3R7ly5fDtt9/qTFffp7fTHtPHjx9j6NCh8PT0hJmZGTw9PREWFqb1mfJJkyZBpVKl+6njf//9FyqVCsWLF0dycrLe5QxJfaw++OADmJqaZmndatWqoWrVqti9ezcuXLiQ7RgqVaqEHj16IC4uDpMmTcp2Orps2rQJ/fr1Q6VKleDo6AgLCwt4e3ujT58+uHTpUrrr/v333+jYsaMy3qyrqyt8fX0xadIkREREaC1/+fJlDBw4EKVLl4aFhQXs7e3x7rvv4scff9RY7ubNm1CpVFi5ciUAoHfv3hrjmk+ePBnXrl2DsbExHB0dERsbqzfGihUrQqVS4ffff8/G0cmeIUOGwNfXFwD4xnoh9fDhQwCAq6trpteJjo6GnZ0dTExMcPv2bb3LtWnTBiqVCgsWLFCmpc0XT506hQ4dOsDFxQXm5uaoUKECvv76a62hCVQqFaZMmQIAmDJlisY1pm/M8t27d6Nly5ZwdHSEpaUlatSogVWrVqW7Xxs2bEDr1q3h6uoKMzMzFC9eHN27d8f58+d1Ln/8+HF07twZHh4eMDMzg52dHUqVKoWgoCBs2bJFY9n0xljfuXMn/P394ebmBlNTUzg6OqJs2bLo3r079u3bl27MuqxduxbNmjWDk5MTzM3N4eXlhT59+uDy5cs6Y1Lfq9Me25IlS6a7jcyuq54GAMuXL0e9evVgb2+vUY7KqFyV0VAbmbm/+/n5oUmTJgCAvXv3apxDr+9rcnIyFi5ciPr168Pe3l4prwwfPhx3797VGcOVK1fQp08feHt7w9zcHDY2NvDy8sL777+P5cuX61wns/lMZn3//fdITk5G//794eDgkK00dEl77kZERGDo0KEoUaKEcm59+OGHePr0abppJCUl4auvvkLFihVhaWkJZ2dndOjQId3yxsWLF9G7d294eXnB3NwcTk5OaNasGdatW6dz+dTUVCxatAgNGjSAg4MDTE1NUaRIEVStWhVhYWFa51fa827z5s1o2LAh7OzsYGtrCz8/v0zlxZm9hwHA48ePMW/ePLRp0wbe3t6wtLSEnZ0datWqha+++grx8fEay6uPu7q87+3trXHevl63uHfvHj766COUL18eVlZWsLW1ha+vL+bPn5+lcqtKpVI6ZYSHh+vcZtpr8uzZs+jcuTPc3d1hbGysUaeJjIzEuHHjULFiRSWmmjVrYsaMGYiLi9PadtrhGhISEjBlyhS88847sLCwQIkSJTB27FjlOEVFRWHUqFEoVaoULCwsULJkSUyePDnfltGJiIjIwISIiIiIcs2ePXvE0tJSAEjp0qUlJCREWrRoIWZmZtK5c2fx8vISAHLjxg1lncaNGwsA2b17t0Za+qarTZo0SQDIpEmTtObNmDFDjIyMBIC888470qlTJ/H395fy5csLAFm+fLnG8osWLVKWr169unTp0kXq168vAASATJ48WWP5lJQUadasmQAQOzs7ee+996RLly7SvHlzZR9PnjypsU6zZs3EyMhIKleuLG3atJGOHTtKjRo1lG3MmTNHaz+WL18uAKRdu3ZSvnx5KVKkiAQFBUnLli2V4zxs2DC5evWqFC1aVDw9PaVTp07SpEkTMTY2FgAyffp0rXRDQ0N1Hgf1Me3Tp494eHiIm5ubdOjQQdq0aSP29vYCQHx9fSUxMVFZ5/79+2JmZibW1tby9OlTnb9Vz549BYBMmTJFmXbjxg0BIF5eXjrXyQp13H379s3W+snJyWJjYyMA5PLly3L//n355ptvZNCgQTJixAhZsWKFPH/+PN00Ro0aJQDkyy+/zPL21ef66tWrJTw8XMzNzcXY2FguXLigsVzfvn31nvMZMTY2FisrK6lVq5Z06NBBAgICpFSpUgJArK2t5eDBgzrXCwsLU87RatWqSUhIiLz33nvKuq9fn+vWrRMLCwsBID4+PhIYGChNmzYVa2trASC9e/dWln38+LGEhoZK6dKlBYA0aNBAQkNDlb/NmzeLiIi/v78AkEWLFumM8e+//1buOampqVrHVd89RB/1dZeZc3Pu3LkCQGxsbCQpKSlL26GCb/Xq1QJAjI2NZefOnZleT31djRs3Tuf8q1evikqlEjs7O417j/qc/vjjj8XMzEzKly8vISEh0rhxY+WeP2LECI20QkNDpWrVqgJAqlatqnGNLV68WFlOnXdNmDBBVCqV1KxZU0JCQqRu3brKPeCbb77RijUpKUk6deokAMTc3Fzq168vHTt2VLZpaWkp27dv11hn586dYmpqqsQUHBwsgYGBUrt2bTE3N5d27dppLK++JkNDQzWmr1ixQlQqlahUKqlTp4507txZAgICpEaNGmJsbKx1LNKTmpqq5FUmJibStGlTCQkJkXfeeUcAiJWVlcZ+7N+/X++xHTlyZLrbyuy66uM+bNgwMTIykoYNG0qXLl2kTp06cvPmTRERneWqtPTl9yKZv79PmzZNWrVqJQDEzc1N4xxKG298fLw0b95cAIiFhYW899570rlzZ/H09BQA4uLiIsePH9eI4b///hM7OzsBIOXKlZMOHTpIx44dpV69emJjYyNVq1bVijsr+Uxm+fj4CAD566+/5OnTp7Jw4UIZMmSIDBs2TBYuXCiPHz/Ocpoir87dgIAAKV26tDg4OEj79u0lMDBQHB0dlf1+9OiRxnq7d+8WAFK/fn1p3ry5WFlZSevWrSUoKEg5ng4ODjp/923btinHp1y5chISEiJNmzZV7hF9+vTRWqd3797K79a8eXPp0qWLtGrVSsqWLSsAlPxYTX3effjhhwJAatWqJV26dJHatWsr59S8efO0tpOde5jIq3tt8eLFpXHjxhISEiLNmjVTym716tWT+Ph4ZXn1NaY+J4KCgjTO27Tlq7179yq/RcmSJSUgIEBatWqlTGvZsqVGuTc9oaGhEhQUpJStdG1TfU32799fzM3NpWTJkko9ZdasWSIicu3aNeUYu7q6SlBQkAQEBIitra0AkBo1akhkZKTGttXnTL169aRx48ZiZ2cnAQEB0rZtW6UM37ZtW4mIiJBy5cop6bZs2VI5XwYNGpSp/SQiIqLChQ3rRERERLkkNjZWihcvLgBk5MiRkpKSosw7d+6cuLm5KQ/bcrNhfcuWLcrDwZ9//llrvXPnzsn58+eVf585c0ZMTExEpVLJqlWrNJb9/fffxczMTHnYqrZ3716lET46OlprG0ePHpUnT55opXXv3j2tZQ8dOiR2dnZiamoqd+7c0ZinfiALQPz9/SUmJkaZd/z4cTExMREjIyOpUKGCDBo0SKNh75dfflEa/tOuJ5JxwzoA6dWrl8ZDylu3bim/75o1azTW69atmwCQ2bNna+3f48ePxdzcXExNTeX+/fvK9PzUsH758mVlv1etWqU8qE375+rqKrt27dKbxqZNmwSANGvWLMvbT9uwLiLy0UcfCQAJDAzUWO5NGtbXrl0rL1680JiWmpoq3333nQCQihUrajRKi4jMmzdPAIizs7P8/fffWmkeOXJEbt26pfz7zJkzYm5uLhYWFrJx40aNZW/evCmVK1cWALJy5UqNeek1/IiI7NixQ2l80kX9EPvrr7/WmJ4XDesHDhxQzpGrV69maTtU8D1//ly5L6pUKvHz85PPP/9cfvvtN62GsrQuX74sKpVKihQponGfVRs5cqQAkLCwMI3p6nMagCxcuFBj3q5du0SlUomxsbHcvn1bY156HdHU1I04pqam8uuvv2rMU18T9vb2EhsbqzFv3LhxAkDq1Kkj169f15i3fv16MTY2FkdHR42OV02aNBEA8uOPP2rF8ezZMzl8+LDO7b/esO7t7S0AZP/+/VrpPHz4UE6cOKF3f1/3/fffK42/aTvGpaamKsfPwcFB63fNzLHVJ6N11b+1nZ2d1jFRy27Delbv7+pGu8aNG+vdn7FjxyqdnNLGk5iYqORf3t7ekpCQoMxTN+h+8cUXWunFxsbK3r17NaZlN59JT0JCgqhUKgEgP/zwgxQpUkSrDGBjYyP/+9//Mp2mWtpyXN26dSUiIkKZ9/TpU6UTZ0hIiMZ66uOtLmumLT/FxcUpHR0GDBigsd6DBw+URtQvvvhCI18/evSo0lictqNaeHi4ABAPDw+N7aidP39ewsPDNaapzzuVSqV1Ha9du1ZUKpWYmJjIf//9pzEvu/ew8+fP67wGIiMjpWXLlgJAZsyYoTU/o+vj/v374uzsLCqVShYsWKBRd3ny5Ik0bdpUAM2OoRnJqHyrvibVHQzSblOtTp06SoeMtGW3R48eKZ1yu3btqrFO2nOmdu3aGvWQmzdvKr995cqVteoUR48eVeoUr//WRERERGxYJyIiIsolq1atUh4k6XqzY/78+XnSsF6tWjWdjWz6qB/2dujQQef8YcOGCQBp0aKFMm3dunUCQIYPH56pbWTkk08+EQDy3XffaUxXP5C1sbGRhw8faq0XEBAgAKREiRISFxenNV/9gPn1B9MZNax7eHhoNcaLiEyfPl0A7bed/v33XwEgZcuW1WqcnTZtmgCQLl26aEzPTw3rhw8fVs5NU1NTady4sRw9elSeP38up06dkjZt2ii/w+XLl3WmcenSJQEgjo6OWd7+6w3rERERyoPxtA+S36RhPT316tUTAHLu3DllWlJSkri6ugoArcYLfTp37iwAlDeuXqc+T2rWrKkxPaOGdRGRihUr6mxAu337tpiYmIiVlZXWFxPyomH94sWLyrlz5MiRLG2H3g4XL15UGkFe/6tWrZp8//33kpycrLWe+r6ivu7VYmNjxdHRUVQqlVy8eFFjnvqc1pdftW7dWukglFZWGtY/+ugjnfPVb/Tu27dPmRYRESGWlpZiYWGh1TFMbciQIQJAvv32W2VahQoVBIDWG5f66GtYt7KyEnt7+0ylkRH1lzN0vWWbmpoqVapUEQAydepUjXl50bD+2Wef6U0jOw3r2bm/Z9SwHhcXp3RK27p1q9b8mJgYpYPlTz/9pExXXweZ7QSR3XwmPffv39coA1SpUkX27Nkj0dHRcvHiRenVq5cAL79Mkfb8z4y0Deuvf8lI5GVHAZVKJUZGRhqNyerjrVKp5NSpU1rr/fPPPwJASpUqpTH9888/T3f/Z82apZTX1NTHLCAgINP7pT7v2rdvr3O+usNb//79NaZn9x6WHnX5y9fXV2+c+q4PdWeQYcOG6Zx/584dMTU1FVdXV63yrT6ZbVh/5513dOYN+/fvF+DlVzIePHigNf/YsWMCIN1z5vUODSIiw4cPT7dOof46UFY6pRAREVHhwDHWiYiIiHLJ3r17AQAdO3bUOUZ1t27dcj2GBw8e4NSpUzAyMkLfvn0ztU7aMQ91Uaezf/9+pKSkAABq1KgBY2NjLFu2DN999x3u37+fqW1FRERg1apVGDNmDPr3749evXqhV69eyrHTN9Z1zZo1UaRIEa3pZcuWBQA0adIEFhYWeuffu3cvU/GpNWvWDFZWVlrTy5cvDwBa46T6+vqiXr16uHLlCv78809lempqKhYuXAgAGDZsWJZiyEuSZjzP4sWL488//0StWrVgY2ODqlWrYuvWrahUqRJevHiB6dOn60zD2dkZAPD06VMkJia+UTxOTk4YO3YsACj/zQlXr17F/Pnz8cEHH6Bv377K+aceJzrt+Xf8+HE8fvwYLi4uCAwMzDDt1NRUbN++HQDQuXNnncuoj+nJkye1xkPNyPDhwwEA8+fP15j+ww8/IDk5Gd26dcvRcXEzKzU1Vfl/9VjIVLiUK1cO//zzD44cOYKJEyeiVatWypjrp06dwuDBg9G6dWut+8KIESMAaJ/Ta9aswdOnT9G8eXOUK1dO5zb9/f11Ttd3j86KrKS9e/duxMXFoUGDBihevLjO9fz8/AAAhw4dUqbVrl0bwMtywYEDB7I9rm/t2rURFRWFnj174vjx4xrXY1bcuXMH165dAwCEhoZqzVepVOjduzeAl/uc14KDg3M0vaze3zPj2LFjePHiBZycnHSeQ1ZWVggJCQGgeQzV58LgwYPx559/pps35FY+k7YMYGlpiZ07d6Jx48awtbVFuXLlsHz5crz33ntISUnRGAM7K6pWrYpq1appTa9cuTKqV6+O1NRU7Nu3T2t+iRIlULVqVa3p+q51dZlW13kMvCrTXrlyRSkb+vj4wNbWFr///jumTp2KGzduZHq/9G1HPf31sczVsnMPS0lJwa5du/D5559jyJAh6N27N3r16oWpU6cC0F+GTs9vv/0GQP/5VLx4cZQtWxaPHz/GlStXspx+etq3bw9jY2Ot6epj1rp1a7i5uWnNr1mzJqpWrYrU1FSl/pBWiRIlUKlSJa3p6jpBRnWKrNYZiIiI6O1nYugAiIiIiN5Wd+7cAQCULFlS53wHBwfY29sjKioq12K4desWAMDd3R329vaZWkf98M7b21vn/NKlSwMA4uPjERERgSJFiqB06dL45ptvMHr0aAwbNgzDhg2Dl5cX6tWrh7Zt26Jjx44wMzPTSGfx4sX48MMPERMTozeW6OhondNLlCihc7qNjU26821tbZXYs0JfenZ2dnrTGz58OA4fPoz58+ejdevWAIBt27YhPDwc1atXR/369TO17VGjRuHJkyda01esWJHJ6LNOfZyAlx0szM3NNeYbGxtj4MCBCAsLw86dO3WmoT42APDs2TPloWV29+eDDz7A/PnzsW/fPmzbtg1t27bVudzFixd1NvY3bNgQ/fr1A/DyYfSwYcPwww8/aDQgvC7t+RceHg7gZaNhZhqMIyIilPU9PT0ztby+hjhdunfvjo8//hibNm3C/fv34e7ujsTERCxevBhA5jtu/PLLL/jll1+0pvfr1w8NGzbMdDxqaX9bJycnAJn7TejtU7t2baWRUERw8uRJzJw5E2vXrsXOnTsxd+5cjB49Wlm+RYsWKF++PI4cOYLjx4+jZs2aAIDvvvsOQPrndHbu0ZmVlbSvX78OANi1a1eG94nHjx8r/z9t2jScOXMG27dvx/bt22FpaYkaNWrAz88P3bp1UxrXMrJgwQK0bdsWq1evxurVq2FrawtfX180bdoUPXr00Lsvr1OXA5ydnTXu5WmpywJv0mkhu/SVq7Irq/f3zMioLAXoPoajR4/GgQMHsHPnTrRu3RqmpqaoWrUq3n33XYSEhMDX11dZNrv5zJIlS3DgwAGt+R9//LHSqKzWoUMHpWNMWkOGDMH27duxf/9+JCYmwszMDAcOHMCSJUu0lm3fvj3at2+vMS294+Lt7Y0TJ04o5ei0MroeExISNKZn9Ds4ODjAyckJkZGRuHPnDooVKwZbW1ssX74cvXv3xvjx4zF+/Hi4u7ujbt26aN26Nbp27aqUN3XFnt50XfuUmf16/R525coVBAYG4ty5czrXA/SXodOjvoc1atQow2UfP36Md955J8vb0EffdZ3Za+n06dM670d5XWcgIiKitx8b1omIiIhyWXoPaXPyjc7svpmWU8LCwtCpUyds3boVBw4cwIEDB7B27VqsXbsWkyZNwv79++Hu7g7g5dthAwcOhLGxMb766iv4+/ujRIkSsLKygkqlwqJFizBw4EC9jZ5GRul/eCmj+VmVnfSCg4MxatQobN++HTdu3IC3t3emGohet2HDBuWhf1q52bBesmRJqFQqiAhKlSqlcxn1dH1fJ0jbYcTR0VH5/+zuj6WlJSZNmoSBAwdi3LhxaNOmjc7lHjx4gJUrV+qcp27EnTt3LhYuXIiiRYti9uzZqF+/Ptzc3JSvHHTt2hX/+9//0m10z0ja61HfG2xpvd55ISNWVlbo378/ZsyYgUWLFmHSpEnYuHEjHj58iEaNGqFKlSqZSufUqVM6j5efn1+2GtZPnDgB4OUDafVD8sz8JvR2U6lUqFGjBv73v/8hNjYWW7duxS+//KLRsK5SqRAWFoYhQ4Zg/vz5WL58OQ4fPoyTJ0+iZMmSejvTADl/z89u2urrvkyZMmjQoEG6y/r4+Cj/X7RoURw7dgx79+7Fzp07cfDgQRw5cgQHDx7El19+iWnTpmXqax3ly5fHpUuX8Ndff+Hvv//GoUOHsH//fvz999/47LPPsHTpUnTv3j3T+5NfWVpaZntdQ5eVMmJlZYUdO3bg6NGj+OOPP3Do0CEcOnQIx44dw+zZszFkyBClLJHdfObAgQM678m9evWCj48PbGxs4OrqisePH2dYBkhKSsKTJ09QrFgxXL16VWe6JUuW1GpYzwxdeXBuXutpBQUFoXnz5ti6dSv279+PgwcPYvPmzdi8eTMmTpyIHTt2oHLlyllON7vl2tcFBwfj3LlzaNu2LcaMGYMKFSrAzs4OpqamSExMzHKZQk19TgUHB8Pa2jrdZdVfJsopb3Jdpyev6wxERET09mPDOhEREVEuUb99evPmTZ3zo6Ki8OzZs0ynp37j+/nz5zrn62qsVL+Fcf/+fURFRWXqrfXixYvj2rVruH79us5PJ6rfZrGwsFDeSFVzc3ND//790b9/fwAv31Tt06cPDh8+jI8//lh54Lp+/XqICMLCwjBmzBitbeT05yUNwcTEBIMHD8b48eOxYMEC9O/fHzt27ICTkxO6dOmS6XT0nT+5ycbGBuXKlcPFixd1vl0OvHozWd9bWxEREQBeNqqnHQrhTfanb9++mD17Nv777z+sXr1a5zJ+fn4ZNoivW7cOwMvPpgcEBGjN13X+qa+ly5cvQ0Qy7BTj4uICS0tLxMXFYdasWXBxcUl3+ewYOnQovv76ayxatAjjxo1TPqGdlY4bkydPzvanfHX56aefAABNmzZVPumamd+ECo+WLVti69atOu8tPXv2xLhx47B27VrMmjVLOacHDx5cIBo/1G8NlytXLsudn1QqFfz8/JRPxcfHx2PFihUYOnQoxo0bh+DgYOUN5/SYmJigTZs2Suej6OhozJ49G1OmTMHAgQMRGBiYYYOZuvyifiNa11vr6rJAVr60kRfepKyU2ft7ZqiPS3qfEU/vGPr6+ipvpycnJ+OXX35Bz549sWDBAgQHB6NJkybZzmdWrFiR4flZs2ZN/PHHHxmWAYBX5QD1cCqZkd5xUZcTPDw8MpVWeooXL46LFy8qx/p1UVFRiIyMVJZNy97eHj169ECPHj0AALdv30ZYWBi2bNmCYcOG6fzs+I0bN3R+qj4n9+nixYs4c+YMihQpgs2bN8PERPPR7puUoT09PXHlyhWMHTsWtWrVetNQc4T6d9H3G6adl9/uR0RERPR2yv81UyIiIqIC6t133wXwshFZ13ipa9asyVJ66odFFy5c0JoXGxurc5zTokWLKuMOLlu2LFPbUT/U1/fQVZ1Oo0aNtB7mvc7Hx0d5y+7UqVPKdPVDTC8vL6114uPjsXHjxkzFmt8NHDgQFhYWWLZsGb7++muICPr27Ztrb+XkpI4dOwKA3k+979ixA8Cr8WBfd/bsWQBQPuecE4yNjfHll18CACZOnKj1ydfMSu/8O3funMa5qlarVi24uLjg8ePHOj+drivWFi1aAHjVkJ9Z6oahjMZZLlGiBNq3b4979+5h4sSJOHToEIoVK4YOHTpkaXs5ZcGCBTh69CgA6OwwQ2+/zHSgUA9RoquBydraGn379kV8fDy+/PJLbNiwARYWFso4yDkls9dYVjVr1gxmZmbYs2cPHj169EZpWVhYYNCgQahSpQpSU1Nx5syZbKVjZ2eHyZMnw8HBAbGxsbh8+XKG63h4eCiN+LrKAiKiTG/SpEm24sot6ZWVHjx4oHxVI62s3t+BjM8h9djmkZGR2Lp1q9b8uLg4rF27FkDGx9DExATBwcFo1aoVgFflqTfJZzKiLgP8/fffOt/yV5cBypUrp3e4gPScOXNG5zl97tw5nDhxAkZGRko5+k2oy7T6vpqiLtOWLVs2w0ZZT09PTJkyBQB0lhMA6O30t2rVKo143oS6DFOsWDGd5fAff/xR77oZnbfvvfcegJw/n96E+pj98ccfePjwodb8kydP4tSpUzl2zhARERFlhA3rRERERLmkY8eOcHd3x82bN/Hpp59qPJi8ePEiPvvssyyl17x5cwAvx5tNO4ZgTEwMBgwYgNu3b+tcb9KkSQCATz/9VGeD9fnz5zUeQI8YMQImJib45ZdftB7O/fXXX/jhhx8AvBwrW+3vv//G77//jqSkJI3lRQTbtm0DoNmIqR4vduXKlRpvlcXHx2PIkCHpvslUkLi4uKBr166IjIzEokWLYGRkhCFDhhg6LMXNmzehUqmgUqm03iQfPnw4HB0d8fvvvyu/udratWuVN5OHDx+uM+1Dhw4BePnmck7q0KED6tSpg1u3bmHTpk3ZSkN9/n333Xca1+X9+/fRs2dPnQ+cTUxM8OmnnwIABgwYgH379mktc/ToUY3xUydNmgQzMzOMHj0aK1eu1Nk4cfbsWa39UDc4pjd2qtqIESMAQBnDfODAgRl2eMlpDx48wEcffaS8Kf/JJ5+gfv36eRoD5Q8LFixAaGiocv2nJSLYtGmT8hZ6SEiIzjSGDRsGIyMjzJ49G4mJiejSpUuOf3I4K9dYVri5uSEsLAwxMTHw9/fHf//9p7VMQkICtm7diosXLyrTZs2apXQ4SOvixYvK26e6OgKlFRsbi9mzZ2uM3a62f/9+PHv2DMbGxpl+Y1adx3/++ec4ffq0Ml1E8MUXX+DUqVNwcHBQvlCTX6jLSl999ZXGV4EeP36Mnj174sWLF1rrZOf+rj6OV65c0Sr7AC87RgwdOhQAMHLkSI035ZOSkjBixAg8ePAA3t7eCA4OVuYtWLAAly5d0krvwYMHOHbsGADNcyG7+UxGunfvjtKlS+Ps2bOYOHGiRrq7d+/G7NmzAegvA2RERDB48GA8ffpUmRYVFYXBgwdDRBAUFJSpceMz0r9/f9jZ2eHEiRP48ssvNTr/nDx5El988QUAaAxLcfLkSfz888+Ii4vTSu/XX38FoP963Lx5s9JhQm3Dhg3YuHEjTExMEBYW9sb79M4778DY2Bj//fcf9uzZoxXfN998o3fdjO59o0ePhoODA2bPno2vv/4aiYmJWsvcuHEj3cb7nNawYUPUqVMHcXFxGDhwIGJjY5V5T548wcCBAwG8zFNy4pwhIiIiygg/BU9ERESUS6ysrPDjjz/i/fffx4wZM7Bp0ybUqlULkZGR2LNnD9q1a4cjR47g1q1byhsk6enUqRPmzJmDY8eOoWLFimjYsCFSU1Nx7NgxmJmZoU+fPjrfSg8MDMTUqVMxfvx4BAcHw8fHB1WrVkVcXByuXr2K8+fPY/ny5UpjY+XKlfHdd99h8ODB6NGjB7755hv4+PggPDwchw4dgohg8uTJaNmypbKNM2fO4MMPP4SdnR1q1KiBYsWKIS4uDidOnEB4eDjs7e01OhL07t0bc+fOxcmTJ+Ht7Y1GjRrB2NgY+/fvR1xcHEaMGIG5c+fmwK9geMOHD1d+l/fff18Zdzon3b9/H4GBgcq/1Q//t27dirp16yrTFyxYgBo1aij/TvugPO3n2oGXnQJ+/vlnBAQEYNCgQfj2229Rvnx5XLt2DSdPngQATJgwQe9Y5+o33du1a/eGe6ftq6++gp+fn8bD1awYN24c/vjjDyxevBi7d+9GjRo1EB0djb1796JUqVIIDAzE5s2btdYbMWIELl26hIULF6Jx48aoXr06ypUrh+joaOVTs7t371YeXNeoUQM//vij8nnc8ePHo0KFCnB1dUVkZCT+++8/3LlzB507d9Z4y7x9+/aYMmUK5s2bh7Nnz8LT0xNGRkYICAjQ+nR9o0aNUL16dZw8eRKmpqYYMGBAto5JZjx58kT5zG9qaiqeP3+Oa9eu4dy5c0hNTYWNjQ2mTZumNCZR4ZOUlIRVq1Zh1apVcHV1RfXq1eHi4oJnz57h/PnzSgee7t27630LvWTJkggICFDeHM7K0AaZ1apVK1hbW+OXX35Bw4YNUbZsWRgbG6NBgwbo3bv3G6U9ffp03L9/H2vWrEG1atVQtWpVlCpVCiYmJrhz5w5OnTqFmJgYbN++XRln/YsvvsDo0aPh4+OD8uXLw9LSEvfu3cOBAweQnJyMnj17aty7dUlMTMTIkSMxevRoVK5cGWXLloWpqSlu3ryJf/75B8DLDnaurq6Z2o+BAwfi0KFDWL16NWrVqoXGjRujSJEiOHHiBC5dugRLS0usWbMm0+nllaFDh2Lx4sU4ceIEypUrh3r16iEmJgZHjx5VvvKh6630rN7fS5QogVq1auHYsWOoXLkyatWqBQsLC7i4uCgdnaZMmYJjx45h165dKF++PJo0aQJbW1scPnwYt27dgrOzM9avX69RBly0aBGGDh0Kb29vVKpUCXZ2dnj8+LFSPmratKlGPpDdfCYjZmZm2LRpE5o0aYKpU6di7dq1qFatGu7evYt///0XqampCA0NxeDBg7P1OwUEBODs2bMoVaoUmjRpApVKhT179iAyMhJly5ZVOuC8KTc3N/z000/o2LEjPv30U6xevRrVq1fHo0ePsHfvXiQnJ6N3794aHUTCw8MREhICS0tL1KhRA56enkhOTsZ///2HS5cuwczMDDNmzNC5vREjRqBLly6YPXs2ypYti2vXruHIkSMAXnagqVKlyhvvk4uLC4YNG4a5c+eiWbNmaNSoEYoVK4ZLly7hxIkTGD9+vNJh4HVBQUHYvXs3unfvjpYtW8LR0RHAywb1cuXKwcPDA1u2bEFQUBBGjRqFGTNmoFKlSnB3d0dUVBQuXLiAa9euoU6dOujevfsb70tmrVmzBk2bNsWWLVvg7e2Nd999F0lJSdi9ezeio6NRo0aNHDtniIiIiDIkRERERJSrTp8+LYGBgeLk5CQWFhZSoUIFmTlzpiQkJIiZmZkYGRlJXFycsnzjxo0FgOzevVsrradPn8qwYcPEw8NDTE1NpXjx4jJgwAB5+PChTJo0SQDIpEmTdMZx+PBh6dKlixQvXlxMTU3FyclJqlatKmPGjJHw8HCt5f/55x8JDg6WokWLiomJiTg7O8v7778vf/31l9ayV69elcmTJ0uzZs2kRIkSYmFhIY6OjlKlShX5+OOP5fbt21rrPH78WIYMGSKlS5cWc3NzKVasmHTv3l2uXLkiy5cvFwASGhqqsY6+6WoZHYPQ0FABIMuXL8/U9IzS2717twCQxo0b65yvVrRoUQEgf/75p95lbty4IQDEy8sr3bTSWzejv9fPqXXr1gkAadWqld60L126JKGhocp54+zsLG3atEl3X06cOCEApEmTJlneF5FX18Dq1av1LtOmTRtlv/T9Puk5c+aMBAQEiLu7u1hYWEjZsmVlzJgxEh0drfd8UNu+fbu0a9dO3NzcxNTUVFxdXaV27doyZcoUiYiI0Fr+xo0b8uGHH0qlSpXE2tpaLCwsxMvLS/z8/GT69Oly9epVrXU2b94sDRo0EFtbW1GpVOnu59ixYwWAdOnSJd19Tu/ekh71dZf2T30PKV++vISEhMgPP/wgUVFRWUqX3j7R0dHyyy+/SFhYmNSuXVvJqywtLaV06dLSpUsX2b59e4bpfP/99wJA6tWrl+5yGZ3T6d3D9+3bJ82bNxdHR0cxMjLSylu8vLwEgNy4cUNn2hndJ37//Xfp0KGDcu90cHBQrpc1a9ZITEyMsuyPP/4ovXv3lkqVKomTk5OYm5uLl5eXvPfee7J582ZJTU3VSFtXXpiUlCQLFy6ULl26iI+Pj9jb2yvHPSgoSHbt2qUzzoysWbNG/Pz8xMHBQUxNTcXT01N69eolFy9e1Ll8RvlmejJaV33/ycidO3ekZ8+eUqRIETEzMxNvb28ZPXq0PH/+PEfv7+Hh4dK1a1dxd3cXExMTnXl4UlKSLFiwQOrWrSu2trZiZmYmpUuXlrCwMLlz547W9rdt2yaDBw+W6tWri6urq5iZmYmHh4f4+fnJypUrJTExUWfc2clnMuPevXsydOhQKVmypJiZmYmDg4M0adJE/ve//2UrvbTn7qNHj2TgwIHi4eEhZmZm4unpKcOHD9eZj2amvJXe+XH+/HkJDQ1V7knq/Vi7dq3Wsvfv35fp06dLmzZtxNvbW6ysrMTOzk4qVKggQ4cO1Xnup71frFu3TurVqyc2NjZibW0tjRo1kl9//VVnXNm9h6WmpsrSpUulZs2aYmNjI/b29tKwYUNlf/Qdi5SUFJk2bZpUrFhRLCws9JYPHz58KBMmTJAaNWoo562Hh4fUr19fJk2aJGfOnNEZry4ZlW8zuibVIiIi5JNPPpHy5cuLhYWFWFlZSfXq1WX69OkSGxurtXxG58yb1imIiIio8FKJZGIQNCIiIiLKcfv27UPjxo1RuXLlbI+dSvnfzp070aJFC5QrVw4XLlyASqUydEiK/v37Y+nSpThx4gSqVauWY+mGhYVh/vz52LJli9Yb1pSzUlJSULp0aeWLEvXq1TN0SERvrGHDhjh48CDWrFmDLl26GDocIsohK1asQO/evREaGooVK1YYOpwcU7JkSYSHh+PGjRu58mUiIiIiIso/OMY6ERERUS56/PixzvHCz549q3x28k0/O0v5V0pKijLG/UcffZSvGtUBYMeOHejatWuONqrfvn0bS5YsgZ+fHxvV88CiRYsQHh6OevXqsVGd3grbt2/HwYMHUaJECY2xp4mIiIiIiIgMjWOsExEREeWic+fOoUmTJqhQoQJKlSoFS0tL3LhxAydOnEBqaipatGiBsLAwQ4dJOWz58uXYt28fjh07hrNnz6Jy5cro06ePocPSoh7vOCdNmTIFSUlJmDt3bo6nTS9dunQJM2fOxIMHD/DHH3/AyMgIs2bNMnRYRNkWERGBsWPH4unTp/j9998BADNmzICpqamBIyMiIiIiIiJ6hQ3rRERERLnonXfewdChQ7F3714cPHgQz58/h62tLerXr4+uXbuif//+MDFhkexts3fvXqxcuRIODg4IDAzEnDlzCs3vvGTJEixZssTQYbzV7t+/j6VLl8LMzAwVK1bE5MmTUb9+fUOHRZRtz58/x9KlS2FiYoJSpUph5MiR6Ny5s6HDIiIiIiIiItLAMdaJiIiIiIiIiIiIiIiIiIjSwTHWiYiIiIiIiIiIiIiIiIiI0sGGdSIiIiIiIiIiIiIiIiIionQUqob1kiVLQqVSYcWKFeku5+fnB5VKhcmTJ+d6TDdv3oRKpULJkiVzfVukX17/DupzbM+ePTmS3ooVK6BSqdCrV68cSa8g2rNnD1QqFfz8/N4onR07dqB379545513YGdnB3Nzc7i7u6NFixb45ptv8Pjx45wJ+DUBAQFQqVRQqVQ4e/ZsrmyjIFDfp2/evGnoULTwOiMqGFi2yp9yuuyTHW9SVlDnT2n/zM3N4eHhgXbt2mHbtm161+3Xrx9MTEzw33//vUH0RFTQ5FT9pDB48eIFSpUqpdxf79y5k6fbV2/3bTJ58uQ8e66VH8ydOxcqlQobN27UmK4+Dhldh+q6nq7yY9oywIgRI9JNZ+bMmcqyJiYmWvPV5aG0f2ZmZnB3d4e/vz9+/fXXDPf1TeSH8lhOYfmKiIiIDKVQNaxT4fU2VR7yytv4cCE9T548QYsWLdCyZUusWLECSUlJaNKkCYKCglC+fHkcOnQIH330EUqVKoUjR47k6Lbv37+P33//Xfn30qVLczT9/KKwPdwhosKrsOWhhUmDBg0QGhqK0NBQtGnTBiYmJti6dSv8/f3x0Ucf6Vxn8uTJMDU1xfDhw/M4WiLKTfm5Q2hBM3r0aB7HAq5Xr16ZepElNzx+/BiTJ0+Gr68vgoKCcnVbP/30ExITE/XOX7ZsWabSqVq1qlKeCAgIgJWVFbZt24aAgIAMG+8Lg8x0TGL5ioiIiAyFDetEVOhFRUWhYcOG2LlzJ3x8fLBv3z7cuHEDW7ZswZo1a/D3338jMjISP/zwA2xsbHD//v0c3f7KlSuRkpKC4sWLAwB+/PHHdCvrZBiBgYG4cOECpk2bZuhQiIgKnFWrVuHChQuoXbu2oUN5I/369cOKFSuwYsUKbN68GVevXsWwYcMAAN988w2OHj2qtY6Hhwf69euHPXv2YOvWrXkdMhFRvrZjxw4sXLgQQ4cONXQoVEBNmTIFz549y/UO3LVq1UJERAS2bNmic/6hQ4dw8eJF+Pr6ZphW+/btlfLEhg0bcOXKFXzyyScAgHnz5uHvv//O0djfRixfERERkaGwYZ2ICr2wsDBcunQJJUuWxMGDB9GoUSOtZczNzTFgwACcOnUK5cuXz9Htq3u1f/311yhVqhSePHmit7JOhmNvbw8fHx+4u7sbOhQiogKnRIkS8PHxgZWVlaFDyVEmJiaYOXMm7OzsAEDvJ1z79u0LAJgzZ05ehUZElO9FR0ejb9++8Pb2xvTp0w0dDhVAz549w4oVK1C8eHG0bt06V7fVp08fAPrfSld/eU69XFYYGRnh888/R6lSpQAA69aty2aUhQvLV0RERGQIbFjPpEmTJkGlUmHgwIF6l/n333+hUqlQvHhxJCcna8zbtm0bGjduDFtbW9jb26NRo0bpNpylHR80JSUFs2fPRvXq1WFjY6P1adE///wTbdu2RZEiRWBmZoZixYqhc+fOOHbsmMZykZGR8PLygkqlwsKFC7W2+eLFC/j4+EClUuGrr74CACxfvhwqlQqtWrXSG+u9e/dgamoKS0tLREREaMWfmpqKefPmoUqVKrCysoK7uzsGDRqEyMhIAEBCQgI+//xz+Pj4wNLSEsWKFcOIESMQExOjd5vHjx9Ht27dUKJECZibm8PJyQmtWrXS+Jw28OrzUXv37gUANGnSRGMsK12fKRMRLFq0CDVr1oS1tTXs7e3RsmVLHD58WGO5a9euwdjYGI6OjoiNjdUba8WKFaFSqbRi0ycmJgYTJkxA2bJlYW5ujmLFiqFPnz64e/dulj+lndGY0LrGoVVvQ+318b/Un+hLSkrCjz/+iG7dusHHxwd2dnawtLREuXLlMHz4cNy7d0/nNtN+ln///v3w9/eHq6srjIyMsGLFCoSGhkKlUqX7VvC6deugUqly5K2369evY82aNQCA2bNnw8nJKd3l3dzcUK5cOa3pa9euRbNmzeDk5ARzc3N4eXmhT58+uHz5crrp7d27F1euXIGzszMCAwPRu3dvANn/HHxqaioWLVqEBg0awMHBAaampihSpAiqVq2KsLAw5feLjo6GnZ0dTExMcPv2bb3ptWnTBiqVCgsWLFCmpf0NT506hQ4dOsDFxQXm5uaoUKECvv76a4iIRjoqlQpTpkwB8PKNgrTnlL7zc/fu3WjZsiUcHR1haWmJGjVqYNWqVenu/4YNG9C6dWu4urrCzMwMxYsXR/fu3XH+/Hmdyx8/fhydO3eGh4cHzMzMYGdnh1KlSiEoKEjrHp3e9bRz5074+/vDzc0NpqamcHR0RNmyZdG9e3fs27cv3ZiJCpOzZ88iKCgILi4usLKyQuXKlTFnzhykpqbq/KTu+fPnMWnSJDRo0ADFixeHmZkZnJ2d0bx582w9cPz3338xZswY1K5dG0WLFoWZmRnc3Nzg7++PnTt3prvu5cuXMWTIEJQrVw5WVlaws7NDhQoVMGTIEJw9exZA5vNQtcyW4dSioqIwfvx4VK5cGdbW1ko5oUGDBpg4cSKSkpI0lt+5cyfCwsJQrVo15T7t4eGBzp0763yzOu0+TJ48Gffu3UO/fv1QrFgxWFpaolKlShr508WLF9G1a1cULVoUFhYWqFq1Kn7++Wed6eobliftp2Nv3LiBHj16oGjRojA3N0fp0qUxfvx4JCQkaKyT12WFjFhYWKBs2bIAgIcPH+pcplq1aqhatSp2796NCxcu5HpMRG+ruLg4fP3116hbty4cHBxgYWGBcuXKYcyYMUpdNK205bfIyEh88MEHKF26NMzNzeHn54fdu3dDpVLBx8dHq/yqFh8fD2dnZ6hUKpw/f15JMzw8HADg7e2tca/XNfxYUlISvvrqK1SsWBGWlpZwdnZGhw4ddN4PcuNZgFpW8xEAePr0KSZNmoRq1arB1tZWyb+/+OKLdOvBmfHBBx/gzp07WLJkCaytrd8orbTHLTk5GTNmzFCOt4uLCzp16oSLFy9mmM7GjRvRsGFD2NnZwdraGg0aNEi3Lh8ZGYlx48ahYsWKsLKygq2tLWrWrIkZM2YgLi5O5zpZqTukzSdPnz6NDh06wNXVFZaWlqhSpQrmzp2LlJSUdPfp8ePHGDp0KDw9PWFmZgZPT0+EhYXh2bNnWstmtZ6vPu4rV64EAPTu3Vvjenj92UVWr+GMLF++HDExMejRoweMjHL3EWflypVRq1Yt/PXXX7h7967GvBcvXmDdunXw8PBAy5Yts5W+sbExqlWrBgBZGhph/fr1aN68OZydnWFqagpnZ2dUqFAB/fv3x5kzZ/Sul9m6PJDx8Ir6nlWlnZ7Z89DPzw9NmjQB8PJ5SdrzKe2zK4DlKyIiIjIQKUS8vLwEgCxfvjzd5Ro3biwAZNKkScq0+/fvi5mZmVhbW8vTp091rtezZ08BIFOmTNGYPnv2bAEgAKR27drSpUsXqVWrlgCQjz76SACIl5eXxjo3btwQAFKiRAkJCAgQMzMzadasmXTp0kWqVKmiLDd+/HgBICqVSho0aCBdunSRatWqCQAxNjaWpUuXaqT7zz//iKmpqVhYWMjJkyc15nXp0kUAyPvvvy+pqakiIhIfHy+urq6iUqnk0qVLOvd74sSJAkB69+6tFb+Xl5d06dJFLC0tpXXr1tK+fXspUqSIAJDq1avLixcvpGHDhmJnZycBAQHStm1bsbe3FwDy3nvv6dzenDlzxMjISABItWrVJDg4WBo2bChmZmZax//ChQsSGhoqbm5uAkBatWoloaGhyt/+/fu14g0NDRVTU1Np2rSpdOrUSd555x0BIObm5vLPP/9oxOLv7y8AZNGiRTpj/fvvvwWAlC5dWjmmIq/Osd27d2ss/+LFC/H19RUAYmNjI23btpWOHTuKu7u7FClSRHr16qV1boqILF++XABIaGhopqbr+p3UNm/eLKGhoco5m/Z4hYaGyuPHj0VE5Pbt2wJA7O3tpW7dutKxY0dp06aNFCtWTACIq6urXLlyRWub6n0fMmSIGBkZSYUKFSQkJERatmwpa9askePHjyvnfnJyss643333XQEgK1euVKbt3r1bAEjjxo11rqPP3LlzBYA4ODjo3V56UlNTlWvfxMREmjZtKiEhIcp5Y2VlJdu3b9e7fo8ePQSADB8+XEReHlcjIyMxMjKSW7duZTme3r17CwCxsLCQ5s2bS5cuXaRVq1ZStmxZASCbN29Wlg0LCxMAMm7cOJ1pXb16VVQqldjZ2cnz58+V6erf8OOPPxYzMzMpX768hISESOPGjcXY2FgAyIgRIzTSCg0NlapVqwoAqVq1qsY5tXjxYmU59X16woQJolKppGbNmhISEiJ169ZVzslvvvlGK9akpCTp1KmTcq3Wr19fOnbsqGzT0tJS63fYuXOnmJqaKjEFBwdLYGCg1K5dW8zNzaVdu3Yay+u7nlasWCEqlUpUKpXUqVNHOnfuLAEBAVKjRg0xNjbWOhZEhdWePXvE0tJSyRdDQkKkRYsWYmZmJp07d1au/xs3bijr9O3bVwCIj4+PtGrVSjp37iz16tVTygEffvih1nZ05W1qzZo1EyMjI6lcubK0adNGOnbsKDVq1FDuL3PmzNEZ+08//STm5uZK/hQUFCSBgYFStWpVUalUSr6c2TxUJOtluJiYGKlUqZKSx/r7+0tISIj4+flJ0aJFBYBWGbV06dJiZmYm1atXl4CAAOnQoYNUqFBBybM2bNigta+TJk1SynVFixaVEiVKSKdOnaRJkybKPX7WrFly+PBhsbW1lXLlyklISIjUq1dP2e+1a9dqpauv7KM+XiNGjBA7Ozvx8vKSTp06SfPmzZXzpX379hrr5HVZQSTjeoQ6n50wYYLeNEaNGiUA5Msvv8zy9olI5O7du1K5cmUBIE5OTtK8eXMJDAxUrs+SJUvKzZs3NdZRl9/ef/998fb2FkdHRwkICJCOHTtKt27dRESUNP/66y+d2122bJkAkCZNmoiIyP79+yU0NFSsra0FgAQFBWnc6y9cuCAir+459evXl+bNm4uVlZW0bt1agoKCxNPTU6mDpM33RHLvWUB28pFz584psbq7u0vr1q3F399fqV9Xq1ZNnj17luXfUkRk27ZtAkAGDBigTFPnI7dv385yemnz/w4dOoipqak0b95cQkJCpFSpUkod+9ChQ1rrqrc7ceJE5Xh27txZqUuoVCrZtGmT1nrXrl1Tzj9XV1cJCgqSgIAAsbW1FQBSo0YNiYyM1Fgnq3UHdT45ePBgsbCwkJIlS0rnzp2lZcuWyjOQ4OBgjecNIq/y8z59+oiHh4e4ublJhw4dpE2bNsozF19fX0lMTNRYL6v1/MePH0toaKiULl1aAEiDBg00roe09c/sXMMZUef3O3fu1DlffRwyyvvV9wpd5Ud1fPv375cFCxYIAPniiy80llm6dKkAkE8//VQ5F42NjbXS0vW8Ma3mzZsLAAkICEg3XrUpU6Yo5bp3331XunTpIm3atJFKlSqJSqXSqjtnpy6fdr3Xy3Fq6uP8+n5l5zycNm2atGrVSgCIm5ubxvk0cuRIrW2zfEVERER5jQ3rOugr6Hbr1k0AyOzZs7XWefz4sZibm4upqancv39fmX769GkxNjYWIyMjWb9+vcY6P/74o6hUKp2Fd3VBHIB4eHjobNTevn270oD2+kOAJUuWCAAxNTWVs2fPasz75ptvBICULVtWoqOjRUTk+++/VyrvERERGst/+umnGg1/aSUmJiqV8OPHj+uMv3Tp0hqVoydPnigPHytXriy1a9eWJ0+eKPOvX78ujo6OAkAOHDigsb0//vhDVCqVuLi4yN69ezXmnTlzRjw8PASA7NmzR2NeRpWAtPF6eXlpHO/k5GTp06ePAJCWLVtqrLdjxw6lUU6XoKAgASBff/11puL58MMPBYBUqFBB7t27p0yPi4uT4OBgJcbcbFhXU29Ln+joaNmyZYskJCRoTE9MTJRPPvlEAEibNm201lPvOwD57rvvdKbdoEEDAaDz4cV///2nVOjj4+OV6dl9WK5u2G7atGmW1lNTXzsuLi4anVVSU1OVSqSDg4M8evRIa91nz54pjQanTp1SpqsrkZ999lmWYgkPD1fuGWnvQ2rnz5+X8PBw5d+XL18WlUolRYoU0TiWaiNHjhQAEhYWpjE97W+4cOFCjXm7du0SlUolxsbGWg/E9FW201Lfp01NTeXXX3/VmKc+n+3t7SU2NlZj3rhx4wSA1KlTR65fv64xb/369WJsbCyOjo4aDwubNGkiAOTHH3/UiuPZs2dy+PBhndt//Xry9vZWHrS87uHDh3LixAm9+0tUWMTGxkrx4sUFgIwcOVJSUlKUeefOnVMe0AOaDet79uyRa9euaaV38eJFJc8/cuSIxrz08rbff/9dI39VO3TokNjZ2YmpqancuXNHY96xY8fE1NRUVCqVzJs3TyN2EZGbN2/KsWPHNKZllIdmpwy3cuVKAV52PHz9IXhKSors2bNHK0/evHmz1gN99XQTExNxdnbWup+q79UAZNCgQZKUlKTM27p1qwAQW1tb8fLyki+++ELjQf6cOXMEgJQpU0Zrmxk1rKsfRqdtKP/vv/+UhqvXG0Lysqwgkn494vz588rD6KNHj+pNY9OmTQJAmjVrluXtExV2qampynXft29fpR4r8rKDpbrcqm78VlOX39TXXlRUlFbaixcvTrchq2bNmgJANm7cqDFdV4ewtNT3HOBlp/K05fO4uDilzJ+2YVkk954FZDUfiY2NVRpLx48frzEvJiZG6ZiftoN9ZkVGRoq7u7t4enpq/Cbq/X6ThnV13ez06dPKvOTkZKVTsZeXl1bdR72eg4ODVmd6db74zjvvaG2zTp06yrnz4sULZfqjR4+Ujntdu3bVWCerdYe0+eSQIUM08uWzZ8+Kq6urznpZ2vy8V69eGvt869YtpVy2Zs0ajfWyW89Xx6nveVt2r+H0xMbGipmZmRgZGWmkl1ZON6yr6/Cvl3UaNGggKpVKrl27lu2G9Xv37imdMiZOnJhuvCIvX4SxtLQUGxsbuXjxotb8mzdvKh19Xt9+Vuvyb9qwntXzMCtlNpaviIiIKK8Vyob1zP69XiD8999/BXjZIP16b+Bp06YJAOnSpYvG9H79+gkA6dy5s86Y2rVrp7PwnrZSuGrVKp3rNmvWTICXb73r0rZtWwEg/fv315rXoUMHJa4TJ04onQJeb0gSedmr2NTUVOzt7TUqiyIi//vf/wSA1KtXT2/8v/32m1aa6rf4VSqV/Pfff1rz1ZXe19/+V1dcdb1hJSKybt06AV6+NZBWVhrWt27dqjX//v37Arx8E/b1hxAVK1bUWTG+ffu2mJiYiJWVlVbPf13xxMbGio2NjQCQP//8UyuGR48eiZWVlc5z0xAN6xkpVqyYzgquet/Ta8hW/466KkYDBw4UAPLJJ59oTM/uw/LWrVsLAAkJCcnSemrqh03z5s3TmpeamipVqlQRADJ16lSt+epG+Zo1a2pMV++/t7e31r0mPep7VGZ7t4uItGnTRgDI6tWrNabHxsaKo6OjqFQqrUq6+jfs0KGDzjTVx/T1e1dWGtb13dd8fHwEgOzbt0+ZFhERIZaWlmJhYaHVIKY2ZMgQASDffvutMk391qauRidd9F1PVlZWYm9vn6k0iAqrVatWKfnN6/moiMj8+fOVfEdfA8XrfvjhBwEgo0eP1pieXt6WHvXD4tc7fbVv314A7U5G6ckoD81OGW7GjBkC6O7gmR3qBpHXy2nqe3WJEiUkLi5Oaz11vla7dm2tPCopKUmcnJwEgEZHLpGMG9Zr1qypM88bNGiQANqdzfKyrCCiu2H92bNn8ueffyp50/jx49NN49KlSwJAHB0ds7x9osJO3ZBcrVo1jYZFtZSUFOVt7LT1S3X5zdTUVGdHLZGX5V5nZ2cxMjLSelv28OHDAkA8PT21vpCR2YZ1lUql0YlW7Z9//hEAUqpUKY3pufUsIKv5iLqu0rZtW53znz9/LkWKFBETE5NMl6fVunbtKgC0viil3u83bVjX9QWa+Ph4pRHvp59+0rldXXW6+Ph45c3atF8U279/vwAvv1D24MEDrfWOHTsmAMTIyEhjf7Jad1Dnk+7u7jrz5W+//VZ5TpWWOj/38PCQmJgYrfWmT58uwMs3ibNCXz0/o4b17F7D6Tl69KhSZtEnbcNuZv4yalgXefXSjfqFjosXLwoA8fPzExHJcsP6ixcvZO/evUpnDGtr60x9ve7Ro0cCQONLFhnJbl3+TRvWs3oeZqXMxvIVERER5bVCOcZ6gwYNEBoaqvfPzc1N53q+vr6oV68erly5gj///FOZnpqaqoxZPmzYMI111OMPde/eXWeaoaGhGcYbFBSkNS05ORkHDx4EAL3jE/ft2xfAy3GKX7ds2TKUKlUKP//8M5o0aYKEhARMnz4ddevW1Vq2WLFiCA4ORlRUFFavXq0x77vvvgOgvd9qJiYmOseXUo9DWaJECVSqVEnv/LTjdz158gT//vsvLC0t4e/vr3N7fn5+AIBDhw7pnJ8RExMTtG7dWmt60aJF4ejoiISEBK1xv4YPHw4AmD9/vsb0H374AcnJyejWrRscHBwy3Pbx48fx4sULuLi46Dxmrq6uaNGiRRb2Jm+cPn0as2fPRlhYGPr06YNevXqhV69eSE5ORmpqKq5evapzveDgYL1pBgYGwtPTE7t27dIYBy8qKgo//vgjjI2NMXjw4Bzfl6y6c+cOrl27BkD3taxSqZQx03Vdh0uWLAEA9OnTR2N6u3bt4OzsjBs3buDvv//OdDw+Pj6wtbXF77//jqlTp+LGjRsZrjNixAgA2ufvmjVr8PTpUzRv3lznmPIA9F6H5cuXBwCtceeyIitp7969G3FxccoYzLroujeox93t1q0bDhw4gOTk5GzFWrt2bURFRaFnz544fvw4UlNTs5UO0dts7969AICOHTvC1NRUa363bt30rvvixQusX78e48aNw4ABA5R8ZuPGjQCAS5cuZSmWiIgIrFq1CmPGjEH//v2V9NQxpk0vJSUFO3bsAAAMGDAgS9vRJ7tlOF9fXwDAjBkzsGrVKkRGRmZqe/fu3cPixYsxcuRI9OvXT9nfc+fOAdB//Jo0aQILCwut6eoy2nvvvac11q+JiYky9uXrY7BmpG3btlrpAfrzFEOVFdKOHevg4IBWrVrhypUr+PHHH/H555+nu66zszOAl+MVJyYm5nhsRG+z3377DcDLurGJiYnWfCMjI7z77rsAdNcFq1evjlKlSulM29LSEgMGDEBqaiq+//57jXnq+u6gQYNgbGycrdhLlCiBqlWrak3PTJk5J58FZDUfUR/zzp0765xvY2ODWrVqITk5GUePHk03rbQ2bdqENWvWoHfv3jrr3jlBV93M3Nxc2Rd940TrqoOYm5sr507a30qdRuvWrXU+Q6pZsyaqVq2K1NRUpYwBZL/u0KlTJ535snpfr1y5ojPvbdasGaysrLSmZ3T+Zbeer8+bXsO6PHz4EMCr/DU9bm5u6T4HbNCgQWZ3Ram/L1u2TOO/r9fr0zNlyhSlPGFjY4PGjRvjxIkTKFKkCLZs2QJPT88M03B1dUXJkiVx5swZjBw5EufPn8/09nOzLq9Lds/DzGD5ioiIiPKadmm2EFA/VNTHz89PKaC/bvjw4Th8+DDmz5+vVAK3bduG8PBwVK9eHfXr19dY/s6dOwAAb29vnenpm65WpEgRnYXPiIgIxMfHp5tG6dKlAeguoNrb22P16tVo0KABoqKi0KZNG3z00Ud64xg+fDj+97//4bvvvsOgQYMAAGfOnMGBAwfg5uamt5HU3d1dZ6XJxsYGwMuHDLrY2toCgLKPAHDjxg2ICOLi4mBubq43VgB4/PhxuvP1cXd31/nAHwDs7Ozw9OlTjZiAl50mPv74Y2zatAn379+Hu7s7EhMTsXjxYgD6Ox28Tn2uqB9I65LevLwWExODHj16YPPmzekuFx0drXN6evtiYmKCIUOG4JNPPsH8+fOVRt+VK1ciJiZGeZieGbqudRcXF8yaNQvAy8ooADx69ChT6aWlvracnZ1hZ2encxl91+Hp06dx/PhxWFhYoGvXrhrzzMzM0K1bN8ybNw/Lli1Ds2bNMhWPra0tli9fjt69e2P8+PEYP3483N3dUbduXbRu3Rpdu3ZVrj21Fi1aoHz58jhy5AiOHz+OmjVrAsi40wyg//pVH4vXr5WsyEra169fBwDs2rVLZ6NMWmnvDdOmTcOZM2ewfft2bN++HZaWlqhRowb8/PzQrVs3pZKfkQULFqBt27ZYvXo1Vq9eDVtbW/j6+qJp06bo0aOH3n0hKkwyyuMcHBxgb2+PqKgojem//vorevfurdWpLS19+YwuixcvxocffoiYmJhMpRcREaEsq6+TUVZltwzn5+eHsWPHYubMmQgNDYVKpULZsmXRoEEDtGvXDv7+/jAy0uwzO2XKFEydOhVJSUl649F3/PTdu7JThsuMrOYpeVlWSKtBgwYoU6YMgJd5yv79+/H8+XMMHjwYZcuWVTptpbcvAPDs2TMUKVIkU/ER0avy3oQJEzBhwoR0l9VVF8yoHjVkyBDMnDkTS5cuxeTJk2FhYYHHjx9j/fr1MDc3R//+/bMde0b3t4SEBJ3zc/pZQFbzEfUx79GjB3r06JHuPqqP+YEDB5TOw2m1b98e7du3x5MnTzB48GAUK1YMs2fPTjfNtJ48eYJRo0ZpTffx8cHHH3+sMc3BwUFvx3b18VKXS16XlbxIfWzTe6ZTunRpnD59WuN3yG7dQd92bG1t4ezsjIiICNy5cwfFihXL9j4Bb17P1+dNr2Fd1OVGfXXxtHx8fLBixQq981esWKF0WMlIkyZN4O3tjQ0bNmDOnDlYtWoV7Ozs0n154HVVq1ZFtWrVAACmpqZwcnJCzZo14e/vD0tLy0yns2rVKgQHB2P27NmYPXs2nJycUKdOHbRo0QI9evSAi4uLzvVysy6f19tj+YqIiIjyWqFsWH8TwcHBGDVqFLZv344bN27A29s7Uw1Q2ZWVAnVWpX37/MKFC4iKioK9vb3OZevWrYvatWvj33//xd69e9G4cWNlvwcMGAAzMzOd673+gDer89NS9+S2sbHR2XM/J2QlHjUrKyv0798fM2bMwKJFizBp0iRs3LgRDx8+RKNGjVClSpUspZdew2BGjYZZ9SZv1n7yySfYvHkzfHx8MH36dPj6+sLFxUU5F+rXr4/Dhw9DRHSun9G53b9/f3z22WdYtWoVpk2bBhsbGyxYsABA1q61lStXak3z8vJSHpbXrFkTq1evxokTJ5CSkpLtN1GyaunSpQBeNgy0bdtWa766EWnTpk149uxZpr56ALx8A6B58+bYunUr9u/fj4MHD2Lz5s3YvHkzJk6ciB07dqBy5crK8iqVCmFhYRgyZAjmz5+P5cuX4/Dhwzh58iRKliypMza17FwvmZWde0OZMmUyfNPAx8dH+f+iRYvi2LFj2Lt3L3bu3ImDBw/iyJEjOHjwIL788ktMmzYNY8eOzXD75cuXx6VLl/DXX3/h77//xqFDh7B//378/fff+Oyzz7B06VK9Xy0hKmyyksfdvXsXnTt3RlxcHMaMGYNu3bqhZMmSsLGxgZGREf766y+0atVKbz7zuuPHj2PgwIEwNjbGV199BX9/f5QoUQJWVlZQqVRYtGgRBg4cmOn0DGH69OkYNGgQfv31Vxw4cAAHDx7E8uXLsXz5cvj6+mL37t2wtrYG8DL/mDx5MmxsbDB//nw0bdoUxYoVg6WlJVQqFcaNG4dp06bp3d+cLMNlRnbSy6uyQlqvd9CNiopCYGAgdu/ejU6dOuH8+fM6G8LUy6o5OjpmOj4ielXea9iwodJorE/FihW1pmVU9/Dw8ECHDh2wbt06/PzzzwgNDcWSJUuQkJCAHj16KJ1xsyO798vceBaQlXxEfcz1vZGdlpeXFwDg6tWrOu+pJUuWRPv27XHgwAE8evQIHh4eaN++vd70OnbsCHNzc+Ut6RcvXuhMt3HjxloN65mR3bwvJ+Rm3UHXfmV1n960nq/Pm17DuqjryFlt5H9TKpUKvXr1wqRJkxAaGooHDx5gwIABWbpm27dvj8mTJ79xLI0aNcLNmzfx22+/Ye/evTh06BD+/PNPbN++HZMmTcLmzZt1dtTP6XM9o2dLuXltsXxFREREeY0N61lkYmKCwYMHY/z48ViwYAH69++PHTt2wMnJCV26dNFavnjx4rh27Rpu3ryps3Jw8+bNbMXh7OwMc3NzJCQk4Pr16zobb9U9gnV9Gnnt2rVYuHAh3NzcUKtWLfz222/o06eP8llVXYYPH47u3btj/vz5qFq1Kn766SeYmJgob7DnNvVbRyqVCsuWLcuTSm9mDR06FF9//TUWLVqEcePGKW9NZeWhrvp3Su+cyOr5oq78Pn/+XOf88PDwLKWX1rp16wAAP//8s87z78qVK9lOG3h5jnfr1g1LlizBqlWr8M477+DSpUuoUKECmjZtmul0Mqrwt23bFh999BGePXuGrVu3IjAwMNNpq3+ziIgIREdH6+wpr+s6TEhIwE8//QTg5SeO0+sZHx8fj59++glDhw7NdFz29vYab5bcvn0bYWFh2LJlC4YNG6bxKUIA6NmzJ8aNG4e1a9di1qxZyvk7ePDgfHWd6aO+N5QrVy7dtxB0UalU8PPzUz4VHx8fjxUrVmDo0KEYN24cgoODM3zwA7zMG9q0aYM2bdoAePlwZ/bs2ZgyZQoGDhyIwMBA5SElUWGUUR4XFRWFZ8+eaUz79ddfERcXh8DAQHz11Vda62Q1n1m/fj1EBGFhYRgzZkym0nN2doaVlRViY2Nx6dIlncPXZNWbluFKliyJsLAwhIWFAQCOHj2K7t274+jRo5gxYwamTJkC4FU+PXXqVJ2fsX/TfDo/yKuyQnrs7e3x888/w8fHB+Hh4Zg9ezbGjx+vc1l1pzlHR0e9X0giIt3U5b127drpfHM5JwwfPhzr1q3Dd999h+7du+sd7s3Q8iof8fT0xMWLF9G3b99Mv4mrbgjPyJ07d/S+NQ4A//zzD4BXwzmVLFky0/fqZ8+e6e2YrC6HeHh4ZCqt9KiPrfpY66Lvd8hO3UHfMF/Pnz9X8pec2K/cqufnxjWsfjM5vS8b5ZZevXphypQp+PXXXwFk7TPwOc3S0hLBwcHKdfr48WOMHz8eixYtQp8+fd7ouY9abj5belMsXxEREVFey/8tJvnQwIEDYWFhgWXLluHrr7+GiKBv3746e6c2btwYAJRGtNetWrUqWzGYmJigYcOGAKC3IUk9zlOTJk00pl++fBkDBgyAkZERfvrpJ6xZswalS5fGpk2bMG/ePL3b7NSpE9zd3fHLL79g6tSpymc2X//UWG4pVqwYqlSpgufPn+OPP/7I0rrqSkB2x1DOSIkSJdC+fXvcu3cPEydOxKFDh1CsWDF06NAh02nUrFkTVlZWePz4MXbu3Kk1/8mTJ8o4r5mlrsCnHXs0LfU4Z7qoKyT6jpl6TD712wlp/fnnn3jy5EmWYtVFPX79d999pzT2ZqWBOTNKly6tdIoZOXJkhmMNPnr0SBmP1sPDQ2l01XUdiogyPe11uGnTJkRGRqJYsWJITk6GiOj8U791p367Pbs8PT2VB2SnTp3Smm9tbY2+ffsiPj4eX375JTZs2AALCwtlbMacklvXYbNmzWBmZoY9e/Zk65P+aVlYWGDQoEGoUqUKUlNTcebMmWylY2dnh8mTJ8PBwQGxsbG4fPnyG8VFVNCpx8xcv369znvAmjVrtKall8+IiM510pNeevHx8To7FxobG6NFixYAoAzxkhnp5aFvUobTxdfXF0OGDAGgeY9Pb38fPXqU5TJFfpUXZYWMuLq6Ko3ps2bN0uokonb27FkAUIZdIaLMe++99wC86iSVGxo0aICaNWvi6NGjGD9+PG7dugVfX1+9Qzzkdh1Tn7zKR9THXN3QmhPat2+vt+6T9ne9ffs2RCTbb/Sm/TqfWmJiIn7++WcArxrs34Q6jT/++EPnUIInT57EqVOnNMYO1yczdYf169frHDZAva9lypTR2ZEiq7Jbz8/oesiNa7hixYowMzPDnTt39Db45pYSJUqgXbt2cHZ2Rt26dVGnTp083X56XF1dMWPGDADArVu38PTp0zdOU31uXbhwQWtebGwsdu/e/cbbSCsr91eWr4iIiCivsWE9G1xcXNC1a1dERkZi0aJFMDIyUiqirwsLC4OxsTHWrVunNUbV2rVr8csvv2Q7jpEjRwIAvv/+e+zatUtj3ooVK7B161aYmppixIgRyvT4+Hh07NgRz58/x4QJE9CsWTPY2dlh3bp1MDc3x+jRo3H06FGd2zM1NcXgwYORnJysfBozr3vvf/HFFwCA3r17Kz2D0xIRHDlyBH/99ZfGdHXP7XPnzuVabOrjPH36dAAvO2DoGl9eHysrK/Tr1w8A8OGHH2pUzhMSEjBs2LB0x4TVpXbt2rCzs8P58+e1Hi6sX78+3Y4UGR0z9fjT3377rcb0S5cu5dhXDCpXroymTZviwoUL2Lp1K+zs7NCzZ88cSTutb7/9FmXKlMGNGzfQsGFDHDhwQGuZxMRELFu2DNWrV9eoTKp723/++ec4ffq0Ml1E8MUXX+DUqVNwcHDQGJdR3VDevXv3dD89HxISAjMzM+WhTEZOnjyJn3/+GXFxcVrz1NeLrgckwMtr2cjICLNnz0ZiYiK6dOkCZ2fnDLeZFbl1Hbq5uSEsLAwxMTHw9/fHf//9p7VMQkICtm7dqtHJZNasWbh165bWshcvXlTexNB3vNRiY2Mxe/ZsneMA7t+/H8+ePYOxsXGOvD1CVJB17NgR7u7uuHnzJj799FONz0VevHgRn332mdY66nxmw4YNuH//vjI9JSVF6cSWFer0Vq5cqfHwNT4+HkOGDNH7Jtinn34KExMTzJ8/HwsWLNB6GBweHo7jx49rTMvofpedMtzmzZuxb98+rU9tJiUlKR0O096z1Pu7aNEiJCYmKtOjoqIQGhqqNZ59QZVXZYWMDBkyBCVKlEBUVBS+/vprncuoz9msvE1PRC+1a9cOvr6++Pfff9G7d2+dZa+nT59i4cKFb9TQ/XqdLr36bl7UMfXJi3xkwIAB8PLywvr16zF27FidDZcPHjzIUsezvPL5558rjW3Ay89Ujx07Fnfu3IGnp2eODC3XsGFD1KlTB3FxcRg4cCBiY2OVeU+ePMHAgQMBvKzTqd/WfpO6w7179zBq1CikpKQo0y5cuKCUoT788MM33icg+/X8jK6H3LiGLS0tUbduXaSmpuLIkSOZWicnbdq0CU+ePMHhw4fzfNvAyzLokiVLdH4KX13/d3R0zNQY9Blp3rw5gJcdGe/evatMj4mJwYABA3D79u033kZa6vPpypUrSEpKSndZlq+IiIgoz0kh4uXlJQBk+fLl6S7XuHFjASCTJk3Su8ypU6cEgAAQf3//dNObMWOGsmydOnWka9eu4uvrKwDkww8/FADi5eWlsc6NGzd0Tn/d+PHjBYCoVCpp2LChdO3aVWrUqCEAxNjYWJYuXaqxfL9+/QSANG3aVFJSUjTmffvttwJAvL295enTpzq39/DhQzE3NxcAUqVKFb1xZRT/7t27BYA0btxY5/zly5cLAAkNDdWaN3fuXDExMREAUqZMGXn//fela9eu0qJFCylSpIgAkLFjx2qss23bNgEgZmZm0rZtW+nTp4/07dtXDh48mKl4RV6dPzdu3NC7TPXq1QWAmJqayv379/Uupz7Hdu/erTH9+fPnUrNmTQEgNjY2EhAQIJ06dZJixYqJi4uLhIaGCgCZOnVqpo/XN998o5x/9erVk+DgYKlYsaKoVCqZMGGC3v0eNWqUABAXFxfp1KmT9O3bV/r27StPnjwREZGNGzeKSqUSAFK5cmUJCQmRpk2biqmpqTRt2lTq16+vcx/17bs+v/zyixJ/WFiY3uUyOqcy8vDhQ/Hz81O25e3tLe3atZMuXbpI06ZNxcbGRgCInZ2dHDlyRFkvNTVVevToIQDExMREmjVrJl26dJFy5coJALG0tJTff/9dWf769evKcTt37lyGcXXo0EEAyLBhwzJcdvPmzco2GzRoICEhIRIcHKzEYmZmJtu3b9e7fvv27ZX9P378uN7lMvoNJ02apPMe+uDBA7G2thYA0qBBA+nVq5f07dtXli1bpiyT0XWmvgZev48nJSVJ165dBYAYGRlJ9erVJSgoSDp37iwNGjRQtpt2/+3t7QWA+Pj4SGBgoHTt2lX8/PyU+0vPnj01tqHrOnv69KmyzapVq0pwcLB06dJF6tWrp/zOEydO1HssiQqTXbt2iYWFhZJ/h4SESMuWLcXMzEw6duwoJUqUEABy9+5dEXl5XafNE99//33p1KmTeHl5iampqYwdO1bnfV9fnv706VPlHuPs7Czt27eXoKAgKVKkiNja2sqIESP05qUrV64UU1NTJd3g4GDp0KGDVKtWTVQqldb9LqM8VCTrZTh1fC4uLtKiRQvp1q2bBAQEKGWf4sWLy+3bt5Xlr1+/Lg4ODsq8oKAgCQgIEHt7e3F3d5c+ffrovFfru4er6bsPq+nLI/RNzyi99Mo4anlRVshMPWLZsmUCQGxtbSUiIkJrfpUqVTKd/xORtrt370q1atUEgFhbW0v9+vUlJCREuR8bGxsLAImLi1PWycw9JK2EhARxc3MTAOLq6irx8fF6l50/f76SR3Xo0EG511+8eFFEMnfPUd+70sqtZwFZzUdERM6ePSslS5YUAOLg4CDvvvuudO3aVdq3by8VKlQQlUolbm5u6caZFerj8XocmaE+biVKlJDAwEAxNTWVFi1aSEhIiJQuXVo5b/bv3693u/roy8OuXbum5A9FihSR4OBgadeundjZ2QkAqVGjhkRGRirLZ6fuoM4nBw0aJBYWFuLt7S0hISHSqlUrMTMzEwASGBgoqampGutllJ/rOz+zW88/ffq0GBkZiZGRkTRv3lx69+4tffv2lS1btijLZOcazsjs2bMFgIwZM0bnfPVxyCjvV98rdF136t9Y17mji/pcNDY21pqXmeeNmXXy5Enl+ZOvr6906tRJOnXqpDyXUqlUsmTJEp3bz2pdPjExUWrVqiUAxN7eXt5//3157733xNXVVYoXL57tcmV690n19sqVKyfdunWTvn37aj3rE2H5ioiIiPIeG9Z1yGxBt2jRogJA/vzzzwy3vWXLFmnYsKFYW1uLjY2N1K9fXzZs2KC30pzZyrSIyPbt26VNmzbi7OwsJiYmUrRoUenYsaNG45+IyI8//igAxM3NTW+jb3BwsFIx06dOnToCQH744Qe9y+Rmw7qIyH///ScDBgyQsmXLioWFhVhZWUmpUqWkVatWMm/ePOWhfFqLFy+WGjVqiJWVlVJxVp8LOdWwrn7I36VLF73LiKRfmXn+/LmMGzdOSpUqJWZmZlK0aFHp0aOHhIeHK5WV1499Rsdr5cqVUqNGDbGwsBA7Oztp2rSp7NixI939jouLkzFjxkiZMmWUCvvr+79v3z5p1qyZuLi4iJWVlVSqVEmmTp0qCQkJWX6wrs/z58/F2NhYVCqV8pBKlzdtWFfbvn279OzZU8qUKSM2NjZiamoqRYsWlRYtWsicOXN0PigXEVmzZo34+fmJg4ODmJqaiqenp/Tq1UsrZnVnhlq1amUqHnVjgaOjY4YPGO7fvy/Tp0+XNm3aiLe3t1hZWYmdnZ1UqFBBhg4dmu7xExH5/vvvBXjZASM92a2Mi7w8Z5o3by6Ojo5iZGSkdd5mt2Fd7ffff5cOHTpI8eLFxdTUVBwcHKR8+fISEhIia9askZiYGGXZH3/8UXr37i2VKlUSJycnMTc3Fy8vL3nvvfdk8+bNWg+ndF1nSUlJsnDhQunSpYv4+PiIvb29WFpaSunSpSUoKEh27dqlM06iwur06dMSGBgoTk5OYmFhIRUqVJCZM2dKQkKCmJmZiZGRkca9Tp0nlitXTiwsLKRIkSLSvn17OXbsmN77fnp52+PHj2XIkCFSunRpMTc3l2LFikn37t3lypUrGeal586dk759+4q3t7eYm5uLvb29VKhQQYYNG6b1IC8zeahI5stwIi8fnn788cfSsGFDKV68uJiZmYmrq6vUrFlTvvzyS41G+7THolu3blKiRAnlHjdo0CB58OCB3nt1QWxYz4uyQmbqEcnJyVKhQgUBIB9//LHGvBMnTggAadKkSZa3TUSvxMfHy8KFC6VJkybKvbNIkSJSrVo1GTp0qFbdPKsN6yIinTt3FgDyySefpLtcSkqKTJs2TSpWrKh0HEt7n8vthnWR3M9HRESio6NlxowZUq9ePaWu4+7uLr6+vjJ69Gg5dOhQhnFmVk40rHt5eUlSUpJMnTpVfHx8xNzcXJycnCQoKEhvw1t2G9ZFRCIiIuSTTz6R8uXLK88mqlevLtOnT5fY2FiNZbNTd0ibT544cUL8/f3F2dlZzM3NpWLFijJ79mxJSkrSWu9NGjSzU88XednRu0GDBmJra6s0zr++/axewxl5+vSpWFtbS7FixSQ5OVnvcXgbG9ajo6Nlzpw5EhgYKGXLlhUbGxuxtraWd955R3r27CnHjh3Tu/3s1OWfPn0qw4YNEw8PDzE1NZXixYvLgAED5OHDh9kuV6Z3HoaHh0vXrl3F3d1d6fz++u/D8hUREREZgkoklwYoe8vt3LkTLVq0QLly5XDhwgWoVCpDh5QnLl++DB8fH9jb2+Pu3buwsrIydEj5RkpKCkqXLo3w8HAcOnQI9erVy9H0k5KSUKlSJVy+fBnHjx9HjRo1cjT9/GrJkiXo378/WrZsiT///NPQ4bzVGjZsiIMHD2LNmjXKuPNERHlh3759aNy4MSpXrowzZ84YOhwqYApCWSEsLAzz58/Hli1bEBAQYOhwiEiPZ8+ewcPDA/Hx8bhx44byCW/K/27evAlvb294eXnh5s2bhg4nx/Tq1QsrV67E8uXL0atXL0OHky8NGzYM3333HbZu3Qp/f39Dh0N5iOUrIiIiMgSOsZ4NKSkpmDRpEgDgo48+KjSN6gAwceJEiAgGDx7MRvXXLFq0COHh4ahXr94bNaofP35ca9y7Fy9eYNiwYbh8+TKqVKlSaBrVY2JiMG3aNACvxhGk3LF9+3YcPHgQJUqUQHBwsKHDIaK30OPHj3WOY3727Fn0798fANC7d++8DosKuIJQVrh9+zaWLFkCPz8/PvQlyuemTZuGmJgYdOrUiY3qRAXEpEmT4ODgoIw3T4UDy1dERERkKCaGDqAgWb58Ofbt24djx47h7NmzqFy5Mvr06WPosHLd1q1bsWXLFpw7dw5HjhxB0aJFMWbMGEOHlS9cunQJM2fOxIMHD/DHH3/AyMgIs2bNeqM0g4KCEBsbi8qVK6NIkSJ49OgRTp06hcjISDg5OWHFihU5E3w+NnPmTJw9exYHDhzA9evX0bp1a7Rs2dLQYb11IiIiMHbsWDx9+hS///47AGDGjBkwNTU1cGRE9DY6d+4cmjRpggoVKqBUqVKwtLTEjRs3cOLECaSmpqJFixYICwszdJhUQBSkssKUKVOQlJSEuXPnGjoUItLh0KFDWLZsGW7cuIG///4bVlZW+OKLLwwdFhFlkqurKyZPnowPPvgAGzZsYEfxQoLlKyIiIjIUvrGeBXv37sWKFStw584dBAYGYtu2bTAxefv7Jpw4cQLLli3D+fPn0bx5c/z1119wcHAwdFj5wv3797F06VLs2LEDFStWxIYNG1C/fv03SvOjjz5CxYoVcf78eWzevBmHDx9GkSJFMHz4cJw6dQrVq1fPoejzr99++w2rVq1CdHQ0evXqhTVr1hg6pLfS8+fPsXTpUmzduhUlSpTADz/8gM6dOxs6LHpL7NmzByqVSuffP//8Y+jwyADeeecdDB06FEZGRjh48CA2b96Ma9euoX79+liwYAF+//33QlGuopxRkMoKS5YsQXJyMqpUqWLoUIhIh8uXL2Pp0qU4fPgw6tati+3bt6NUqVKGDouIsmDEiBEQETaqFyIsXxEREZGhcIx1IiIiynF79uxBkyZNMHz4cPj6+mrMa926NVxcXAwUGRERERERERERERFR1vG1ICIiIso1jRo14psjRERERERERERERFTgvbUN66mpqbh37x5sbW2hUqkMHQ4REREAQETw/PlzFCtWDEZGhWNElufPn8PS0jJbn/lmfk5ERPlVYczTs4v5ORER5VfMz4mIiCgr3tqG9Xv37sHT09PQYRAREel0+/ZteHh4GDqMXNe7d2+8ePECxsbGaNSoEWbOnIlatWrpXT4hIQEJCQnKv+/evYsKFSrkRahERETZUljy9DfB+jkREeV3zM+JiIgoM97ahnVbW1sALwtFdnZ2Bo6GiIjopejoaHh6eir51NvKzMwMQUFBaNOmDVxcXHD+/HnMmjULjRo1wqFDh1C9enWd602bNg1TpkzRms78nIiI8pvCkqfnBNbPiYgov2J+TkRERFmhEhExdBC5ITo6Gvb29oiKimLFnYiI8o3CnD9dvXoVVapUwbvvvos//vhD5zKvv7GufshRGI8XERHlb4U5T88qHisiIsqvmEcRERFRVry1b6wTERFR/lKmTBm0a9cOmzZtQkpKCoyNjbWWMTc3h7m5uQGiIyIiIiIiIiIiIiLSz8jQARAREVHh4enpicTERMTExBg6FCIiIiIiIiIiIiKiTGPDOhEREeWZ69evw8LCAjY2NoYOhYiIiIiIiIiIiIgo09iwTkRERDnu8ePHWtNOnz6NrVu3omXLljAyYhGEiIiIiIiIiIiIiAoOjrFOREREOa5z586wtLRE/fr1UaRIEZw/fx6LFi2ClZUVpk+fbujwiIiIiIiIiIiIiIiyhA3rRERElOPat2+Pn376CbNnz0Z0dDRcXV3RoUMHTJo0CWXKlDF0eEREREREREREREREWcKGdSIiIspxw4cPx/Dhww0dBhERERERERERERFRjuAAp0REb4mEhAS0b98eP/zwg6FDISKi/3f48GE0a9YMJ06cMHQoRERERJSHFixYgKCgICQlJRk6FCIiIiLKIWxYJyJ6Szx69AjXrl3D/PnzDR0KERH9v4ULF+LRo0dYtWqVoUMhIiIiojz0/fff4/Lly4iKijJ0KERERESUQ9iwTkRERESUS5KTkwEAKSkpBo6EiIiIiIiIiIiI3gQb1omIiIiIiIiIiIiIiIiIiNLBhnUioreESqUydAhERERERERERERERERvJTasExERERERERERERERERERpSPfN6xPnToVKpUKlSpVMnQoRJRPREZG4smTJxARQ4dCREREREREREREREREhUC+bli/c+cOvvzyS1hbWxs6FCLKJ3744Qc0btwYTZo0wZdffmnocPIVdjQgIsp/eG8mIiIiIiIiIiJ6O+TrhvVRo0ahbt26qFWrlqFDIaJ84tixYwAAE1NjHD9+3MDRUEGRnJyMf//9FwkJCYYOhYgKGZVKBYAN7ERERERERERERAVdvm1Y37dvHzZs2IA5c+YYOhQiykdu3boFGwcrOBa1x61bt5CammrokKgA+OWXX9C3b18sWLDA0KEQUSGlbmAnIiIiIiIiIiKigilfNqynpKQgLCwM/fr1Q+XKlTO1TkJCAqKjozX+iOjtEhcXh/v378OhiB0ci9ghISEBDx48MHRY+QYbbfQ7deoUAGDXrl2GDYSIiIiIiIiIiIiIiAqkfNmwvnDhQoSHh+Pzzz/P9DrTpk2Dvb298ufp6ZmLERLlvkePHmHXrl38dGwa4eHhEBE4FrGDg5sdAODatWsGjir/4LmSMR4jIiIiIiIiIiIiIiLKjnzXsB4REYGJEydiwoQJcHV1zfR6n3zyCaKiopS/27dv52KURLlvzJgx+OCDD3Dy5ElDh5JvXL16FQDgWNQeTkXtAQDXr183ZEhUwPCtfiIiIiIiIiIiIiIiyg4TQwfwuvHjx8PJyQlhYWFZWs/c3Bzm5ua5FBVR3jt+/DgAIDIy0sCR5B+XL18GADi728PK1lJjGlFm8I11IiIiIiIiIiIiIiLKjnzVsH7lyhUsWrQIc+bMwb1795Tp8fHxSEpKws2bN2FnZwcnJycDRkmUt/iG7StKw3oxR5iamcDU3ASXLl0ycFRUkPB6IiIiIiIiIiIiIiKi7MhXn4K/e/cuUlNTMXz4cHh7eyt/R44cweXLl+Ht7Y3PPvvM0GES5Sm+YfuSiODcubOwc7GBmYUpVEYquBRzxNWrVxEfH2/o8KiA4PVEREQFwaVLl/hVHiIiIiIiIiKifCZfvbFeqVIlbN68WWv6+PHj8fz5c8ydOxelS5c2QGREZGi3b9/Gs2dRKFvDS5lWxMsZ9288xoULF1C9enUDRkdERESUc4KDg6FSqXDmzBlDh0JERERERERERP8vXzWsu7i4oH379lrT58yZAwA65xG97fjp6peOHTsGAHAvVUSZ5l7KFaf3XMTRo0fZsE6ZwuuJiIgKCn5lhfKDc+fOYfLkyTh+/DgePHgAKysrVKhQAaNHj4a/v7+hwyMiIiIiIiLKU/nqU/BEpI0PVV86dOgQAMCjrJsyrVhpN0D1ah5RRng9EREREWVeeHg4nj9/jtDQUMydOxcTJkwAAAQEBGDRokUGjo6IiIiIiIgob+WrN9b12bNnj6FDICIDSkhIwP79++Hgagt7V1tluoWVGdy9XXHy5ElERkbCycnJgFFSQcA31omIiIgyr02bNmjTpo3GtGHDhqFmzZqYPXs2BgwYYKDIiIiIiIiIiPIe31gnyufYEPiyc01sbCxKVfXUOh6lq5ZAamoq/vjjDwNFl3/wXMkY31gnIiIiejPGxsbw9PTEs2fPDB0KERERERERUZ5iwzpRPseGQGD9+vUAAJ/apbTmla3hBWMTI6xfv77QH6vCvv/pUXc6YOcDIiIioqyLiYnBkydPcO3aNXzzzTfYvn07mjVrpnf5hIQEREdHa/wRERERERERFXRsWCeifO3ixYs4cuQIPMq6wcHVTmu+pY0FSlUtgatXr+Lw4cMGiJAKAnWnA3Y+ICIiIsq6kSNHwtXVFWXKlMGoUaMQGBiI+fPn611+2rRpsLe3V/48PT3zMFoiIiIiIiKi3MGGdaJ8rrC/Ybt48WIAQPVmFfQuU71peQDADz/8wIZTIiIiIqIc9sEHH2DHjh1YuXIl3nvvPaSkpCAxMVHv8p988gmioqKUv9u3b+dhtERERERERES5gw3rRPlcYW4oPnfuHP766y+4eTnD452iepdzKeYI78oeOHHiBA4cOJCHERIRERERvf18fHzQvHlz9OzZE9u2bcOLFy/g7++vt65ibm4OOzs7jT8iIiIiIiKigo4N60SUL4kIvv76awBAPf/qGb65X6dNVahUKnz99ddITk7OixCpACnsX34gIiIiyknBwcE4evQoLl++bOhQiIiIiIiIiPKMyZusnJiYiJ07d+LixYuIiYnBhAkTAADx8fGIjo6Gi4sLjIzYdk9EWbdjxw4cPXoU3pU9UKx0kQyXdypqj/J1S+P84atYv349unTpkgdREr0dmJ8TEREVfHmZn8fFxQEAoqKiciQ9IiIiIiIiooIg27XqrVu3okSJEvD398eoUaMwefJkZd6ZM2fg7u6OtWvX5kSMRIVaYXzTNi4uDjNnzYSxiTHqB9TI9Hp12lSBuaUZ5s+fj8jIyFyMkAqawjykQkaYnxMRERV8uZWfP3r0SGtaUlISVq1aBUtLS1SoUOFNwiYiIiIiIiIqULLVsH7w4EEEBwfD3Nwcc+fORdeuXTXm165dG2XKlMHGjRtzJEiiwqwwNgguXrwYD+4/QLUmPrB3scn0epY2Fqjdpgqio6Mxb968XIyQ6O2Q1/n51KlToVKpUKlSpRxJj4iIiHI3Px84cCCaNWuGKVOmYMmSJfjiiy9QpUoVnDhxAl988QVsbDJfViciIiIiIiIq6LL1KfjPP/8cDg4OOH78OFxcXBAREaG1TK1atXDkyJE3DpCICpfbt29jxYoVsHWyRo3mFbO8fsX6ZXDhn2vYuHEjgoODC1UDXmH8ugG9mbzMz+/cuYMvv/wS1tbWb5wWERERvZKb+Xnnzp2xdOlSfP/994iIiICtrS1q1qyJr776CgEBATkRPhEREREREVGBka031o8cOYJ27drBxcVF7zKenp548OBBtgMjopeSk5MNHUKemjlzJpKSklC/XQ2YmmW974+RkREadagJAJg2bRpSU1NzOsR8iw3rGeMx0pSX+fmoUaNQt25d1KpV643TIiIioldyMz8PCQnBjh078ODBAyQlJSEyMhI7duxgozoREREREREVStlqWE9ISICdnV26yzx79gxGRtkewp2I/l98fLyhQ8gzR48exe7du1G8jBtKVfbIdjrupYqgbA0vnDlzBn/99VcORpi/FcZhA7KKx0hTXuXn+/btw4YNGzBnzpw3SoeIiIi0sX5ORERERERElDeyVbMuVaoUjh49mu4yhw8fho+PT7aCIqJXEhMTDR1CnhARzJ49GwBQv131N36zuO771WBsYoQ5c+YgKSkpJ0LM99honDG+sa4pL/LzlJQUhIWFoV+/fqhcuXKGyyckJCA6Olrjj4iIiPRj/ZyIiIiIiIgob2SrYT0oKAgHDx7E8uXLdc6fNWsWzp49i86dO79RcESEQtMo/Pfff+Ps2bMoW8MLrh5Ob5yerZM1KjV8B3fv3sXmzZtzIEJ6G7Dzgaa8yM8XLlyI8PBwfP7555laftq0abC3t1f+PD09s71tIiKiwoD1cyIiIiIiIqK8ka2G9dGjR6N8+fLo168fWrRogV27dgEAxowZg0aNGmHs2LGoVq0ahg0blqPBEhVGhWGM9dTUVCxYsAAqIxV8W2f8Rmtm1WhWAaZmJvjhhx8KzZv/lD6+sa4pt/PziIgITJw4ERMmTICrq2um1vnkk08QFRWl/N2+fTtb2yYiIiosWD8nyn2pqam4ceMGYmNjDR0KEREREREZULYa1m1sbLB//36EhIRgz549OHDgAEQEs2bNwqFDh9CpUyfs3LkT5ubmOR0vUaGTkpJi6BBy3Z49e3D58mW8U7MkHFzTHx8yKyxtLFCp0Tt49OgRfvnllxxLlwouvrGuKbfz8/Hjx8PJyQlhYWGZXsfc3Bx2dnYaf0QFGe87RDnnxYsXaNeuHdasWWPoUPIV1s+Jct/ChQsREBCAoKAgQ4dCREREREQGZJLdFR0dHfHTTz9h3rx5OHr0KCIjI2FnZwdfX1+4ubnlZIxE9BYTESxevBhQATWbV8zx9Ks29sF/+y5h6dKl6NChA0xMsn3bo7cA31jXllv5+ZUrV7Bo0SLMmTMH9+7dU6bHx8cjKSkJN2/ehJ2dHZyc3nzoB6L8jPcdopxz4cIFXL9+HdOmTUPXrl0NHU6+wvo5Ue66c+eOxn+JiIiIiKhwylYLU9OmTdGgQQN8/vnncHZ2RuvWrXM6LiL6f2/7A/l//vkHZ8+eRelqJeBQJOffTLWytUD5uqXx3/7L2L59O/z9/XN8G5T/qd8Y5ZujmnIzP7979y5SU1MxfPhwDB8+XGu+t7c3RowYgTlz5uTYNomIiAoj1s+Jcl98fLzy/yLy1tfTiYiIiIhIt2w1rB85cgR169bN6ViISAcjo2yN2FBgLFmyBMDL8dBzS7Um5XHu4FUsXboU77///lt/TEk/PgDTlJv5eaVKlbB582at6ePHj8fz588xd+5clC5dOle2TUREVJiwfk6U+5KSkpT/T05OhqmpqQGjISIiIiIiQ8lWw7qPjw/Cw8NzOhYi0uFt/nT56dOn8e+//6JEeXe4euTe56BtHa3xTq2SuPjvNezZswdNmzbNtW1R/sY31jXlZn7u4uKC9u3ba01Xv6Guax4RERFlHevnRLkvMTFR+f+EhAQ2rBMRERERFVLZem0zLCwMW7Zswfnz53M6HiJ6jbGxsaFDyDULFy4EANRsXinXt1WjWQWoVCosXLiQjatE/4/5ORERUcHH/Jwo96VtWE/7/0REREREVLhk61XYUqVKwc/PD3Xr1sXAgQPh6+sLNzc3nZ/Yfffdd984SKLC7G39bPnp06dx4MABeLxTFO6lXHN9ew5F7FC2hhcuHL+A3bt3v5VvrfMz5/ql/XQjvWKI/HzPnj05kg4RERG9xPo5Ue5L25jOugURERERUeGVrYZ1Pz8/qFQqiAi+/vrrdBtzUlJSsh0cEb2dRET5HLRv68p5tt1arSrhyslwzJs3D40bN36rvwZAmqKjowEAqampBo4kf2F+TkREVPAxPyfKfXxjnYiIiIiIgGw2rE+cOJFvRhLlkbfxs+X79u3DsWPH4F3ZA+7euf+2upqDqx3K1ymN84ev4pdffkFQUFCebTsvvI3nSk6JjIwEALx48cLAkeQvzM+JiIgKPubnRLmPDetERERERARks2F98uTJORwGEemTnJxs6BByVEJCAr766isYGRmh7vtV83z7tVtXxpUT4Zg7dy6aN28Oe3v7PI+B8t7Tp08BAFFRUUhJSeHXCv4f83MiIqKCj/k5Ue6Lj4/X+f9ERERERFS4vJ2DNxO9RRISEgwdQo5atmwZbt++jSqNy8HRLe8bta3sLFGrVSU8ffoU3377bZ5vnwxD3bAuInj+/LmBoyGiwohfFSEiIiq40tbL2bBORERERFR4ZeuNdbWYmBj88ssvOHXqFKKjo2FnZ4dq1aqhffv2sLa2zqkYiQq1t6lh/fLly1i0aBFsHKxQq2Ulg8VR5d1yuHz0Bn7++We0atUKvr6+BoslJ3H8cN3i4uI0rqOnT5/CwcHBcAHlQ8zPiXIfP9NMRLmN+TlR7uEb60REREREBLxBw/rGjRsxYMAAPHv2TOMNHJVKBQcHByxevBgdOnTIkSCJCrPY2FhDh5AjEhMT8emnnyI5ORl+nRvCzMLUYLEYGxuhSZe62DjnT0yYMAHr16+Hra2tweLJKWxY1y06Ojrdfxd2zM+JiKigYAcV/ZifE+UeEUFcXJzy77T/T0TZ07NnTxQvXhzTpk0zdChEREREWZKtT8EfOnQIISEhiImJQb9+/bBmzRrs3r0b//vf/9C/f3/ExsYiJCQEhw8fzul4iQqFlJQU5f/flob1OXPm4OLFi6hQrwxK+BQzdDgo4umEmi0q4u7du/jiiy/eik/0JicnGzqEfOnFixca/+an4F9hfk5ERFTwMT+nnBQeHo6HDx8aOox8JS4uTqMTc0xMjAGjIXo7nDx5Etu2bTN0GERERERZlq031r/88kuYm5vj4MGDqFq1qsa8zp07Y8iQIahfvz6+/PJL/PrrrzkSKFFhkrYx/W2otO/atQurV6+Go5sdGrSvYehwFLVaVMLdyw/x+++/o1atWujYsaOhQ3ojSUlJhg4hX1JfQyoTgSSr3prOKjmB+TkRERUkb0NHyNzA/Jxyyr1799C2bVsAwKlTp2BsbGzgiPIHdX3CxMwcyYkJWh13iYiIiIio8MjWG+uHDx9G586dtSrtalWqVEGnTp1w6NChNwqOqLBKW1Ev6JX28PBwfPrppzA1M0Gr0IYwNcv2CBQ5zsjYCM171IeljTmmTZuGs2fPGjqkN5KYmGjoEPIldUO66v9HH3gbOqvkFObnREREBR/zc8opT58+Vf6fw0y9ov7ilZ1LUY1/ExERERFR4ZOthvXY2Fi4ubmlu4ybmxvfCiTKprRjQBfkSntMTAyGDx+OmJgY+HWuDSd3B0OHpMXW0RotejRAcnIyPvjgA0RERBg6pGzjWH+6xcfHAwBUZi//zeP0CvNzIiKigo/5OeUUlUpl6BDyJXX93KGIu8a/iTKLHVWIiIiI3h7ZalgvWbIkduzYke4yu3btQsmSJbOTPFGhl7YxvaBW2kUE48ePx/Xr11HVzwdla5Q0dEh6ebxTFHX9q+Hhw4cYOXJkgf2kOt/E1k19XIzMXn4+lg+VX2F+TkREVPAxP6ecYmRkpPP/C7uoqCgAgGNRT41/E2UWhzIhIiIientkq6bUqVMnHD9+HKGhobh3757GvPv376NXr144fvw4OnfunCNBEhU2aSvqBbXSvnTpUuzcuRMeZd1Qr201Q4eToWp+PihT3QvHjx/H7NmzDR1OtqTtkMGK+yuvGtZf/psN668wPyfKOykpKYYOgYjeUszPKaekfWOdDeuvqD+R71SshMa/iTKLb6wTERERvT2yNdjx2LFj8ccff2D16tX4+eefUaZMGbi5ueHhw4e4evUqEhMTUbt2bYwdOzbLaZ87dw6TJ0/G8ePH8eDBA1hZWaFChQoYPXo0/P39sxMuUYGTtqL+7NkziEiB+izfv//+i2+//fblZ9Z7NoCRcf5/KKNSqdCkcx1EPniGH3/8EVWrVkXr1q0NHVaWpP26QWxsLKytrQ0YTf6hvp6MrETj35S7+TkRaeIDVcosdo6jrGJ+TjnF2NhY+f+CVP/MbZGRkQAAe5eiMDW3VP5NlFnM24mIiIjeHtlq7bKyssK+ffswefJkeHh44Pz589i9ezfOnz8PDw8PTJkyBXv37oWlpWWW0w4PD8fz588RGhqKuXPnYsKECQCAgIAALFq0KDvhEhU46oY/U3MLJCUlFahPfD979gwff/wxoAJa9moISxsLQ4eUaabmJmjduxFMzU0xZcoU3L1719AhZUnaBmM2Hr/y5MkTAICxtWj8m3I3PyciTXxjnTKLnTAoq5ifU05J27BOr0RERAAArOwdYW3vyPoEZRnLgURERERvj2y9sQ4A5ubmmDhxIiZOnIjnz58jOjoadnZ2sLW1faOA2rRpgzZt2mhMGzZsGGrWrInZs2djwIABb5Q+UUGg7gHvXLwkHly/iKdPn8LGxsbAUWXO1KlT8fjxY9Tzrwa3Es6GDifLHFzt0CioJv5e8w/Gjx+PZcuWFZi3NZ49e6bx/x4eHoYLJh+5ffs2AMDYBlCZvvo3vZRb+TkRvaR+kJqcnGzgSKigSNuwXtC+WkSGw/yccgIb1nV7/PgxAMDG0QXWDs54cP0CUlNT+bl8IiIiIqJCKEdqAba2tihevHiuVdqNjY3h6emp0WhE9DZTv23sXLwkgFc95PO7Q4cO4Y8//kBRb1dU8ytv6HCyrVwtb3hX9sCxY8ewbds2Q4eTaWnfUufnCV+5dv0ajMwBlTFgZCm4desWkpKSDB1WvpTb+TlRYaRuUOd9hzIrbcM6317XxE4GmcP8nLKLDeu6PXr0CCojY1jZOcDG0QUpycmsb1GW8I11IiIiordHthrWDx48iI8++ggPHjzQOf/+/fv46KOP8M8//2Q7sJiYGDx58gTXrl3DN998g+3bt6NZs2Z6l09ISEB0dLTGH1FBpW4gdfn/hvWCUGlPTU3FzJkzoTJSoXGwL1RGBffBp0qlQsPAmjA1M8E333yDxMREQ4eUKWk7H0VFRRkukHzkyZMneHD/AYxsXjZMGNsIkpKScPnyZQNHlj/kRX5OVNglJCQAQIHJS8jwXn9jnV7h8dCN+TnlFDas6/bgwQPYODrDyMgYts5FAAAPHz40cFRUkLCjHBEREdHbI1sN67Nnz8avv/6KokWL6pzv7u6Obdu24Ztvvsl2YCNHjoSrqyvKlCmDUaNGITAwEPPnz9e7/LRp02Bvb6/8eXp6ZnvbRIYWEREBC2sbpdJeEMbL3r9/P65evYpytbzhXMzB0OG8MVtHa1Rq+A4eP36MrVu3GjqcTEnboYidi146fvw4AMDETjT+e+zYMYPFlJ/kRX5OVNjFx8cDeNXATpSRtI3HfBBPmcH8nHIKG9a1paam4tGjR7D7/7q5rZMrAOjtyEKkC99YJyIiInp7ZKth/ejRo2jYsGG6y7z77rtv1CP+gw8+wI4dO7By5Uq89957SElJSfdNn08++QRRUVHKH8fQpYLsyZMnsLJzgrW9I4BXY7rlZ+vXrwcAVGtacD8B/7qqjcvByNgIGzduNHQomRIbG6vz/wuzAwcOAABMHEXjv+rphV1e5OdEhV3c/zesx8XFGTgSKij4xjplFfNzyikcbkHbkydPkJSUBDsXdwCAvevL/96/f9+QYVEBw45yRERERG+PbDWsP3r0CMWLF093maJFi+LRo0fZCgoAfHx80Lx5c/Ts2RPbtm3Dixcv4O/vr/fhkrm5Oezs7DT+iAqipKQkREZGwsbJBTaOL3vDv8m1lBdiY2Nx+PBhuBR3hJObvaHDyTFWdpYoXtYNZ8+eLRBvJKRttGEDzstrac+ePTAyB4xtXk4zMgOMbQVHjx7l5/KRN/k5UWEXGxMD4OUwR0SZkfatNr7hRpnB/JxyCt9Y13b37l0AgL2L28v/uhbVmE6UGcnJyYYOgYiIiIhySLYa1h0cHHDr1q10lwkPD4eNjU22gtIlODgYR48e5bi49NZ78OABRAT2LkWVT8Hfu3fPwFGl79y5c0hMTESJ8u6GDiXHeZUvBgA4ceKEgSPJWGJiIqBK8/+F3L///otnz57B1CUVaV++MXVNRUpKCnbs2GG44PIJQ+TnRIVJQkLC/7F33+FRVekDx793+kzKpPeEkITeBEQFFBVBReyLZfWnrq51bSusa++VtaKCBSkqIEVAeu+99xJKSGjpZdLbzP39MUxIpKXMzJ1Mzud58pBMuecdIHPm3vec96WqqgqwJ9bF7mOhPkRiXWgoMZ8LziJ2rJ/r5MmTAJjD7eeFAWH2RSyiSqLQECKxLgiCIAiC4D0alVi/6qqrmDlz5gVPJI4fP86ff/5Jnz59mhRcbY7dl2KHoeDtHBfFzGFR6AxGfAOCPf6k/fDhwwAERwUqHInzOfrFHzlyRNlA6qG6uhpJrar5vqWbNWsWANqwumX3dGEySDB79mwlwvIoSszngtCSFBUV1XxvtVpFmw6hXmrP4WI+F+pDzOeCs4gFYOdKS0sDIDAiBgC9yQejn/mSi1kEoTbHQktBEARBEASh+WtUYn3o0KGUlpbSt29ffv3115reUunp6fzyyy/07duXsrIyhg0b1uBjn688XVVVFb/++itGo5GOHTs2JmRBaDZSUlIACI6KAyAoqhUnT56k/EyPVk/k+L31C/RROBLn8w+y7+xpDqUzrVarSKyfYbFYWLpsKSqTjNqv7n0qPWgCbezYsaPm962lcuV8LgjCuQtCCwoKlAlEaFZqV50RFWiE+hDzueAsIrF+LkcCPTA8pua2wIhYTp48KaqKCPUmEuuCIAiCIAjeQ9OYJ/Xr148vv/ySYcOG8eijjwL2kmGOkzCVSsWIESPo169fg4/91FNPUVhYSL9+/YiOjiYjI4OJEydy8OBBvvjiC1G+TvB6ycnJAITGJdr/jE3g+P7tHD16lE6dOikZ2gUVFxcDoDdqFY7E+XRnXlPtXYeeymazIensfRFb+kWeWbNmUVVZhSG2bhl4B12ETHUeTJs2jVdeecX9AXoIV87nQssiyzJHjx4lKioKk8mkdDgeIy8vr87PBQUFl+yDLAgisS40lJjPBWex2WyXflALc+zYMXslucCQmtuCImM5fXgvp06dIi4uTsHohOaioqJC6RAEQRAEQRAEJ2lUYh3gxRdf5Prrr+eHH35gy5YtWCwWAgICuOKKK3j66afp3Llzo4573333MWbMGL7//ntyc3Px8/OjZ8+eDB8+nNtvv72x4QpCs7F//350BiMB4fYL72HxbQB7H3NPTaw7Vl+rNWqFI3E+1Zkd4J6eqLZardhsNrQaLVZa9o51m83GlClTkFSgCz//rhttiIxKD7Nm/cnzzz/fohOBrprPhZZlzZo1PPvss3Ts2JEpU6YoHY7HcCTWzaGRWLLTyc3NVTgioTmoXaXIkysWCZ5FzOeCM7Tkc4jzsdlsHEtNJTCyVZ3+88HRrQB7tTmRWLcvsCwvL8doNCodiscSiXVBEARBEATv0ejEOkDXrl0ZNWqUs2IB4P777+f+++936jEFobkoLi7m6NGjxLS/DJXKnqSOTOwAwK5du7j33nuVDK9Fclw+8fSyiI4dbaozO9Zb8g63devWcfz4cXQRNlQXKKIgSaCLsFKUVsy8efO455573Bukh3HFfC60LI4E8v79+xWOxLPk5OQAEB7fFkt2es3PgnAxtZPppaWlCkYiNDdiPheaSpSrruvUqVNUlJcTEtO6zu3B0fEAHDlyhOuuu879gXmYDz/8kKlTp/Lqq6/y4IMPKh2Ox6i9UEUk1gVBEARBELxHo3qsC4LgGjt27MBmsxHdrkvNbcFRrTD6mdm2bZuCkQnS+eqJexDHhXe1QVvn55ZowoQJAOijL17KUhclI6ngtwm/ibKXgtBEKpX4SHk+WVlZAIS3blvnZ0G4mNpzeFlZmYKRCILQ0tRO/nn6wmJ3OHToEGBvz1ZbaKy9bdvhw4fdHpMnWrlyJQAHDx5UNhAPU3uhnJjPBUEQBEEQvEeDroJeKPFQUFDASy+9RLdu3ejWrRvPPfecuHAoXFJFRQW333EHl112GTfeeCOFhYVKh6S49evXA9CqU8+a2ySViriOPTh16hRpaWlKhdZynUmoe3ri1dEDXmPSgSQ1i57wrpCcnMz69evRBNhQ+178sSodaMNsHEs5xtq1a90ToIcQ87ngbGq197UCcYaMjAwAotp0rvOzIFxM7cR6SUmJgpEInk7M54Kz1U4Eit3rtRLrcYl1bvcPCUdv9CE5OVmJsDyOYxGG2JVdl1goJwiCIAiC4J3qnVj/5ptv0Gq1LFmypM7t5eXl9OvXj2+++YY9e/awZ88eRo0aRd++fVtsYkeon7S0NI6lpGC1WklPT2/xJ6WyLLNs+XL0Jl+i23apc19i994ALF++XInQLslxAu2NPdbVZ3qse/qFpfz8fABUBjVqvabm55bml19+AUAfU78dNvoY+wXpsWPHuiwmT+Ou+Xzfvn3cc889JCQkYDKZCAkJoV+/fsyZM8dZL0XwICKxfn4ZGRmo1GoiE9vX/CwIl1L7QnxLrkBzPp5eQcidxPm54Aq1k38iEXi2xU14fJs6t0uSRFh8G44dOyb+nmppye3IzkcslLswT9+4IAiCIAiCcDH1TqyvWrWKsLAwBg4cWOf20aNHs3fvXjp27Mjy5cvZvHkzQ4YMISUlhW+++cbpAQve4+TJkwD4hkTU+bml2rFjB+mnT9P2imtRazR17kvqeTVqjZa5c+d6ZEk+xwmjRud9iRVJJaHRqj3+wnZ2djYAGqMOtVFT83NLcvr0aebNm4faR0YTVL/fE7UPaIJsbNu2jZ07d7o2QA/hrvk8LS2NoqIiHnnkEUaMGMFbb70FwO23385PP/3klNcieA6tVqt0CB7p1KlT+AeHozOYMPkHcurUKaVDEpqB2kma2rtHBaE2cX4uuEJxcfF5v2+p9u3bj39wGCb/wHPuC2/dDpvN1uI3CNQmEut11U6mi8R6XbX7zwuCIAiCIDQ39U6s7969m2uvvfac26dMmYIkSUycOJHrrruOyy+/nN9//53o6Ghmz57t1GAF73L8+HEAglq1A+DEiRNKhqO4adOmAdD5mpvPuc/g40eby6/h0KFD7Nq1y92hXZLFYkGlUqEzeGdiRW/SY7FYlA7jojIzMwFQm7RoTFqKioo8fjGAs40fPx6bzYY+zkZDNrQZ4uyr5ceMGeOiyDyLu+bzW265hYULF/LOO+/wxBNP8OKLL7JixQq6devGl19+6YyXIngQkVg/V2VlJVlZWfiH2hcQmsMiOX36tNihI1yS2DF6YZ64wFQp4vxccAWRWD8rIyOD7OwsIhI7nPf+qKSOgP13UbDz9Cpv7iYq0FyYSKwLgiAIgtCc1Tuxnp2dTWJi3b5SVVVVbN26laSkJLp27Vpzu1qt5qabbqrpRyUI5+PoFx7S2l4eNTU1VcFolJWZmcnChQsJiU0gpn238z6m+8C7AJgwYYI7Q6uX3NxcDL56ry3PafTVk5ubq3QYF3X69GkAtL46NL56ANLT05UMya1yc3OZPn06KgNoQxt20V1jBrVZZuXKlRw+fNhFEXoOJedztVpNbGwsBQUFTjmeIHiykydPIssyAWHRAASERdck2wXhYmr3qBW7/+ry1s+ajSHOzwVXqL2YuLCwUMFIlOeoZhXdpvN5749q06nO41oyR5JUzFl1iR3rFyYS64IgCIIgNGf1TqyXl5efU4pwz549VFZWctVVV53z+PDwcLEiU7ioY8eOIUkqzJFxaPRGjh07pnRIipk4cSLV1dX0uuW+C14wjGnfjYiE9ixZssTjdvfn5GTj429QOgyXMfkbKC4u9uhdY47EusZXj8ZXB9CiSg7//vvvVFZWoo+1Nmi3uoMh1r6DdPz48c4NzAO5ez4vKSkhJyeHo0eP8tVXX7FgwQJuuOGGCz6+oqKCwsLCOl+C0Bw5WtwEhp9JrIdHAaJCj3BptRMTtZPsgtixXps4PxdcoXZiPT8/X8FIlLdt2zYAott1Oe/9/sHh+IeEs23bthb/3lRZWXHmT5FYr00k1i9MJNYFQRAEQWjO6p1Yj4yMZP/+/XVuW79+PZIkccUVV5zz+MLCQoKDg5seoeC1jh07hsEchEqtwRQYSlpaGlarVemw3M5isTBlyhT8gsLo2HfgBR8nSRJX3v4gNpuNsWPHujHCi6uoqKCkpBSjnxcn1v2MgGdfXDp58iQqnRqVTo3WT19zW0tQVlbG5MmTUWlBF964i1qaIBm1SWb+/Plev5vU3fP5sGHDCA0NJSkpif/85z/cddddfPfddxd8/CeffILZbK75io2NbfTYgqAkRwI9IMKeWA8Mj6lzuyBcSO1SuqKsrnAh4vxccIXa5zstvcLQli1b0BmMhMe3u+BjYttfRl5eHikpKW6MzPOUl1ec+bP8Eo9sWURi/cLE5xtBEARBEJqzeifW+/Xrx9KlS1m9ejVgT2SMHj0agJtvPrcn9K5du4iJiXFSmIK3sVgs5OXl4RMUBoBPUChVVVU1u25bksmTJ1NaWkqvwfej1ly8R23by/sRFBnHrFmzyM7OdlOEF+fY1aA36RWOxHX0JvsOcE+9uCTLMidPnUTjo0OSpJpS8C1lx/r8+fOxWCxoo2xI6sYdQ5JAF2OjurqaqVOnOjdAD+Pu+fzf//43S5Ys4ZdffmHQoEFYrdaL7mZ57bXXsFgsNV8iCSk0VzWJ9TCxY11omNoXm8XuP+FCxPm54Ap5eXk133t6KyxXys7O5ujRo8S074Zao7ng4+I69QBg48aN7grN41RVVdVskBCJ9bpqV7zz5Op3ShA71gVBEARBaM7qnVj/73//i1qtZsCAAfTo0YOEhAT27NnDbbfddk5vt9zcXDZs2MDVV1/t9IAF7+Dop24KDK3zZ0srB19RUcHEiRMx+vrT9fpbL/l4SaXiilv/TlVVFRMnTnRDhJfmOCFSq+v9dtLsqM68Nk89+bNYLJSWlNbsVNeeKQXfEhaqyLLMpEmTQAJ9lK1Jx9KFyai0MHXqVK9OZLh7Pm/fvj0DBgzg4YcfZu7cuRQXF3PbbbddsGSmXq/H39+/zpcgNEeOqiEBYVF1/hSJ9bN2797Nzz//jM3WtPdvb1P784b4uxEuxF3z+ZYtW3juuefo1KkTPj4+xMXFce+994p+7V4qJyen5vuWnFhfv349APFdzq3+UFt8l151Ht8S1d6JXVxcrGAknqd2+w2RWK+r9medlli5UhAEQRCE5q3embBOnToxZ84c4uLi2LlzJ7m5udx9993nLUn9448/Ul1dzU033eTUYAXvcfz4cQBMAcFn/gypc3tLMW/ePPLz87lswF3oDMZ6Pafj1TfiYw5i2rRpHtEnUaWyv41484Vf2WZPADpeq6c521/dnlBX6TWoNKoWkVjftWsXhw4dQhtiQ6Vr2rEkNWgjbOTn57N8+XLnBOiBlJ7PhwwZwpYtW8QFecHrnTx5EqOfGb3JBwCfgGA0Wl2LeG+ur6effpoRI0Zw+PBhpUPxKLUvMHvqoj5Bee6az4cPH8706dO54YYbGDFiBE8++SSrV6+mR48e7N271xkvRfAg2dnZqHX2xbq1k+wtzZo1awBIuOzKiz7OLyiU0NgENm3e3GJ3axcVFdV8X1xc3OL7zddWO5nuCdduPEntzzfis44gCIIgCM1Ng7JEAwcO5MiRI2RmZlJcXMwff/xBUFDQOY/797//TX5+PjfeeKPTAhW8i6NEtdFsT6wbzEF1bm8JHDttVWo13QfeWe/nabQ6LhtwJ4WFhSxYsMB1AdaT4z2grMh7LySUFtlPiD21L2VGRgYAGh97ZlmSJNQ+uprbvdm0adMA0EU55wKOLtK+QGTKlClOOZ6nUnI+d1xgcrSREARvJMsyp0+fxj8kouY2SZLwD4mo2ckunL0YL/ps1iWSEkJ9uWM+Hzp0KGlpaXzzzTc8/vjjvPnmm6xZs4bq6mo+/fRTZ7wMwUPIskxWdjY+gWGoNFqysrKUDkkRVVVVrFu3nsDwGIIi4y75+ITuvakoL2fz5s1uiM7zFBYW1nxfXV0tdmbXUlFh7z2PJFFe4b3XSxqjdjJdfA4UBEEQBKG5adT2y9DQUHS6C28NNJlMmM1mJElqdGCCd3Mk/Ax+AXX+bAmJQIddu3aRnJxMm17X4hsY0qDndut/Gyq1msmTJyt+8VWn0xESGkJBVtGlH9xMWbKL0Gg0hIQ07N/JXRwXvTSms+/LGh8deXl5Xl3S3GKxsGDhAlQmGY3ZOb8HaiNoAm1s3bqVlJQUpxzTk7lyPj/fxdiqqip+/fVXjEYjHTt2bPAxBc+l9FzkaQoLCykrK8M/JLzO7f4h4eTn55+90CoIlyB+t4T6cOV83qdPn3OO3aZNGzp16sSBAwcafDzBcxUWFlJZUYHez4zex79FnZvXtnXrVoqLi0js2bdej0/qYW+xsGLFCleG5bEKCgou+nNL5vi8p9arqSgXn/1qq51MF4l1QRAEQRCaG8+sayx4PUdZOZ2PHwBagwmVWk12draSYbnV5MmTAegx8K4GP9c3MIQ2va7l4MGD7Nq1y9mhNVjXLl0pyi+hxOJ95c2qq6xkn8ynY8eOaLVapcM5L8fvk9p0Nj610f59Xl6eIjG5w59//klVZRW6SBvOXMfl2P3u7bvWXe2pp57ihhtu4L333uPnn3/mww8/pGvXrmzfvp0PP/wQX19fpUMUnEhcEKsrPT0dAP+gsDq3+wXbf87MzHR7TELzUTv5KRYqC55IlmUyMzMvuui0oqKCwsLCOl+CZ3PMXQZfMwa/AHJzc1vk/O5oCZXUo36J9aikjviYg1i5apVXt0e7EMf5pv7MIu/8/Hwlw/EojsS6SqemqqpKLJarpfYGgJb4PiMIgiAIQvMmEuuCIvLz81FptGjO9G+TJAmtwafFnIRlZWWxaNEiQmMTiGnfrVHH6HHj3QBMmDDBmaE1Sq9evQBIO+B9fWNPHsrAZrVx+eWXKx3KBdUk1g1nE+sao6bOfd7GarUyadIkJDXoIpx7gUIbLKPS2xP3xcXFTj12S3LfffehUqn4/vvveeaZZ/jyyy+JiYlh1qxZDB06VOnwBCcTvyt1ORLnfn/dsR4cXud+wa4lJiIupnYyXaUSp2u1iYUGnmHixImcOnWK++6774KP+eSTTzCbzTVfsbGxboxQaIzTp+3ncgb/AAx+AdhsthZXDt5ms7FixQqMfmZi2nWp13MklYrEHn3Jyc5m9+7dLo7Q8ziu4QSFmwHvXtjdUI7ksUqrxmaziV7itdROrItKToIgCIIgNDfiSo2gCIvFgtZgqnOb1mjCYmkZOxl++eUXqqur6Tno3kZfIIxp15Xw1u1YsmQJqampzg2wgQYMGADAkR3HFY3DFY7sSANwao9pZ3NczFAbNDW3qc4k2b11scrSpUs5ffo02nAbKs2lH98QkgS6KCulpaVMnz7duQdvQe6//36WLFlCRkYGVVVV5OXlsWTJEm6//XalQxNcwGKxKB2CR3GUz/X7S6sXx451x65AwU4k1utSq9Xn/V4QPMHBgwd59tln6d27N4888sgFH/faa69hsVhqvk6cOOHGKIXGOHXqFAAG/yAM5sA6t7UU+/btIzMzk6QefVGp63+S0faKawFYtmyZq0LzWI7zzcAIc52fhVqJdZ19Lhc7s8+qnUz35vZ1giAIgiB4J5FYFxRRWFiIRm+oc5tGb6S4uMjry2Olp6czZcpU/EMi6HR145O1kiTR586HsdlsfPfdd06MsOEiIiLo2bMnJw9nYMnxnl2L5aWVpOw+QXx8vEf3g87Pz0dSSTUn7ABqvabmPm8jyzJjx44FCfQxrknG6KJkJA38+uuv4gKIINRD7fcab5/H66OmFHxIRJ3bHTvWW2rf2gsRO7jqqp1MFzvW6xLvL8rKyMhg8ODBmM1m/vjjj4su/NDr9fj7+9f5EjybY/GD0RyE0RwMwPHj3rdw+mKWLl0KQJte/Rr0vFadeqA3+rB06dIW9z7l2KEeJBLr53CcR6q09vdKkUA+q7y8/LzfC4IgCIIgNAfiSo3gdrIsU1xcjEZvrHO7RmfAarVSVlamUGSuJ8syw4cPp6KinGvufQK1pmk9u5Muv4botp1ZtGgRGzZscFKUjXPvvfeCDPs3HFY0DmdK3pxCdZWVe+9tfGUBd8jLy0Nl0NSJ0bF73RsvbKxdu5b9+/ejDbGhNl768Y2h0oAu0l7+8s8//3TNIILgRbKzs2u+F310z5bT9f9rKfgzPzvuF+xEYr2u2sl0sWNd8BQWi4VBgwZRUFDAwoULiYqKUjokwckcSXSjORhTQMtLrMuyzNJly9AZTcR3blgbMLVGS2KPPpw8eZLk5GQXReiZanash9sXz4hS8GdVVFSABNKZxLooeX5W7et+3nwNUBAEQRAE7yQS64LblZeXU1VVde6OdYM9Q+bNF+T//PNPli1bRlzHHnTsO7DJx5MkiYGPDkOlVvPmm28qmkQdMGAAISHB7N9wlKqK5r/D12a1sXtNMkaj0eNLV+fn59fsUHdwJNZzc3OVCMllZFnmhx9+AMDQyrWlg/UxNiQ1jB49WuxaF4RLqN0zXOzGtu/60+j0+J5JTDiYQyOQJJUoifwXYgdXXbUXynnywj6h5SgvL+e2227j0KFDzJ0716MrOQmNl3LsGHpfMxqdHlNgKADHjh1TOCr3OXToEMfT0ki4rDcanb7Bz2/by14OfsmSJc4OzaMVFBSgUqnwD/Kt+Vmwq6ioQKVWoVLb53KxM/us2sl08fciCIIgCEJz06jEusViYffu3ZSWlp73/pKSEnbv3u3VCVKh8RwnWuf0WD/zszfusAXYuXMnH370EUZff2555nWnXSgNa5XENfc+SVZWFsOGDVPs4rROp+Pvf3+AirJKDmxKUSQGZ0rZfYKivBLuvPNOzGaz0uFcUEVFBYWFhaiNdasfOH72tsT6hg0b2L17t323uo9rx1Lp7LvW09PTmT17tmsHU4iYzwVnqd2DtaX1Y/0rWZZJTU0jMDwa6S9lvNUaLebQCFJSmv886UxiB1ddtcsIt7SSwkLjuHI+t1qt3HfffWzYsIFp06bRu3fvpoYreKCSkhLST5/G90zLEq3BhN7Hn8OHvaca2aU4EuLtrryuUc9v3e1KtHoDixcvblHv3RaLBYOPDr2PvuZnwa60tBRJq67ZsS52Zp8ldqwLgiAIgtCcNSqx/v7779O3b1+sVut577darfTt25ePPvqoScEJ3iknJwcAvcmvzu26Mz877vcmhw4d4rnnnqO6uppbn3unpseqs1wx+H7aXXk9W7Zs4fXXX1espOp9992HwWBg18qD2Kyu3U3sSrIss2PFAVQqFQ899JDS4VxUVlYWABpT3cS65kxivfYu0uZOlmW+//57wPW71R30sfZd6z/99JNX7loX87ngDFVVVZw+01McaPG7sTMyMiguLiIkNuG894fEJpCTk+O1CwkbQ+xUqqv2e/KF3p8FoTZXzufDhg1j9uzZDBo0iLy8PCZMmFDnS/AOhw4dAsAnJLLmNp+QCE6dOkVRUZFSYbmNLMssWrQIrd5IQrerGnUMrd5AQvc+pKamtqgFCRaLBb1Jh96gRZIksWO9luLiYiSNCpVGVfOzYFd7IVhJSYmCkQiCIAiCIDRcoxLrCxcuZODAgfj5+Z33fn9/f2666Sbmz5/fpOAE75R+5uK73q/uLmDDmZ/Ta12c9wb79u3jscceo7CwkEFPvkrrrlc4fQxJpWLwM28Q27E7ixYtUmznutlsZsiQIRTll3BkZ/Ptx3fqSCbZJ/IYOHAgsbGxSodzUY6doRrfuuUKJbUKjUnrVTtHt27dys6dO+271X3dM6ZKB9oIG6dPn2bhwoXuGdSNxHwuOMPp06exWa34R8QBkJaWpnBEytq/fz9gryhzPo7bDxw44LaYPJ0oBV9X7QWS3rioS3A+V87nO3fuBGDOnDk89NBD53wJ3sExd/mFRdXc5hcWDbSM+So5OZnU1FQSe/RB+5eWdQ3R4ar+ACxYsMBZoXk0WZbPJNb1SCoJvUkndqzXYrFYUOs1qM60bRN/N2fV3qV+oWorgiAIgiAInqpRifXjx4/Tpk2biz4mMTGR48ebb2JNcB3H/wujuW7fUaM5CPCunW4rVqzg0UcfpaiomFueeYNO19zssrE0Oj1DXv4f8V16sXz5ch5//HFFdsM99NBDqNUqdizf32xL4O1YZr+w9NhjjykcyaU5fp+0fuf2AdT46TmdftprEhbjxo0D7LvI3ckQYwMJxo4d22z/T1+ImM8FZ0hNTQUgpHU7JEmq+bml2rVrFwBRSZ3Oe39kYsc6jxPEjvW/qv33IcrkC/Xhyvl85cqVyLJ8wS/BOzjmJPOZRXL27+0LjHfv3q1ITO40Z84cADr2Hdik4yRcdhV6ky/z5s3DZmu+Fdzqq7S0lOrqagw+OoAzifUCZYPyEOXl5ZSXl6PWq1GfSayLakVn1U6mi1LwgiAIgiA0N41KrEuSdMmLPBUVFaJ0oXBejrJoPkFhdW53/OwNZdNkWWbs2LG8+OKLVNvgzqEf0enqm1w+rlZv4O7/fErHvgPZsWMHDzzwQE1ZP3eJiopi0KBbyD1dwPEDp906tjNkncjjRHIGvXv3pmPHjkqHc0mO3xddgPGc+3QBRmxWG8eOHXN3WE6XlpbGmjVrUJtlNP7uHVtlAG2ojSNHjrBlyxb3Du5iYj4XnMHRL9w3JAqDf5BXvOc0xdatW1GpNUQktD/v/dFtOiNJEtu2bXNzZJ6ldkLOWxaAOUvtC8yiPKpQH2I+F5pClmW2b9+OzscPg39gze3myFYAbN++XanQ3KKqqoq5c+di9DPTuuuVTTqWRqen3ZXXk56e7nXnDefjSBQbzvRXN/joKSiwiEU3QHZ2NgBqk66mbZvjNqHu5xvxWUcQBEEQhOamUYn19u3bs3Dhwgt+WLbZbCxYsIB27do1KTjBO+3ZuxetwVTnpB1AozdiNAezd+++Zn0iVlpayssvv8xXX32FX1AoD7wzkqQefd02vkarY/C/3uLqIf/k5MmTPPjgg24vYf3Pf/4TgO1L97t1XGfYsWwfAI8//rjCkdTPwYMHkVQSWvO5JQt1QaaaxzR3U6dOBUAfrczOD8e4kydPVmR8VxHzueAMjkS6T1AoPkFh5OTkUFhYqHBUyigqKmL//v1Etel0wVKyBl8/wlq1YceOHS16p3btBJ9IrNdVu5+x6Mcq1IeYz4WmOHHiBJmZmQRGJyBJUs3tOpMvPkFhbNu+vU6LCm+zatUq8vLy6NxvEGqNpsnH63r9YAD++OOPJh/L0+Xl5QFg9LF/5jH5GqiqqhJzF5CRkQGAxqRFbbLv6M/MzFQyJI9SexGh2LEuCIIgCEJz06izhr///e8MGzaMxx57jK+//hqz+WyvbIvFwosvvsiRI0f4/PPPnRaoUkpKSvjxxx89ZgXllVdeyY033qh0GI2WlZXFiePHCWndoc5Ju0NAVDzpB7Zx9OhRkpLO35vUkx0/frzm/39sh8u4/YX38TEHXvqJTiZJEn3u/gehrZKYN+oDXn75Zfbv388LL7yAxgkXCy4lKSmJ66+/nhUrVnD6aBZRiWGXfpIHyMu0cHT3Cbp27UqvXr2UDueSKioq2LdvH7ogEyrNueukDKE+AOzYsYM77rjD3eE5TVVVFXPmzLH3Ow9WZtGNxh/UvjIrVqwgPz+fwED3/167QkuazwXXOXbsGJJKjcEchCkoFI4dIDU1la5duyodmttt2LABq9VKfJeLzyHxXXqRmXqILVu2cM0117gpOs9SO0njzQmbxigsLESlU4NNbrGLVISGEfO50BQbNmwAIDA28Zz7AmOTOLlrPXv27KF79+7uDs0tJk2aBEDX629zyvEiEzsSGpfIkiVLyc7OJjQ01CnH9UQ5OTkAmPztiXWjn6Hmdj8/P8Xi8gSnTp0CQOOrR3OmVL7jNqHuLnWxEEMQBEEQhOamURm25557junTp/PLL78wa9YsevXqRXR0NKdOnWLLli0UFBTQr18/nnvuOWfH63ZTpkyp6evrCWbNmsVVV12Fv7+bayE7yZo1awAIanX+HoBBrdqQfmAbq1evbnaJ9fXr1zNs2H8oLi6i56B7uO7v/3LKivemaNPzah76YDQzv3ydcePGcfDgQT777LM6F9tc5cknn2TFihVsXbyX25/p7/LxnGH7kn0g22M/38IPT7N7926qqqowhwWd935dgBGVTt3syxCuWbOG/Px89DE2pEbVWXEOXbiNsqPVzJ8/nwcffFC5QJyoJc3nguscS03FGBCMSqXGFGi/eNxSE+urVq0CILF7n4s+LrF7HzbNmciqVatabGK99o51UZ66rry8PFR6Ddjkmt2Agl1VVZXSIXgkMZ8LTbF27VoAglu1Pee+4Pi2nNy1njVr1nhlYv3gwYNs2bKF1l2vIDgq7tJPqAdJkuh50xAWjh7O77//zgsvvOCU43oix65sX7O9UprvmfZkmZmZtG7dWrG4PIEjia711aHSqNCYtJw8eVLhqDxH7WS6p2xkEgRBEARBqK9GZf20Wi1Lly7lzTffZPTo0SxZsqTmPn9/f15++WXef/99tFqt0wJVgizLTJ8+HYCqTjcia5R9Per0ZCqyjjB//nzuv/9+RWNpLEdJ8tCE8/euDo5vh6RSs2DBAh577DF3htYkv//+O59++ikqtYbB/3qLTld7TlWB4Kg4HvrgJ+Z//yEbNqzhwQcfZNSoUcTFOefCwYV07tyZvn37sm7dOtKPZRPZ2rNX6hdkFXJ4exrt27enX79+SodTL46LYKao8y+0kVQSxkg/jqcd58SJE8TGxrozPKeZM2cOYE9sK0kbJlOWYo/HWxLrLWU+F1zHYrFgKSgg5My8bgoIASAtLU3JsBRRVVXFypUr8Q8JJ6zVxRcHRrXthMk/gGXLl/P666+jUim4akghIrF+frIsk5+fj9qsFYn187hUH/GWSsznQmOVl5ezYcMGfEMizmnVBhAYk4hKo2X16tVemSAeO3YsAL0GO/f6Sse+A1kzdTSTJ0/mn//8Jz4+Pk49vqdwJI99g+yvzy/Qt87tLZnjs7D2zG5+jZ+e0+mnqaqqEu/F2NveSHoZuUKq0wJHEARBEAShOWj0VTyDwcDnn39OXl4ee/bsYe3atezdu5fc3FyGDx+OXq93ZpyK2LFjB8ePH8caEo/sGwQGP0W/rDGdQVLx559/Kv1X0ygnTpxg06ZNBMQknPekHUBrMBGS0IGDBw+yb98+N0fYcLIs88033/Dxxx9jMgfy97e/86ikuoPeaOLOf39I7zsfJi0tjYceeoj9+13f//yZZ54BYMvCPS4fq6m2Lt6LLMs8/fTTzWK3OsDq1auRNCoMERcus2eKNtc8tjnKy8tj5cqVqH1l1L7KxqLSgSbIxr59+zh06JCywThRS5jPBddx7LwxBQQDYDzz54kTJxSLSSmbN2+msLCQtr2uveQ8olKpSep5DTnZ2ezcudM9AXqY2r2gL9QXuiUqLCyksrLS3pPVqKWsrEzs5KpFJNYvTMznQmNs2LCBiooKQlp3OO/9aq2OoNgkkpOTvW63bUpKCgsXLiS8dTtadb7cqcfW6PT0HHQPRUVFNaXmvZEjeRwQaj8fNZ/5MzU1VamQPMbx48eRNCrUJnsSXetvwGa1iUUHZ1gKLUgakNT2hbqCIAiCIAjNSZO3x6jVajp16kSfPn3o2LEjarXaGXF5hNmzZwNgC/WQElZaAzZzJPv27ePo0aNKR9Ng48ePR5ZlYrr2vujjHPd7Ugn+85FlmeHDhzN69GiCIuP4v/d+JDLx/BckPIGkUnHNvU9w0xP/Jb+ggMcee4w9e1yb8O7WrRtXX301Jw9lcOpIpkvHaoq8DAuHtqfSvn17+vdvHmXr09LSOHLkCKYo//P2V3cwxQYAsHz5cjdF5lxz5syhuroaXYSyu9Ud9BH25M+MGTMUjsT5vHk+F1zHkUA3mO0tKfQ+fqjUmhZ50XD+/PkAtO9dv3mkw5nHOZ7X0ogd6+eXmWn/vKQ2atGcuRiflZWlZEgepby8XOkQPJ6Yz4WGWLZsGQChSZ0v+JjQpE51HustRo4ciSzLXD3kMZcsrO5x492Y/MyMGzfOaxOHhw4dwsdsQm+09xAPijDX3N6SybJMWloaWj99zf8trZ99cdPx48eVDM0jWK1WiouKUWllJI3stb8fgiAIgiB4r5ZXd7KeKioqWLRoEbLeB9kvTOlwalhD44GzpZGbi7S0NKZPn44pMJSwi5y0AwTGJuIfHsuiRYs8etf6t99+y8SJEwlrlcQD74zEPyRc6ZDqpdv1t3H78+9SVlbO008/zeHDh106nqOX46Z5uzx2R9qm+btAhueff77Z7FZfunQpAD5xARd9nMaoxRDuy9atW5tdOdnq6momTpyIpAZtuGf839EEy6j0MHPmTFGyThA421vT4BcAgCSp0PuZOX06XcGo3K+0tJSlS5cSEB5NZOL52938VWzH7vgGhrBw0SIqKytdHKHnsdls5/2+pXMk1jU+OjQ+9kSF4/dMoEX+rgiCq1RVVbFixQoM/oH4hUVf8HEhCR2RJFXN+Yc32L17N4sXLyaqTWcSLrv4wv/G0hlMXHn7/1FUVMSYMWNcMoaScnJySE9PJyw2qOY2nUFLQJg/e/bsadFzu8Viobi4uCaZDmdLwrfEqk5/VVhYiCzLSFqQtFBQUKB0SIIgCIIgCA1Srx7r/fv3R5IkfvnlF2JiYuq9o1OSpGa7qnnVqlUUFxdji+oIHpRokwOiQKNj7rx5vPDCC82iJ6csy3z00UdYrVaS+g5CukTMkiSRdM0tbP/jRz748EMm/PYbGk29/qu6zZ9//mnfqR7Vinte/RKTf4DSITVIuyuvx2q1Mm/k+zz73HNM/v13goKCLv3ERujUqRMDBw5kyZIlpO49ResuMS4Zp7EyUnM4tuckPXr04JprrlE6nHpbsmQJkkqq2ZF+MT6tAinPLGb58uUMGTLE9cE5yeLFi0lPT0cXbUPlIW8BkgS6aCulKaVMnTqVf/7zn0qH1CAtcT4XXMuRBDT4mmtuM/gGkHsqherqao+bv11l6dKllJaW0v2W++u9QEulUtPx6hvZPGcSq1evZsCAAS6O0rOIxPr5pafbF6VofHTINvuiMpFYP0sk1u3EfC44g6OFSVyPay46d+mMPgTGJrBz504yMzMJD28eC8ovRJZlPvvsMwCuf/BfLl1Y3f3Gu9m+eAYTJkzgnnvuITY21mVjudvWrVsBiIgPqXN7RHwIBzencPjwYdq1a6dEaIpzVG7S1E6s++nq3NeS5efnA9hLwWtlSvNLqaysRKfTKRyZIAiCIAhC/dTraufKlSuRJInS0tKan+ujuez8PJ+5c+cCYA1upXAkf6FSYw2MITMjhW3bttGrVy+lI7qkyZMns2HDBkISOhBSz11cgTEJRHTowb692xk9enRNr25PkJyczPvvv4/Rz8yQl/+Hj/n8/eI9Xcc+AyjMTmf1lJ945ZVX+PHHH122UOOFF15g+fLlbJizg7gOkag1nlGSUpZl1s/aDsBLL73UbN6zTp8+zb59+zBF+6PWX/pt3LdVILmbT7BkyZJmk1ivrq5m1KhRIIE+xrMSLvpImcoTMHbsWO677z58fRVu/t4ALXE+F1wrOzsbAH2txLre1x9ZlsnLyyMszHOq/rjSjBkzkCSJztfc3KDndel3C5vnTGL69OktOrEuSsGfdfr0aQC0vvqaxLq4CH9WdXW10iF4BDGfC86wePFiAMKSulzysaFJXcg7foSlS5fy4IMPujo0l5o7dy47d+6k3VX9iW576dfeFBqtjuse/Bezvn6L4cOH891337l0PHdav349ANFtI+rcHtM2goObU1i/fn2LT6xrfc8mijW+9iS7Y55vyXJzcwGQdCCd+TiYl5dHRETERZ4lCIIgCILgOeqVRbPZbFitVtq2bVvzc32+GnORbMuWLTz33HN06tQJHx8f4uLiuPfee93ao8lisbBmzRpspkAwmS/9BDezhcQDMG/ePGUDqYedO3fyv//9D53Jl/Y3/K1BF3PaXns7Bv9Avv/+e9auXevCKOuvqqqK1157jaqqKm7911sEhEcpHVKTXHn7/5HU82o2btzI77//7rJx4uPjuf/++ynILmLvOteWnm+IIzuPk5Gaw0033cRll12mdDj15thp5NOqfos6ND469CE+bNq0icLCQleG5jSzZs0iLS0NXaQNtUHpaOqSNKCLsVJYWMj48eOVDqdB3DmfCy1DTk4OSBJag6nmNp3J9+x9LUBKin2xY3yXXphDIxv03ODoVkS37cK6detqdiq3FLUTpOI95qyTJ08C9gvwjhKyjtsEkVh3EPO50FTV1dUsW7Ycva8Z/8hL76IOS+qMJEk1yfjmymKx8Pnnn6PVG7j+wX+5Zcy2va6lVefLWbVqFcuXL3fLmK5mtVpZtWoVPmYjodF1z0nj2kciqSRWrFihUHTKq93WxUGlU6PSqGrua8kciXWVTkalrXubIAiCIAhCc+BxdcSHDx/O9OnTueGGGxgxYgRPPvkkq1evpkePHuzdu9ctMSxatIjq6mpsIR62W/0M2S8UWWdi0eLFVFRUKB3OBaWmpvL8889jtdnoPOgB9D5+DXq+1mCky60PIak1DB02jAMHDrgo0vqbOnUqhw8f5rIBd9K625VKh9NkkiRx0+P/xehn5rvvRtaU5HKFZ555BrPZny0L91BaVOayceqrsryK9bN2oNPreOmll5QOp0GWLl0K0qX7q9fm0yqg5gKIpyspKeGbb75BUoOhlWftVnfQR9t7rY8fP16U6BVatLy8PHRGnzptXlpaYn3atGkAdOt/e6Oe363/7ciyzB9//OHMsDxe7QSfKAV/1vHjx1FpVKiNGtQmLZJaxfHjx5UOy2OIxLAgOMe2bduwWAoITeqEJF36spDO5Is5ujU7duxo1vP7V199RV5eHn2H/BP/YPeUtJckiYGPvoRao+Xjjz+muLjYLeO60tatW8nLy6N15xgkVd3NEwYfPVGJYTWtA1qirKwsANSms4l1SZJQm7Qt9u+kNsffj0oHkl6uc5sgCIIgCEJz4HGJ9aFDh5KWlsY333zD448/zptvvsmaNWuorq7m008/dUsM9jLwEjZPKwPvINljKy4q8tgk2cmTJ3n88ccpKCig/Q1/IzA2sVHH8Q+LptPN91NeVsaTTz7JkSNHnBxp/VVUVPDjjz9i8PHlmnseVywOZ/MxB9L37kcpLi7il19+cdk4ZrOZF1/8d01CW2lbFu2hxFLKE48/QXR0tNLh1FtOTg47duzAEO6H2qCt9/Mcu9ubQ1/NMWPGkJeXhz7WispD26xJajC0tlJRUcHXX3+tdDiCoJjcvDx0proL5xw/5+XlKRGSW5WXlzNr1ix8A0NI7NG3Ucdof9X1GH39mT59OlVVVU6O0HPVTpCKXch2siyTmpqKxmxAkiQkSULrpyc1NRVZlpUOzyOIRRjeT5ZlCgsLverLE9/bly5dCkBYYud6PycssTOyLDfbncgbNmxg+vTphMW34fKb3dseKygyjt53PkxmZiZfffWVW8d2hfnz5wOQ1P3818zadG+FLMssWLDAnWF5DMfiE42x7vm62qglNy+3xc9ljsUFkt6+WB0Qi9UFQRAEQWhW6tVjffXq1Y0eoF+/fg16fJ8+fc65rU2bNnTq1MktO5bT0tLYsWMHNnME6IwuH6+xrKHxqNMPMHv2bG688Ualw6nj+PHjPPbPf5KZmUmbfoOJ6nR5k44XltSZ9gP+xoElf/DYY48xevRoRXp1LV26lPz8fK66/f8w+nlei4Cm6Nr/NtbNGMf0GTN49tln0Wrrn7BtiLvvvpuZM2eyZ9se2l4eT1x7ZUrpZ5/IY/eqZOLi4nj00UcViaGxli9fjizLDdqtDqDzN6ALMLJ27VpKS0sxmUyXfpICTp48yfjx41EZQB/j2UkEbZiM+pTMvHnz+Pvf/063bt2UDumS3DmfC96vqqqKQouFoLi6fdS1Rh+gZSTW58+fT1FREX3uHoJaU6+P1efQ6PR0vvYWtsybzLJly7j55ob1aW+uaifTRWLdLjMzk7KyMnwjgmpu05oNFKXlk5ubS0hIiILReYaWnoxw8Ob5/MUXX2y2idsL8fc3M2/eXAICApQOBbAvXli5ciVaowlzdHy9nxeS2JFDq2azcuVK7rnnHtcF6ALFxcW88+67qNRqbnnqdVTqxs3ZTXHl7f/HoS2rmDp1KgMGDKB3795uj8EZysvLWbRoEX5BPkS2Dj3vYxK6xbFmxlZmz57NP/7xD/cG6AEcn4HVhrr/z9QGLTarjYKCAoKCgs731BbB0f5IpQfOLBxsaS2RBEEQBEFo3up1NnHdddc1qDd2bc4o1yfLMpmZmXTq1OmCj6moqKhTFr2xfYRnzZoFgC20daOe7zZGMzafINasWUN2djahoec/oXG35ORknnzqKfJyc2nTbzBxPZxz4SaqUy8ADiydzqOPPsrIkSPp3r27U45dX2vWrAGgU79Bbh3XHTRaHR1638D2xTPYt2+fy/qNq9Vq3n33Xe69915WTdvCfS/fgq4Bu66dwWq1sWLKJmRZ5t1330Wv17t1/KZyrPr3ja9ff/XafFoHkr/jNMuXL+fWW291dmhOMWLECKqqqjAlWZHUSkdzcZIExkQrxTs1fPbZZ/z222+NnivdRen5XPAujl6IjtLvDnoff8D7S8HLssykSZNQqdV0u6FxZeAdug+4i63zpzBp0qQWk1ivnSAVyVI7R2UmXYCh5jZdgIGSNPt9IrEu/q84eOt8XlJSwqpV9kUDtsAYhaNxDin/FIWFFjZs2MCgQZ5xHnno0CEyMjKI6NADlar+H7iN/oH4hkSycdMmysrKMBo9dyPCX3322Weknz5N3789RlirJEViUGs0DHrqdX576wneeustZsyYgb+/vyKxNMXKlSspKSnh8r6dzykD72Aw6YjvFM3hXYc5ePAg7du3d3OUysrLy0Ol0yCp6xYJVRvtl2Dz8/NbdGL95MmTSGqQtGfLqJ48eVLRmARBEARBEBqiXon1t99+W9FkwcSJEzl16hTvv//+BR/zySef8N577zVpnOrqav7880/Q6JrFibwtNAFb6lZmzZrF448rX5p8y5YtPP/885SUltL+hruJ7uLcHuRRnXqh1ujYt2gyTzzxBF988QXXXnutU8e4mIMHD2Lw9ScoMtZtY7pTdNsubF88gwMHDrgssQ7Qtm1bnnjiCX744Qc2zt1JvyG9XDbW+exYto+cU/kMGTKEXr3cO3ZTnT59mm3btmGM9ENjaniNdL/WQeTvOM2cOXM8MrG+a9cuFi5ciNpfRhvq2bvVHTRm0Iba2LVrF4sXL+amm25SOqSLUno+F7yLo4yjI5HuoPf1r3O/t9q6dSvJycm0730DfoFNS3gGhEeR2KMvO7atZe/evXTuXP/SvM1V7eSeJyf63Ono0aMA6ALOJqt0gfbvjxw5wlVXXaVIXJ5KluUWO6d563y+Y8cObDYr1uhOWGO6KB2OU0gleWj3LmbLli0ek1h3LBgPiW94sjOkdXtSt6xgy5YtHl/9wGHZsmXMmDGD8NbtuOqOhxSNJTy+DX3/9hhrpo7m448/dlu7Q2eyt06Etj3jL/q4tpe35uiuE8ybN6/FJdZzc3NRG85dtKLW2y/B5uXlkZjYuHaJzZ0sy6SmpaIyykgSoAVJY68eKgiCIAiC0FzUK7H+7rvvujiMCzt48CDPPvssvXv35pFHHrng41577TWGDh1a83NhYSGxsQ1LgDp2f1vD20IDVm4rxRbcCo7v5I/p03nsscdQqVSXfpKLLF26lP++8gpWq43OtzxAeJuuLhknvF03NHoDe+ZN4IUXX+S9d9/lzjvvdMlYf1VWVobe6OOVF7EAdEZ7afDalR9c5cknn2T58uXsXXeIhG6xxLSJcPmYADmn8tm6eB8RkREMGzbMLWM60+TJk5FlGb+kxiVwtP4GDOG+rF+/ntTUVOLj450bYBPIssyXX34JgDHBSnP6NTO0tlGVo2LEiBH079/fZa0UnEHJ+VzwPo6SjQb/uhU0tEYfVBqt15d0/PXXXwHoNehepxyv5833cGTbWn799Vf+97//OeWYnqx2z2FP7D+shEOHDgFnk+lwNsnu2M0uCOC98/n27dsBsPl5RjU2Z5BNAaDWsGPHDqVDqbFu3TokSSKoVZsGPzc4vh2pW1awbt26ZpFYz87O5t333kOj03Prv95qdNsWZ7rytgdI2bmBefPm0a9fP2655RalQ6o3i8XCunXrCI0NIiDs4rvt49pHojfpWLhwIUOHDvXa6yh/ZbPZyMvLQxt8bkUH9Zme646qTy1RVlYWZaVlNQvpJQlURpnU1FSsVitqtedfixUEQRAEQVAuE1sPGRkZDB48GLPZzB9//HHRD1h6vR5/f/86Xw01ZcoUAGxhzWTlqEaLNTiOUydPsmHDBsXCmDFjBsOGDUNGotudj7osqe4QHN+O7n97ErVOz1tvvcUvv/zi0vEcwsLCKMrLoqqi3C3juVt+hr30ljvaCmi1Wj788EPUajUrJm+istz1F9St1VaW/74Rm9XG+++9j6+v76Wf5EGKior4Y/p0NEZto8rAO5g7hAPw22+/OSs0p1izZg3bt29HG2JDY1Y6moZRG0EXZePEiRPMnDlT6XAEwW1OnDgBgNFct5SlJEkY/AM5fvy4EmG5xaFDh1i5ciUx7bsRmdTRKceM69id8Pi2LFq0qObv1puJxPq5jhw5gqRRofE726ZG629AUkkcPnxYwcgEwT327NkDgOwTrHAkTiSpsPkEcfToUUpKSpSOhtLSUnbu3IlfeAxag6nBz/ePjEOjM7B+/XoXROdcNpuNN954g4L8fK5/8FmCo1spHRIAKrWGwc+8hc5o4oMPPuT06dNKh1Rvq1evprq6msRucZd8rFqjpnXnGDIyMti3b58bovMMhYWF9gSx8dzF1iKxfrY6j9p0tkKdyiRTWVnZrH4XBEEQBEFo2ZqcWF+3bh0jR47kk08+YeTIkaxbt84ZcWGxWBg0aBAFBQUsXLiQqKgopxz3Qo4fP866deuw+YUim5pPVscWbl9lPnnyZEXGnzhxIu+88w4ag4nuQ54iKNY9/crMEbH0vOdfGPwC+Pzzz/n++++RZdeWju7Vqxc2q5XkTStdOo4SZFnmwPqlSJJEz5493TJmhw4dePLJJynKK2Hj3J0uH2/7sv3knMrnnnvuoXfv3i4fz9nGjRtHUWEh/p3Cz+nV1hA+cQFozQamT5/uMeXWbDYb33zzDUhgiG+evVMNcTYkNfzwww+UlzfPxTeums8F7+W4MGYKCjvnPp/AUCwWi9f2WR89ejSAU0vKSpLEVXc+hM1m4+eff3bacT1VZWXleb9vqaxWK0eOHkFnNtTZ1SepJLRmA0eOHnH5Z93moPbfgei3fn7NdT6XZZl9+/cjG/xA47nVfxpD9glClmUOHjyodChs3bqV6upqglq1bdTzVSo1ATEJpKamkpGR4eTonOu3335jw4YNJPW8mssG3Kl0OHUEhEcx8NGhFBcX8eqrr1JdXa10SPWyevVqAFp3qV/rxPjO0XWe1xI4WiFpTOe+jzlu8/Z2SRfjqM6jqrXPQe0j17lPEARBEATB0zU6O7N+/XratWtHv379eOGFF3jjjTd44YUX6NevH+3bt2/SDury8nJuu+02Dh06xNy5c+nY0Tk7gS7m999/B8Aa3vByaEqSfQKx+QazatUqt+9umjRpEp9++il6XzM9hjyNf1i0W8f3CQql573PYAoIYdSoUfzwww8uHe/ee+9Fp9Ox9o8xlJcUuXQsdzuwfinpRw9w8803ExHhnrLsAE888QRt27Zl77rDnDriupPLvPQCti1pviXgT506xa+//orGR4e5/bkJrIaQVBLBPaKxWq18/vnnHnGRfunSpSQnJ6MNs6H2UTqaxlHpQBdtIzs7u6b6SXPhyvlc8G4HDhxAozdg8As45z7f0EgAj0giONvBgwdZuHAhEQkdaN31Cqceu+3l/QiJTWDWrFkcO3bMqcf2NGVlZef9vqU6efIklRWVdcrAO+gCjZSWlIqdXFAn+WS1WhWMxPM09/k8MzOTosJCbKbGV2byVLKP/TUlJycrHAls2rQJgKCYxlfpC4qzL6bfvHmzU2JyhYMHDzJixAh8A4O5+YlXPLIMeaerb6JDn4Hs2LGjZsGeJ7PZbGzYuAG/IB8CQv3q9ZyYNhGoVCqPf/9xJseCE7WP7pz7NGdu8/RFKa504MAB4GwyHUB9Jsm+f/9+JUISBEEQBEFosEYl1vft28eNN97I4cOHGTBgAB999BHjxo3j448/ZuDAgRw6dIibbrqpUR+KrFYr9913Hxs2bGDatGlu2VlaUlJiL9+rMyEH1m/lrSexRbRFlmW37lqfM2cOn3zyCXpff3oMeQqfIGX64Bn8Aug+5EmM5mBGjRrFxIkTXTZWREQEjz/+OIU5Gcz7/iNs1uaxqvxSso8fZdHPn+Hj68vzzz/v1rG1Wi3vv/8+KpWK1dO2YK12/gVS2SazatoWbFYb77z9Dj4+zStzK8syb7/9NhUVFQT1jEGlaXoHD1NcAMZIP1auXMnChQudEGXjWa1WRo4cad+t3qp57zzTx9qQNDBmzBhKS0uVDqdeXDmfC96toKCAY8eO4R8ee96L1eYIe4lQT+op6wyyLPPFF18AcO3fn3L6hXpJpaLfvU9itVr56quvnHpsT1O7JHJzec90JUcP9fMm1s/0WXdUiWjJKioqzvt9S+cN87nj/3dzqh5XX7LR/po84Xd406ZNqDVazJGNL4seGJtYcyxPVFFRwSuvvkpVVRW3PP0GJv8ApUO6oBsfG4p/SAQ//vhjTSsET3X48GEsBRai24TX+/OPzqAlrFUwe/bsaTFz/cmT9hZ7Wl/9OfepjVoktarmMS3R3r17kbSgMpy9Te1nT7K3pJYBgiAIgiA0b43K0Lz//vtUVlYyf/58Fi1axKuvvsojjzzCK6+8wsKFC5k/fz7l5eW8//77DT72sGHDmD17NoMGDSIvL48JEybU+XKFP//8k5KSEqrDk0Dl0W3nz8sWGAs6E9OnT3dL37atW7fy1ltvoTWY6H7345gClO2BZ/A10/1vT6D39Wf48OGsXLnSZWM9+eST9O3bl6Pb1zF35AdYm0nJtgvJOn6EKR+9SHVlOR99+CGxsbFuj6FTp0488MAD5GcVsnOl83c2HtySQvqxbAYNGsTVV1/t9OO72u+//87mzZvxiQvAt7VzdvBIkkRon3hUGjUffvihoivmlyxZQkpKCrpwG+pzcwnNikoD+mgr+fn5TJ06Velw6sWV8/mWLVt47rnn6NSpEz4+PsTFxXHvvfeKEn9eYsOGDciyTOAFWsCYo+KRVOpm0YO1IZYuXcrGjRtJ7N6HVp1c0zolsUcf4jr1YMWKFaxdu9YlY3iC2p9Zi4uLFYzEMzjeGx1J9NocyXZP2O2qtKKiovN+39K5cj53l9TUVAB7KXgv43hNSrdhys/PJzk5GXNUPCqNptHH8QkKR2fyZePGjR5R/eqvRowYQcrRo/QcdA/xXXopHc5F6U2+DH7mDWw2G6+99ppHV3DZtm0bAFGJDaugFpUYitVqZdeuXa4Iy+M4fs+1/ucm1iVJQuOnIy0tzSN/d1wtPz+f48ePo/azUXtthkoDKqPM7t27RZsXQRAEQRCahUZlkVeuXMmQIUO4+eabz3v/zTffzJAhQ1ixYkWDj71z507AviP6oYceOufL2Ww2m32Xs0qNLbTx5dAUpVJRHZZISUkJs2bNculQGRkZDB06FFmGrrc9jE9QuEvHqy+jfyDd7ngUlUbLK6+8WnNhxtnUajVffvklvXr14uDG5cz44lUqSpvnxeC0fduY/MHzlBUX8t5773HDDTcoFsuzzz5LcEgw25fso8TivIsJleVVbJq3C5PJxMsvv+y047rLvn37+Pzzz9EYtYT0buXUnZFaPz3BV8RQWFjIf//7X0X6+smyzJgxY0ACfZx3nEDrY2QkDfz666/NomewK+fz4cOHM336dG644QZGjBjBk08+yerVq+nRowd79+5tauiCwhYvXgxASEKH896v0ekJjE1k7969nDp1yp2huUxRURGffvopGq2OGx5+0WXjSJLEgEf+jUptX/zkrTu8aifTS0qa52cpZ3IkzXVBpnPu05+5zRtbKzRUfn7+eb9v6Vw5n7tLzQ5Og+/FH9gcqdTIOpPiu1RrysDHnX9RXH1JkkRgbBJZWVke17Zkx44dTJgwgeDoVvS77ymlw6mX2A6X0Wvw/aSlpfHdd98pHc4Fbd++HWh4Yj0ywf54R2Le2x05cgQk0Pobznu/LsBIYWEh2dnZbo5MeY5KVhr/cxcVaMwyRUVFNRV8BEEQBEEQPFmjEusWi4XWrVtf9DGtW7fGYrE0+NgrV65EluULfjnb2rVrOXHiBNbgVqA9d0Vpc2ELSwKVmkmTJrlshafVauXVV18lPz+fNtfdTkD0xf8PuJtfaBQdBt5DaWkJ//nPy1RVVblkHJPJxMiRI7nuuus4tmsTE9/9F/kZzaeUlyzL7Fj6J9M+HYa1soJPP/2Uu+66S9GYfH19efGFF6mqrGbzgt1OO+6O5fspLSrn8ccfJzRUmXYFjVVYWMiw//yHqqoqQq9pjcaodfoYfm1C8G0dxI4dOxS5iLN161YOHjyINrT571Z3kDSgi7T3Wl+0aJHS4VySK+fzoUOHkpaWxjfffMPjjz/Om2++yZo1a6iurubTTz9tbMiCB8jJyWHlypX4hkTiG3zhBXYR7bsDMGPGDHeF5lKff/45WVlZ9Ln7HwSER7l0rJCY1lx524OcOnWKb775xqVjKaWwsBAAg4+ekpJSl31uay727duHxqQ973yvNmlRGzQeXcbbXWonIrKyshSMxLO4cj53l8zMTABkXfNq21Rfss5EZmamorsxHVVkguLaNPlYwa3aArBu3bomH8tZKisrefvtt0GSGPTUa2h1zef6ztVD/klQVCt+++03jywJL8syW7ZswTfAhF9gw35HI1uHIqkktm7d6qLoPIcsyyQnJ6P1N1ywhZu+BVeh2bJlCwCagPMn1ms/RhAEQRAEwZM1KrEeFRXFxo0bL/qYTZs2ERXl2ouOzjBp0iTA3qe8WdPqsQbHkZaWxoYNG1wyxIQJE9i2bRthbboS3eVKl4zRVOFtuxLd5SqSkw/y/fffu2wco9HI119/zT/+8Q9yTh7j1zef4Mh2z7mocCFVlRUs+PETloz9ArO/P2PHjuWWW25ROiwAbr/9dtq0acPBzSnkZxU2+XilRWXsXpVMWFiYS6pduJIsy7z55pucOnmSwG6RmKL8XTKOvSR8K7T+BsaMGePSNgrn40i26aO8Y7e6g+P1NIdkoivn8z59+qDT6erc1qZNGzp16sSBAwcafDzBc0yYMIHq6mpiuvW+6OPC2nRBZ/Rh8pQpbmlV40qrVq1ixowZhLduxxW3/t0tY/a+6xFCYlozceJEj+1j2xR5eXkABIbb57iWvPs4MzOTjIwM9KHnT1ZIkoQ+1IeTJ0+Sk5Pj5ug8y+nTp2u+T09PVzASz+IN5+fZ2dkgqUCju/SDmyOdkerqagoKChQZ3mazsWbNWvQ+fviGRjb5eEFnEuue1LLkl19+ITU1lR43/Y2opE5Kh9MgGp2em5/4L7Is88EHH2C1WpUOqY7k5GTy8vKIaRvR4CpqOoOWsNggdu3a5fWtX44fP05hYSH64HOrzzjog+1zfUus4LVhwwYkNajP0/FDE2hPrF9qLhMEQRAEQfAEjUqs33777axcuZK33nqL8vLyOveVl5fzzjvvsGLFCu644w6nBOkqJ06cYN26ddj8QpFNAUqH02S2cPvKc1f09j1x4gTffvstOpMf7W+4y6klqZ2tTb/BGM1BjBk71qWrgNVqNcOGDePTTz9FslUz4/NXWfvHGGQP7QllyU5n4rvPsHf1Arp06cLUqVO57LLLlA6rhlqt5oUXXrCvhl/Y9FX6O5btp6qymqeffhqD4fxl2DzVr7/+yooVKzBG+RPYzbUXQFVaNeHXJ6LSqHjt9dfddpG6oqKCJUuWoDLKqF2zbkAxKgNoAm1s3bpV0f719eHu+VyWZTIzMwkJCbngYyoqKigsLKzzJXiOjIwMJk6ciN7XTGSHi/cYV2u0xPa4hkKLhbFjx7opQufLy8vj7bffRqPVMfiZN1CpG9+XtiE0Wh23PPMGKrWaN9980+t+FzIzM+0LvGKCan5uqTZv3gyAIezCvaUNYfby2C1hx9+FFBYWUlRUhKS3X3w/ceKEwhF5Dm84P8/Pz0fW6sGDzzObQtbYd08rlVjft28fOTnZBMW3Q5IadRmoDr2PH/7hMWzevNkjkqVZWVn8+NNP+AYEc/Xf/ql0OI0S064rXa69hQMHDjBz5kylw6nD0UaiVYfGnZu26hhNdXW1Ry3EcAVHa0vHnH0+jkV0jrLoLUVGRgZHjx5FbbZxvrcglR5UJplNmzZRUVHh/gAFQRAEQRAaoFFnVG+99RatW7fm448/Ji4ujltvvZV//vOf3HrrrbRq1YoPPviA1q1b89Zbbzk7XqdynKzYwpppb/W/kH2CsPkEsXLlSqfuZpFlmQ8//JCKigraXnc7WsOFV996ArVWR/sb/obNauW9995zebm9wYMHM3HiRGLj4lg/YzzTP3/F4/qup+3bxq9vPE5W6mHuvfdexo8fT0REhNJhnePaa6+lU6dOHNmZRn5m40tVlhaVsW/9UaKiorjzzjudF6AbJCcnM2LECDQmLeH9WiOpXH9xUR9oJPiqOIqLinj99dfdskNi9+7dVFRUoA2WvfL6qTaoeZSyc/d8PnHiRE6dOsV99913wcd88sknmM3mmq/Y2FinjC00nSzLDB8+nPLychL73IRKc+kEc+xlfTH4BTBu/HiP68NaH7Is8+6775KXl0e/+58mJMa9bXAiWrfj6iH/JCMjg48++sitY7taamoqvoEmAsL8a35uqRzlmS9WocYUZQY8q+yyuzkS6dozu9qU7lftSbzh/NxisSCrvXS3OtTsxFdqkdSSJUsACE103k7u0MROVFdXu73q1fmMGjWKivJyrrnvSfSm5ttO4Jr7nkSrNzJy1ChKS0uVDgewfxaaN28eGp2GuA6Nq3aQ2M3+eX7evHnODM3j1CyUC7/wQjm1XoMu0MiOHTuorKx0V2iKW758OQDa4Au3+NQGy5SVldX8PQqCIAiCIHiqRiXWg4OD2bhxI4888gjFxcXMnz+fcePGMX/+fIqKinj00UfZuHEjQUFBzo7XaWw2G3PmzAGNDluQ91y0t4UmYLPZnHrCsmTJEtavX09wfDvC2nRx2nFdKSguiYj23dmzZw9//PGHy8dr27Ytk3//nX79+pGycyMT3nmGgqzTl36iG+xaPptpnw6juqKM999/n7feeuuc8syeQpIknnrqKZBhx/LGl4retTKZ6qpqHn/8cbRa5/cmd5WqqipeffVVe1/1vvGoDe6L3S8xGJ/4QLZu3cqECRNcPp6j9J2jl5q3UZ95XZ5e4s+d8/nBgwd59tln6d27N4888sgFH/faa69hsVhqvsSORM8xb948li5dSkBMAhEdutfrOWqtjrbX3U5VZSWvv/56s+uj/eeff7JixQpadb6cnjf9TZEYrrjtAaLbdmH+/PksWLBAkRicLTc3l6ysLIIjAwiOCgBosS0iqqqqWLVqFRpfHdqAC1fY0QUZ0fjoWLFiRbP7PXIWx3yg9pWRtPaSu4KdN5yfl5SWgpsqgihBPvPalEiW2mw2Fi5ciEZvIDjOeS3wwtp0BVB8bjp16hR//vknIbEJdLrmJkVjaSrfgGB6Db6fnOxsj2krtXnzZtLS0kjsGotW37jz08BwM2FxwaxZs9pr23jIssz69etRG7XoLjKfAxij/CkvL6/Z4d4SOBb3XDyxbqvzWEEQBEEQBE/V6BpgISEhjB07FovFwq5du1izZg27du3CYrEwZsyYi5Z59QS7d+8mIyMDa2AMqNRKh+M0tuBYkFQsXLjQKccrLS1l+PDhqNQa2l1/h0eXgP+rpGsGo9EbGDFiRE0fT1fy9/fnm2++4dFHHyX3VCoT33mGrLQjLh/3QmRZZu0fY1j082cEmM2MHTuWu+66S7F46uvaa68lKSmJQ1tTKcpveE/eirJK9q0/TGhoqEeXuzyfSZMmceTIEfzbhWKKNrt1bEmSCO3dCrVBy8iRI11ektdRVcNRztXbqOzVPptFL1x3zOcZGRkMHjwYs9nMH3/8gVp94XlXr9fj7+9f50tQXkpKCu+//z4avYGOA+9pUBnZ0MRORHbsyd69e/nqq69cGKVzpaenM3z4cAw+vtzy9OtIqqaXzm0MlUrN4H+9ic5g5KOPPmoW7yuX4ih/Gh4fQmhMEGqNqsWVRHXYsGEDRUVF+LQKvOjnbEmS8IkLwGKxtNidXI5EusoIKoPMiZMnXF6ZqjlpzufnsixTVVnpVefl5zjz2pQocbxt2zbS09MJS+pSr2oz9WUKDME/PIa169YpOjf9+uuvWK1Wet/xECov+D90+c33oDMYGT9+PNXV1UqHw7hx4wDock3TFmV0uaYtVquNX3/91RlheZz9+/eTk5ODKcr/ktfNHOf6q1atckdoisvIyGDbtm1ozLaa8+TzUfvbz6OXLl0iysELgiAIguDRmnyFUKvV0qVLF/r27UuXLl2aze5QR48oW1CMwpE4mUaPzT+MvXv3kp2d3eTDjRo1iqysLFr1uh6jOdgJAbqP3sePhD43UVhY6LYL+Wq1mqFDh/LWW29RWpjP7+8/x6lD7t+xKssyy375mvUzxhMX14qJEyd6VD/1i1GpVPzzn//EZrOxa+XBBj9/3/ojVJZX8cgjj3jszvzzycnJYeSokagNWoJ6RCsSg1qvIahnNGVlZXz55ZcuHctxouyEFo8eSTpzTe+vfU49mavmc4vFwqBBgygoKGDhwoVERTWuN6OgHIvFwnPPPUdZWRkdBt6D0dzwHY/trr8Tn+BwfvvtN2bNmuWCKJ3LUQK+pKSEGx5+Eb+gUEXjCQiL4toH/oXFYuHDDz9UNBZncJQzj2kbgUarJiI+lH379pGfn69wZO73559/AuCXcOnP2b4J9t+95vA75AqOHesqo4zKKFNVWUVWVpbCUXme5nh+frYNUfNZxN1gZz70KpEodVRwi+x0udOPHdmpFzarVbH3pbKyMmbNno1/cBjtrrxOkRiczeDrR5frbiUzM5PVq1crGsv27dtZt24dse0iCItr2vWgpO6t8A/2ZerUqWRkZDgpQs/huMboExdwyccaw31R6dSsWLECWfbOhea1zZ49G1mW0UZc/LVKEmjDbRQVFbNs2TI3RScIgiAIgtBwTUppVFRUMHPmTN5++21efPFF3n77bWbOnNksVhZu3LgRVGpk/3ClQ3E6m9ne92rTpk1NOk5ycjK/TZiAKSCE+Muvc0Jk7hfT5Sr8wqL5888/2bZtm9vGvffee/nss8+orqxg2qdDOXlwt9vGlm02loz9gu2LZ9CufXt+/fUXYmKa1wKSm266iYjICA5sPEp5af37jlmrrexZnYyvry9DhgxxYYTON3bsWMpKywjsHoVar1wZTL+kYPTBJhYsWMCRI66ruBAcbL8wY6v0zguotjP/bT15d1htrprPy8vLue222zh06BBz586lY8eOTopYcJfKykr+/e9/c+LECeKvvIGwpM6NOo5aq6PrbQ+jNZh477332LJli5Mjda758+ezfv16Err3puPVnlFW9rL+txPXsQfLli2r6VPZHNlsNlauXInJz0BYjD1R3KpjFLIst5idWw55eXmsWLECXaARfbDpko/Xh/igCzCydNlSLBaLGyL0LI6e6io9qM5U2T116pSCEXme5nx+7vUUyp3l5OSwePFifEMiMEe2cvrxI9pdhlqnZ8qUKbUWSLjP8uXLKSkupst1t6LyolYC3frfBpxdfKUEm83GZ599BsAVt3Rr8vHUahW9bupCZWUlI0aMaPLxPIksyyxZsgSVRoUx6tLVtiS1ClOMmRMnTpCcnOyGCJVTXV3NtGnTkNSgC730G6Euwl6JZsqUKa4OTRAEWLtRMAAA+VxJREFUQRAEodEanVifPXs2rVq1YsiQIXz44Yd8++23fPjhhwwZMoT4+Hh7/3IPVVZWxsGDydh8gryy3Jzsb99VtWvXrkYfw2az8eGHH2KzWmnX/06nloxzJ0mlov0Nd4Mk8cEHH7i1J+VNN93El19+ga26ij/+9x9OJrs+uS7LMkvGfcnOZbPo2LEjY37+uSaB2ZxotVoefuhhqiqr2b+h/sndw9vTKCks45577sHHx8eFETpXdnY2U6ZMQeurx7+Nsv9ekiQR1D0aWZb5/vvvXTZO69atAbAWuWwIRVmL7AsG4uPjlQ2kHlw1n1utVu677z42bNjAtGnT6N27t5MjF1zNarXy+uuvs3XrVsLbdiPhqgFNOp4pIIQutz6E1SbzwgsveOyFxMLCQv73v/+h1RsY+I+hHtMGR1KpGPjYMNQaLZ988okifXqdYffu3eTk5BDfKRpJZf+7bd3FvgCwpe1OmjVrFtXV1fi3q19FBEmS8GsTQlVlFbNnz3ZxdJ4nIyMDlV5GUtlLwQNe26u3MZrz+fnZFjHevHPT/tpUbm4rMmXKFKqrq4np1scl85lGbyCyQ0/S09MVWfTlmDfa977B7WO7UkhMa0JjE1i/fr1i8/2ff/7J3r17adMznvAm7lZ3aNsznrC4IObOncv27dudckxPcPjwYVJSUjDGmFFp63eN0Tc+EMBpbRw91fLly8nIyEAbYaup6nYxaiNogmxs376dffv2uT5AQRAEQRCERmjUWd2yZcv429/+RkFBAY899hjjx49nwYIFjB8/nkcffZT8/Hzuvvtuj91Nc+zYMWw2K7IpUOlQXEI2mkGSOHz4cKOPMXv2bHbu3El4224ExbVxYnTu5x8eQ0zXqzh69Ci///67W8fu378/X3755Znk+ssuLQsvyzJLx3/FzmWz6NChAz/99BNms3v7dDvTXXfdhY+Pib1rD2GzXrp/pizL7F6djFqt5oEHHnBDhM4zbtw4KisrCegWqVgP39qM0f7oQ31YsmQJR48edckYvXv3RqVSUZWr/Ot1haoc+4XLq6++WuFILs6V8/mwYcOYPXs2gwYNIi8vjwkTJtT5EjybLMt8+umnLFq0iICYBDreeG+D+qpfSGBMAh1vuo/ikhKefvrpmh2onuTHH38kLy+PPnf/A3NohNLh1BEcFceVtz9IRkYGY8eOVTqcRlm6dCkACV1ja24zh/gRHBWgaALB3Ww2G1OnTUOlUdWUeK8Pv6RgJLWKqVOntojysQ6yLJOZlYl0psuP6syfohS8XXM/P5ckCa1OB7ZLf+Zvtmz23dx6/UUaDDtZaWkpv/8+GZ3Jh4gOPVw2Tmz3viBJjB071q3vSzabjY0bNxIYEUNwVJzbxnWXpJ5XU1FRwc6dO90+dn5+Pl9+9SU6g5Y+t3V32nEllcQ1f+sFEm7f+OBK8+fPB8C3df3nc2OUGZVOzfz587F56XufLMuMGTMGJNBH1/816mPs7yPN9bOuIAiCIAjer1FXSN955x2MRiNbt25l9OjRPPzww9x00008/PDD/Pzzz2zevBmDwcA777zj7HidwrGzQTY0nx2tDaJSI2tNnD7duB0cZWVljBgxArVWR1K/wU4OThkJvW9EazQxatQoCgoK3Dr29ddfzxdffIG1spJpw4e5JLnuSKrvWDKT9u3bM3r06GadVAfw9fXlzjvvoriglNR9ly7zmZmWQ86pfAYMGEBEhGclQi4mJyeHqVOnovXV45fY8L7FriBJEoHdIpFlmR9//NElY5jNZq6++mqshRLVXlbN1loGVTkq2rRpQ1JSktLhXJQr53PHRcA5c+bw0EMPnfMleLYffviByZMn4xsaRbfbHnFq5Zrwtl1pe93t5OTk8OSTT5KTk+O0YzdVamoqEydOIjAihssH3at0OOd15W0P4h8cxrhx4zh9+rTS4TSILMv20ucGLdFt6rZjSugSS2VlJevXr1coOvfauHEjJ0+cwCchCLWu/r9far0G3/hAUlNT2bp1qwsj9CxlZWVUVVYh6ewX2yWt/U93f673VM39/BzAaDCCzf39x91FOvPajEaj28acNm0aFksBMd36otZoXTaOKSCEsKTO7N27195yz02OHTtGUVERMe2bXqbcE0W36wqgSGL9q6++wlJg4YpBXfExO/f/bHhcMJ36tOHIkSNesdjWZrMxf/58VDo1puj6X4NRaVT4tAokPT1dkX9jd1i7di379+9HG2JD3YD/RpoAGbWfvbx+SkqK6wIUBEEQBEFopEYl1nfs2MF9991H587n77PZtWtX7r33Xo8t7VRYWGj/Rq1TNhAXkjU6iooKG/XcqVOnkpOTQ1yPfhh8m3dy1kFrMNH6igGUlJTw66+/un38/v3788UXn9dKru9x2rFlm40l476sSar//PPPzT6p7nDvvfakxr56lIPft97+mPvuu8+lMTnbDz/8QEVFBQFdIzxit7qDKdqMPsTea33//v0uGeOpp54CoCxFjTdtuis/pgLZ/vo8pYT0hbhyPl+5ciWyLF/wS/BcU6dOZdSoUZgCQrjszsfQ6A1OHyO2Wx9aXzmAEydO8K9//YuSkhKnj9EYX3/9NVZrNdc9+KxLkxBNodUb6Pf3Z6isrOTbb79VOpwGSU1N5fjx48S2j0StqVsPNL5TNGB/72gJpk2bBoC5bf3KwNfmKB3fkvqPFhXZe8c4yshKZ9YiFBcXKxSRZ2nu5+cAvr4+SFbv2L16Xmdem6+vr1uGKy8vZ9z48Wh0BmK69XH5ePG9+gP2qi/u+pznSLiFxXn2QtbGCotLBOwLCNxp586dzJw5k9CYQDpf7ZrqhVcN7obRz8D3339PRkaGS8Zwl507d5Keno5Pq0BUmoadzzsq1jh2vHsTWZYZNWoUAIZWDduRL0n258iyzA8//OCK8ARBEARBEJqkUVkck8lEaOjFLwKFhYVhMpkaFZTbeHjCo0kkGlVOymazMWHCRNQ6PXE9PLuEcUNFd7kSncmPKVOmUFlZ6fbxb7jhhrPJ9U+HcfpI0/tFybLMsl9HsHPpn3To0MGrkuoACQkJdO/enZPJGZRYLlwatqqiipRdJ4iNi+Xyyy93Y4RNk5KSwrRp09CaDfglhSgdTh2SJBHc097z9vPPP3fJBbKuXbty8803Yy2UqDjhHe/HlVkSVdkqunfvzsCBA5UO55K8Zj4XnGb58uV89NFH6H38uOyux9D7+LlsrNZXDSC6y1UcOHCAoUOHKl4OdPv27SxbtozYjt1J6tFX0VgupcNV/YlI6MDcuXObVf/J1atXA9CqY9Q594XEBOJjNrJmzRqvLYnqkJ2dzfIVy9EHm9CHNLyClj7UB12gkWXLlpGbm+uCCD2P47N7TWL9zFlsRUWFQhF5Fm+Yz81mM1K1+8/R3ObMa3PXudq0adPIzckhpntftAbX75L3C4siJKEj27ZtY/PmzS4fD6ip2uLvYW1bnMUnIBi1VufW6jQ2m42PP/4YgH5DeqFy0cJvvVFH79suo6ysjC+//NIlY7jL3LlzAfBrQFsXB2O4HxqTjgULFyj+OdjZVq9ezd69e9GG2lA3olioJsi+a33hwoVNanMpCIIgCILgCo36lDxgwICa/ogXsnTpUo9NKtSsEvfiE3epuhI/v4ZfDN+9ezcZGemEt+2GRu++MnXuoNJoiOzYk8LCQjZs2KBIDDXJ9apK/hj+H7KOX3on9sWsnvIj2xfPoJ2XlH8/n9tvvx1Zljmy8/gFH5O67xRVldXcduttHr9D2MFms/Hee+9hs9kIvjwGSeV5cRsj/THFBrBlyxbmzJnjkjHefPNNQkJCKE9VU5XveX8HDWEthrLDaoxGIx999JHLLkQ5U3OfzwXn2rdvH//9739RabR0u+NRjOZgl44nSRLtrr+D0MROrF+/ng8//FCxagayLPPFF18AcN0D//L4uURSqbjuwWcA+PLLL5tNFYg1a9aABK06nJtYlySJuA5R5OXlNavFAo0xffp0bFZbzc7zhpIkCf92oVRXVzNz5kwnR+eZqqvPlAh3/GpKf7m9hfOG+TwwMNC+q/tML3JvI1XZF4EEBga6fKyysjJ+HjMGjc5AXHf3LZZPuGoAACNHjnTLvFRWVgaAzuC5C0aaQpIkdAYj5eXlbhtz3rx5HDhwgHa9WhPeyrULv9v1bE14q2AWLFjAnj3Oq+jnTlVVVSxavAiNSYshvOHX3ySVhE/rQAothV7VCkeWZUaOHAkSGOIbt1hSOvNcWZb5/vvvnRyhIAiCIAhC0zTqqv/nn39OVlYWDz/8MCdOnKhz34kTJ3jooYfIycnh888/d0qQzhYVZb+YJ1V4aelAmxWpspTo6OgGP3Xbtm0AhLRu7+yoPEJwfDvg7OtUwg033MAnn3xCRWkJ0//3CkX5jestu2Ppn2yaPZGEhAR++vFHr0yqg71HvSRJHNtz8oKPObbXft+NN97orrCabNq0aWzfvh2f+EB8YgOUDueCQq+KQ6VVM3z4cJf0QTabzXz55ZdoNVpK96uxNtO3ZVs5lOzVgFXio48+IjY2VumQ6qW5z+eC82RlZfHcc89RWVlJp0EP4BfW8M8QjSGpVHS6+X78w2OYMWMGv/32m1vG/atly5axe/du2l3Vn8iE5vEZKK5DdxK692bz5s2sW7dO6XAuyWKxsHXrVsLjgjH6nr+9QHxH+/+7FStWuDM0t6quruaPP/5ApVPj27rhu9sc/BKDUWnVTJ06FavVOxORtdW8xr8k1lvCa68Pb5jPQ0LOJPGqypQNxFWqyvDz80Ov17t8qGnTppGXm0ts975o3Zh09guLJjSxEzt27GDTpk0uH69mEZzsxVVO3LhwrqqqipEjR6LWqLnyFtf3rZdUEn1u7wHAN9984/LxXGHjxo0UWgrxiQ9q9EJ5v9beVw5++fLlHDhwwL5bvQlvQZpAGbW/vdd6cnKy8wIUBEEQBEFoonol1vv371/n66GHHiIwMJCJEyeSmJhIUlISffv2JSkpicTERCZNmkRAQAAPPfSQq+NvlMTERNRqNVKxd5ZOlEoLQJZp165dg5976tQpAEyBYU6OyjP4BNlfl+N1KmXQoEEMGzaMorwsZo94C5u1YbttTh/ex7LxXxMcHMyoUaMICmr8hVlPFxwcTLdu3Ug/lk1l+bnl0WSbzInkDGJiYkhISFAgwoZLSUnhs88+Q23QEHJlnNLhXJTGR0dQz2gKCwt58803XbL7pHv37gwfPhysEiW7Nc0uuW4rh+LdGmwV8Morr3j0bjBvm88F56iqqmLo0KHk5OSQ1O9Wty+uU2t1dL39EfS+Zr744gu3lZB1qKqq4uuvv0alVtPv3ifcOnZTXXv/00gqFV9//bXHl09fvnw5VquVhK4XXngU0y4CrU7DokWLms0u/IZaunQpmZmZNYnxxlJp1fgmBJGenu7VCxEcHCVyHXk0Ryn4lrpj3Rvn8/DwcACkSu9MrKsqy2peoyuVlZUxZuxYNHoDsd2vcfl4f9X6Svuu9VGjRrn8fdyx+7+0sMCl4yjFWl1NWXGhW6ocgH23+qlTp+jUJwnfAPcsyIhMCCWufSQbN25k586dbhnTmRYtWgSAb+vG/xvpgk1o/fWsWLnCK9qb1PRFlxreW/2vpFo73n/88UdnhCcIgiAIguAUmvo8aOXKlRe8r7q6mpSUFFJSUurcvmvXLo8to2kwGOjWrRvbt++AqnLQnn/XTHOlKrD34Lryyisb/FzHRSuVuvEX+jyZSm3/L+8J/asefvhhDhw4wLx589g0ZxK973y4Xs+rqqxg7qgPAHvZ2sZUJmhurrjiCnbu3EnGsWzi/lI+Nje9gIrSSq4cdKXHvufUVlVVxauvvkpFRQUR1yeiMWqVDumS/NuFUnrSwrp165g0aRIPPvig08cYOHAg77//Pm+//TYluzWYOlej8Xf6ME5nLYOS3Rps5fDcc8+55O/GmbxtPhec48svv2TXrl1EdOhB7GXK9BbX+/jTZfD/sX3aD7z88sv88ccfl+wX7CwzZ84kLS2NHjfeTWBEjFvGdJbQ2AQ6X3Mze1bNZ968edx2221Kh3RBM2fOBAmSure64GO0Og2tu8RwaFsqO3fupHv37m6M0PVkWWbcuHEggblj0xNs5o7hFCZnM3bsWG644Qavfq8uKSmxf+M4RVH/5fYWxhvn84gIe59sqaIE2c897/9uY62G6oqa1+hK06dPJy83l9ZX3uCW3up/5RcWVbNrfcuWLVxxxRUuGysmxj5n555Oc9kYSspLt7dCc7xOV7LZbIwbNw61RkX3/h1cPl5tPQd25vjBdMaNG8eIESPcOnZTVFVVsXz5cjQ+OvQhjWgifoYkSfi0CqRgTwbr16/n+uuvd2KU7rdmzRoOHjyINqxpu9UdNAFnd60fOXKEpKSkph9UEARBEAShieq1Y91mszXqy5NL89l3FMqocr3sJEyWUeWkYTSa6N27d4Of7tj5XFFS6OzIPILjdXnCDm9JknjjjTcIDQ1j46wJFNezJPy2hdMoyDzFI488Qs+ePV0cpWfo0cNeIi4z7dwqE5lp9r+35nLx/dtvv+XAgQP4tQ3Bp5V7dh80lSRJhPaNR23U8sWXX3Do0CGXjHPnnXfyySefgFVFyW4NVXmee/EXoLoISnbak+pDhw7lqaeeUjqkS/LG+VxomlWrVjFhwgR8gsNp3/8uRZMu5sg4kvrdSl5eHq+99ppbdmCXlpYyatQodEYTve/6h8vHc4Wrh/wTjVbHN99+67E7nfbt28eOHTuIax+FX+DFLz537G2/YDphwgR3hOZWy5YtY//+/fjGB6H1a3o5aJ3ZgE+rQPbs2cPq1audEKHnKigoAEClse+AlSSQNGdvb2m8cT6vSR5WeN9iCUcLOlcviK6qqmL8+PGodXpi3dhb/a/ir7wBgNGjR7t0nE6dOgH2am7e6PThvcDZ1+lKa9euJSUlhTY94vExu7dnfWRCKOHxIaxYsYLjx4+7deym2LJlC0VFRfi0Cmzy52ffM9cFli1b5ozQFCPLcs3vvSHOOZ/jJensscaMGeOUYwqCIAiCIDRVo3qse4Nbb70VnU6H+vguVFlHUWWneMWXOmUTUkUxgwffgsnU8BOitm3bAlCYceISj2yeCjPtvbjbtGmjcCR2fn5+PP/8c1RVlLFl/pRLPr6qopzNcyYREBjIk08+6YYIPUOHDvZV89mn8s65L/tkPgAdO3Z0a0yNsXHjRsaNG4fObCCkV/Powe2gMWoJ6xtPVWUV//3vf12WvBk8eDAjRoxAq9ZRsldNZaZnJter8iVKdmmgWuLdd9/l0UcfVTokQWiw3Nxc3nrrLVQaLZ1veQC1Vqd0SMR0601IQkc2bdrkln7r48ePJzc3lytufQAfc/NY7PRXfsFhXD7oXjLS0/n999+VDue8fvjhBwAuu/7SbQYiE0IJiwtiyZIlHD582NWhuU1FRQVff/01kkoiqHvUpZ9QT0Hdo0CyV57whIpMrpKeng6AVKvQmKSXOX36tEIRCc4WG2v/bCyVFykciQuU2xPrjtfoKvPmzSMzM5PoLle5tbf6X/mHRRMU14aNGzeyb5/rkt5ms5mOHTtyfP8OKkq9b0HGke3rARq1YaKhHJ+5ul3r3nZADt2ubY8sy0ycOFGR8RvD0YbFJy6gycfSBZvQ+OhYuXJls25xsm3bNnbu3Ik22Ia68Zv4z6EJklH7ysyfP1/xto6CIAiCIAjQghPrAQEB3H333UiyDc2xLWhSNnvFlzonFZVKxcMP16+s+F/16tULgNzUZGf+dXuM3GMHgcaVyXeVW2+9lZCQEPaumk915cWTlQc3Lqe8pIi/338/vr6+bopQeUFBQYSEhpCXYTnnvrwMC2q1mvj4ePcH1gAlJSW89dZbSCqJsH4JTeqrqhRTjBlzhzCOHj3K999/77JxrrvuOsaMGYOfrx+lB9WUn/Ss5HpllkTpHjVatY6vvx7B3/72N6VDEoQGk2WZDz74gPz8fJKuHoRvsOvL09aHJEl0GDgEvY8fI775hqNHj7psrJycHMaPH49vQDCXD7rXZeO4w5W3P4jRz8xPP/2ExXLuXKmkTZs2sXLlSqKTwolOunT5c0mSuGJQV2RZ5vPPP/eaXuvff/89aWlp+LcPQ+vvvDZUugAj/u3CSElJ8er+o45djGrD2f8PKoNMYWFhi9217m0iIyPRaDRIZ5LQ3sSxWKBVqwu3wmgqWZb59ddfkVQqYrsr09altlaXXwe4vvrIwIEDsVmrObixee/0/asSSx6puzbRvn17l5eCT05OZuPGjcS0jSA4KsClY11IQpcY/IJ8mDFjhsd9jjkfWZZZuXIlar0GQ1jTr8tIkoQp1ozFYmH37t1OiFAZjh3leiftVneQJNDH2iuv/PLLL049tiAIgiAIQmPUq8f6hZw8eZIVK1Zw+vTp8+5elCSJt956qylDuNRLL71Ez549m/WK0POJjY2ldevWjXpuWFgYXbt2Zc+evVSWFqMzeU/ytrqygpxjB2jVqhWJiYlKh1NDq9Vyxx13MGbMGI7t2UKbnhcu23dg/VIA7rrrLneF5zESWiewZcsWqqusaGolpQuyComLi0Or9exe5V999RUZGRkEXhaFPli5HSRNFdQzmtKTFsaNG8fAgQNdVprwsssu47fffuPJp54k62gWcqUNQ2sbSrcGrTglUXZEja+fL6NGjmo2LQgupbnP50LDzZs3j2XLlhEYm0hMN9fvhGoIndGH9jf8jV2zx/PGm28y4bff0Gia9JH1vMaMGUNZWRk3PvAcOgX60DqT3uRL7zsfZvlv3/LLL7/wwgsvKB0SAGVlZbz//vtIKom+d/aod6nU2HaRtOoYxfr165k/fz6DBw92caSutWXLFsaNG4fWX09QD+ftVncI7hlN2SkLo0ePpnfv3l7ZKujQoUMggarWRyi1D1Tn2u9zZR/n5qQ5z+cajYa4uDhSjp8EWUbxD31OJJXZ25ElJCS4bIwtW7Zw+PBhItp3x+Brdtk49RUYm4hvSCQLFixg2LBhhISEuGScO++8k5EjR7J98Qy6Xn+boi1tnGnXstlYrdXcc889Lh/r559/BuCy693bW702lVpFt2vbs3bmNiZOnMi//vUvxWKpjyNHjpCRkYFvQhCSyjn/50wxARQezGb16tU1rfCak+TkZNauXYvGbEPj7/zja0NlVMdgxowZPP300x7R3lEQBEEQhJar0VcpX375ZUaMGFGnT5ssyzUnMo7vPfXEHcBkMnHzzTcrHYbHufXWW9m9ezfpB7bTqmc/pcNxmqzDe7BWVXLbbZ53wn3jjTcyZswYDm1aecHEellxIcf3badbt25ERka6OULlxcXFsXnzZoryigkMt18sKi+tpLykwqW7P5zh0KFDTJs2DV2AkcAunrErtLFUGjWhfeM5vTCZ4cOH88svv7js9ykxMZGJEybyxJNPkHosFdkGxkTlkuvlJyTKU9SEhoby008/kZSUpEwgTuYN87nQMFlZWXz8ySdodHo6DLwHSfK8AkYhCR2I7NiTfXu3MW7cOJ544gmnHj8zM5OpU6cSEBZFl2tvceqxlXLZDXewZd5kJkyYwEMPPURgoPKl7b/++muOHz/OZdd3ICS6/vFIksQ1f7uc00cX8NFHH9GzZ08iIprn/Hnq1CleeuklZAnCrm6NSuP8ijUqrZrQq+NJX3SIl156iSlTpnjVZ8Wqqir279+P2kem9tuV2s++e3337t0isY53zOeJiYmkpKRAVTnomveCp9qkskK0Op1Le6xPnjwZgJhufVw2RkNIkkRMt94cXDaD6dOn89RTT7lknJCQEAYNGsScOXM4sn3dRRepNxcVpSVsWzgNs9nMrbfe6tKxDh06xKJFiwiLCyK2nbLzbIerEtm+dB+//fYbDzzwAAEBAYrGczFr1qwB7BXdnMUY4YekUbFmzRr+/e9/O+247jJ+/HgA9HGuqTQkSaCPsVJ2pILff/+dZ5991iXjCIIgCIIg1EejrqSOHj2aL774guuvv54//vgDWZZ55JFH+P3333n66afRaDTcc889LF++3NnxCm5wyy23oNPrOb1nE7Ls3BJOSjq1eyMqlYo77rhD6VDO0aFDByIjIzm6Yz026/krKKTsWI/NZqV///5ujs4zxMXFAWDJPtt3sTDH/r2r+xU21VdffYXNZiO4VwyS2vMSWA1ljPDDp1UgO3bscPn7fEREBOPHjadN2zZUnlJRdkSFElWBHUn1iIgIfvnlF69Jqov5vOWRZZl3332XosJCkq4ZjNFf+eTrhbTpdxsGXzOjRo0iOdm5LWomT55MZWUlve98GLULdsMrQaPTc+VtD1JWVsa0adOUDocVK1YwadIkgiLNXDGoa4Of7x/ky9V39aCoqIhXX321WVaYKigo4Jl/PYPFYiHkqjinlIu9EGO4H8FXxpGfn8+//vWvZlFKt74OHDhAeXk5av+6HwA0Z37etm2bEmF5FG+Zzx2fr6Qy7/n/iyyjKreQ0Lo1arVrWkHl5uayfPly/EKj8I/wnPOiiPbd0ej0TJ8+HZvNddcVnnzySVQqFWumjsZms176CR5uy/wplBUX8uijj2Iyua7SmSzLfPHFF8iyzBWDuim++UCr09BjQCeKi4td2nbMGdatWwcSmKKcl1hXaVQYI/w4dOgQmZmZTjuuO6SnpzN//nzUPjKaQNedrOsiZFRamDRpEqWlpS4bRxAEQRAE4VIaleH56aefiI+PZ8GCBTUlqePj47nvvvsYOXIkixcvZubMmWRnZzs1WME9zGYztw4eTGlBjtf0Wrekp1GYeYLrr7/eI3c8SZJE//79KS8p4sTBXed9zKGt9lXR119/vTtD8xg1ifXcs30XLTmu71fYVI6SaMZIP0zRypdldJbgntEgnS0d6NKxgoMZN3Yc7dq1o/K0ivI09y5OqEi3J9UjIyP55ZdfPH4hR0OI+bzlmTZtGmvWrCG4VVuiOnv2Dk+twUj7gUOorq7mtddeO29Z48aoqqpi2h9/YDIH0qHvQKcc01N0vnYQepMvkydPqbNr1d1OnDjBG2+8gUar4caHr67TwqUh2l+RQFL3Vmzbto1vv/3WyVG6VmlpKc888wzHUo4R0DkC/zauKYNcm7ldKOaO4Rw5coRnn33Way46r1+/HuCci/UqHah8ZLZu3UplZaUSoXkMV8/nxcXFvPPOO9x8880EBQUhSVLN7kRnatOmDQBSqRcl1itKwFpd89pcYe7cuVitVqI6X6F4crQ2tVZHeLvLSE9PZ9OmTS4bJz4+nrvuuoucEynsXjHXZeO4Q1FuFpvnTiIsLIy///3vLh1r8eLFrF+/nrgOkcS194wqJ536tiEgzJ/Jkydz4MABpcM5r5KSErZv344+xAe1wbmLM03R9hrqjnmvuZg4cSI2mw19rGury0lq0EVZKSwsZPbs2a4bSBAEQRAE4RIalZ04ePAgN998MyrV2afX3kVy7bXXMnjwYD7//POmRygo4qGHHgIgdctKZCW2hzpZ6pYVADz88MMKR3JhN9xwAwCHNq86577K8jKO7dpEQmIirVu3dndoHsGRzKy9Y93SDHasO8oymjt53oKOptD6G/CJDWDv3r3s27fP5eOZzWZ++OEH4uLiqEhTUZHunouGVbkSZYfUBAUFMXr0aKKinN8bV0liPm9ZUlJS+N///ofWYKLDjfd41MX3Cwlu1ZaYbn04fPgwX3/9tVOOuWPHDiwFBXTsMxCNVueUY3oKncFE+6v6k52dxf79+xWJoaysjJdeeomioiKuvacXQRGNX1QmSRLX3XsFAWH+jB07lmXLljkxUtcpLy/nueeeY+/evfi1CSGop+vKP/9VcK8Y/BKD2bVrF//+97+dtiBFSWvWrAEJtAHnnpNoA2XKy8vZunWrApF5DlfP5zk5Obz//vscOHCAbt26NTneC2nXrh0AUmm+y8ZwN6m0ADj72lxhzpw5qNRqwtu57t+msSI7Xg7g8iTYc889h4+PD2umjqasqPkuzFg+4TuqKyv497//7dLd6vn5+XzyySdotGquubuXy8ZpKLVaxbVDemGz2Xj77bepqqpSOqRzbNy4kerqapcsmncc01FqvjkoLS1l+vTpqPT2Puiupouyt4WZOHGiV1yrFARBEASheWr0tr/a/Y58fHzIzc2tc3+7du3ckmwRXCMpKYkBAwZgOZ1K7rGDSofTJAWnU8lJOcDll19Ojx49lA7ngnr06EFwcDCHNq86p4Rdys4NVFdWcNONNyoUnfLi4uKQJImCrMKa2/Kz7In1+Ph4haK6OJvNxpIlS9D46GpWn3sTv7ahgH23gzuEhITw008/ERAYQNlhNdUFrh3PWgKlB9UYDHq+//57j66M0BRiPm8ZKioq+O9/X6GiooIOA4ag92k+70lJ19yCT1A4EyZMYPXq1U0+3saNGwFo3e3KJh/LEzle14YNG9w+tizLfPjhhyQnJ9O5bxva9Wr6YkCdQcvNj16DVqfh9TdeJzU1temBulBVVRVDhw5ly5Yt+MQHEtq7lVsXsUiSRGjfeHxaBbBhwwb+85//eGRior5ycnLYs2cPGrMN6TwbA7XB9vLSK1eudG9gHsiV83lkZCTp6emkpaXx2WefNSXMi4qJicFoMtUko72B6swigfbt27vk+IcPHyY5OZng1h3QGlyXiG0s/4hYTAEhLFu2zKVVNEJCQnjuuecoK7Kw8nfPLiN+ISm7NpG8aQU9evRwaW91WZb54IMPyM3N5cpbumEOcV2bksaIbhNOx95JHDx40CNLwtf0V3fB+b3W34DWT8/69eubzdw9b948iouL0UVakdxQWE6lA22ojdTU1JrP9IIgCIIgCO7WqI890dHRnDx5subnxMTEc0p77d27Fx8fn6ZFJyjq+eefR61Wc2jVbKorm+duF1t1NcnL/wTgpZdeUjaYS1Cr1QwYMIASSx6nDu2tc59jF/vAgd5VsrYhDAYDkZGRNcl0gILMQvR6vUeW9wc4evQoFosFY6Rfs9gZ2lDGCF8kleTWXWLR0dGM+HoEGrWG0gMabC6q/CpboXS/GrkaPvroYzp27OiagRQm5vOW48svvyQ5+SDRXa8iNKmT0uE0iFqjpfMtf0el0fLGG2+QlZXVpOM5+lYGR3vnYpngKHvrlKb+PTXGrFmzmD17NuGtgul7p/MWMwZFmLn+/ispLSll2LBhHrsLW5Zl3n33XdasWYMpxkz4Na2RVO6f/yWVRHi/BIzR/qxcuZIPPvig2e7qWrnSXj1LG3z++NVmkLSwfMXyZvsancHV87m7Pm+rVCo6tG+PqswCXtArG0AqcW1iff78+YC9n7knkiSJ8HaXUVZWxqpV51Zmc6b777+fDh06sGflPE4c2OnSsZytsryMJWO/QK3W8Oabb7r03HHmzJksWbKEqKQwuvZzXSWFpuhze3f8g30ZM2aMR1UkkWWZNWvWoDZo0Ie45vzIFGOmpKSEXbvO3yLQ08yYMQMk0EW6bw7WRdkX1c2cOdNtYwqCIAiCINTWqMR6375966wMvOOOO9ixYwdPPfUU8+bN47XXXmPBggX069fPaYEK7peQkMBjjz1GmSWP5BV/NsuLVUfWLaA4J5377ruPrl27Kh3OJfXv3x+AlB1nd5nZrNUc272Z2NhYkpKSlArNIyQmJlJiKaWirBLZJpOfVUhCQkKdspee5OjRowAuO+lWmkqjRhtg4MjRI24dt0ePHvznP//BVgmlB1W44q2p7IgKa6nEP/7xD2704koRYj5vGVauXMmkSZPwDYmgTT/X7YByJd+QSNr0u5WCggJee+01bDZbo49VVlYGgEard1Z4HkWjMwC4vb/2iRMn+Pjjj9Ebddz4yNWoNY3rq34hSd1b0blvGw4dOuS0tgDONmrUKGbPno0hzIfw6xKR1Mp9PpHUKiKuT0Qf4sPMmTP56aefFIulKZYvXw6ANuT8k70kgSbIRmZGpmLtDzyBJ87nFRUVFBYW1vmqjw4dOoAse82udakkn8jIyDoVBZxFlmUWLVqERqcnON4zE6RATYn6hQsXunQcjUbDO++8g0qlYtHPn1Fd5aIVuC6wfsY4LNnpPPbYo7Rp08Zl46SkpPDJJ5+gN+kY8GAfRRZ/1YfOoGXgQ30AeO21V7FYPKO8/6FDh8jKysIYbXbZ4gdTjL0cvDOqNLlaSkoKe/fuRRNkQ+XG7kpqP1D5yCxdtpSioqJLP0EQBEEQBMHJGnW156GHHiIxMZG0tDQAXn75ZS677DJGjx7N7bffzvDhw2nVqpVLy8QJ7vHMM8/QtWtXMg5s5/g2164wd7ZTezdzYsdaEhISGDp0qNLh1EuPHj3Q6/Wk7j27Kjvz2CEqSovp06ePV+56bojExEQA8jMLKcovobqyuuY2T+Qo36bkhXVXk9QqrNXu31H0wAMPcN1111Gdr6LSyf3Wq3IlKjNUdOrUiRdeeMGpx/Y0Yj73frm5ubz99tuoNFo6DXoAtUardEiNFt3lSkITO7F582Z+++23xh8n2t7vOu/0cWeF5lHyTtt/n2NiYtw2pqMXallZGdfe0wu/QNcsKOtze3cCI8xMnDiR7du3u2SMxlq7di0//PADWn8DEf2TUGmUn/tVGjURNySh9dMzcuTIc3Ywe7ri4mI2btyI2ldGZbjw43Rnku7Lli1zU2SexxPn808++QSz2VzzFRsbW6/nOaoEOXZ6N2uVZUhVZXTq5JpKMQcOHODEiROEJHT06PndJygM35BI1q5dS0lJiUvH6tSpEw888AB56cfZNHuiS8dylqy0I2ydP5XYuDiefPJJl41TVVXFK6+8Qnl5OdfffyW+AZ7XOqC28FYhXDGoCxkZmbz33nsesdHDUXXBJ9b5/dUdjBF+qDQql1d4cAbHYhldmHv/bSQJdGE2qiqrWLFihVvHFgRBEARBgEYm1q+77joWLFhQ02/W19eXjRs3MnXqVD7++GMmTZrEnj17vLYfbUui1Wr5+uuvCY+I4MjaBZzc5f5+nY2RcXAHB5fNIDAwkO+++w6TybNPGh0MBgMdO3Yk+/iRmvL7p4/ad994cn94d3Ek0fMyLORlWOrc5okMBvtVYGt58+iP1hi2imr0evfv+pQkiXfeeQc/Pz/Kj6mxOakqsFwNZYfVaLQaPvroI7Raz71I6QxiPvd+//vf/8jPzyfp6kH4BocrHU6TSJJE+wF/Q+/jxzfffFun7HFDdO7cGYBjezY7MzyPcWzPFuDs63SHWbNmsXXrVhK7xZLU3XXvFxqdhv73X4mMzHvvvecx/Udzc3N5/Y3XkdQqwq9LQG3wnLlDY9QSfm0CSPDqq69SUFCgdEj1tmLFCqqqqtCGXLxChSZQRlLD4sWLPSLxogRPnM9fe+01LBZLzdeJEyfq9TxHEloqyb3EIz2fVJIH4LLE+uLFiwEIa+P5ldnC2nShsrLSLcnC559/noiICDbO+o289Pr9v1OKbLOxeMzn2GxW3n7rrZrzR1f4/vvvOXjwIB17J5HQpX4LXZTWvX9HopPCWbJkCXPmzFE6HFatWoWkkjBGOb+/uoOkVmGM8iclJaXe75tKWbp0KZKKC7ZrcSVtqFhUJwiCIAiCcpy2lUKr1TJkyBBeeeUV7r//ftGP1YuEhoby8+jRBAeHkLziT1I3r/Doi1Ynd29k36Ip+Pn68uOPP9Z7d4SnaNu2LTarlfzMUwDknEytub2lO7tj3UJ+pucn1i+//HIkSaLsdP1KXzY3VYXlVBVWcMUVVygyfkhICC+//LI9GZ7inOms/LgKWwU8+cSTHv1/y5XEfO49du7cyfz58zFHxhHTrbfS4TiFzuhDm2tvo7Kygi+//LJRx+jXrx8BgYHsXDKTyvIyJ0eorPLiInYvn014eDi9e7vn3zw/P58vvvgcnUHL1Xf1dPl44a1C6NK3LSkpKfz6668uH68+vv32W/Lz8gnqGY0+yPMWc+pDfAjqEU1OTg7fffed0uHU29y5cwHQXmInnKQGTbCNtLQ09uzZ447QmgWl53O9Xo+/v3+dr/qIj4/HaDJ5xY511ZnEumMXvjPJsszixYvR6PQEtXJd6XBncST/HYsBXMlkMvHqq69ira5i6fivPPraxe6Vczl9ZB+DBw/mqquuctk4Bw4cYOzYMZhDfOl7R/NZsC+pJPo/cBU6g5bhw4eTk5OjWCy5ubns2bMHQ7gvap3GpWOZYgMAzy4Hf+rUKQ4fPow60Ibk3O4/9aI2gsoks379eioqnLTKXhAEQRAEoZ4alYlISEjgm2++uehjRo4cSUJCQqOCEjxPfHw8v/76C5GRkRxdv5Dk5X9is7q//PPFyLKNI2sXkLx8JkFBQYwbN87eo6+ZcZRutWSlA1CYnV7n9pasdevWgL0UfH6WPVntye8zgYGBXHbZZZSlF1Ge49qyh0oo2JcJwLXXXqtYDHfccQedO3emKktFdRPXL1jLofKUiqioKB577DHnBOjhxHzu3Rw9ldv0uxVJUr4stbOEtemKOTKOJUuWkJKS0uDnG41G/u/BBykrLmTtH2NcEKFyVk35kcryMh555BG3VdwYPnw4FkshV97SFR+ze5LKV9zSFZO/kVHfjyI1NdUtY15IcnIyM2bMQBdoxNw+TNFYLsbcMRxdgIFp06Zx5MgRpcO5pJSUFNavX4/aLKM2Xvrxugh74mzixOZR+tnZvGk+V6vVdOzQAVWZBazVSofTJFKx63asN5cy8A4+QaH4hkSwxg3l4AH69+/PtddeS+qeLRzZusbl4zVGeXERq6f8hI+vL//5z39cNo4sy3zyySdYrTauvfcKtHrXJoWdzS/Qh6sGd6OwsFDRxWFr165FluWaHuiuZIr2/D7r69atA0AbpNzCFW2wTHl5Odu2bVMsBkEQBEEQWqZGXWVNTU29ZBnBgoKCmh5vgneIi4tj4sSJdOjQgVN7NrLzz7FUlZcqHRYA1ZUV7Jk7gbStK2ndujUTJ0ygXbt2SofVKKGhoQCUWHLP/JmHn5+fS8vCNRe+vr4EBwdTmFOEJbsItVpd0yvXUzl6dGdvSEO2ee5uiYYqzy6mMDmbNm3aMHjwYMXiUKlUvPLKK/aYUpuWOKxIUyHb4N///rci5e2VIOZz75Wbm8u6deswR7bCHOldpfwlSSKuRz/g7I7Whnr44Ydp3bo1W+dPIXXPVmeGp5gj29axa9ks2rdvz/333++WMefOncu8efMIjw+hU1/37ZjUG3X0+9vlVFZU8uqrryq6U+nHH39ElmWCe8UgqSTF4rgUSSURdHksNputZtGNJxs9ejQAhpiLl4F30ATIqH1lFi5cqPhiCyV423zeuXNnkGWk0ma8a12WUZXkERsbh9ns/ESco7dxWFvPLwPvENamK5UVFaxcudLlY0mSxH//+1+0Wi3LJ35HdVWly8dsqHUzxlFWZOGZp58mJCTEZeOsWbOGHTt2kNA1lpg2ES4bx5U69kkiODKAmTNnKvY+tnbtWuBs0tuVNCYtumATW7ZupazMM6srrVljX7CiZGJdc2Zsx7+NIAiCIAiCu7hs+5LFYmkxiYmWJDQ0lPHjxzNgwADyTxxhy+/fUZybqWhMZZY8tk0dRfbRffTu3ZsJEyY0693dQUFBAJQWFtT86bhNgNjYWArzSrDkFBMZGYlG49kr7i+//HLuvPNOKnNL7cl1Dy5FWF9VRRVkrkhBkiTefvttxfuQX3bZZfTt25fqfBXVlsYdw1YOlZkqEhMTuemmm5wbYDMn5vPmaefOndhsNkISnF9+1hMEt26PJKkavUPFaDTy6aefotFomPPtO+SePu7kCN0r+/hR5o36AJ1ez6effuqW9+Xdu3fz7rvvojfqGPBAb1Qq91ZFSOgaS4erEtm3bx/vvvsuNlv9ErDOdOTIEZYsWYI+1AdjpOv6rTqLKdoffbCJhQsXcuzYMaXDuaDt27czd+5c1L4ymnr2bZUkMLSyYbPZ+OSTT7zi85azNaf5vHPnzsDZHd/NUkUJVFfQubPzd6vbbDYWLFiARm8kOK75tAsLb9cNgPnz57tlvLi4OP7v//4PS1Y6OxbPcMuY9ZWfcZIdS2YQF9eKBx54wKVjjR8/HoBeN3dx6TiupFKpuPzmLthsNn777Te3j2+1Wlm/YT0aXz1as3s2PJiizVRVVrJlyxa3jNcQFRUVbNq0CZVJRqXg/g+Nv4ykPpvkFwRBEARBcJd6Z6T+WoIoNTX1vGWJrFYrJ06cYOLEiaIntJcymUx88cUX/PDDD3z//fdsmzKSTjf/nZAE95ddzz9xlD3zJ1BVVspDDz3E0KFDPT7ReimBgYGAPaEuyzL/z959hzdVvn8cf5+kbbpbSktbRhllbxFZIlMFBw5QEUXWTxFxgoqKAoILBRwgU5GhXwcCDpbKVNkgexdKKaW0pXuPJOf3R20VaaAtSU6S3q/r4oI26XnuHtp8cs6z8rIyqFbXeQcKWFt4eDgHDhwgNzOPFk3CtS6nXF577TWio6M5dOgQeoOeoBtroyiOO6vtaox5RVxcH4Uxt3iWYNu2bbUuCYAnnniCbdu2UXBBh1tAxTtXCi7oQC0+jr07h+xN8rxqSEhIAMC7mu1mQGlJ7+aOwS+AixcvVvoYzZs3Z8qUKYwfP54VH7zMo5Pn4hPgfAPZslKSWP7BOArzc5kxYwaRkZE2b/P48eM89dRTFBYVcufj3QkI8bN5m2W5pX970hIyWL16NT4+PowfP96ur+Gff/45ANXahDtFriuKQrU24SRsOsPChQt5++23tS7pCjk5OUyYMAEAr0YmKnJa3aqruAWZ2b59OytXrmTAgAE2qtIx2DvPP/30U9LT04mPjwdg1apVxMXFAfDss89adVZ2ydLpSo7zdqyX7K/eqpX1OzP/+usvEhISqNniJnROdO3rHRiMf2gdtm3bRkpKCtWrV7d5m48//jgrV65kx49Lad3zbgzevjZvszz+/P5zzCYTY8a8YNPBcBcuXGDPnj3UbhxG9fBAm7VjD/Vb1sKvmg9r1qzh5ZdftutAoRMnTpCZkYl/42C75b13TX/SD11k586ddOvWzS5tlteuXbvIy8vDUEfbQWyKDtyqmYmJiSE6OtoptjsRQgghhGso91VYjx49St9AKorCkiVLWLJkSZnPVVUVRVGYOnVqhQvKzs5m2rRp7Nq1i927d5OWlsaiRYsYNmxYhY8lbEen0zF69GgaNWrE+PHjObhqCY1uuYs6N3S124VG/NE9nNi4Eje9nilTpnD//ffbpV1bK1kGLic9hfzsTEzGIpsuDedsQkNDy/y3I/P29mbOnDkMGTKE6CPRmIvMBHeMcOglY8tSlJnPxQ2nKcrM58knn+TRRx/VuqRS7dq1o3nz5hw7fgxzgRldBe6zqCYoTNAREhLC7bffbrsiHYS98lxoq2SQmWoyaVyJ7ahm83XfjO7Xrx9xcXHMmTOH794dw8Ovf4K3f6B1CrSD7LRkvn3nebJSk3jppZe47bbbbN7mnj17ePbZZ8nNzaX3o52p26ymzdu0xM1dz10je/DT7I1899135OTkMGXKFLvM2I+JiWHdunV4BHnbZVlYa/GuE4hHoBerV6/mySefpE6dOlqXVEpVVd58801iY2Mx1DHjVsFFABQFvBuZyd6n491336VFixY0bdrUNsU6AHvn+fTp0y9bgnnlypWsXFk8C3jw4MFW7VivXbs2gYGBpOWk4KwppmQXb+tVMvvemn7++WcAwpq1s/qxbS2sWTtObTnP2rVreeyxx2zenr+/P48//jgzZsxgz9rv6PrA/9m8zWtJij3NiR0badmyJb1797ZpWxs3bgSg8Y31bNqOPeh0OhreUJf9m46xa9cuu3Y279y5E8Cuq9N4hvigc9Oxa9cuu7VZXr/++isA7sH2Xy3ov9xDVIqSi2t66qmntC5HCCGEEFVEuTvWJ06ciKIoqKrKlClT6N69Oz169LjieXq9nqCgIHr27EmzZhWfwZycnMyUKVOIiIigTZs2dtl/S1TebbfdRu3atXnmmWeI+mM1eZlpNO52N4oNZwupqsrZnes5u2sjgYGBfPLJJ7Rr53w3FSwJCgrCw8ODzOREMi4VzzgMD3eOmdn28O+ZDfaY5WAtAQEBLFy4kNGjR3P8+HGMuUWEdq+Pzk2vdWnlkp+cQ8KG05jyixg9ejSjRo3SuqTLKIrCww8/zMSJEylMUPCsW/7R80WXFFQjPPDAA5ova28P9spzoa2SzrLslARCaaNxNdZXlJ9LQXYGEW2vfybgqFGjyM7OZunSpSx7bywDX/8YL1/HX9Y7JyON794dQ1pCHE8++SRDhw61eZurVq1i4sSJqJi5fejNRLaJsHmb12Lw8uDep3uz5rPfWb16NYmJiXz00Uc22dP432bOnInZbCaorXPMVi+hKArV2tYkccsZZs2axQcffKB1SaVmz57NL7/8gj5AxbN+5W7W6zzBq4mJnCOFjH56NF//72vCwpxzT+FrsXee23PvekVRaNGiBdu2bQNjAbg5xxL2/6bkpKLT6a0+uCMnJ4dff/0Vr4AgAmvVs+qx7SGsSRtO/7malStXMnjwYLu8fj788MMsWbKEv9Z9T/s7HsLTR5tVVkpsX7kYKF7pwdbff0mHcEQz17ifULd5TfZvOmb3Wdx79+4FwDPMfj87il6HoYYvUVFRZGRk2Px9TXllZWWxfv1v6LxU9Nr+KgHgXr14OfiffvqJkSNHotc7x/0VIYQQQji3cnesv/nmm6X//v333xk+fDhDhgyxekHh4eFcvHiRsLAw9u7dy0033WT1NoR1NWvWjK+//prRo0dz6sA2ivJyaH77Q+hs8IZWVc2c3PQTFw7vpE6dOsyfP9+hZtpYg06no3bt2lxMjCMtsXh5RWfeM97aSpbKBwgMDNSukEoIDg5m0aJFjB07lu3btxO/7iRhvRvi5u2hdWlXlR2TyqWtMWAuzgJHXVq1T58+TJ06lfyEXAwRxnIvH1uQoENRFJdZ9eJa7JXnQlutW7fG3d2d5LMniOzSR+tyrC757AkAbrzxxus+lqIovPTSSxQWFvLtt9/y3bsvMHC8Y3eu52Sk8e3bz5FyIYbhw4fz9NNP27Q9VVWZM2cO8+bNw9PbQN8RPagZWcOmbVaEwcuDe0b1ZOPXO9mzZw+DBw9mzpw5NnuPeODAAdavX49nDV+86wTapA1b8qkbiCHYh3Xr1vHYY4/ZZKnqivr222+ZP38+Oi/waVGxJeD/y726imekiUtnLvHUU0/xxRdfXPb+0VW4ep63bNmSbdu2oWSnogY6WaegakaXm0ajRg3x8vKy6qHXrVtHXl4eDW7ohqI43/ZF7l4+BEe24PSpQxw6dIg2bWw/+M/T05Nhw4Yxffp0/vp1OTf3H27zNi1JjjvLqd2/06ZNGzp37mzTtsxmMwcOHCCwhj/eftb9OdRKaN3q6N10HDx40G5tmkwm9u/fj3uAJ25e9h2E7RXqS158Jn/99Re9evWya9uWrFixgry8fDwbmK8rq61F0YN7DTMXLlxgy5YtNl8FQgghhBACoFJXYps3b7bZRbvBYHDZWQWuLDQ0lMWLF9OuXTsSTx7gyLqvMVt5+VlVNXN8/QouHN5J06ZN+eqrr1yuU71E/fr1ycvK4PzxAwCyV9S/+Pv/09HhKKO2K8LHx4dPP/2UAQMGUJCSy4XVJyhIztG6rDKpqkrawXgSt0Tj6eFZWrej8vb2pm/fvpjzwZRRvq8x5YEpQ6FTp05VcmUIW+a50Javry/dunUj+1I8mX8P0nIl8Uf2AMUDaqxBURTGjx/PwIEDSYqJ4rt3XyA/O8sqx7a2nIw0vnvneVIuxDBs2DDGjBlj0xlvJpOJSZMmMW/ePAJC/Oj//G0O1alews3DjduH3Ey7W5sTExPD4MGDOXnypNXbMRqNpXuTV7+ptlPNVi+hKArVbyoetPnOO+9g0njLiOXLl/POO++g8wCfVkZ0Vui38KytYqht5vTp0zz++ONkZJTzjYGTcsU8Lxnw4Yz7rCt5WWAylu4Vby2qqvLdd9+h6HTUbOG8ExBqteoEwLJly+zW5oMPPkhAYCD7f11BUUG+3dr9r92rvwFg5MiRNs+P8+fPk5WVRWiE86zydi16Nz3Va1bjxIkTFBUV2aXNM2fOkJOTg2eor13a+zfPGsVt2nMgwdXk5OTwxaIvUNzAEK7t/ur/ZqhtBgXmzJmj+XsaIYQQQlQN1zXEef/+/YwbN4577rmHW2+9tfTz586dY9myZaSm2u8iuKCggMzMzMv+CPvy8/Nj3rx5dOrUiUunj3D0129RzdbZc0lVVU5u+pGLx/bSqlUrFi5cSFBQkFWO7YgiIyMBOLFzEwANGzbUshyH4uv7zwWtn58DrD1WCe7u7kyaNIlx48ZhzjcS/8tJcmLTtS7rMqrJTNLWGFL3x1OrVi3+97//ccstt2hd1jXdfffdABQmlS/eipKKb2j169fPZjU5A0fKc2E9Dz30EACx+/7UuBLrykyMI/1CNF27drXqADtFUXj99ddLO9eXTX2Rgtxsqx3fGvKyMlj27gskx51l6NChjB071qY35lVVZcKECfzwww+E1q3OgOdvJ7CG487kV3QKne5qS4+HOpCalsrw4cOJioqyahvLli3j5MmT+DUKxjPE/jfZrcUr1A/fyOocPXq0dJ9sLSxdupTJkycXd6q3NqK34qRKzwZmPGqaOXXqFMOGDSMpKcl6B3dQrpTnJZ3Supw0jSupOCWneH91a3esHzx4kBMnThAS2QKDxsuZX49qtRvgHRTCL7/8Qlqaff5/vb29eWTQIHKzMjjyxzq7tPlfWWnJHN+2nsjISLtcVx07dgyAkDqudd8kpE4QhYWFnDlzxi7tlXRqa5H5hmAfUBSH6VhfsGABaalpGOqYUMq9/qnt6b3BI7Q473/44QetyxFCCCFEFVDpjvVx48bRvn17pk+fzurVq9m8eXPpY6qq8sgjj/Dll19apcjyeO+99wgICCj946ozmR2dl5cXM2fOpH379iSdOsSp339GVa9/JOvZneu5cHgXzZs3Z/78+ZfNWnZFjRo1AiA/O5PAwECn2kvc1v7dse7j46NhJddHURQee+wxZs2ahYe7gYRNp0k/lqh1WQCYCozEr48i+0wKbdu25euvv3aawR033ngjoaGhFF3SoV5jXI+qFnfAGwwGh1laTwuOlufCejp37kzTpk1JOnWQ3PRkrcuxmpjdxYPORowYYfVjl8xcHzBgAAnRx1n+wcsU5udZvZ3KKMjNYdl7Y7l0PprBgwfz4osv2ny22/z581m1ahVh9UO456leePo4xz7HzTs35NZHu5CVlcVTTz1Fenq6VY6bkpLCrFmz0BvcqH5jLascU0vV29dG56Hn448/tto5Ki+z2cyHH37ItGnT0Bn+7lS38ts6RQGvhmYMtYpnrg8ePJjo6GjrNuJAXC3Pg4ODCQ0Ndc4Z638PBrB2x/o33xTPdq7dpotVj2tviqJQu3UXCgsL7TqwZ+DAgXh4ePDXL8utNgGgIg6s/wGTycjQoUPtstrJ0aNHAQip7WId639/PyUDB2yt5Dx6Btv/3oPOXY9HoCfHjh3TfCb20aNHWbx4MTovMNRynNnqJTzrmVHcYMaM6SQmOsZ9FSGEEEK4rkqNMVy0aBHTp0+nX79+vPPOO3zzzTdMnTq19PF69erRoUMHfv75Z55//nmrFXs1r732GmPHji39ODMzUzrXNeLl5cWsWbMYNmw4Jw/uwDswhDo33Fzp4108vo+zuzYSERHB3LlznXaWckX8uxOzUaNGTrnMqK14e3uX/tuZO9ZLdOvWjaVLljB69GiSd5/HnG+k2g01Nfs/N+YVcfG3UxSm5dG3b1/efvttDAbn6EgB0Ol03HHHHSxevBhjmoJ7dcsX/eYcMOcq9OjTwyV+lirD1nmenZ3NtGnT2LVrF7t37yYtLY1FixYxbNgwq30Pn376Kdu3b7fa8RzRrbfeWqlOZEVRGDVqFC+88AIxuzbRvM9DNqjOvrIuxXPpzFFuuOEG2rdvb5M2dDodEydOpKioiJ9//pmfPplA/xffQ+9m3301/62osICVM14lMeYUDz74IOPGjbN5TkRFRTFv3jz8gny48/+64W7Q7vuvjMY31iMrLYddaw4yY8YM3nrrres+5vz588nOzia4UwR6T+c6H2Vx83KnWtuapOw+z4IFCxg3bpxd2s3NzeW1115j06ZN6L3/Xv7d0zZtKQp4RppRPFQunr3Io48+wrRp0+natattGtSII16fW0Pz5s1J3LwZivLB3UY/JDag5KTi7u5O48aNrXbM5ORkfvvtN3yDwwisVd9qx9VKeLN2nNn+C9999x3Dhg1Dr9fbvM3q1atz11138cMPP3Du6F/Ua2W/5fSNRYUc3PQzgdWqceedd9qlzcOHD6PoFIJrV7NLe/ZSI6K4Y/3w4cP079/f5u0dPXoUnZsO9wBtXoMMwT5kRSVz7tw5zbYIzM3N5bXxr2E2m/FpbEKx/a9rhekM4BlpIvtkDuPHj2fBggV2eV0RQgghRNVUqY71OXPm0KxZM1asWIGbmxseHh5XPKdp06Zs2LDhugssL4PB4FSdP67O19eX2bM/5eGHHybqj9X4BodRrU5khY+TmXSBExtW4Ofvz5w5c1x6+fd/i4yM5M033yQhIeGyZRzF5R3rXl5WXC9UQ82aNePrr7/miSee4Nyhc5iNJqrfVMfunevGnELifz1FUWY+gwcP5uWXX0anu64dQzTRt29fFi9eTOGlq3esF17SlT6/qrJ1nicnJzNlyhQiIiJo06YNW7Zsuc6KL3fu3Dnmz59f/IHegdYjtCaTkcOHD3PvvfdWavWSnj170rhxY6JO7Kdex954Bzr3Cihnd20E4KmnnrLpa6ROp2Py5MlkZmayZcsW1s1/j7tGT9Bk0JPZbGLN7Lc4f/wAd9xxB6+//rpd6li0aBEmk4luA9o7zUz1/7qhVzPOHIjl559/5umnnyYsLKzSx7p48SLff/897v4G/BuHWLFKbQU0DSHzeBLffvctQ4cOJTQ01KbtxcbG8sILLxAVFYVbNTPezcxW2VP9ahQFPCNUdJ4mck7m8PTTo3nhhTEMGzbMZQavOuL1uTU0b96czZs3o+SkogbW1Lqc8lHN6HLTadysKe7u1vvh/umnnzAajdRq3dklfm7dDJ6ENbmBC4d3sn37drttOTVw4EB++OEH9m/40a4d66f2/EFuZjoPjxhhl/tWRqORo0ePUj08EHcP13qPHBQWgLuHG4cOHbJ5W0ajkejoaNwDvVB02vzeeVQrvudx6tQpTTrWVVXlnXfe4Wz0WQy1zbgHOt5s9RIeoSrGFDO7d+/ms88+Y9SoUVqXJIQQQggXVal32MeOHeOJJ57Azc3yl4eGhlaJfeyEZaGhoXz88ccMGzaMo798Q4dHX8DDu/z7UhkLCziy5n+oZhPTPviAunXr2rBax6IoCgMGDNC6DIf07850V+lYBwgPD2fx4sWMHDmSqGNRoChUb1/bbjfOjHlFxP96kqLMAkaOHMkzzzzjtDftmjdvTs2aNbmYFI9qNqNYGBtQlKzg6enpcrPWKsLWeR4eHs7FixcJCwtj79693HSTdW9gent7o9PpMHl4U9SsF3h4X/uLnEl+Nh7HN+GOsdI3YXU6HaNGjWLs2LGc27OZZrc9YOUi7Sc7OYFLp4/Qpk0bOnXqZPP23NzcmD59Ok888QT7t60noEZNbnnwcZu3+19bvp7LqT2/06lTJ9555x27zL4xGo1s2LCBgBA/Ipo5SWdWGXQ6Ha1uaczmb3exYcMGBg8eXOljrVixAqPRSI02dTS7uW4Lik5HYOtwLm2L4YcffrDpTeg//viDV14ZR3Z2Dh61zHhFmrHnWw2PGio6LyO5R9348MMPOXz4MG+99ZZLrFrjqtfnJUupKzlpTtOxruRlgtlEs2bNrHZMVVVZuXIlencPwpq0tdpxtVarVUcuHN7JDz/8YLeO9RYtWtCiRQuO79tGTkYqPgH2Gbh/aNMqAB54wD7vw06fPk1BQQEN67reKo46nY6QiCBOnz5Nbm7uZQPvre38+fMUFhbiV027rQgNf3es22tP+f9avnw5P//8M3o/Fc/69t9CoSIUBbwamzFl65gzZw6tWrXi5psrv3qmEEIIIYQllZqK6ObmRmFh4VWfEx8ff9leyKJqatOmDS+88AIFOVmc2LiyQvutR/2+iryMFB5//HF5MyxK/btzydPTeZaELI/g4GC++OILIiMjyTiaSPrhBLu0ayowcnF9FEWZBTz55JM8++yzTtupDsUDU3r16oVqBGNG2d+HKa94Gfibb77Z5X6OKsLWeW4wGK5rhui1hISEMGrUKJT8bDyO/IYuJRYqkDMOS1XRXYrG4+hvUJjL2DFjrus9Ve/evYmMjOTi8b/Iy0yzYqH2VbK3+qhRo+z2GmUwGJg5cyYREXXZ8cMSjm791S7tljiw8Wf2rv2Ohg0b8uGHH1p19uPVnD59mry8PGo3CnXqPACo3bj4NejgwYPXdZxff/0VnZsOn7qBVqjKsfjWq4ai1/Hrr7b5+TabzcydO5enn36anLwcvJua8G5o3071Em5+4NvOiFugmfXr1zPokUHExMTYvxArc9Xr85LOaZ0T7bNesie8NTvWjxw5QmxsLCENW+JmcJ33rX41auIbHM6WLVvIzMy0W7v9+/fHbDJxdOtvdmkv49JFYo/to2PHjnbbrrBkX/AaEc69UpElNepUx2w2c/LkSZu2c/78eQDc/bVbucfdv/h3PjY21u5tHz58mHffexedB/g0N1kcsO5IdO7g3dwIisq4ceO4cOGC1iUJIYQQwgVV6m1Rq1at2LRpEyaTqczHc3Nz2bBhAzfeeON1FSdcw2OPPUaHDh24dOYoSacPl+trUmNPE390D82bN+epp56ycYXCmfy7U8EVO0QDAwNZsGABNWvWJHXfBbLOpNi0PdVsJnHLGQpTcxk0aBBPP/20Tduzl27dugFgTC37rn3J57t37263mhyRI+Z5QUEBmZmZl/25mlGjRjFhwgQMOhW309txO74ZJcd5O4+VrGTcjm7ALXo33h5uvPvuu9c1yxaKZ/aMHDkS1Wzm3J7NVqrUvnJSk0iMOkSLFi3sPtguMDCQuXPn4O/vz6+ffUD86aN2aTf2+H42LP6QatWqMXv2bPz8/OzSLkB0dDQA1cMD7damrfgGeuPh6V76PVVGTk4OMTExeIb5oXNzvf06de56PGv4cPr0afLz86167NzcXMaMGcOcOXPQeYFvWyMeodoOgNJ5gE9rM4baZs5Gn2XQoIf5888/Na3pejlinltDcHAwISEhTpXrSk46ULyCkrVs2lQ8sCy0cWurHdNRhDZuQ1FREdu3b7dbm3369MHd3Z1jdupYP7ZtPQD33HOPXdqDf3I8yAVyvCxB4QGA7Wdxl3TKuvtp17Gu93ZH0SnExcXZtd309HTGjBmD0WjEq6kJnRPdenHzA89GJjIzMxk7duw1B54JIYQQQlRUpTrWR4wYwalTpxg1ahQFBQWXPZaZmcmwYcNISEjgiSeeqFRRn376KW+//TZffPEFAKtWreLtt9/m7bffJiMjo1LHFNrR6XS8+eabeHgYOP37akxFV39TazaZOLn5R3R6PVOmTLHb7CzhfMraP9IV1KhRgwULFuDr58el7efIv5Rts7aSd8eRdzGL3r178+qrrzr9zMQS7dq1w2AwUJRW9vdT8vnOnTvbsyyHY+s8r4z33nuPgICA0j/XmtmjKAoPPfQQP/74I7169UKXlYT7kV/RR++CIut2ENlUQS7609txP7YBXU4Kd955Jz///DP9+vWzyuH79OlD/fr1uXh0L3kZzjPzr0T0zvWgqjbfW92SiIgIZsyYgWo28eNHb5Bj43OYmZLIz59MRKcofPLJJ9Ssad8lkE+fPg1AtbAAu7ZrC4qiUC3Un5iYGIxGY6WOkZpa/P+t93Ld96R67+L3VCXfqzWkpqYyYsQINm3ahFs1M743GNE7yIRpRQGvSDPezUzk5OXwzDPP8MMPP2hdVqU5Yp5bS/PmzVEKc6Go4NpPdgBKbio6nZ5GjRpZ7Zjbtm1D5+ZOtToNrXZMR1G9flOg+Hu0l4CAALp27UrSuShSL563eXsndm7CYDDQu3dvm7dV4uLFiwAEVHeQF10rC6hePNgwPj7epu1cunQJ0Db/FUVB7+VOSoptB9z/m6qqTJw4kcTERDzrmXCv5nwrghnCVDzCzRw7doxPPvlE63KEEEII4WIq3bH+8MMPs3DhQkJCQli4cCEAHTp0oFatWixfvpyhQ4dWev+o6dOnM2HCBObOnQvAypUrmTBhAhMmTCAtzXlGq4t/1KlThxEjhpOfnUHs/q1XfW78kd3kpl3i4YEDadKkiZ0qFM7IlQdd1K1blxnTp6OokLg5GmNekdXbyDydTOaJJBo3acy7776LTucEa7uVk8FgoF27dphzFMz/OXWqCqYMHfXq1bPpMuXOwNZ5XhmvvfYaGRkZpX9KlmC8ltq1a/PJJ5+wcOFCmjRpgv7SWTwOrUWXFO3Yy8OrKrqEU3gcXos+JZbWrVvz5Zdf8v777xMaGmq1ZvR6PaNHj8ZsNnFmu32XM79eGQnnSTp1iFatWpWuRqGFTp06MXbsWLLTkvnpk4mYKtlJey3GokJ+/GgCuZnpjB8/nhtuuMEm7VzNoUOHQIHgWtXs3rYthNQOorCwkBMnTlTq60sG8pkLy54N7ApKvrd/b7lzPZKTk3nsscc4evQoHuFmfFqZ0Tng2zaPGio+rY2gNzNx4kT+97//aV1SpThinltL06bFHa9KrhPcB1BVdLkZREY2sNrvUmFhIadOncKvRi30bg74S3SdfINDcfPwLF263F5uu+02AKL2/GHTdtISL3Ap9gxdunTBx8fHpm39W8mKTwYv1xyIbvAp/r5svYWAowys03u52bVj/ccff2Tz5s24VTNjqOPA11HX4BVpRu+tsnTpUnbv3q11OUIIIYRwIZXuRfn666+ZP38+9evX58KFC6iqyt69e4mIiGDu3Lmls80rIyYmBlVVy/xTr169Sh9XaGv48OFUq1aN83/9gbGw7BkHZqORmD2b8fL25sknn7RzhcLZuLm5aV2CTXXp0oWxY8dizC0k6Y9oVLP1LmoLUnNJ2RmLn78fMz+Zibe3t9WO7ShKljs1/WefdVM2qEZo3769FmU5HFvmeWUYDAb8/f0v+1MRHTp04LvvvmPChAn4enrgdnY3blHbwGj9wSnXragAt5N/4HZuH4F+vrz99tt89dVXtG3b1ibN9enTh1atWpF48gBpcZVfFtueVLOZU1t+AuDll1/WfFWNxx57jDvuuIO4Ewf547v5Nmlj05ezSIg+zv33369JJ1hGRgZ79+6lRp0gl7khX+vvfdZLllOuqBo1ahAUFERBcg6qIw/UqSRVVSlIziE0NJTq1a9/P97c3FyeeeYZYmNjMdQx49VIm/3Uy8vNH3zaGtEZYOrUqTbba97WHC3PreWfjvV0bQspj4JsMBVZdX/1uLg4TCYTPkE1rHZMR6IoOnyq1yi9B2Qv3bt3R6/Xc+bADpu2E/338Xv27GnTdv6r5P2SiutlFkDJt2XrgeE5OTnF7bhruw2Mzl1Pfn5+pVfeqYjU1FSmTZuG4gbeTRw7v69F0YNXMxMoMHny5CtWdBFCCCGEqKzrehf6xBNPcPDgQbKzs4mLiyMzM5OjR49Kh6gok7e3N0OGDKGoII/4I2WPFk2MOkRBdgYPDxxIUFCQnSsUzkbrDhZ7GDJkCL179ybvYhZphy5a5ZjmIhOJv0djNpqZ+t5UatWqZZXjOpp27doBYMz8T8f63x+XPC5cL8/1en3p8vAdOnRAlxaH+7ENUJCrdWn/yM/C/dh6dBkX6d69Oz/99BP33nuvTV/XFEXh9ddfR6fTcWLjSkyOONjgP+IO7iAz4Tx33323JjO3/0tRFN58800aNGjAnjXfErXXuvsyH9u+gQMbfqRp06a8/vrrmuTczz//jMlkouENde3etq3UbRqOh6c7P/30U6VuSiuKQrdu3TDmFJIb53rbUuXEpmPKK+KWW26xyvHee++94pnqYWY86zvHTXm9N/i0NKK4wWvjXyMmJkbrkirF1fIcKF3BzBn2WS/ZX92aq66VrNhn8PGz2jEdjbuXL0VFRWRn2277q//y9/endevWxJ86QkGe7d4fxhzaAxQPmLankoGp+Tmuubd0fk5xB2lAgG23rMnNLf7ZcISOdfinHlv67LPPyMrKwrOeCZ12W8tbjZsvGGqZiY2NZfny5VqXI4QQQggXYZXhnV5eXtSsWRNfX9fcv0lYz4MPPoiHwcCFI7vKHJF+4fAudDodgwYN0qA6IRyPoihMmTKFmjVrknYwnryErOs+ZvKuWIoy8hk+fLimyyrbWsuWLdHpdFd0rJd83KZNGy3KcmiuluehoaEsWLCAxx57DCUvA4/jGyH/+n+HrpeSm47HsY0o+dmMGjWKmTNn2m0wWYsWLXjsscfITbvE6T/X2qXNyspOSeD0tnVUq1aNl19+WetySnl7e/Phhx/i6enJuvnvkZmcaJXjpl48z2+ff4CPry8ffvih1ZYRrgiTycTXX3+Nm7uepjc1sHv7tuLm4UbTDg1ISkpiw4YNlTrG8OHDAUg7EG/VFWS0pprNpB2MR1EUhg0bdt3H27NnDz/++CN6XxWvxs7RqV5C7wteTUwUFRbxzjvvOPXqBK6U57Vr18bHx8cpZqzr/l6uvmSWvXU50S9TBWk1WLp9+/aYzSbio47Y5Piq2cyFU4eoV6+eVbf3KY+SgdMZl7R/32sL6X9/XzVr1rRpOyUznBW9tr9/ir741m1hoW0HSqSmprJs2TJ0nuAR7rwZ+F+GCDOKGyxcuJCiIscfWCyEEEIIx+c6G+oKpxAQEEDvXr3ITb1E9qXLZ9/mZaaRER9D586dCQ8P16hCIRyPv78/H3zwATpFR9KfZzEVVH4JuKzoVLJOp9CqVSueffZZK1bpeLy8vGjcuDHmbOWyLbZNWQr+Af7UqVNHu+KE3ej1esaNG8fYsWOhIAePYxvRXTyJLilamz/xx3E/vgnFWMCECRN4+umnbb6M5X89++yzNGrUiLiD20k8ddCubZeXsbCAI2v+h9lYxJQpUxxuFZvIyEhef/118nOyWPXpm5hN17c0p8lYxKpZb1KYn8fkN9/U7PVp69atxMXF0aRDAzx9XGCa0r+0uqUJiqLw9ddfV+rrGzRowD333ENBSi7pRxOsXJ120g8nUJiaR//+/alb9/pXKZg7dy4AXo1NTtWpXsK9uopbkJmdO3eyb98+rcsRFHe6NmnSBF1+JphNWpdzVSWd/9acsV4yOKKoIM9qx3Q0RQV56HQ6PD097dpuyepV8VG22d89Jf4c+TnZmqy4UzK441Jcqt3btodL54v3G7fNIJZ/FBUVoegUzVfKK+nYt3Wn8Nq1ayksLMRQ24TiQneLde7gEWrm0qVLbN++XetyhBBCCOECyr1BcYMGFZ+1oigKZ86cqfDXCdd22223sW7dOpLPHsOvxj8jjJPPHgfg1ltv1ao0IRxWmzZteOaZZ5g5cybJO2MJ7V7x12RjTiHJO8/h5e3F+++/j7u7uw0qdSwtW7bkxIkTmHOKZ6KZjWDOU2jVrpXmN0i0Yu88//TTT0lPTyc+Ph6AVatWERcXBxR38Np6CccSw4cPx9/fnylTpuAWu98ubVri5u7Oex98QN++fTVp32Aw8OGHHzJw4MMcX78c78Bg/Go4zpYQqmrm6C/fkpOaxPDhw+nRo4fWJZXp3nvvZceOHaxdu5Zdq76m831DKn2sbcu/IDHmFAMGDKBPnz5WrLJi1qxZA0CLLg01q8FWAoJ9qdMkjP3793PhwoVKbYMybtw4duzYQfKBi3iF+eEZ4tyzgfOTskk7eJHQsFBeeuml6z5edHQ0e/bswa2aGTcnXbVaUcAzwkx2qo7vvvuOG2+8UeuSLKpK1+dNmjRh3759KHkZqD6ONdDq35TcdELDwqz63qZkoFVu2iWrHdPR5KUlEx4ebvdrk+bNmwOQcPakTY6f+PdxW7RoYZPjX03btm0BiD+TROtu1hvo4SguRl/Cx8ebRo0a2bQdo9GIonOAa8a/a7D1HusbNmwABdxruM5s9RLuoWYKLuhYv3493bt317ocIYQQQji5cnesx8TEoNfrcXMr95cIUaaOHTui0+lIi4umfsd/Pp8eFw3Yf/8xIZzFiBEj+PPPP9m/fz8+EYH41i//jUVVVUnaehZzoYnxE8ZXmdnaJTfMTNkKel8VU5Zy2eerInvn+fTp0zl37lzpxytXrmTlypUADB482G4d6wADBgygZcuWnD592m5tlqVZs2aV6hCxpnr16vHBB+/z7LPPcvDnJbQfOBpPv0BNaypx+s+1JEcfo0uXLjz33HNal2NRyZ71+/btY9uKL2jQthOh9RpX+DgXTh1h1+qvqRMRwbhx42xQafkdOnQIb38vgmtW07QOW6nTNJzYExc5fPhwpTrWAwICeO+99xg5ciSJW6KpdXcz3Lycc5CaMbeQxC1n0Ck6pr431SpLhv/000+A8y8fq/cHnbfKho0byMrKws/PMUcJVKXr88aNi19bldx0x+1YNxaiFObSpHF7qx7W29ubunXrciEhFtVsRrHzKje2lpeRSkFOJs27dLz2k60sKCiIkJAaJJ+PtsnxL8WdBay7gkF51axZk9q1a3PhdCJms9nuqyPZUk5GHqkJGXTt2tXmr39FRUWlndpaKunct+WM9aKiIg4fPozeR0XnnG9trkrvC4obshqNEEIIIayiwu9Ce/TowYgRI7jvvvuqxGxHYX3+/v7Uq1+f2PMXUFW1dNZoZmIcwcEhNt8nSwhnpdfreeedd+jfvz/Ju8/jFe6P3rN8L+NZp1PIu5hFjx49uPfee21cqeNo1qwZAMZsBQ9UTNmXf74qs1eex8TE2OzYldGkSRNNbnA6ou7du/PSSy8xbdo0Dv60iHYPjMLd00vTmmL3/Unsvj9p2LAh06dPd/gOI39/f95++20ef/xx1i2YypC3FqDTl79mY2EB6xZMBVXl3Xfewdvb24bVlqMeoxE3d72mNdiSu0fx/8313Jju2LEjY8aMYcaMGSRuPk347U3QuTlXh4XZaCZh0xmMuUW8+uqrtG9//R2BRUVFrFq1CsWteDl1Z6YoxUvG5p8tYu3atQwcOFDrkq6qKlyfl+S2I++zrvy9v7ot3mN07NiRZcuWkZEQS2DNelY/vpZSzp0Cir9HLTRsGMmOHTsoKsjH3WDdpehTLhQPLNVqMGWXLl1YtmwZl86nElo3WJMabOH8yeItBW+++Wabt1VYWFi6v7mW7LHHekJCAoWFhXgEOXeGW6IooPcxc+HCheL3uw5+jSGEEEIIx1bud4jHjh3j+eef58CBAzz88MPUrFmTMWPGcPjwYVvWJ1xUk8aNMRbmU5CdARTfWM7PTKNJk4rP9BKiKqlTpw7PPPMMprwiUv6KK9fXmPKNpO6Nw8fHhzfeeKNKLYEeGRmJTqfDnFP8sSmn+Huvyh2rkufi34YMGcLgwYPJTk7g4M+LMRXZ7obdtVw8vo+oP1ZTo0YN5s6d67CzRP+rY8eODBgwgKSYKPasXVahr9358/9IjT/H4MGDS5dt1VKjRo3ITMkmJT5d61Js4uyR4twsmf1aWUOHDqVfv37kJ+VwaVsMquo8N6FLVrApSM7h/vvv55FHHrHKcVetWsWlS5fwCDO7xL6sHmEqig4WL15k86V3K6sq5XnDhg1RFMXBO9bTget/fSlL7969AUg8dcjqx9Za0qlDKIpCz549NWm/bt26AKQnXrD6sdMunicoKAh/f3+rH7s8Sjqezx2L16R9W4k9Ufz92KNjPT8/3yGWgi/ZYz0/P99mbaSmpha35ZrjswBQPMBsNpOenq51KUIIIYRwcuW+7dG0aVOmT59OXFwcK1asoHPnzsyePZu2bdvSvn175s6dS0ZGhi1rFS6k5AI2Nz0FgLyM4r8jIiI0q0kIZzF48GAaN2lCVlQyBSm513x+6oF4TAVGnn32WUJDQ+1QoePw8vKidu3amHKL486cq2AwGKhdu7bGlWlH8lz818svv8xdd91FRnwMh9f+D7PJZPcakqOPc/y37/EPCGDBggWEhYXZvYbrMXbsWIKCgtjxw2Ky0pLL9TXpSfHsXvU/wsLCeOaZZ2xcYfkMHjwYgA3/20FRgWN2JlbWsZ1nOHcsnvbt21/34CpFUXjzzTe58cYbyT6bSmo5B7ppTVVVUnafJycmjQ4dOjBhwgSrDLbLy8tj7ty5KDow1DZboVLt6TzAPdxMXNyF0u1LHE1VynMvLy/q1q2LLjcDHHQgiy071m+66SaqV69O0skDmB10oEdl5GWkkBZ3hvbt21OjRg1Naii5/5Bm5Y51s9lEelK8pttvderUCTc3N84dd52OdbPJzPkTCdSuXZt69erZvL2s7Cx0Htqv5KP7e8Wd7Oxs27XhQtsFXIter/3/qRBCCCGcW4XfOen1eu677z5+/vlnzp8/z7vvvktOTg5PP/00NWvWZPDgwcTGxtqiVuFCSpZ7L8hKByD/778rs9+lEFWNXq9n3MsvA1xz1npRZj6ZJy9Rv359HnroIXuU53AaNGiAWgjmouKO9fr161epGweWSJ6LEjqdjrfeeotu3bqRcvYEx35bhqrar3MsLS6aw2v/h6engblz5hAZGWm3tq3F39+fF154gcL8PP78bn65vub3r+diLCrkpZde0nwJ+BI333wzDz/8MCnxafw0ZyO5WbabGWVPh7ee4vdluwkIDOCtt96yyjE9PDz45JNPaNCgAelHEkk/kmCV49pS+uEEMo4n0bhxYz766COrLRu+dOlSEhIS8KhtRmewyiEdgmeEGcUNZs6cSVZWltblWFRV8rxx48ZgLICiPK1LKZOSm4HBYCgdQG5N7u7u3HvvvRTm5ZB05ojVj6+VC4d3AzBgwADNaijp+Lb2jPWslEuYTUZNJw54e3tz0003cel8KjkZjvl7U1EXz16iIK+Qbt262XwVNpPJRFZmFjqD9kuG6//u3E9LS7NZGwEBAQCottvGXXPmQgWdToevr6/WpQghhBDCyV1Xz0JoaCivvPIKx48fZ/369QQFBfHNN99w4MABK5UnXFXJTLT8v5eCL8gq/ruqzaYVorI6duzIzTffTF58JvnJORafl3YkAVSVZ5991mX33byWktkMpgwF1YRNbng6O8lz4e7uzowZM2jXrh2JJw9wassquyxvnZV0gUOrlqBX4JNPPqF169Y2b9NW7r33Xpo2bcrRP3/l0vnoqz734pnjnNy9hRtuuIHbb7/dThWWzyuvvML9999PUmwK389Yx/mTjt9hbElBXiEb/redP1fsJah6EPPnzbfqiiUBAQHMnz+f0LBQUvbGkXEiyWrHtraM44mk7rtAeHg4c+fOtdrSxPn5+Xz55Zfo3Is7ol2JzgMMdUxkZGQ47Kz1/3LlPC+ZCe6Qy8GrZnR5GTRq1MhmMyEfeOABFEUh7uAOmxzf3sxGIxeP7qFatWrcdtttmtVR0rFu7RnraQlxlx1fKyVL7JdsheLszh4u/j569Ohh87aSkpJQVRU3Hw+bt3UtJTUkJibarI2aNWvi5uZWunWaq1HV4kH2tWrVqrL3RYQQQghhPdc9ZW/Pnj089dRTPPDAA1y4cIGaNWtW6SV2RfmUdqxnFo+4LZmx7mxLvwqhpf/7v/8DimeglcWYW0T26RTq169fujdjVVSSSUXpymUfi8tJngtPT08+/fRTmjRpStzB7ZzdtdGm7eWmJ3Pgxy8wFxXy/vvv07lzZ5u2Z2s6nY7nn38eVVXZtuKLqz536/KFADz//PM2n3FVUW5ubkyePJmXXnqJgpwiVs3bxJZlu8nPKdC6tAo5eziO7z5Yy6m9MbRp04av//c1LVq0sHo7YWFhfP7Z51SvXp3knbFkRpVvKwB7yjh5ieRd5wkJCWHhwoVWXXJ506ZNZGRk4B5uRnHBlVU9aqooelixYoXWpZSbq+Z506ZNAVBy0rUtpCz5WWA22WQZ+BJ16tThlltuISM+hqxLzr+0d2LUIQrzchgwYAAeHtp1XNapUwedTkfqReuu6pCWcB7QfkBv7969URSF0wfOaVqHNZjNZs4cPE9AYAA33XSTzds7f774/9Dd1wE61v2Kl4Ox5eojbm5utGzZElO2guo6O06UMucWz8Zv06aN1qUIIYQQwgVUqmM9OTmZjz76iNatW9OpUye++OILevfuzZo1azh37hzt2rWzdp3CxZR2rP9nKfjw8HCNKhLC+bRv357mzZuTez4dY96Va7ZlnUlGNasMHjy4Si99XvK6Ysoo7rwq2YpCSJ6LK/n5+TFv3lxq16nD2Z3ruXB4l03aKcjJ4sAPCynMzWbChAmazlazpptvvplWrVoRtecPizfpE2OiOHtwFx07duTGG2+0c4XloygKQ4cO5ZtvvqFx48Yc23Gab6au4diO05jNjj0rOT0pkzWfbWHdF39QkFPEM888w+LFi2362l+vXj0+//xzAgMDubQtxqFmrqcfTSR5xzmCqgexcOFCq8+eTE9PB0Dv55j7Xl8vnRsoBpWMjHStS7mqqpDnTZo0AUDJS9e2kDLo/u7sL6nRVgYNGgRA3MHtNm3HHuIObken02m+VZWHhwe169QhJS7GqsdNjjsLoPn2NjVq1KBDhw7En04iI9l2+3PbQ9zJBHIycunbpy9ubrZfnv3o0aMAeFTTfrsed18DOjcdx44ds2k7Xbp0ARWK0hxr0Kc1FKUUf0/OPpBXCCGEEI6h3D0tZrOZ1atX079/f2rVqsWLL74IwIwZM7hw4QLLly/njjvuqNKdN6L8vLy8qB4cTOq5U0T9uZbEkwfw8PAgJCRE69KEcBqKojBgwABUs0r2mZTLHlNVlayoFDw9Pbnzzjs1qtAxlGwxYcpWLvu4qpI8F9cSHBzMgvnzCQoK4uSmH0g+e9yqxzcVFXLo58XkZaTy9NNP88ADD1j1+FpSFIX/+7//Q1VV9q77vszn7F23DIARI0bYs7RKadq0Kd999x0vvfQSilnHlmW7Wf7hr8RFOd7y8Pk5BWz78S++fX8t547F06lTJ1auXMmTTz5plxvwDRs2ZPHixQQHB5O8M5a0Qxftsp2CJaqqknownpQ956lRowaLFy2mfv36Vm/Hx8cHAFOW692EBzAXglqg4O3to3UpV6hqeR4aGoqfv39pJ7YjKVme3tYd6126dKF27doknjyIscB598zOSoonM+E83bt3d4iB9Y0bNSI3M43s9JRrP7mcLp2PRq93s8nrbkX1798fgCPbTmlcyfU59Gdx/QMGDLBLe/v37wfAs4b2r/+KTsEj2IeoqCiysrJs1k7JQNeiJNfL9KJLOtzd3e2yjYAQQgghXF+5r7Jr167Nvffey5YtW/i///s/du/ezaFDh3jhhRcIDg62ZY3CRbX8eznO2L9+B6B58+Yuc+NHCHvp06cPer2e7HNpl32+MD2fosx8evbsia+vr0bVOYb/ZlRVzyzJc1EederUYc6cOXh6enJk7ddkJlpnb07VbObIuuLjPfDAAzz55JNWOa4j6dGjB+E1a3Js668U5OVe9lheVgYndm6iQYMGTjNjxs3NjaFDh7JmzRr69+9PSnw6P8/ZxOoFW0hNyNC6PExGEwe3nOB/767i4O8nqVW7FjNnzmTBggXUq1fPrrVERkayZMkSwsPDSd13gZTd5zXpXFfNKsm7YknbH0+tWrVYsmSJzTp3brvtNmqE1qAwTofJuSdDXkFVIe+MDtUEQ4cO1bqcK1S1PFcUhWZNm6LkZ4HJsdYpVnKL34fbcil4KN5y5IEHHsBUVEjiqUM2bcuW4o/uBuDBBx/UuJJizZs3ByAh+oRVjmc2m0g8e4pGjRpqusx9idtuu40aNWpwfOcZp9vWpURyfBqxx+O58cYbadasmc3by8nJYdu2bXgEeuLmrf3/IYB3TX/MZjNbtmyxWRsNGzakSZMmGFN1mK9cEM9pmbKLB9l369YNf39/rcsRQgghhAso9/SNhIQE3N3dadOmDTExMUycOPGaX6MoCmvWrLmuAoXrmjZtGtHR0aUf2/vmpxCuICCgeI+5nTt3YswtLL3wz40tvsFXlfdWLxEYGIiiKKWdG9WrV9e4Im1JnovyatGiBdOnT+fZZ5/l0M9LaP/w03j6BV7XMaP+WE1y9HG6du3K66+/7nD7i1uDXq/nwQceYObMmZzctZnWPe4qfezY9vWYigp58MEHne57Dw4OZvLkyTzyyCNMnz6dnTt3cv7kRVp0bshNfVvh5etp13pUVeXs4Th2rNpPRnI2AQH+vPDKGAYOHIi7u7tda/m3iIgIvvrqK0aNGkXU8ShMeUXUuKU+it4+g0fNRjNJf54l51waTZo0Yd68eTbtZPX29mb8a+N54YUXyDnkhndLI24ucM9aVSH3pI6iJB0tW7Z0mA7Af6uKed6kSRN2796NkpeB6usg7+dUFV1uOrVr18bPz8/mzd1zzz3MnDWL+KN7qdWqo83bszaz0UjiyQOEhoYWLzvtAFq2bAlA/KkjNGx383Uf71LsGYoK8kqPqzV3d3dGjBjB1KlT2b/pGJ373aB1SRW2e13xQJKRI0fapb0NGzZQWFhIUAvH2ULMt34QqfsusHr1avr162ezdu655x6mTZtG0SUFQ03X2OalMLH4Pdg999yjcSVCCCGEcBUVWhexqKiI33//vdzPd7YbhsK+vLy8aPH3rHUhROV169aNnTt3kncxC7/I4puMufGZ6HQ6h7lhpSW9Xo+fnx+ZmZlA8WCEqk7yXJRXt27dGDduHFOnTuXQz0u48aGn0LtXbubOhcO7OH9gG40aNWL69Ol2WZ5bK3fffTczZ87k2LbfLu9Y37YevV7PHXfcoWF116dJkyYsWLCArVu3Mn36dI5siyJq/zk63tmG5p0j7bL6UFpiBn+u/Iu4Uwm4ubkxZMgQRo4c6TCv7zVq1GDx4sU8//zz7N27F2NeEWG9GqI32PZn3pRvJGHTafKTsunYsSMff/yxXVat6d27N++++y5vvPEGOYfc8GpkwiPUeW/Gmwsh94QOY5qONm3aMHv2bPR6vdZllamq5XnJTFUlJ9VxOtaL8qEo3y6zaAFCQkLo0rkzW7duJTc9Ge9A51qdIDnmBEX5edz96CMO83vVpk0b9Ho3zp84aJXjnT9efJx27dpZ5XjW8MADD7B06VIO/XGS5p0bERDsPCuaxUUlEHPkAjfddJNdVvsxm8188cUXKDoF30gHeZ0B3P0MeIb5sX37do4fP26z15w777yTGTNmUJioYqhpskkb9qSqxcvA+/v7c8stt2hdjhBCCCFcRLnv7pw9e9aWdQghhKikDh06AJCXUNyxbjaaKbiUQ4vmLewyc8YZBAYGkpmZiZubG56e9p1V6Wgkz0VFPfLII0RHR7Ns2TKO/fY9Le98pMKdM2kXznJy849Uq1aNTz/9tHRfZlcVHh5Ou3bt2H/gALmZ6Xj7B5KZksjF08e4+eabnX7lDEVRuOWWW+jUqRPLli3j008/5Y/lezixO5pegzoRFGabDm6Tycz+jUfZ+9tRzCYz3bp145VXXiEiIsIm7V0Pf39/5s+fzxtvvMG6deuIX3uCsNsa4e5rsEl7RVkFXNwQRVFGPv369WPy5Ml2nbnfr18//P39eeWVV8g5kYMxw4xXpBnFMfrNys2YDrkn3DAXQM+ePZk6dSre3t5al1Wmqpjn/3Ssp13jmfZTUkvTpk3t1uZdd93F1q1bSTx5kPodnWt1qsSTB4DizjtH4e3tTatWLTl0+DAFuTkYvK/vPUrMkT3AP9dojsBgMDB27Fheeukltv6wlzsf7+4UA21MRhN/rtiLTqdj3Lhxdqn5119/JTo6Gr9GwTbL7MoKahNOfEIWs2fP5tNPP7VJG8HBwbRv357du3djLgSdY6yEX2mmbDAXQO87e2u6opEQQgghXEu5O9br1q1ryzqEEEJUUqNGjfDx8SH/UvHmpoWpuahmlRtucL5l/mylpBPPx8fHKW4i2ZLkuagoRVF49dVXiY6OZu/evcT+VZu67buX++vzszM4suYr9DodH3/8MTVrOs6ymrbUq1cv9u3bR/SBHbTsdgdn9m0v/byrcHd359FHH6Vv377MmDGDVatW8f2MdXS6uy2tuzWx6uttxqUsflu6lUtxaYSGhvL666/Ts2dPqx3fFjw8PJg6dSphYWEsWrSI+LUnCbu1IYYg63bUFqTkkrAhCmNeEU888QTPPvusJlnXvXt3vv/+e8aOHcuJEycwZSp4NzWhd4KJkaoZ8s/pKDivQ6/T89JLYxgyZIhDv2eoinler149DAYDeQ7VsZ4K/LNPtz307NkTD4OBpKhDTtWxbioqJOXsCRo0iLT5fvQV1aVLFw4cOEDs0b9odFO3Sh/HWFjA+WMHiIyMJDQ01IoVXr/bb7+dLl26sH37dqIPnieyreMNSvuv/ZuOk5aYyaOPPmqXwSuZmZm8//77KHod1VqH27y9ivIM88Mr3I/ff/+djRs32mzbt1tuuYXdu3djTFfwqOG8K9AAGNOKc7xbt8r/XgshhBBC/Jd9NvsTQghhMzqdjhYtWlCUkY+5yERBSg6Aw+zr5wi8vLwAHHbWmRCOzt3dnRkzZhAaGsqZbetIi4su19eZTSaOrP2awtxsxo0b51DLotpayXKTMYeLZ66dPbQbcM0be9WrV+fdd99l9uzZBAZWY9uP+/h18VaKCoxWOf7Zw3F8/+EvXIpLY8CAAfz4448O36leQqfTMXbsWF599VVMeUVc/OUUeYlZVjt+XkIW8b+cxFxg4o033uC5557TtDO4Tp06fPXVVwwePBhTjkL2fjfyzyuoDnxf3pQL2Qf0FMTqqFWrFkuWLGHo0KEO3aleVbm5udG0aVN0eRlgdowlinV/d/Lbayl4KB4oekvXrmQnJ5CTeslu7V6v5LPHMRmL6NPndq1LuUJJZp/ev/26jnP++AGKCvIcMusVReGNN97AYDDw58q95OcUaF3SVaUmZPDX+iOEhobyzDPP2KXNadOmkZKSQrW24bj7OdZsdSj+PwzuXBdFr+Ptt98mPT3dJu20bdsWAFOW8+dgyffQpk0bjSsRQgghhCuRjnUhhHABTZs2BRUK0/MoSM3753MCKF7+EJDl34S4DkFBQcyYMQOdTsfRX76lKD/3ml9zdtcGMuJjuPPOO3n44YftUKXjqF+/PiEhNYg9ug+z2UTciQNE1K1LWFiY1qXZTLdu3Vj+/XI6dOhA9KHz/PjpBnKz8q7rmIf+OMm6RX+gV9yYNm0ab775pl32Dbe2Rx99lA8++ABMKgnro8iNz7juY+bGZZCwIQo9OmbMmMHAgQOtUOn1MxgMvPLKK8yfP5/qQcHkR+vJPqDHdO2XDLtSVcg/r5D9lxumLIX77ruPFctXyM13B9e8eXNQzSi51/87ZA1KbiqhYWF23+KjT58+ACRFWWdfcHtIPHUIKJ457WiaN29OcHAw0ft3oJrNlT7O6X3bgOIVPBxRnTp1ePbZZ8nNymfrj39pXY5FZrOZzd/uxGQ0M3HiRLvk/qpVq/jxxx8xVPcmsIXjvlfz8Pek2g01SU5OZvz48Ziv4+fVksjISABM1/cWziGY8hT8/f0JCQnRuhQhhBBCuJByLwUvhBDCcTVs2BCAwvR8CtPzcHNzc8g9Z7Xi4VG8OZxOJ+PJhLgebdq04bnnnuOjjz7ixKYfaHXnoxafmxF/jnN7NlOnTh0mTpxY5WZ/KopC+/Y3sm7dOmIO7SE/J5sbb79N67JsLjg4mHnz5vHuu++yfPlyfvp0I/eM7o1PgFeFj7Vvw1F2rjlIjRo1mD17ttMPGOvbty/e3t6MGTOGhI1nCLu1Id7h/pU6Vm58Bgmbz+Dh5s7MmTPp0qWLlau9fl26dOHHH3/kvffeY82aNWT/5YahrglDbRVF4zg2ZUPuKT2mLIXq1aszadIkp1kFoaorWZHJ/ehvmAM13lrEWIBSmEerljfbvenu3btjMBhIPHmQeh16O3zGGgsLSIk5SWRkZOl1iyPR6XT06NGD5cuXE3/6KLUat6rwMVSzmdN/bSWwWrXSGb+OaPDgwfz6668c3nuYhm3rUq9FLa1LusLB30+SeC6Ffv362WX2f1RUFFOmTEHnoSe0RySKzrF/nwJbhJKfkMWff/7JwoULeeKJJ6x6fF9fXzw9PSkqdP6edbVQoUbdGlqXIYQQQggXIz0MQgjhAurVqwdAUWY+xqwC6tSpg5ubjJ0qUTJT3RYj+oWoaoYOHUq7du1IOnWIS9HHynyO2WTk+IblKIrCu+++i4+Pj52rdAwlHUD71q8EoFWrit+od0bu7u5MnDiR//u//yMtKZOf522q8JKzB38/wc41B6lZsyZLly51+k71Et26dWP27Nm46fUkbjpDflJ2hY+Rl5hF4qbiTvW5c+c6ZKd6iYCAAKZOncrHH39cPHv9rJ7sfXqM6drUoxoh74yOrH3Fs9Tvvvtup9paQECHDh1KM0WXHq/tn+wUFEWhR48edj8P3t7e9OzZk5zUJLIvxdu9/Yq6dPoIZmMRd955p9alWFSyX/WpPX9U6usvRh8nK/USPXv0QK/XW7M0q9Lr9bz99tt4eHjw+7Ld5OcWal3SZdKSMtmz7hDVg6vzyiuv2Ly9jIwMnn3uOfLz86nRtZ5DLgH/X4qiUKNrfdx8PZg1axZ//FG5n9mrHd9gMKC6wqWzGTw9PbWuQgghhBAuRnpdhBDCBdSqVTzToDAtD1O+kdq1a2tckWORmepCWI9er+fNN99kwIABnNryM9UjGqP7z0Ce8/u3kZOaxKOPPurQs7ZsraQzOHr/DgCaNGmiZTl2pSgKzz//PCaTicWLF7Pmsy3cM7o37h7Xvvw49VcM237cR2hoKIsWLaJmTY1npVpZp06d+Pijj3nuuedI2HSaWnc1K/eN/KLMfBI3nUGv6Pnkk0+46aabbFytdfTu3ZsOHTowa9Ysvv32W7IPKriHmPGsb0Zf8cUMKkxVoTBBoSBGj7kQIiIimDBhAp06dbJ948KqwsLC2Lp1K0VFRVqXAhS/xyzZcsje+vXrxy+//EL80b00qeF4s47/Lf7oHhSleDCLo+rYsSM+vr5E7fmDHo+MrvAqAFF7/gT+6aB3ZA0aNODpp5/mo48+YusPe7n1UccYoGU2m9n0zU6MRSYmTZxEQECATdszmUy8/PLLXIiLo1rbmvhEVLNpe9ak93QjrGdD4tedYNy4cXzzzTfUr1/fasc3u0SvejFVVbUuQQghhBAuRnoahBDCBQQHB6PX68m/lAPg0nv4VoYjzxoRwhnVr1+fwYMHk5+ZRtyhHZc9ZizI49yezQQEBDJ69GiNKnQMDRo0uOzjkj0rqwpFURg7diz33XcfiedS2PDVdlTz1W9uxp9JYtM3O/H392fBggUu16leolu3bkyaNAlTvpGEjacxF5mu+TXmQhMJG09jKjAyefJkbr7Z/stPXw8/Pz/Gjx/PN998ww033EDRJR3Ze93Ii9ahGm3XblGaQvY+PXmn9HjoPXn22WdZuXKldKo7MTc3N7y8vBzij1ad6lC83UJoaCgJJ/ZhLKzYqiD2lJ2SSPqFs3Tp0sWhX9Pd3d3p3q0b6UnxXIo9U6GvVVWVqL1/4OPj4zSvLUOHDqV169ac2htD9KHzWpcDwIFNx0mMSebuu++2y0ois2fPZseOHfhEBFKtTbjN27M2Q3VvQm6uR05ODmPGjCE3N9dqx87Py0dxgUtoRY9Vz4sQQgghBEjHuhDCCTVp2oTg4GCty3AoOp2O6tWrYy4ovjMdEhKicUWOpaRj3dH3nxTCmTz++OP4+PgQu+8PzMZ/esXiDu2kqCCPxx//P/z9K7d/tKsIDg7m1Vdf5YEHHmDChAlVckl8RVGYOHEinTp14uzhOHb/csjiczNTs/ll0Z/oFB0ff/zxFQMTXM3999/P4MGDKUzPI3n3tTs1knfFUpiRz/Dhw+nXr58dKrSNFi1asGTJEqZNm0ZYaDgF53Vk7Xaj4IJi1WVnTTmQfVhHziE95hwd999/P2vXrGXkyJGadoYKYS1ubm489NBDGAvyuXhsr9blWHR+f/FM7oEDB2pcybWVzDaP2vtnhb4u5UIMqRfPc8sttzjN64ter+edd97BYDCwZdlucjK07XxMOp/K7l8OU6NGDV599VWbt/fnn3/y2Wef4e7vSUjXek57nehbP4iA5qGcOXOGt99+2yrHzM/Pp6ioCMXNBWZ6u6lkZmZqXYUQQgghXIx0rAshnM7X//uatWvXal2Gw6levXrpv4OCgjSsxPGULAUvy8AJYT3+/v48+OCDFGRncin6KACq2cyFQzvx8fHhwQcf1LhCx/Doo48yadIkHnroIa1L0Yy7uzszZsygbt26/LX+KGePxF3xHJPRxK+L/iQ/p4CJEyc6zRLn12vs2LE0b96crKhkcs6nW3xe9rk0ss6k0KpVK5577jn7FWgjiqLQt29fVq1axZgxY/Dy8CHvtJ7sv/QUpV1f54bZCLmndWT95YYxVUfHjh1ZtmwZU6ZMkYGHwuU89NBDeHp6EvvX5YPcHEV+ZhoJx/dRr149unfvrnU519S1a1c8PDyI2luxPatLOuKdYRn4f6tXrx7jxo0jP6eA9V9tx2zSZvnvgrxC1i/dhmpWee+992y+BHx2djZvTp6MolcI7dEAfTm2qXFk1dvXwhDiw6pVq/j999+v+3gZGRkAKM59WoDi7yEjI0PuAwghhBDCqqRjXQjhdDw8PPDyssOGnE7m3zNDbX0zwtmUzEBw1pkIQjiqBx54AICLx/cBkHbhLPlZ6dx1111Vcna2sMzf35+PP/4Yg8HA5m93kZuZd9nju9Ye4lJcGg888AD333+/RlXan7u7O++99x7u7u6k7D6P2Xhlp4bZaCZl93k8PDx47733cHNzgTvdfzMYDIwYMYJ169YxaNAg1Hw9OYf05BzTYa7gytYl+6hn73Gj8IKO+vXqMXv2bD777DOaNm1qm29ACI0FBgYyaNAg8rPSuXB4l9blXOHs7o2YTSZGjRpVOtDVkXl7e9OlSxeSzp0m49LFcn9d1N4/cXNzo2vXrjaszjYefPBBbr/9duJPJ7FzzUG7t6+aVTZ+vYOM5CyeeOIJOnToYPM2Z82aRVJiItVah2MI8rZ5e7am6HTUuLkeil7hrbfeuu6lz0tmeCvu1qhOW4qbitFoJC8v79pPFkIIIYQoJ8e/shFCCFEufn5+Zf5b/ENGqgthXXXr1qVp06akxUZhKirk0pnimet9+/bVuDLhiBo2bMjLL79Mfk4B237aV/r5S3GpHNxyonTmXFXToEEDBg8eTFFWARknkq54PONYIsacQoYOHUrdunU1qND2qlWrxvjx41m2bNll+68XXirfgDhzEeQc1ZF7sngf9bFjx7JixUq6desmg+qEyxsxYgR+fn6c3bWBonzH2Us4Kyme+KN7adSokVO9L+jRowcAp//aVq7nZ6UlkxB9gg4dOuDr62vDymxDURSmTJlC/fr1ObD5OMd3VWx/+eu1c80BYo5coEuXLowePdrm7aWmpvL98u9x9zcQ2DLM5u3Zi0egFwEtwkhMTGT16tXXdaySjnlX2WMdkI51IYQQQliVdKwLIYSL+PfsUJkpWja5uS6E9d1yyy2YTSbS42NIi43C29ubtm3bal2WcFAPPvggrVu3JmrfOZJiUwDYseoAqqoyYcKEKrsizciRI/Hz9yfjaOJls9bNRhMZRxMJDAzk8ccf17BC+2jSpAlLlixhypQpeLh7kntMT+4p3VX3XjdmQPZeN4wpOrp06cLqVasZPnw47u4uMNVOiHIIDAxk9OjRFOXncnqrY2yXpZrNnNz8A6gqr776Knq98/TQlSxZf3pf+TrWo/dvB/7pkHdGPj4+zJ49m4DAALYs201MGVu22MLB30+wf9NxGjRowLRp0+zyc7Jy5UqKCosIaBaKonetW6IBzWqg6BS+/vrr6xpQXlhYCICiOP+gdOXv/+KS70kIIYQQwhpc612kEEJUYd7e/yxjV1U7Jizp2LEjer2e++67T+tShHA5N954IwDR238jJzWJNm3aSIeWsEin0/HCCy8AsH/zcS7FpRJ3KoGbb77ZLsu/OipfX18eHjgQU14R2dEppZ/POp2CqcDII488clnOuzJFUbj//vtZ/v1yWrRoQeFFHbnHyu5cL0pTyDnshs7sxksvvcTcuXMJDQ21f9FCaGzgwIE0bdqU+CN7SI2N0roczh/YRsbFWO6++26ne20PDg6mZcuWnD9+gMJyrABwZv8OAKfYQ/5q6tSpw5zZc/A0ePLb0m2cP5lg0/aO7TzNth/3UaNGDebOnXvZtma2tGPHDlDAL7K6XdqzJzcvd7xrB3DmzBmSk5O1LschlAwNkAH2QgghhLAm6VgXQggX8e/O9Kpy8728+vXrx969e6vEbD8h7K1Vq1b4+PiQmXgegJtvvlnjioSja9++PY0bNybmSByHfj8JwKOPPqpxVdobNGgQOp2OrDOXd6zr9DoeeughDSvTRt26dVm0aBFdunShKEVH7nEd/56AZ8yE3CN63HTuzJo1i6FDhzrFHs5C2IK7uztvvfUWbm5uHPttGUV5OZrVkp18kTPbfiGoenWn3d6jW7dumE1Gzh3ee9XnGYsKOXfkLyIjI6lZs6adqrOd1q1bM3PmTHQ6PesW/s75k+XfZ74iju04zZbvdhMUFMRnn31m13N39uxZ3H0N6DycZxWFivCoVnxPICYmptLH8PT0BEA1uUBntKn4r5LvSQghhBDCGuTOgxBCuIh/XyzKheOV3NzctC5BCJfk7+/Phg0b+OWXX1i/fj1DhgzRuiTh4BRFoW/fvpiMZk7uPYufnx+dO3fWuizNhYSE0KFDB/ITszHmFFKUVUBBcg5dOnehenXXm1lXHl5eXsycOZObbrqJomQdhYnFN/lVE+SecANVYdasWXTt2lXjSoXQXtOmTXn22WcpyM7k2G/fo15tDwUbMRYWcGTt15hNRt55+22qVatm9xqsoeQ15eyh3Vd93oWThygqyOOWW26xR1l20alTJz6d9Sk6nZ61n//O2cPWXRb+4O8n2LKsuFN94cKFNGjQwKrHv5ac3BwUN9e9Faq4Fw8YyM7OrvQxSn5vzUVWKUlT5kIFnV6Hn5+f1qUIIYQQwoW47rtJIYSoYgwGQ5n/FkIIW/P19aVWrVqEhYXJUouiXPr06UNwcHUMBgMPPPCADH76W69evQDIjc8kLz7zss9VVQaDgXfffRdfP1/yz+hRjVBwXsGcB8OHD5dVMoT4l2HDhnHzzTeTfPY4Mbs327VtVVU5vv57clKTGDZsmFMPeGnevDmBgYGcPbT7qntVRx/cBUCXLl3sVZpddO7cmXlz52Hw8OSXxX9y6q+z131MVVXZ88thtv24j9DQUBYvXkzDhg2tUG3F1K9Xn6Ksguvag9yRFWXkA1C/fv1KHyM8PByDwYA5x7nf06sqmHMV6tWth17vmisUCCGEEEIb0rEuhBAu4t+d6R4eHhpWIoQQQlxdREQEmzdvYe/evYwdO1brchxGx44dAchLyCIvIQsonj1Y1YWFhfF/I/6vuFM9QaHwoh5/f39GjRqldWlCOBSdTsfUqVOpWbMm0Tt+49Lpo3ZrO2b3JpKiDtO+fXuef/55u7VrC3q9nk6dOpFx6SJpCZZnbJ87sheDpyft2rWzY3X2cdNNN/H555/j5+fHhv/t4Mi2qEofS1VVtv+0jz2/HqZOnTosWbLkujp+r0fLli1RjWZyYtI0ad+WTAVGcmPT8Q/wp3bt2pU+jl6vp3Xr1piyFcwFVizQzkyZoBqhTZs2WpcihBBCCBfjkB3rBQUFvPLKK9SsWRMvLy86duzI+vXrtS5LCCEc2r8706VjXTgCyXMhhKiY+vXr4+PjQ2FKLgUpuQQEBFzXzXFXMmDAAHQ6Hfln9JgL4d5778XLy0vrsqoEyXPnEhgYyKxZs/Dy9ubor9+SmXTB5m0mnjpI9I7fqFmzJh9++KFLrEJSskVJzOE9ZT6ek5FK0rnTtL/xRpddLaxVq1YsXrSY6tWr88fyPRzccqLCx1DNavHX/n6Shg0bsnTpUmrVqmWDastn+PDhuLu7k7rvAmajSbM6bCH98EVMBUZGPjHyun8H77rrLgAKLjjkbeNyKYgrrr3kexFCCCGEsBaHfIc0bNgwPvzwQx599FE++eQT9Ho9d955J1u3btW6NCGEcFju7u6l/9bpHPLlXVQxkudCCFExiqLQpEkTCtPzKMrMp3HjxrK9wt+qVatG8+bNSz+WJeDtR/Lc+TRu3JhpH3yAajJy6KdF5GfabnZuenwMx35dho+PD7Nnz3bafdX/q6Rj/dyRvWU+fu7IX5c9z1U1atSIJYuXEBYWyraf9lWoc101q/y+fA9Ht5+mefPmLFq0iODgYBtWe221atXi0UcfpSirgIRNZzAbzZrWYy0ZJ5JIP5JIzZo1efjhh6/7eHfeeSe1a9emIE6HMdMKBdpZ4SWFomQdN9xwAzfddJPW5QghhBDCxThcz8vu3bv59ttvee+995g2bRojR45k06ZN1K1bl3HjxmldnhBCOKx/d6wLoTXJcyGEqJw6deqU/jsiIkLDShxPs2bNSv/dtGlTDSupOiTPnVf37t159dVXKcjJ4sCPX1CUn2v1NnJSEzn082IUVD7++GNN9sy2lfDwcOrWrUvssf2YTcYrHi/pcK8K23XUrVuXL75YRGhocef6sZ1nyvV1O1Yf4NiO4k71zz77jMDAQNsWWk7PPfccvXv3Ji8+k4TNpzEVXvn/6yxUVSXjeBLJO2OpXr068+bNs8oKCl5eXkyePBmdoiP3sJtTda4XJSvkndDj6elZ/D3IpAMhhBBCWJnDvbtYvnw5er2ekSNHln7O09OT//u//2PHjh2cP39ew+qEEMJxucKSi8J1SJ4LIUTl/HuJ3Jo1a2pYieP59/kICgrSsJKqQ/LcuQ0aNIgRI0aQk5rEwZ+XYDIWWe3YBdmZHPhxEUX5ebz11lsu2cHcqVMnCnKzSTh78rLPq6rKuSN7qVatGo0aNdKoOvuqU6cOCxcupFq1avz+/W5iT8Rf9fmHt57iwObjNGjQgAULFuDv72+nSq/N3d2dadOm0aNHD/IuZBL38zHyLjpRz/HfjHlFJGw6Q/KuWKoFVeOLL76w6t71HTp04P333wezjpxDbhTEK6iq1Q5vdaoZ8mMUco7p8TR4MXfuXKueDyGEEEKIEg7XC7N//34aN258xZvuDh06AHDgwIHLZnGUKCgooKCgoPTjzEzne1MshBDXQ2asC0cieS6EEJXz72VyQ0JCNKzE8fx7tqMskW8fkufO7/nnnycpKYnVq1dz4McvCAi98v+rMpJjTpCfmcYLL7xAv379rHJMR9OpUye+++47tn6/kBr1/ulANxbkk5mSxB133FGlZsPWrVuXOXPmMHTYUNZ/uZ1mHSMp66XYZDRzZGsU1YOrM3/+fAICAuxf7DW4u7vz0Ucf8fnnnzNv3jzifz2Fb2R13Lyc45paVVWyz6Riyi+iU6dOvPXWW4SFhVm9nb59++Ll5cX48ePJjMqk6JIZvZ/Vm7EKY5qCKVshPDyc6dOn07p1a61LEkIIIYSLcriO9YsXLxIeHn7F50s+Fx9f9qjY9957j8mTJ9u0NiGEcGShoaGALBsrHIPkuRBCVE5kZGTpvxs0aKBhJY6n5D1Oq1atNK6k6pA8d346nY4pU6aQlpbGtm3bSI+LttqxBw8ezIgRI6x2PEdz00034eXlRczhPcQc3nPF4z169LB/URpr2bIlb056k9dff50Dm49bfJ6HhwcfffiRTTp7rcXNzY1Ro0Zxyy238Nprr3H2zFmtS6oQDw8PXn71VQYNGmTTAR7du3fnxx9/5M033+SPP/7AmG6zpq5b//79efnll/H19dW6FCGEEEK4MIfrWM/LyytzPyBPT8/Sx8vy2muvMXbs2NKPMzMzyxw5L4QQrqpFixasXbuWatWqaV2KEJLnQghRSe3atWP16tXodDp5/fuPDh06sHr1apnJb0eS567B3d2dOXPmEBUVhdFonf2kvby8XH7wT0BAAGvWrCEpKemKxzw9PV3++7ekX79+3HjjjaSlpVl8Tmho6GUrsDiyFi1asGLFCk6fPo3ZbNa6nHILCwujevXqdmkrJCSETz/9lOjoaPLz8+3SZkX5+/tLzgghhBDCLhyuY93Ly+uyJeNKlLxx8/LyKvPrDAZDmRf8QghRlciFpHAUkudCCFF5devW1boEhyXnxr4kz12HTqejSZMmWpfhdEJCQmQwTxlq1qxJzZo1tS7Datzd3WnWrJnWZTg0RVEuW1VHCCGEEKKqcrjNoMLDw7l48eIVny/5nCu9cRdCCCFcleS5EEII4fwkz4UQQgghhBBCiH84XMd627ZtOXXqFJmZmZd9fteuXaWPCyGEEMKxSZ4LIYQQzk/yXAghhBBCCCGE+IfDdaw/8MADmEwmFixYUPq5goICFi1aRMeOHWWZYyGEEMIJSJ4LIYQQzk/yXAghhBBCCCGE+IfD7bHesWNHHnzwQV577TWSkpJo2LAhS5YsISYmhoULF2pdnhBCCCHKQfJcCCGEcH6S50IIIYQQQgghxD8crmMdYOnSpUyYMIEvv/yStLQ0WrduzerVq+nWrZvWpQkhhBCinCTPhRBCCOcneS6EEEIIIYQQQhRTVFVVtS7CFjIzMwkICCAjIwN/f3+tyxFCCCEAyaeKkvMlhBDCUUlGlZ+cKyGEEI5KMkoIIYQQFeGQM9atoWS8QGZmpsaVCCGEEP8oySUXHddmdZLnQgghHJVkevlJngshhHBUkudCCCGEqAiX7VjPysoCoE6dOhpXIoQQQlwpKyuLgIAArctweJLnQgghHJ1k+rVJngshhHB0kudCCCGEKA+XXQrebDYTHx+Pn58fiqJoWktmZiZ16tTh/PnzsqTQf8i5sUzOjWVybsom58UyRzo3qqqSlZVFzZo10el0mtbiDCTPnYOcG8vk3Fgm56Zscl4sc7RzI5lefo6U5+B4P0uOQs6LZXJuLJNzY5mcm7I52nmRPBdCCCFERbjsjHWdTkft2rW1LuMy/v7+DvGG0RHJubFMzo1lcm7KJufFMkc5NzIKvvwkz52LnBvL5NxYJuembHJeLHOkcyOZXj6OmOfgWD9LjkTOi2VybiyTc2OZnJuyOdJ5kTwXQgghRHnJMDwhhBBCCCGEEEIIIYQQQgghhBDiKqRjXQghhBBCCCGEEEIIIYQQQgghhLgK6Vi3A4PBwKRJkzAYDFqX4nDk3Fgm58YyOTdlk/NimZwbYQ3yc2SZnBvL5NxYJuembHJeLJNzI6xFfpbKJufFMjk3lsm5sUzOTdnkvAghhBDCmSmqqqpaFyGEEEIIIYQQQgghhBBCCCGEEEI4KpmxLoQQQgghhBBCCCGEEEIIIYQQQlyFdKwLIYQQQgghhBBCCCGEEEIIIYQQVyEd60IIIYQQQgghhBBCCCGEEEIIIcRVSMe6EEIIIYQQQgghhBBCCCGEEEIIcRXSsS6EEEIIIYQQQgghhBBCCCGEEEJchXSsCyGEE8rNzaV///7873//07oUIYQQQlSS5LkQQgjhGiTThRBCCCGqBulYF0IIJ+Tt7c2GDRvIzc3VuhSHdMcdd/D111+Tl5endSlCCCGERZLnVyd5LoQQwllIplsmeS6EEEIIV6KoqqpqXURVoaoqmzdvpqCggK5du+Ln56d1SXaxb9++Cn9Nu3btbFCJ40lNTa3w1wQFBdmgEseVm5tLnTp1ePXVV3n55Ze1Lseh3HnnnYSFhfHFF19oXYrDadKkCVFRUfj6+nL//ffz2GOP0bt3bxRF0bo04QIkz8tP8twyyXNRQvLcMslzYUtVNc9BMt0SyfNrkzy/Osn0skmeCyGEEMKVSMe6jbz++uts376dzZs3A8UX7bfffjubNm1CVVUiIiLYuHEjkZGRGldqezqdrtxvllVVRVEUTCaTjatyDBU5NyWqyrn5t9DQUCZNmsTo0aO1LsWhREdH06dPHwYOHMioUaOoXbu21iU5lD179vDVV1+xbNkykpKSCAsL45FHHuHRRx+lbdu2WpcnnITk+T8kzy2TPC8fyfOySZ5fneS5sAbJ88tJppdN8rx8JM8tk0y3TPJcCCGEEK5COtZtpGnTptx77728//77AHz//fcMHDiQd955hzZt2vDkk0/So0cPvvzyS40rtb0lS5ZU+GuGDh1qg0ocz5tvvlnhC/dJkybZqBrHNXr0aE6cOMHGjRtlRPO/+Pn5YTQaKSwsBMDNzQ2DwXDZcxRFISMjQ4vyHIbZbGb9+vV89dVX/PTTT+Tk5NCsWTOGDBnCI488Ijc7xFVJnv9D8twyyfPykTwvm+R5+Uiei+sheX45yfSySZ6Xj+S5ZZLp1yZ5LoQQQghnJx3rNuLn58eHH37IE088AcCgQYM4fPgwR44cAeDdd99l7ty5nD9/XssyHZ7ZbCYuLo6wsDA8PDy0Lkdo5I8//mD06NEEBwfzxBNPUK9ePby8vK54XlVYnvDfhg0bVq4bGYsWLbJDNc4hPT2dJ598ku+//x4onpXSo0cPxowZw1133aVxdcIRSZ5bh+S5AMlzSyTPK07yXFSU5Ln1SKYLyXPLJNMrRvJcCCGEEM7ITesCXJWbmxsFBQVA8dJpGzduZMiQIaWPh4aGkpycrFV5TuPSpUvUr1+f9evX06tXL63LERrp0aNH6b///PPPKx6vSssT/tvixYu1LsFpbN26la+++orly5eTmppKy5YtGTJkCO7u7nzxxRfcc889vP7660yZMkXrUoWDkTy3DslzAZLnlkiel5/kuagsyXPrkUwXkueWSaaXj+S5EEIIIZyZdKzbSMuWLfnqq6949NFH+eGHH0hJSblstOW5c+cIDg7WsELn4cqLKuzbt6/CX1MVR33LaG5RGceOHeOrr77im2++ITY2lho1ajB06FAee+yxy/Zwe/755xk5ciSzZ8+WC3dxBclz65E8v5zkuRDlI3kurEHy3LpcNdMlz8tH8lxUhuS5EEIIIVyFdKzbyMSJE+nXr1/pxfnNN99Mz549Sx9fs2YNN910k1blCQfRvn37cu9JVpVHfVeF/fyuR1xcHPv37ycjIwOz2XzF4/+ejVNVtG3blsOHD2MwGLj33nuZM2cOffr0QafTlfn8nj178vnnn9u5SuEMJM9FeUiel4/k+dVJnl9J8lxYi+S5KA/J8/KRPL82yfTLSZ4LIYQQwpVIx7qN3Hbbbezbt4/169cTGBjIwIEDSx9LS0ujW7du3HvvvRpWKByBjPQW1yM/P5+hQ4eyYsUKzGYziqKUzh759w2hqnbRDhAYGMiCBQt48MEH8ff3v+bz7733Xs6ePWuHyoSzkTwX5SF5Lq6H5LllkufCWiTPRXlInovrJZleNslzIYQQQrgSRXXVNbyES0hMTCQ8PJwNGzbI/m3/YTabiYuLIywsDA8PD63Lsbn8/HxWrFjBvn37yhz1rSgKCxcu1Kg6bYwdO5ZZs2bx9ttv07lzZ3r06MGSJUsIDw/n448/Jj4+nqVLl9KyZUutS7Wr/Px8FixYQNu2benWrZvW5QghkDy/GslzyXPJ87JJngvhmCTTyyZ5LnkOkullkTwXQgghhKuRjnXh0OSi3bLExERq1qzJ+vXrXf7cnDt3jp49exITE0NgYCAZGRkEBQWRnp6OyWQiODgYX19foqOjtS7VriIiIujbty8LFiwgJSWFkJCQy35XevXqRZMmTZg7d67Gldqft7c3n3zyCU888YTWpQghkDy/GslzyXPJc8skz4VwPJLpZZM8lzwHyXRLJM+FEEII4UrK3sxGVJhOp8PNzY3CwsLSj/V6/VX/uLnJSvzi+lSVcTEvv/wyGRkZ7Ny5k1OnTqGqKt999x3Z2dm8//77eHl58euvv2pdpt0lJSXRoUMHALy8vADIyckpfXzAgAGsXLlSk9q01qJFC2JiYrQuQzghyXOhBclzyXPJ87JJnovKkjwXWpA8r9p5DpLplkieCyGEEMKVyJWjlUycOBFFUUovxks+FkJcv02bNjF69Gg6dOhAamoqUHzTwmAw8PLLL3P8+HFeeOEF1qxZo3Gl9hUaGkpKSgpQPAK8WrVqnDx5kn79+gGQmZlJfn6+liVq5p133uGRRx6hZ8+e3HrrrVqXI5yI5LkQtiN5XjbJc8skz0VlSZ4LYTuS55ZJppdN8lwIIYQQrkQ61q3kzTffvOrHonJ8fX2ZNGkSDRo00LoUoaHc3Fzq1asHgL+/P4qikJGRUfp4586deemllzSqTjsdO3Zk69atvPLKKwD069ePadOmER4ejtls5qOPPqJTp04aV6mNTz/9lKCgIPr06UP9+vWpX79+6YyBEoqi8NNPP2lUoXBUkue2IXkuQPLcEslzyyTPRWVJntuOZLqQPLdMMr1skudCCCGEcCXSsS7sKjY2ltjYWLp27Vr6uYMHDzJjxgwKCgoYNGgQ9913X+ljPj4+TJo0SYNKhSOJiIggLi4OADc3N2rVqsXOnTvp378/AMeOHcPT01PLEjXx3HPP8f3331NQUIDBYOCtt95ix44dPPbYYwBERkYyc+ZMjavUxqFDh1AUhYiICEwmE6dPn77iOTJrSYjKkzwXlSF5XjbJc8skz4WwPcl0UVGS55ZJppdN8lwIIYQQrkRRq8omUDa2dOnSSn3dkCFDrFyJY7vvvvvIzs5mw4YNACQmJtKsWTMKCwvx8/MjKSmJ77//vvSCTFiWmJhIeHg4GzZsoFevXlqXY1OjR49m586d7Nu3DyheyvG9995j2LBhmM1mvvzyS4YMGcLnn3+ucaXaM5vNHD58GL1eT9OmTWWvSCEqSPK8fCTPrUfyXPK8LJLnQlwfyfPyk0y3DslzyXNLJNOFEEIIIVyLdKxbiU6nq/DXKIqCyWSyQTWOq2bNmjz//POly2JNmzaNiRMncuTIEerXr0/fvn3Jzs5m+/btGlfq+KrShXtsbCx79uzh7rvvxmAwkJ+fzzPPPMOKFSvQ6/XcfffdzJw5E39/f61LFUI4Ocnz8pE8tx7Jc8lzIYT1SZ6Xn2S6dUieS54LIYQQQoiqQYZJWsnZs2e1LsEppKamUqNGjdKPV69eTffu3YmMjASgf//+jB8/XqvyhIOKiIggIiKi9GNPT08+//zzKjcC/o8//qjU13Xr1s3KlTiP33//nTVr1nDu3DkA6taty1133UX37t01rkw4Ksnz8pE8F5UheV5M8rziJM9FRUmel59kuqgoyfN/SKZXjOS5EEIIIVyBdKxbSd26dbUuwSmEhISUvoFOT09n586dTJ06tfRxo9GI0WjUqjyn4uvry6RJk2jQoIHWpQg76dGjR4X2HVNVtcrOvCksLGTQoEH8+OOPqKpKYGAgUPy6M2PGDO6//36++eYb3N3dtS1UOBzJ8/KRPLceyfOqR/K8/CTPRWVJnpefZLp1SJ5XTZLp5SN5LoQQQghXIh3rwq5uvfXW0iXBtmzZgtls5r777it9/NixY9SpU0e7AjUUGxtLbGwsXbt2Lf3cwYMHmTFjBgUFBQwaNOiyc+Xj48OkSZM0qNT2pkyZUuGvURSFCRMm2KAax7F582atS3AakydP5ocffuCll17ixRdfJDQ0FICkpCRmzJjBtGnTmDJlCm+99ZbGlQrhnCTPLZM8/4fkedkkz8tP8lwI25NML5vk+T8kzy2TTC8fyXMhhBBCuBLZY92GEhISWLhwIfv27SMjIwOz2XzZ44qisHHjRo2q00ZiYiL9+/dnx44deHh48P777/P8888DUFBQQK1atXjkkUeYOXOmxpXa33333Ud2djYbNmwAis9Vs2bNKCwsxM/Pj6SkJL7//nv69++vcaW2J3siiutVv359evTowaJFi8p8fNiwYWzZsoWYmBj7FiackuT5lSTPLZM8/4fkubhekufCmiTPyyaZXjbJ839InovrJXkuhBBCCFciM9Zt5NChQ/To0YO8vDyaNGnC4cOHad68Oenp6Vy4cIHIyMgqOeo7NDSUbdu2kZGRgZeXFx4eHqWPmc1mNm7cWCXPC8Du3btLb2AALF26lLy8PI4cOUL9+vXp27cv06dPrxIX7v+9ySVERV28eJGOHTtafLxjx458++23dqxIOCvJ87JJnlsmef4PyXNxvSTPhbVInlsmmV42yfN/SJ6L6yV5LoQQQghXIh3rNvLqq6/i6+vLgQMH8Pb2pkaNGnzyySf06tWL77//nqeeeor//e9/WpepmYCAgCs+5+XlRZs2bTSoxjGkpqZSo0aN0o9Xr15N9+7diYyMBKB///6MHz9eq/KEAxoxYsQ1n6MoCgsXLrRDNY6ldu3abNmyhVGjRpX5+O+//07t2rXtXJVwRpLnVyd5fiXJc1FRkueWSZ4La5E8vzbJ9MtJnovKkEwvm+S5EEIIIVyJdKzbyLZt2xg3bhwRERGkpqYC/4zyffDBB9m6dSsvv/wyv//+u5Zl2tzSpUsBeOyxx1AUpfTjaxkyZIgty3JIISEhnDt3DoD09HR27tzJ1KlTSx83Go0YjUatyhMOaNOmTSiKctnnTCYTFy9exGQyERISgo+Pj0bVaWvo0KFMmjSJwMBAxowZQ8OGDVEUhaioKD7++GO+//57Jk+erHWZwglInheTPC8/yXNRUZLnlkmeC2uRPP+HZHr5SJ6LypBML5vkuRBCCCFciXSs24jZbCY0NBSAwMBA9Hp96QU8QKtWrarECNVhw4ahKAoPP/wwHh4eDBs27JpfoyhKlbtoB7j11luZOXMm/v7+bNmyBbPZzH333Vf6+LFjx6rkEnxQvB/Xfy9O/0tRFM6cOWOnihyDpf3HioqKmD9/Ph9//DHr16+3b1EOYvz48Zw5c4YFCxbw2Wefle4LaDabUVWVoUOHygwTUS6S58Ukz8tP8twyyfOySZ5bJnkurEXy/B+S6eUjeW6Z5LllkullkzwXQgghhCuRjnUbqV+/PmfPngVAp9NRv359NmzYwEMPPQTA9u3bCQwM1LBC+yg5ByX7tJV8LK40depUTp06xUsvvYSHhwfTp0+nfv36ABQUFLBs2TIeeeQRjavURvfu3csc9X3u3Dm2bdtGy5YtueGGGzSqzvG4u7vzzDPPcOzYMZ555hnWrFmjdUl2p9frWbx4MWPHjmXt2rWls03q1q3LnXfeSevWrTWuUDgLyfNikuflJ3lumeR5xUieS54L65E8/4dkevlInlsmeV5xVT3TJc+FEEII4UoUVVVVrYtwRS+99BI///wzp06dAuCjjz7ixRdfpFevXqiqypYtW3jxxRf54IMPNK5UOJqMjAy8vLxKb3QA5OXlcerUKerUqUNQUJCG1TmegwcP0qdPH7766ituvfVWrctxKPPnz+ell14iKytL61LsLjY2lpCQELy8vMp8PC8vj0uXLhEREWHnyoSzkTwXlSV5XjGS55ZJnkuei+sneS4qS/K8YiTPr66qZrrkuRBCCCFciXSs20haWhrR0dG0bt0ad3d3VFXlnXfeYcWKFej1eu6++27Gjx9/2cWZEKJyJk2axOrVq/nrr7+0LsWhPPDAA/z5558kJiZqXYrd6fV6vvzyS4uzSL777jseeeQRTCaTnSsTzkbyXAj7kTwvm+S55Lm4fpLnQtiP5LllVTXTJc+FEEII4UpkKXgbqVatGjfeeGPpx4qi8MYbb/DGG29oWJX99erVq8JfoygKGzdutEE1jmXp0qUAPPbYYyiKUvrxtVS1ve3KIzQ0lGPHjmldht1NmTKlzM+np6fzxx9/sG/fPl599VU7V+UYrjVmrKioqHRfNyGuRvK8mOS5ZZLn1iN5fjnJc8lzYT2S5/+QTC+b5Ln1VNU8B8l0SyTPhRBCCOFKZMa6nUVHR1NQUECzZs20LsUuevToccXeW+WxefNmG1TjWHQ6HYqikJeXh4eHR7kuIhRFkRG8/5GSksLtt99OZmYmUVFRWpdjV5Z+ZqpVq0ZkZCSPP/44TzzxRKV+B51RZmYm6enpANSrV49PPvmEe++994rnpaenM378eA4dOkRsbKydqxSuQvK8fCTPyyZ5fiXJ8ytJnkueC9urankOkumWSJ5bR1XOc5BM/zfJcyGEEEK4KulYt5GZM2eyfft2vv3229LPDR8+vHTU8w033MDatWupUaOGViUKjZ07dw6AunXrXvbxtZQ8vyqxNKsiPT2dEydOUFhYyJdffsmgQYPsXJlwJJMnT7Y4Q+C/VFXl7bffZvz48TauSjg7yXNxLZLn5Sd5LspD8lzYguS5uBbJ8/KTPBflIXkuhBBCCFclHes20rp1a3r27Mknn3wCwK+//sodd9zBk08+SatWrXjjjTcYNGgQs2fP1rhSIRxfWbMqFEUpHfU9YsQImjZtqlF1wlHs2LGD7du3o6oq48aNY9CgQbRr1+6y5yiKgo+PDzfeeCPt27fXqFLhTCTPhbAeyXNRHpLnwhYkz4WwHslzUR6S50IIIYRwVbLHuo2cO3fusuXkli1bRv369Zk7dy4ACQkJfPnll1qV5xCysrLIyMjAbDZf8VhERIQGFQlHtWXLFq1LcAiVXRatqvw+de7cmc6dOwOQk5PDgAEDaNmypcZVCWcneX5tkueivCTPi0meX53kubAFyfPykUwX5SF5/g/JdMskz4UQQgjhqqRj3Ub+uxDAb7/9dtleQvXq1SMhIcHeZTmEuXPn8uGHHxIdHW3xOVVhnzJLy6ddjaIobNy40QbVCGdQr169Su3FVhV+n/5r0qRJl32ckZGBr68ver1eo4qEs5I8t0zyvJjkuagoyfPykzwX1iJ5fnWS6ZLnonIk08tH8lwIIYQQrkQ61m2kcePG/PDDD4waNYpff/2V+Ph47rjjjtLH4+LiCAwM1K5AjcybN4+nn36aPn36MGLECF5//XXGjBmDp6cnixcvJjQ0lOeee07rMu3CbDZX+AKsquzcULLXYUUNGTLEypU4li+++OKynxmz2cwnn3zCuXPnePTRR2nSpAkAJ06c4Ouvv6ZevXpV5vepLHv37uWNN97gjz/+oLCwkN9++41evXqRnJzM//3f/zFmzBh69OihdZnCwUmel03y/B+S55ZJnpdN8rxiJM+FNUieWyaZXkzy3DLJc8sk08tP8lwIIYQQrkL2WLeRb7/9lkceeYSAgABycnJo3LgxBw4cwM2teCxDjx498PLyYt26dRpXal8tWrQgIiKCdevWkZKSQkhICBs2bKBXr15kZGTQvn17Ro0axYsvvqh1qUJDOp3uis+VXKz+9yXr3xexVW3U9zvvvMOXX37Jtm3bqF69+mWPXbp0ia5duzJs2DBee+01jSrUzvbt2+nVqxe1atWid+/efP7556WvNVD8GhweHs4333yjcaXC0Umel03yXJSH5Hn5SJ5bJnkurEXy3DLJdHEtkuflJ5leNslzIYQQQriSK98dC6t4+OGH+fXXXxk2bBivv/46mzdvLr1oT01NJSgoiJEjR2pcpf2dOXOGfv36AeDu7g5AYWEhAAEBATz++OPMmTNHs/qEYzh79uxlf/bv30+rVq3o2rUry5Yt4+DBgxw8eJDvvvuOm2++mdatW7N//36ty7a7efPmMXLkyCsu2AFCQkJ44oknSveNrGrGjx9Ps2bNOHbsGO++++4Vj/fs2ZNdu3ZpUJlwNpLnZZM8F+UheV4+kueWSZ4La5E8t0wyXVyL5Hn5SaaXTfJcCCGEEK5EloK3odtuu43bbrvtis8HBQWxcuVKDSrSXkBAAEajEQB/f3+8vb05f/586eN+fn5Vem+7EllZWWRkZGA2m694LCIiQoOK7Ktu3bqXffzmm28SEhLCb7/9dtkI+FatWjFgwABuv/12PvroIxYtWmTvUjWVkpJCbm6uxcdzc3NJSUmxY0WOY8+ePbz33nsYDAays7OveLxWrVryWiPKTfL8SpLn5SN5LnleHpLnlkmeC2uSPC+bZPq1SZ5LnpeXZHrZJM+FEEII4Upkxrqwq5YtW3Lw4MHSjzt16sTcuXO5cOEC58+fZ/78+TRu3FjDCrU1d+5cGjVqRGBgIHXr1qV+/fpX/KmKfvzxR+6///4y97zT6XT079+fn376SYPKtNWpUyc+/vhj/vrrryse27t3L5988gkdO3bUoDLtubu7l3njq8SFCxfw9fW1Y0VCuBbJ86uTPC+b5HnZJM8tkzwXwvYk0y2TPC+b5LllkullkzwXQgghhCuRGes2dOjQIWbNmsW+ffvKHN2sKApnzpzRqDptDB48mHnz5lFQUIDBYGDy5MnceuutpaO83d3dWbFihcZVamPevHk8/fTT9OnThxEjRvD6668zZswYPD09Wbx4MaGhoTz33HNal6kJVVU5ceKExcePHTt2xd5uVcGnn35Kjx496NChA506daJRo0YAREVFsXPnToKCgpg1a5bGVWqjU6dOLF++nBdeeOGKx3Jycli0aBHdu3e3f2HCKUmeX0ny3DLJc8skz8smeW6Z5LmwJsnzskmml03y3DLJc8sk08smeS6EEEIIl6IKm9i8ebNqMBjUsLAw9e6771YVRVF79+6t3nzzzapOp1NbtWqlDhs2TOsyHcKZM2fUjz/+WJ01a5Z68uRJrcvRTPPmzdW+ffuqqqqqycnJqqIo6saNG1VVVdX09HS1YcOG6vTp07UsUTNDhw5V3dzc1BkzZqg5OTmln8/JyVGnT5+uurm5qUOHDtWuQA0lJCSoL7zwgtqkSRPV09NT9fT0VJs0aaKOGTNGvXjxotblaWbnzp2qwWBQ77zzTvXLL79UFUVRP/zwQ/Wzzz5TmzRponp7e6sHDx7UukzhBCTPy0/yvJjkuWWS55ZJnpdN8lxYi+R5xUimS55fjeT51UmmX0nyXAghhBCuRDrWbeSWW25RmzVrpmZkZKiXLl267CJs586darVq1dS1a9dqXKVwJAaDQZ09e7aqqqqakZGhKoqirlu3rvTxqVOnqg0aNNCqPE2lp6er3bp1UxVFUT08PNS6deuqdevWVT08PFRFUdSuXbuqaWlpWpcpHMzGjRvVxo0bq4qiXPanYcOG6pYtW7QuTzgJyXNRUZLnlkmei8qQPBfWIHkuKkry3DLJc1EZkudCCCGEcBWyFLyN7Nu3j8mTJ+Pv709aWhoAJpMJgI4dO/Lkk08yYcIE7rjjDi3L1FR2djZpaWllLhFWsuxcVRIQEIDRaATA398fb29vzp8/X/q4n58fCQkJWpWnqYCAAH7//Xd++ukn1q1bx7lz5wDo27cvd955J/369StzfzdRNamqSlZWFl26dOHkyZMcOHCAqKgozGYzkZGR3HjjjfLzIspN8vzaJM8vJ3lumeS5qAjJc2FNkuflI5n+D8lzyyTPRUVIngshhBDC1UjHuo24ubnh5+cHQGBgIO7u7iQlJZU+3qBBA44dO6ZVeZrJz89n8uTJLFy4kJSUFIvPK7nJUZW0bNmSgwcPln7cqVMn5s6dy5133onZbGb+/Pk0btxYwwq1d++993LvvfdqXYZDOX78OIsWLSI6OrrMm2CKorBx40aNqtNGYWEhQUFBvPvuu4wbN462bdvStm1brcsSTkryvGyS55ZJnl+b5PmVJM+vJHkurEny3DLJ9LJJnl+b5HnZJNMvJ3kuhBBCCFcjHes20rBhQ6KiooDiN81Nmzblhx9+4NFHHwVgzZo1hIWFaVmiJkaPHs2SJUu47777uOWWW6hWrZrWJTmMwYMHM2/ePAoKCjAYDEyePJlbb721dGaAu7s7K1as0LhK4Ui+/PJLhg8fjru7O02aNCnz96ms2SauzmAwEBYWhsFg0LoU4QIkz8smeW6Z5LmoKMnzskmeC2uSPLdMMr1skueiMiTTryR5LoQQQghXo6hV7R2dnUycOJEvvviCmJgY3NzcWLJkCcOHDycyMhKAM2fO8N577/HKK69oXKl9BQYGMnDgQObPn691KU4hOjqaVatWodfruf3226vsiHhVVVmwYAELFy4sHfX9X4qilC7VV1VERkYSFBTEunXrCA4O1roch/LGG2+wbt06duzYgYeHh9blCCcmeV42yfOKkTwvJnleNslzyyTPhbVInlsmmV5+kufFJM8tk0wvm+S5EEIIIVyJzFi3kQkTJvD888+j1+sBGDp0KHq9nhUrVqDX63n99dcZNmyYtkVqQFEU2rVrp3UZTqNBgwY8//zzWpehuXHjxvHhhx/Stm1bBg8eLLMo/hYfH89LL70kF+xlaNWqFT/++CMtWrRg2LBh1KtXDy8vryue179/fw2qE85E8rxskucVI3leTPK8bJLnlkmeC2uRPLdMMr38JM+LSZ5bJpleNslzIYQQQrgSmbEu7GrYsGHk5OTw/fffa12KQ8vOzi5zLy6gdOm5qqRGjRr06NGDZcuWaV2KQ+nYsSO33347b731ltalOBydTnfN5yiKUuX2ihTCWiTPy0fy/HKS52WTPLdM8lwI25NMvzbJ88tJnlsmmV42yXMhhBBCuBKZsW5jJpOJv/76i5iYGADq169Pu3btSkfKVzUTJkzgoYceYuTIkTz55JNERESUeS6CgoI0qE5b+fn5TJ48mYULF5KSkmLxeVXxQiMvL49bb71V6zIczocffsiDDz7IHXfcQZcuXbQux6Fs3rxZ6xKEi5E8v5zkuWWS55ZJnpdN8twyyXNhbZLnV5JML5vkuWWS55ZJppdN8lwIIYQQrkRmrNvQ4sWLee2110hKSiod2awoCiEhIbz77ruMGDFC4wrt79+jVBVFsfi8qnhxOmLECJYsWcJ9993HLbfcYnE5taFDh9q5Mu3dd999hISE8Nlnn2ldikO55557iIqK4tSpUzRv3rzMm2CKovDTTz9pVKEQrkHy/EqS55ZJnlsmeV42yXMh7EPyvGyS6WWTPLdM8twyyXQhhBBCCNcnHes2Mn/+fJ566inatm3Lk08+SePGjQE4efIk8+fP59ChQ8yePZtRo0ZpXKl9vfnmm1e9WC8xadIkO1TjWAIDAxk4cCDz58/XuhSHEx8fT58+fRg0aBBPPvkk1atX17okh1CvXr1r/j4pikJ0dLSdKnIcqampxMXF0bp16zIfP3z4MLVr15b9AMU1SZ6XTfLcMslzyyTPyyZ5bpnkubAWyXPLJNPLJnlumeS5ZZLpZZM8F0IIIYQrkY51G2nQoAF16tRhw4YNuLu7X/ZYUVERvXr14sKFC1XuzbSwrFq1akydOpUnn3xS61Icjp+fH2azmfz8fAA8PT3LHPWdkZGhRXnCAQ0dOpSTJ0+yc+fOMh/v0qULzZo1Y+HChXauTDgbyXNRUZLnlkmei4qSPBfWInkuKkry3DLJc1FRkudCCCGEcCWyx7qNJCQk8OKLL15x0Q7g7u7Oww8/zLhx4zSozLHk5eUB4OXlpXEl2rv33nvZsGGDXLiXYcCAAeWaRSFEiU2bNvHUU09ZfLxfv37MmzfPjhUJZyV5Xj6S5/+QPLdM8lxUlOS5sBbJ8/KTTC8meW6Z5LmoKMlzIYQQQrgS6Vi3kRtuuIFTp05ZfPzUqVO0bdvWfgU5kNjYWCZNmsTatWtJTk4GIDg4mLvuuotJkyZRt25djSvUxoQJE3jooYcYOXIkTz75ZJl7cQEEBQVpUJ22Fi9erHUJDi8rK4uMjAzMZvMVj0VERGhQkbYuXbpEcHCwxcerV69OUlKSHSsSzkry3DLJ87JJnlsmeX5tkueXkzwX1iJ5fnWS6VeSPLdM8rx8JNP/IXkuhBBCCFciHes2MmvWLO666y4aNGjAyJEjS0d75+XlMW/ePJYtW8batWs1rtL+Tpw4QdeuXUlPT+e2226jWbNmpZ9funQpq1atYuvWrTRp0kTjSu2vUaNGAOzfv/+qy1+ZTCZ7lSScwNy5c/nwww+vumxlVfyZCQ8PZ//+/RYf/+uvvwgJCbFjRcJZSZ6XTfLcMslzURmS52WTPBfWInlumWR62STPRWVJpl9J8lwIIYQQrkQ61q2kdevWV3xOr9czduxYxo0bR82aNQGIj4/HaDQSHh7OsGHDOHjwoL1L1dSrr76KTqdj//79tGrV6rLHjhw5Qu/evXn11Vf54YcfNKpQOxMnTpTl1K4hLi6O/fv3Wxz1PWTIEA2q0s68efN4+umn6dOnDyNGjOD1119nzJgxeHp6snjxYkJDQ3nuuee0LlMT9913H7Nnz+aOO+7gnnvuueyxn376iUWLFl11KTpRdUmel4/kuWWS59cmeX45yXPLJM9FZUmel59ketkkz69N8vxKkullkzwXQgghhCtRVFVVtS7CFfTo0aNSF12bN2+2QTWOq1q1arz44ou88cYbZT7+1ltv8eGHH5KWlmbnyoQjy8/PZ+jQoaxYsQKz2YyiKJS8dP37966qjfpu0aIFERERrFu3jpSUFEJCQtiwYQO9evUiIyOD9u3bM2rUKF588UWtS7W7jIwMunbtyrFjx2jTpg0tW7YEim8OHjx4kGbNmrF161YCAwO1LVQ4HMnz8pE8F5UheV42yXPLJM9FZUmel59kuqgoyXPLJNPLJnkuhBBCCFciM9atZMuWLVqX4BSKiopKl90ri7e3N0VFRXasyHHl5eUBXPV8VRXjx49n5cqVvPPOO3Tu3JkePXqwZMkSwsPD+fjjj4mPj2fp0qVal2l3Z86c4emnnwbA3d0dgMLCQgACAgJ4/PHHmTNnTpW7aIfi73/nzp188MEHrFy5kuXLlwMQGRnJhAkTePnll/Hx8dG4SuGIJM/LR/K8/CTP/yF5XjbJc8skz0VlSZ6Xn2R6+Uie/0Py3DLJ9LJJngshhBDClei0LkAUS05OpkGDBuzYsUPrUmzqhhtu4PPPPycjI+OKxzIzM1m4cCHt2rXToDLHEBsby/DhwwkNDcXX1xdfX19CQ0MZMWIE586d07o8zSxfvpzhw4fzyiuv0KJFCwBq1arFrbfeyurVqwkMDGT27NkaV2l/AQEBGI1GAPz9/fH29ub8+fOlj/v5+ZGQkKBVeZrz8fFh8uTJHD58mNzcXHJzczl8+DBvvvmmXLQLm5E8lzwHyXNLJM/LJnl+dZLnQgtVJc9BMv1qJM/LJnlumWS6ZZLnQgghhHAVMmPdQZhMJmJiYkpHQbuqyZMn07dvX5o2bcrw4cNp3LgxACdPnmTJkiWkpKRU2QuwEydO0LVrV9LT07ntttto1qxZ6eeXLl3KqlWr2Lp1K02aNNG4UvtLSkqiQ4cOwD8zBHJyckofHzBgAFOmTGHu3Lma1KeVli1bXrYPZKdOnZg7dy533nknZrOZ+fPnl/6OVWUXL14kKSmJhg0bygW7sDnJc8lzyXPLJM/LJnlePpLnwp6qSp6DZLolkueWSZ5bJpl+bZLnQgghhHB20rEu7KpXr16sXbuWl19+malTp172WNu2bfnyyy/p2bOnRtVp69VXX0Wn07F//35atWp12WNHjhyhd+/evPrqq/zwww8aVaid0NBQUlJSgOKlCKtVq8bJkyfp168fUDyTIj8/X8sSNTF48GDmzZtHQUEBBoOByZMnc+uttxIREQEULz23YsUKjavUzk8//cQrr7xCVFQUAOvXr6dXr14kJydz2223MWnSJO677z5tixTCSUmeWyZ5bpnkedkkz69O8lwI25JML5vkuWWS55ZJplsmeS6EEEIIV6GoqqpqXYSAxMREwsPD2bBhA7169dK6HLtISEgoXT6tbt26hIWF/X97dx4dVWG4ffy5CQQzmGCAiBD2sIUtIhBBQPZNkQYQrSLILtIUpISlWGWxVlzKLjthM5ZfbaVIICiEsLTigigGEhBEeCkRkLCF1Szz/uExmmYmmZCZuTOT7+ecnmPuvTnnqUf8enMnMyYvMldISIgmTpyoP/3pTzbPv/LKK5ozZ44uXbrk5mXme+KJJ3Tz5k1t3rxZkjR06FAlJiZqzpw5ys3N1cSJE9WiRQt9+OGHJi8134kTJ7R582b5+/urR48epfbV8Js3b1Z0dLTatm2rHj16aMaMGfn+/dqnTx/5+/tr06ZNJi+Fr6Hn9Jye20fPHUfPf0LPYZbS2HOJpv8aPbePnhcPTafnAADAt/Ab6zDNfffdV6pv1P9XVlZW3tuo2WKxWJSVleXGRZ5j3Lhxeu+99/Je9f3KK69o3759Gjx4sCQpPDxcCxYsMHmlZ6hbt67Gjx9v9gzTzZo1Sw8//LCSk5OVkZGhGTNm5Dvftm1bLVu2zJxxgI+h5/nRc/vouePo+U/oOeBeNP0X9Nw+el48NJ2eAwAA38KDdbjUunXrJEmDBw+WYRh5XxdlyJAhrpzlkVq0aKGVK1dq5MiRqlChQr5zV69e1apVq/TAAw+YtM5c7du3V/v27fO+rlGjhtLS0pSSkiJ/f381atRIZcrwrzP84tChQ5ozZ47d81WqVNH58+fduAjwbvTccfTcPnqO4qLngPPRdMfQc/voOYqLngMAAF/Cf+nCpYYOHSrDMPTb3/5WAQEBGjp0aJHfYxhGqbtpl6SZM2eqV69eatSokYYNG5b39mBHjx7V2rVrlZGRobffftvklZ7Dz89PkZGRZs9wKz8/PxmGUazvMQxD2dnZLlrkuSwWi65fv273/IkTJ1SpUiU3LgK8Gz13HD0vHnruGHpuGz0Hio+mO4aeF09p7LlE0x1FzwEAgC/hwTpc6rvvvpMkBQQE5PsaBXXp0kVbt27VpEmTNHv27Hzn7r//fq1fv16dO3c2aZ177dmz546+7+GHH3byEs/y8ssvF/umvbTq3Lmz1q5dqxdeeKHAubNnz2rFihXq06eP+4cBXoqeO46e/4Ke20bPHUfPAeej6Y6h57+g5/bRdMfQcwAA4EsMq9VqNXsEpEuXLql///6aM2eOWrRoYfYcmOzs2bM6deqUJKlWrVql7nPuivuqb6vVKsMwlJOT48JV8CZHjx5VmzZtVLt2bQ0cOFAvvfSSYmNjVbZsWS1btkxWq1X79+9X7dq1zZ4KH0PP8Wv0nJ6jZOg5zELP8Wv0nJ6jZOg5AADwJTxYd6Hz58/rzJkzunnzpu6++27Vq1dPFovF7Fmmqlu3rubNm6e+ffvaPJ+QkKBx48bpxIkTbl4GT7J79+47+r6OHTs6eYlnOn36tPz8/BQWFiZJunXrlhYvXlzguurVq+uJJ55w9zyPcfjwYY0fP17Jycn6deo6deqkt99+WxERESaugzeh5wXRcziCnheOnjuGnsNZ6LltNB1FoedFo+lFo+cAAMBX8FbwTpaRkaHZs2drw4YNSk9Pz3fOz89Pbdq00cSJExUdHW3OQJOdPHlS165ds3v+2rVrea8E93Xr1q2TJA0ePFiGYeR9XZTS8Nl2pekGvLhSUlLUokULzZs3TzExMZKk69evKzY2tsC1/v7+ioiIULNmzdw90yM0adJEO3bs0KVLl3T8+HHl5uaqbt26Cg0NNXsavAA9Lxw9/wU9t4+e20fPHUfPURL0vGg0/Sf03D56Xjia7hh6DgAAfAW/se5Ep06dUocOHZSenq6IiAhZLBalpqYqKytLw4cP1+XLl7V7926dPXtWo0aN0tKlS82e7HZ+fn6Kj4/XU089ZfP8xIkTtWbNGmVkZLh5mfv9/HZqN2/eVEBAgPz8/Ir8Ht5ODTExMUpMTNSxY8fy/pnJyMhQaGio4uPj9dBDD0mScnNz1alTJz322GNatGiRmZMBr0PPi0bPf0HPcSfoOeB69NwxNP0n9Bx3iqYDAACULvzGuhPFxsbq1q1bOnDggJo3by7pp/+YfvLJJ/Xdd98pMTFRubm5evPNNzVt2jS1atVKI0eONHm1682fP1/z58+X9NON5wsvvKAXX3yxwHVXrlzR5cuX9fTTT7t7oim+++47SVJAQEC+r/GL+Ph4Va5cWT179pQkZWZmatCgQQWuq1WrlhYuXOjueaZITk5W//79bf6gp0qVKqpVq1be108//bQ++OADd87zGElJSTpw4IAmTZqUdywuLk4zZszQ7du39fTTT+utt96Sv7+/iSvhqei5bfTcNnpeNHpeED13DD1HSdBz+2h6QfS8aPTcNppeNHoOAAB8CQ/WnSgpKUmxsbF5N+2SVKlSJb3++uuKiorS8ePHVa9ePU2ZMkUHDhzQ4sWLS8WN+7333qsmTZpI+ult5sLCwvI+d+pnhmGofPnyatmypcaOHWvGTLf79c2Vra9Lu61bt2rIkCH68MMP8479+OOPSkhIUJUqVXTXXXdJkqxWq7Zs2aJHHnlEvXv3Nmuu25w8eVKNGjXKd6xMmTKKjIxUUFBQvuN16tQpFW/baMuMGTPy/ZlKSUnRc889p+bNm6tevXpasGCB7rvvPk2ZMsXElfBU9Nw2em4bPS8cPbeNnjuGnqMk6Ll9NL0gel44em4fTS8aPQcAAL6EB+tOdPv2bQUHBxc4XqFCBVmtVp07d0716tWTJHXv3l0JCQnunmiKp556Ku9t5Tp37qw//elP6tq1q8mrPE/dunU1b9489e3b1+b5hIQEjRs3TidOnHDzMnOsW7dOUVFR6tatW4Fz8fHx6tKlS97XUVFRWrNmTam5cc/Nzc33dYUKFfTll18WuM4wDJXWT/tIS0vTgAED8r5ev369goODtXfvXlksFo0ZM0br1q3jxh020XPb6Llj6Hl+9Nw+el40eo6SoOf20fSi0fP86HnhaHrh6DkAAPAlRX9oFBzWokULvfPOOwU+YysuLk7+/v5q0KBB3rFr164pMDDQ3RNNl5yczA27HSdPntS1a9fsnr927VqpemXzJ598okceecSha/v27atPP/3UxYs8Q/Xq1XXw4EGHrj148KCqV6/u4kWe6fr16/l+kLpt2zb16tVLFotFktS6detS9ecJxUPPi0bP7aPn+dFz2+i5Y+g5SoKeO4am20bP86Pn9tH0otFzAADgS3iw7kTTp0/X/v371bhxY02dOlUzZ85Ujx499Prrr2vYsGEKDQ3Nu3bPnj1q1qyZiWvNsWHDBg0dOtTu+WHDhunvf/+7+wZ5GMMw7J77/PPPdc8997hvjMnOnj2rGjVq5DsWGBio8ePHq2bNmvmOh4WF6dy5c+6cZ5ru3bsrPj5e58+fL/S68+fPKz4+Xt27d3fTMs9So0YNff7555Kk48eP69ChQ+rRo0fe+YsXL6pcuXJmzYOHo+dFo+eFo+e/oOe20XPH0HOUBD13DE23j57/gp7bR9OLRs8BAIAv4cG6E3Xv3l3/+te/VKZMGb3xxhuaOXOm9u/fr8mTJ+vtt9/Od+2AAQP0xhtvmLTUPHPmzCn0P5YDAwM1d+5cNy4y1/z581W3bl3VrVtXhmHohRdeyPv61/+rVKmS5s2b5/ArxH3BXXfdVeA3BCwWi+bOnZv3lo0/u379ugICAtw5zzSxsbHKyspS165dtX//fpvX7N+/X926dVNWVpYmTpzo5oWeYdCgQVq+fLn69u2rnj17KiQkRL/5zW/yzn/xxRf5fksJ+DV6XjR6nh89t4+e20bPHUPPURL03DE0/Rf03D56bh9NLxo9BwAAvoTPWHeyPn36qE+fPrp06ZJu376tKlWq2HyV86BBg0xYZ76jR49q+PDhds9HRkbqb3/7mxsXmevee+9VkyZNJP30VnNhYWEKCwvLd41hGCpfvrxatmypsWPHmjHTFPXr19fevXsVExNT5LV79+5V/fr13bDKfLVr19aGDRv01FNP6cEHH1S9evXUtGlT3X333bp27ZoOHTqk48ePKzAwUO+++67q1Klj9mRTvPjii/rxxx+1detW1axZU2vWrMn7jZKLFy9q165dGj9+vLkj4dHoeeHoeX703D56bhs9dww9R0nR86LR9F/Qc/vouX00vWj0HAAA+BQrTPPf//7X7AluFxQUZH3ttdfsnn/ttdes5cuXd+Miz9GpUyfrjh07zJ7hMf785z9by5Yta923b1+h133yySfWMmXKWP/85z+7aZln+Pbbb62jR4+2hoWFWQ3DyPtftWrVrKNGjbIeO3bM7Ile5ccff7Tu3r3bevnyZbOnwAvR84LoOT3/GT0vHD13LnqOkiiNPbdaabo99Dw/el40mu489BwAAHgyw2q1Ws1+uO8r2rZtq+XLlxf52Wy5ubmaN2+eZs6cqStXrrhpnWfo2LGjLl++rM8//7zAW4Pdvn1brVu3VoUKFbR3716TFsJTZGZm6v7779cPP/ygl19+WYMGDVLVqlXzzn///feKj4/XK6+8osqVK+urr75SUFCQiYvNk5mZqatXryooKEjBwcFmz/FK586dU7Vq1bR9+3Z16dLF7DkwGT0vGj2Ho+i54+h5ydFz/Bo9dwxNhyPoefHQ9JKh5wAAwJPxGetOdPLkSbVs2VKTJ0/WzZs3bV7z6aefqmXLloqNjVXHjh3dvNB8U6dO1aFDh9S5c2dt3rxZJ06c0IkTJ/TBBx+oU6dOOnz4sKZOnWr2TFNs2LBBQ4cOtXt+2LBh+vvf/+6+QSYLCgrShx9+qFq1amny5MmqXr26KlWqpFq1aqlSpUqqXr26Jk+erBo1amjbtm2l+qY9KChIYWFh3LCXEK8zw8/oedHouX30PD967jh67hz0HD+j546h6bbR8/zoefHQ9JKj5wAAwFPxYN2Jjhw5ouHDh+uvf/2rmjRpoi1btuSdu3LlisaMGaN27drp4sWLev/99/XBBx+YuNYcvXv31qpVq3To0CFFR0erfv36ql+/vqKjo5WamqoVK1bo0UcfNXumKebMmaNy5crZPR8YGKi5c+e6cZH56tWrp6+++krx8fH67W9/q9q1aysgIEC1atXSk08+qXfeeUdfffVVqfr8NgCuR8+LRs/to+cF0XMAZqDnjqHpttHzgug5AAAAIPFW8C6wb98+jR49WqmpqerXr5+6d++u6dOn6+LFixo/frxmzpwpi8Vi9kxTXb16Vdu3b9e3334rSQoPD1ePHj1K9auaK1SooNdff11jxoyxeX7ZsmWaOnWqLl265OZlgO87d+6cqlatqh07dvBWc8hDz4tGzwui54B56DlsoeeOoen50XPAPPQcAAB4sjJmD/BFbdu21ZdffqmYmBgtX75cGzduVEREhHbs2KGmTZuaPc8jBAcHa8CAAWbP8ChWq1WXL1+2e/7SpUvKyspy3yAPdPv2bR04cEDnz59Xu3btVLlyZbMnAfBh9Lxo9Lwgel40eg7Anei5Y2h6fvS8aPQcAAAApRFvBe8C2dnZeu2117Ru3TqFhISoWrVq+uabb7RmzRpdv37d7HkeITMzU4cOHdLevXu1Z8+eAv8rjVq0aKG//e1v+vHHHwucu337tt599121aNHChGWeYcGCBapataratWun/v376+uvv5YkXbhwQZUrV1ZcXJzJCwH4GnpeNHpeED0vHD0H4G703DE0PT96Xjh6DgAAgFLLCqfas2ePNSIiwmoYhvXpp5+2njt3zpqZmWn9/e9/b/X397fWrFnTumnTJrNnmubChQvW3/72t9ayZcta/fz8rH5+flbDMAr8dWm0detWq5+fn/Whhx6yfvDBB9Zvv/3W+u2331o3bdpkbdOmjdXPz8+akJBg9kxTxMXFWQ3DsD711FPW1atXWw3DsCYlJeWdHzhwoLV79+4mLoS3O3v2bIF/rlC60fPC0XP76Ll99ByuRs/xv+h50Wi6bfTcPnoOV6PnAADAk/FW8E40YsQIrVmzRnXr1tVHH32kbt265Z1bsGCBhgwZotGjR6tfv37q27evFi1apLCwMBMXu9+oUaO0efNmjRs3Th06dFBISIjZkzxG7969tWrVKo0fP17R0dF5x61Wq4KCgrRixQo9+uij5g000V//+lf95je/0bvvvquMjIwC51u2bKkFCxaYsAyAL6LnRaPn9tFz++g5AHei546h6bbRc/voOQAAAEozHqw70TvvvKM//vGPeumll1SuXLkC51u1aqX9+/dr/vz5mj59uho3bqwrV66YsNQ8H330kSZMmKA33njD7CkeaejQoerfv7+2b9+ub7/9VpIUHh6uHj16KCgoyOR15jl+/LjGjRtn93zFihVt3tCjdLpx44aeeeYZDRgwQIMGDXLoeypUqKDVq1erSZMmLl4Hb0DPi0bPC0fPbaPnKA56jpKi546h6fbRc9voOYqDngMAAF/Dg3Un+uqrrxQREVHoNX5+fpowYYIef/zxQm9EfJXFYlHt2rXNnuHRgoODNWDAALNneJR77rlHFy5csHs+NTVV9913nxsXwZNZLBbt2LFDvXv3dvh77rrrLj377LMuXAVvQs+LRs+LRs8LoucoDnqOkqLnjqHphaPnBdFzFAc9BwAAvsawWq1Ws0f4ovT0dG3btk1paWm6evWqgoKC1LhxY/Xq1UvVqlUze55p/vCHPyglJUXbt283e4rHyszM1KlTp3Tp0iXZ+uP58MMPm7DKXMOHD9fOnTv11VdfKScnR6GhodqxY4e6dOmiw4cP68EHH9Tw4cN5uznkeeSRR3TfffcpLi7O7CnwcvTcNnpeNHpeED1HcdFzOAs9t4+mF46eF0TPUVz0HAAA+BIerDvZzZs3FRsbq5UrVyo7O7vAjVfZsmU1cuRIvfXWWwoMDDRppXk+/vhj/f73v1doaKhGjx6tGjVqyN/fv8B1DzzwgAnrzJWRkaGYmBj985//VE5OjqSfPr/NMIx8f/3zudIkPT1dDz74oKxWqx577DEtX75czzzzjHJycvTPf/5TVatW1WeffabKlSubPRUe4sSJE+rZs6eefPJJjRkzRtWrVzd7ErwMPS8cPbePnttHz1Fc9BwlRc+LRtNto+f20XMUFz0HAAC+hAfrTpSdna0ePXpo165d6ty5s4YMGaLIyEgFBQUpMzNTBw8e1Lp165ScnKzOnTvro48+snnD6sv8/Pzy/vrnG9JfK803p/3799fmzZs1btw4dejQQSEhITav69ixo5uXeYbz589r2rRpev/993X58mVJUlBQkAYMGKDZs2fr3nvvNXcgPEpQUJCys7P1448/SpLKlClT4LM1DcMolZ+jiaLR86LRc/voeeHoOYqDnqMk6LljaLpt9Lxw9BzFQc8BAIAv4cG6Ey1cuFDjx4/X22+/reeff97udUuXLtXYsWO1YMECxcTEuHGh+dauXevQdaXxs5TuvvtujR07Vm+88YbZUzzK7du39eGHH6p27dpq3ry5JOmHH35Qbm6uQkND8/0gCPjZ0KFDbf5g8H+tXr3aDWvgbeh50ei5ffTcNnqOO0HPURL03DE03TZ6bhs9x52g5wAAwJfwYN2JoqKiFBYWpo0bNxZ5bXR0tM6cOaPPP//cDcvgDe69917NmDFDY8eONXuKR7Farbrrrrs0f/58jRkzxuw5AEoBeo6SoOe20XMA7kbPURL03DZ6DgAAgNKOl5I6UVpamnr16uXQtb169dKRI0dcvAje5JlnnnHohz6ljWEYql+/vi5cuGD2FAClBD1HSdBz2+g5AHej5ygJem4bPQcAAEBpV8bsAb7EMAzl5uYW6/rSZvjw4UVeYxiGVq1a5YY1nuXxxx/X7t271atXL40ePVo1atSw+Rl/DzzwgAnrzDVt2jT94Q9/0MCBA9WwYUOz58CL/Pe//9WXX36pK1eu2Pz385AhQ0xYBU9Hz4tGz+2j5/bRc9wpeo47Qc8dQ9Nto+f20XPcKXoOAAB8AW8F70RRUVGqWrWqNm3aVOS10dHRSk9P12effeaGZZ6jdu3aBX5gkZOTo++//145OTkKDQ1V+fLldeLECZMWmufXn0Vm64c6VqtVhmEoJyfHnbM8wrhx45SUlKRvvvlGnTp1Uu3atRUYGJjvGsMwNH/+fJMWwtPcunVLzz77rP75z38qNzdXhmHo59z9+s9XafzzhKLR86LRc/vouX30HMVFz1ES9NwxNN02em4fPUdx0XMAAOBL+I11J3rmmWc0YcIELVmyRM8//7zd65YuXarNmzdr7ty5blznGU6ePGnzeFZWlpYtW6Z58+Zp+/bt7h3lIVavXm32BI+1aNGivL9OSkqyeQ037vi1adOm6f3339err76qtm3bqlOnTlq7dq2qVq2qefPmKT09XevWrTN7JjwUPS8aPbePnttHz1Fc9BwlQc8dQ9Nto+f20XMUFz0HAAC+hN9Yd6Ls7Gx169ZNe/fuVdeuXTV48GBFRkYqKChImZmZ+vrrr7V+/Xrt2LFD7du3V1JSksqU4bUNvzZ27FidOnVKW7ZsMXsKAC9Ws2ZN9erVS8uXL1dGRoZCQ0O1Y8cOdenSRZLUpUsXNWzYUEuWLDF5KTwRPS85eg7AGeg5SoKeOwdNB1BS9BwAAPgS7hqdqEyZMtq6dasmTJiguLi4Aq/ctVqt8vf314gRIzR37lxu2m2IjIzU+vXrzZ4BD3Xo0CFt3bo177cq6tSpo969e6tp06bmDoPHOX/+vKKioiQp720Jr1+/nnd+wIABmjVrFjfusImelxw9R2HoORxFz1ES9Nw5aDrsoedwFD0HAAC+hDtHJ7NYLFq2bJlefvllJSYmKjU1VZmZmQoKClJERIR69+6t6tWrmz3TY23fvl0Wi8XsGaYYPnx4kdcYhqFVq1a5YY1nuX37tp577jmtX79eVqs17/PucnNzNXXqVA0aNEgrV65UQECAyUvhKapUqaKMjAxJP/17OSQkREePHtVjjz0mSbp69apu3bpl5kR4OHpeMvS8cPScnsMx9BwlRc9LrrQ2nZ7bR89RXPQcAAD4Eh6su0hYWJhGjhyZ9/WRI0f03nvv6S9/+YsaNWqkoUOHKjg42MSF5pg1a5bN45cvX9aePXt04MABTZ061c2rPMPOnTtlGEa+Yzk5Ofr++++Vk5Oj0NBQlS9f3qR15poyZYrWrVunsWPH6ve//73Cw8NlGIaOHz+uBQsWaMmSJapYsaLmzZtn9lR4iAcffFD//ve/NWXKFEnSY489pjfffFNVq1ZVbm6u5s6dqzZt2pi8Et6AnttGz+2j5/bRcxQXPYez0HP7aLpt9Nw+eo7ioucAAMCX8BnrTrRo0SItWLBAH3/8sSpXrpx3PCEhQY8//riysrL089/uunXr6pNPPsl3XWnw8yuZ/1dISIjCw8M1cuRIjRo1qsANbGmWlZWlZcuWad68edq+fbvq1Klj9iS3q1y5sh599FGtXbvW5vnBgwcrMTFRFy5ccPMyeKp///vfeu+99/TGG2+oXLlyOn36tLp166Zjx45JksLDw5WQkKCGDRuavBSeiJ4XjZ4XHz2n5yg+eo6SoOeOoenFQ8/pOYqPngMAAF/Cg3Un6tGjh/z9/ZWYmJh3LDs7W2FhYbp27ZoWL16sVq1aacuWLXrxxRcVExOjuXPnmrgY3mTs2LE6deqUtmzZYvYUt6tQoYJmz56t559/3ub5JUuW6I9//KMuX77s3mHwKrm5uUpJSZG/v78aNWrE52jCLnoOV6Ln9BwlQ8/hKHoOV6Ln9BwlQ88BAIC3sv3SZNyR1NTUAm9dlJycrB9++EETJkzQs88+qyZNmmjy5Ml64okntHXrVpOWuk/FihX1j3/8I+/rWbNm6dChQyYu8l6RkZHas2eP2TNM0bNnT3344Yd2z2/btk09evRw4yJ4Iz8/P0VGRqpp06bctKNQ9Lwgeu489Jyeo2ToORxFz22j6c5Bz+k5SoaeAwAAb8V/uThRRkaGatSoke9YUlKSDMNQv3798h1v166d3n//fXfOM8W1a9d048aNvK9nzJihevXqqWnTpiau8k7bt2+XxWIxe4YpXnnlFT3xxBPq37+/fve736levXqSpGPHjuntt9/WqVOn9H//93+6ePFivu+rWLGiGXNhgjv9odbDDz/s5CXwBfS8IHruPPScnsM+eg5noue20XTnoOf0HPbRcwAA4Mt4sO5EVapU0dmzZ/Md27t3rywWiyIjI/MdDwgIUEBAgDvnmSI8PFz/+Mc/1KFDBwUHB0uSrl+/XuAG63+VxhuuWbNm2Tx++fJl7dmzRwcOHNDUqVPdvMozRERESJJSUlK0adOmfOd+/jSLxo0bF/i+nJwc14+DR+jUqVOxPvfRarXKMAz+GYFN9Lwgeu44em4fPUdR6DmciZ7bRtMdQ8/to+coCj0HAAC+jM9Yd6LHH39cKSkp2r9/v4KCgnT48GHdf//9+s1vfpPvrdYkKTY2VomJiTp8+LBJa91j/fr1GjZsmIr7j1lp/I9pPz/bn8wQEhKi8PBwjRw5UqNGjSrWzYmvmDFjxh39/54+fboL1sAT7d69+46+r2PHjk5eAl9Azwui546j5/bRcxSFnsOZ6LltNN0x9Nw+eo6i0HMAAODLeLDuRCkpKWrdurXuueceNWnSRF988YVu3Lihffv2qWXLlvmuDQ8PV5cuXbRixQqT1rrP0aNHtWvXLp07d04zZsxQv3791Lx580K/hxsuAIBZ6Llt9BwA4E3ouX00HQAAAADuDG8F70TNmjXTzp079eqrr+rEiRNq06aNYmNjC9y079q1SxaLRQMHDjRpqXs1bNhQDRs2lCStXr1azz77rPr27WvyKvNVrFhRy5cv1+OPPy7pp7ea69+/P59tBwAmo+e20XPb6DkAeCZ6bh9NL4ieAwAAAHAEv7EOj3bhwgVFRUUpPj5ebdu2NXuOUwUEBGjlypUaMmSIpJ/eau6dd97R008/bfIywPsNHz68yGsMw9CqVavcsAYAPQdwJ+g54Hl8ten0HHAdeg4AAHwJv7EOj5aTk6OTJ0/q5s2bZk9xuvDwcP3jH/9Qhw4dFBwcLEm6fv26Ll68WOj3VaxY0R3zAK+2c+fOAp/7l5OTo++//145OTkKDQ1V+fLlTVoHlD70vCB6DhSNngOex1ebTs8B16HnAADAl/Ab6/Bo586dU9WqVbVjxw516dLF7DlOtX79eg0bNkzF/SOYk5PjokWA78vKytKyZcs0b948bd++XXXq1DF7ElAq0POC6Dlw5+g5YB5fbTo9B9yPngMAAG/Eb6wDJhk8eLCioqK0a9cunTt3TjNmzFC/fv3UvHlzs6cBPqts2bKKiYlRamqqYmJitGXLFrMnAfBy9BxwP3oOwNnoOeB+9BwAAHgjHqwDJmrYsKEaNmwoSVq9erWeffZZ9e3b1+RVgO+LjIzU+vXrzZ4BwEfQc8Ac9ByAM9FzwBz0HAAAeBM/swcA+Ml3331XrJv2CxcuqG7dutq3b58LVwG+afv27bJYLGbPAOCD6DngPvQcgKvQc8B96DkAAPAm/MY64KVycnJ08uRJ3bx50+wpgMeZNWuWzeOXL1/Wnj17dODAAU2dOtXNqwCgIHoO2EfPAXgLeg7YR88BAIAv4cE6AMDnzJgxw+bxkJAQhYeHa+nSpRo1apR7RwEAgGKh5wAAeD96DgAAfAkP1uHRAgIC1LFjR4WEhJg9BYAXyc3NNXsCgF+h5wDuBD0HPA9NB1Bc9BwAAPgSHqzD7c6fP68zZ87o5s2buvvuu1WvXj27n6UUEhKi5ORkNy8EAABFoecAAPgGmg4AAAAAjuHBOtwiIyNDs2fP1oYNG5Senp7vnJ+fn9q0aaOJEycqOjranIEAvNr/+3//746+r2bNmk5eAvg2eg7Aleg54D40HYCr0HMAAODLeLAOlzt16pQ6dOig9PR0RUREqFq1akpNTVVWVpaGDx+uy5cva/fu3RowYIBGjRqlpUuXmj0ZgJepXbu2DMMo9vfl5OS4YA3gm+g5AFej54B70HQArkTPAQCAL+PBOlwuNjZWt27d0oEDB9S8eXNJP706/sknn9R3332nxMRE5ebm6s0339S0adPUqlUrjRw50uTVALxJXFxcvhv33NxczZ8/X6dOndKgQYPUsGFDSdKRI0f07rvvqnbt2ho3bpxZcwGvRM8BuBo9B9yDpgNwJXoOAAB8GQ/W4XJJSUmKjY3Nu2GXpEqVKun1119XVFSUjh8/rnr16mnKlCk6cOCAFi9ezE27AwICAtSxY0eFhISYPQUw3dChQ/N9/eqrr+rWrVs6fvy4KlWqlO/cjBkz1L59e509e9aNCwHvR89dg54Dv6DngHvQdOej58Av6DkAAPBlfmYPgO+7ffu2goODCxyvUKGCrFarzp07l3ese/fuOnr0qDvneZTz58/ryy+/1Mcff6yvv/5aN27csHttSEiIkpOT1aJFCzcuBLzD0qVLNXr06AI37ZIUGhqqUaNGacmSJSYsA7wXPXccPQecg54DrkHTHUPPAeeg5wAAwJfwYB0u16JFC73zzjsFPispLi5O/v7+atCgQd6xa9euKTAw0N0TTZWRkaFJkyapRo0aqlq1qlq1aqUOHTqoRYsWqlChgjp06KB//etfZs8EvEpGRkahP/i6ceOGMjIy3LgI8H70vHD0HHA+eg64Bk23j54DzkfPAQCALzGsVqvV7BHwbdu3b1fv3r0VHh6ufv36KTAwUP/5z3+UlJSkESNGaPny5XnX9u/fX5cuXVJycrKJi93n1KlT6tChg9LT0xURESGLxaLU1FRlZWVp+PDhunz5snbv3q2zZ89q1KhRWrp0qdmTAa/QpUsXff311/rwww/VsmXLfOf279+vnj17KjIyUjt37jRpIeB96Ll99BxwDXoOuAZNt42eA65BzwEAgC/hwTrcIiEhQVOmTFFaWpok6Z577tFzzz2nWbNmqWzZsnnXxcfHq0GDBmrdurVZU91q4MCB2r17t3bs2JH3+XYZGRl68sknVbZsWSUmJio3N1dvvvmmpk2bpmXLlvHZdoADUlNT1alTJ2VkZKhNmzaqX7++JOnYsWP65JNPVLFiRe3atUtNmjQxeSngXei5bfQccA16DrgOTS+IngOuQc8BAIAv4cE63OrSpUu6ffu2qlSpIsMwzJ5juooVKyo2NlbTpk3Ld/yLL75QVFSUjh49qnr16kmSnnzySR07dkwHDhwwYyrgdc6dO6fZs2crMTFRp06dkiTVqlVLjzzyiCZPnqz77rvP5IWA96Ln+dFzwHXoOeBaNP0X9BxwHXoOAAB8RRmzB6B0CQkJKfKaM2fOKCwszA1rzHf79m0FBwcXOF6hQgVZrVadO3cu78a9e/fuSkhIcPdEwGtVqVJFc+fO1dy5c82eAvgcep4fPQdch54DrkXTf0HPAdeh5wAAwFf4mT0Avq9t27ZKSUkp8rrc3FzNmTNHjRs3dsMqz9CiRQu98847ysnJyXc8Li5O/v7+atCgQd6xa9euKTAw0N0TAQCQRM8LQ88BAN6EpttGzwEAAAAUhd9Yh8udPHlSLVu21AsvvKCZM2favPn89NNPNWbMGB08eFB9+vQxYaU5pk+frt69e6tx48bq16+fAgMD9Z///EdJSUkaMWKEQkND867ds2ePmjVrZuJawLukpaVp9erVOnHihC5duqT//eQTwzCUlJRk0jrA+9Bz++g54Dr0HHA+mm4bPQdch54DAABfwWesw+WuXLmiKVOmaMWKFapVq5YWLlyoRx99NN+5lStXKiwsTPPnz1d0dLS5g90sISFBU6ZMUVpamiTpnnvu0XPPPadZs2apbNmyedfFx8erQYMGat26tVlTAa+xfv16DRs2TGXLllXDhg3tvsVlcnKym5cB3oueF46eA85HzwHXoOn20XPA+eg5AADwJTxYh9vs27dPo0ePVmpqqvr166fu3btr+vTpunjxosaPH6+ZM2fKYrGYPdM0ly5d0u3bt1WlShUZhmH2HMCrhYeHq2LFikpMTFTlypXNngP4FHpeOHoOOA89B1yLpttHzwHnoecAAMCX8GAdbpWdna2YmBgtX75chmEoIiJCGzZsUNOmTc2e5hXOnDmjsLAws2cAHi8wMFBz5szR888/b/YUwCfR85Kh54Bj6DngejT9ztFzwDH0HAAA+BI/sweg9MjOztZrr72mdevWKSQkRNWqVdM333yjNWvW6Pr162bPM0Xbtm2VkpJS5HW5ubmaM2eOGjdu7IZVgPdr3ry50tPTzZ4B+CR6XhA9B1yDngOuRdPzo+eAa9BzAADgS3iwDrfYu3evmjdvrunTp6tfv35KS0tTWlqaxowZo3nz5qlx48b64IMPzJ7pdidPnlTLli01efJk3bx50+Y1n376qVq2bKnY2Fh17NjRzQsB7zRnzhytWrVKH3/8sdlTAJ9Cz22j54Br0HPAdWh6QfQccA16DgAAfAlvBQ+XGzFihNasWaO6detqyZIl6tatW77z+/fv1+jRo3Xw4EH17dtXixYtKjVvp3blyhVNmTJFK1asUK1atbRw4UI9+uij+c6tXLlSYWFhmj9/vqKjo80dDHiJvn376tixY/rmm2/UuHFj1axZU/7+/vmuMQxDmzZtMmkh4H3ouX30HHANeg64Bk23jZ4DrkHPAQCAL+HBOlyuXLlymjRpkl566SWVK1fO5jW5ubmaP3++pk+fLsMwdOXKFTevNNe+ffs0evRopaamql+/furevbumT5+uixcvavz48Zo5c6YsFovZMwGvUbt2bRmGUeg1hmHoxIkTbloEeD96XjR6DjgXPQdcg6YXjp4DzkXPAQCAL+HBOlwuLS1NERERDl17+vRpjRs3Ths3bnTxKs+TnZ2tmJgYLV++XIZhKCIiQhs2bFDTpk3NngYAAD13ED0HAHg6ml40eg4AAADAFh6sw63S09O1bds2paWl6erVqwoKClLjxo3Vq1cvVatWzex5psnOztZrr72m1157TYGBgbJYLDp37pzGjRunmTNnqnz58mZPBAAgDz23jZ4DALwNTS+IngMAAACwhwfrcIubN28qNjZWK1euVHZ2tv73H7uyZctq5MiReuuttxQYGGjSSnPs3btXzz33nI4cOaKnnnpKc+fOlcVi0bRp07R48WKFhYVp4cKF6tu3r9lTAa+UmZmpK1euKDc3t8C5mjVrmrAI8F703D56DrgWPQeci6bbRs8B16LnAADA2/FgHS6XnZ2tHj16aNeuXercubOGDBmiyMhIBQUFKTMzUwcPHtS6deuUnJyszp0766OPPpK/v7/Zs91ixIgRWrNmjerWraslS5aoW7du+c7v379fo0eP1sGDB9W3b18tWrRIYWFhJq0FvMuSJUs0Z86cQj+nLScnx42LAO9Gz+2j54Dr0HPA+Wi6bfQccB16DgAAfAUP1uFyCxcu1Pjx4/X222/r+eeft3vd0qVLNXbsWC1YsEAxMTFuXGiecuXKadKkSXrppZdUrlw5m9fk5uZq/vz5mj59ugzD0JUrV9y8EvA+P//7pGfPnnr44Yf14osvasKECbrrrru0Zs0aValSRePGjdPQoUPNngp4DXpuHz0HXIOeA65B022j54Br0HMAAOBLeLAOl4uKilJYWJg2btxY5LXR0dE6c+aMPv/8czcsM19aWpoiIiIcuvb06dMaN26cQ38fgdKuSZMmqlmzphITE5WRkaHQ0FDt2LFDXbp00ZUrV9SqVSuNGTNGEydONHsq4DXouX30HHANeg64Bk23jZ4DrkHPAQCALylj9gD4vrS0NI0YMcKha3v16qVJkya5eJHn+PVNe3p6urZt26a0tDRdvXpVQUFBaty4sXr16qVq1aqpRo0a3LQDDvr222/1u9/9TtJPnw8pST/++KMkqUKFCho5cqQWL17MjTtQDPTcPnoOuAY9B1yDpttGzwHXoOcAAMCX8GAdLmcYhnJzc4t1fWly8+ZNxcbGauXKlcrOztb/volE2bJlNXLkSL311lsKDAw0aSXgXSpUqKDs7GxJUnBwsCwWi06fPp13PigoSGfPnjVrHuCV6Hnh6DngfPQccA2abh89B5yPngMAAF/iZ/YA+L5GjRpp27ZtDl27bds2NWrUyMWLPEd2drYeffRRLVmyRO3bt1dcXJwOHDigY8eO6cCBA1q9erXat2+vJUuWqE+fPsrJyTF7MuAVmjZtqoMHD+Z93aZNGy1ZskRnzpzR6dOntWzZMjVo0MDEhYD3oef20XPANeg54Bo03TZ6DrgGPQcAAD7FCrjY/PnzrX5+ftbFixcXet2SJUusfn5+1vnz57tpmfkWLFhgNQzDob83hmFYFy5c6KZlgHeLi4uzRkVFWW/dumW1Wq3Wf//739a77rrL6ufnZ/Xz87OWK1fOmpCQYPJKwLvQc/voOeAa9BxwDZpuGz0HXIOeAwAAX2JYrf/zvlaAk2VnZ6tbt27au3evunbtqsGDBysyMlJBQUHKzMzU119/rfXr12vHjh1q3769kpKSVKZM6fiUgqioKIWFhTn02WzR0dE6c+aMPv/8czcsA3zPiRMntHnzZvn7+6tHjx68Ih4oJnpuHz0H3IeeAyVH022j54D70HMAAOCteLAOt7hx44YmTJiguLi4Ap/lZrVa5e/vr2HDhmnu3LkqX768SSvdLygoSG+99Zaee+65Iq9dunSpJk2apMzMTDcsAwCgIHpuGz0HAHgbml4QPQcAAABQFN9/yTE8gsVi0bJly/Tyyy8rMTFRqampyszMVFBQkCIiItS7d29Vr17d7JluZxhGgR9iFHU9AABmoee20XMAgLeh6QXRcwAAAABF4TfWYaojR47ovffe0/fff69GjRpp6NChCg4ONnuW20RFRalq1aratGlTkddGR0crPT1dn332mRuWAd7Fz8+v2D/YMgxD2dnZLloElC70nJ4DzkDPAfOV5qbTc8A56DkAAPBl/MY6XG7RokVasGCBPv74Y1WuXDnveEJCgh5//HFlZWXp59d3LFiwQJ988km+63zZM888owkTJmjJkiV6/vnn7V63dOlSbd68WXPnznXjOsB7vPzyy/zGCOBi9Nw+eg44Bz0H3IOm20bPAeeg5wAAwJfxG+twuR49esjf31+JiYl5x7KzsxUWFqZr165p8eLFatWqlbZs2aIXX3xRMTExpeYGNTs7W926ddPevXvVtWtXDR48WJGRkQoKClJmZqa+/vprrV+/Xjt27FD79u2VlJSkMmV4PQwAwP3ouX30HADgTWi6bfQcAAAAQFF4sA6Xq169ukaNGqXp06fnHdu+fbt69uypadOm6c9//nPe8UGDBmn//v06evSoGVNNcePGDU2YMEFxcXEFPs/NarXK399fw4YN09y5c1W+fHmTVgLe4fTp0/Lz81NYWJgk6datW1q8eHGB66pXr64nnnjC3fMAr0bPC0fPAeeh54Br0XT76DngPPQcAAD4Il5aC5fLyMhQjRo18h1LSkqSYRjq169fvuPt2rXT+++/7855prNYLFq2bJlefvllJSYmKjU1VZmZmQoKClJERIR69+6t6tWrmz0T8HgpKSlq0aKF5s2bp5iYGEnS9evXFRsbW+Baf39/RUREqFmzZu6eCXgtel44eg44Bz0HXI+m20fPAeeg5wAAwFfxYB0uV6VKFZ09ezbfsb1798pisSgyMjLf8YCAAAUEBLhznscICwvTyJEj874+cuSI3nvvPf3lL39Ro0aNNHToUAUHB5u4EPBsy5YtU61atTR27NgC5+Lj4/XQQw9JknJzc9WpUyctW7ZMixYtcvdMwGvRc8fQc6Bk6DngejS9aPQcKBl6DgAAfJWf2QPg+1q1aqW1a9cqMzNTknT48GF99tln6tmzZ4HPIzty5EipevX3okWL1KBBA124cCHf8YSEBN1///2aMWOGli5dqhdeeEEPPPBAgesA/CI5OVn9+/eXn1/BtFWpUkW1atVSrVq1VKdOHT399NNKTk42YSXgvei5ffQccB56DrgeTbeNngPOQ88BAICv4sE6XG769Ok6deqU6tevr65du6pdu3YyDEN//OMfC1y7cePGvFetlgYffPCBwsPDVbly5bxj2dnZGjFihPz9/RUXF6eUlBTNnj1bp06d0quvvmriWsCznTx5Uo0aNcp3rEyZMoqMjFRQUFC+43Xq1NGpU6fcOQ/wevTcPnoOOA89B1yPpttGzwHnoecAAMBX8WAdLtesWTPt3LlTLVu2VHp6utq0aaOtW7eqZcuW+a7btWuXLBaLBg4caNJS90tNTVWbNm3yHUtOTtYPP/ygCRMm6Nlnn1WTJk00efJkPfHEE9q6datJSwHvkJubm+/rChUq6Msvv1Tr1q3zHTcMQ1ar1Z3TAK9Hz+2j54Bz0XPAtWi6bfQccC56DgAAfBGfsQ63eOihh7Rly5ZCr+nUqZNSUlLctMgzZGRkqEaNGvmOJSUlyTAM9evXL9/xdu3a6f3333fnPMCrVK9eXQcPHnTo2oMHD5aat7QEnIme20bPAeeh54B70PSC6DngPPQcAAD4Kn5jHTBRlSpVdPbs2XzH9u7dK4vFosjIyHzHAwICFBAQ4M55gFfp3r274uPjdf78+UKvO3/+vOLj49W9e3c3LQPg6+g54Dz0HIBZ6DngPPQcAAD4Kh6sAyZq1aqV1q5dq8zMTEnS4cOH9dlnn6lnz54qUyb/G0ocOXKEV/AChYiNjVVWVpa6du2q/fv327xm//796tatm7KysjRx4kQ3LwTgq+g54Dz0HIBZ6DngPPQcAAD4KsPKh9gApklJSVHr1q11zz33qEmTJvriiy9048YN7du3r8Dn24WHh6tLly5asWKFSWsBz5eQkKCnnnpKN27cUL169dS0aVPdfffdunbtmg4dOqTjx48rMDBQ7777rvr27Wv2XAA+gp4DzkXPAZiBngPORc8BAIAv4jfWARM1a9ZMO3fuVMuWLZWenq42bdpo69atBW7ad+3aJYvFooEDB5q0FPAOffr00cGDBzVy5Ehdv35dGzdu1Pr167Vx40Zdu3ZNI0aM0FdffcVNOwCnoueAc9FzAGag54Bz0XMAAOCL+I11AIDPyszM1NWrVxUUFKTg4GCz5wAAgDtAzwEA8H70HAAA+AIerAMAAAAAAAAAAAAAUAjeCh4AAAAAAAAAAAAAgELwYB0AAAAAAAAAAAAAgELwYB0AAAAAAAAAAAAAgELwYB0AAAAAAAAAAAAAgELwYB0AAAAAAAAAAAAAgELwYB0AAAAAAAAAAAAAgELwYB0AAAAAAAAAAAAAgEL8f7ll9YHFInEKAAAAAElFTkSuQmCC",
"text/plain": [
""
]
},
"metadata": {},
"output_type": "display_data"
}
],
"source": [
"fig, axes = sccellfie.plotting.create_multi_violin_plots(mt_adata,\n",
" features=gam_tasks,\n",
" groupby='celltype',\n",
" n_cols=4,\n",
" order=trajectory,\n",
" stripplot=False,\n",
" fontsize=12,\n",
" wrapped_title_length=45,\n",
" w_pad=None,\n",
" ylabel='Metabolic Score',\n",
" save='GAM-Violin-Control.pdf',\n",
" )"
]
},
{
"cell_type": "markdown",
"id": "developmental-shark",
"metadata": {},
"source": [
"### Heatmap\n",
"\n",
"We can also visualize an aggregated value for each ordered cell-type label. In this case we aggregate the single cells into cell types by using the Tuckey's trimean."
]
},
{
"cell_type": "code",
"execution_count": 16,
"id": "bored-finnish",
"metadata": {},
"outputs": [],
"source": [
"agg = sccellfie.expression.agg_expression_cells(adata=mt_adata,\n",
" groupby='celltype',\n",
" agg_func='trimean',\n",
" gene_symbols=gam_tasks\n",
" )"
]
},
{
"cell_type": "markdown",
"id": "physical-saudi",
"metadata": {},
"source": [
"We order the cell types as defined in the trajectory:"
]
},
{
"cell_type": "code",
"execution_count": 17,
"id": "opposed-concord",
"metadata": {},
"outputs": [],
"source": [
"agg = agg.loc[trajectory, :]"
]
},
{
"cell_type": "markdown",
"id": "theoretical-world",
"metadata": {},
"source": [
"and perform a min-max normalization per metabolic task (min value across cell types will be 0, while max value will be 1)"
]
},
{
"cell_type": "code",
"execution_count": 18,
"id": "desperate-offering",
"metadata": {},
"outputs": [],
"source": [
"df_plot = sccellfie.preprocessing.min_max_normalization(agg.T, axis=1)"
]
},
{
"cell_type": "markdown",
"id": "separate-boutique",
"metadata": {},
"source": [
"#### Marsilea\n",
"\n",
"Then, we can use **seaborn** or any plotting library to visualize this information as a heatmap. Here, we use **marsilea**, which allows us to visualize additional information.\n",
"\n",
"To install it, run:\n",
"``pip install marsilea``\n",
"\n",
"You can read more on its [library documentation](https://marsilea.readthedocs.io/)"
]
},
{
"cell_type": "code",
"execution_count": 19,
"id": "stylish-trade",
"metadata": {},
"outputs": [],
"source": [
"import matplotlib.pyplot as plt\n",
"import seaborn as sns\n",
"import glasbey\n",
"\n",
"import marsilea.plotter as mp\n",
"import marsilea as ma"
]
},
{
"cell_type": "markdown",
"id": "waiting-ordinance",
"metadata": {},
"source": [
"#### Data preparation for marsilea"
]
},
{
"cell_type": "markdown",
"id": "gorgeous-destruction",
"metadata": {},
"source": [
"We first generate the annotation of each cell type to indicate in which menstrual cycle they are present:"
]
},
{
"cell_type": "code",
"execution_count": 20,
"id": "indoor-default",
"metadata": {},
"outputs": [],
"source": [
"cell_groups = {'Basal' : ['SOX9_basalis'],\n",
" 'Proliferative / Folicular' : ['SOX9_functionalis_I', 'SOX9_functionalis_II'],\n",
" 'Secretory / Luteal' : ['preGlandular', 'Glandular', 'Glandular_secretory']\n",
" }\n",
"\n",
"cell_groups = {cell : phase for phase, cells in cell_groups.items() for cell in cells}"
]
},
{
"cell_type": "markdown",
"id": "stuffed-shoulder",
"metadata": {},
"source": [
"Then, we annotate each metabolic task by their major groups. We can find this information in the scCellFie's metabolic database:"
]
},
{
"cell_type": "code",
"execution_count": 21,
"id": "rolled-option",
"metadata": {},
"outputs": [],
"source": [
"sccellfie_db = sccellfie.datasets.database.load_sccellfie_database(organism='human')"
]
},
{
"cell_type": "markdown",
"id": "french-living",
"metadata": {},
"source": [
"We generate a dictionary mapping this information into each task. Additionally we rename some of them to keep specific capital letters (e.g. ER and Golgi)."
]
},
{
"cell_type": "code",
"execution_count": 22,
"id": "political-elder",
"metadata": {},
"outputs": [],
"source": [
"task_dict = sccellfie_db['task_info'].set_index('Task')['System'].to_dict()\n",
"task_dict = {k : v.capitalize() for k, v in task_dict.items()}\n",
"for k, v in task_dict.items():\n",
" if v == 'Processing in the er':\n",
" task_dict[k] = 'Processing in the ER'\n",
" elif v == 'Processing in the golgi':\n",
" task_dict[k] = 'Processing in the Golgi'"
]
},
{
"cell_type": "markdown",
"id": "balanced-average",
"metadata": {},
"source": [
"These are the inputs, including data and some annotations for the plot"
]
},
{
"cell_type": "code",
"execution_count": 23,
"id": "unnecessary-stupid",
"metadata": {},
"outputs": [],
"source": [
"data = df_plot.values\n",
"cell_labels = np.asarray(list(df_plot.columns))\n",
"task_labels = np.asarray(list(df_plot.index))\n",
"cell_cat = np.asarray([cell_groups[cell] for cell in df_plot.columns])\n",
"task_cat = np.asarray([task_dict[task] for task in df_plot.index])"
]
},
{
"cell_type": "markdown",
"id": "confident-combining",
"metadata": {},
"source": [
"We generate a color palette for the metabolic task annotations:"
]
},
{
"cell_type": "code",
"execution_count": 24,
"id": "accompanied-professional",
"metadata": {},
"outputs": [],
"source": [
"total_task_cat = sorted(set(task_dict.values()))\n",
"task_palette = glasbey.extend_palette('tab10', palette_size=len(total_task_cat))\n",
"task_colors = {t : task_palette[i] for i, t in enumerate(total_task_cat)}\n",
"cell_palette = ['#FFABA4', '#D8A3B7', '#EDAF5C']"
]
},
{
"cell_type": "markdown",
"id": "gorgeous-shark",
"metadata": {},
"source": [
"And specify the size of the legend"
]
},
{
"cell_type": "code",
"execution_count": 25,
"id": "automatic-potato",
"metadata": {},
"outputs": [],
"source": [
"plt.rc('legend',fontsize=28, title_fontsize=32)"
]
},
{
"cell_type": "markdown",
"id": "opposed-worse",
"metadata": {},
"source": [
"Finally generate the plot:"
]
},
{
"cell_type": "code",
"execution_count": 26,
"id": "sound-mileage",
"metadata": {},
"outputs": [
{
"data": {
"image/png": "iVBORw0KGgoAAAANSUhEUgAAEaUAAAf9CAYAAAB3KZLFAAAAOXRFWHRTb2Z0d2FyZQBNYXRwbG90bGliIHZlcnNpb24zLjkuMSwgaHR0cHM6Ly9tYXRwbG90bGliLm9yZy/TGe4hAAAACXBIWXMAAA9hAAAPYQGoP6dpAAEAAElEQVR4nOzdd5zcVb3/8fd3+vae3fRNT0gjPaGFHqRFKZaLxIASFCx4vaL+VAS8XkFR9F4VBaQrIlUsQCgJIQnppPeySXaT7b1N//2xZsjszszO7M7sbJLX8/HYB3u+55zv+czMd85MgO87ht/v9wsAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEmmZBcAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAOg/CKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQQCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCAUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQAChNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAAEJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIsyS4A6LGX/pTsCoD+44abYhq+57k3E1QIAOBUNu7zV8Q0vva1mxNUCQAAwOkh95PPJrsEAAAAAAAAAAAAAAAAAAAAAACAHjEluwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQP9BKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIIBQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABAAKE0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIAAQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgglAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEEAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAggFAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEAAoTQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgABCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYTSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQQCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCAUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQAChNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAAEuyCwCAfueGm8L3vf+2VFXZd7XglDLyk/NlTU8J2Xd89TY1HiwL2Vc0b7KyRg0O2ddwoEzlH26LugZ7bqYyRwxSSn6WrOmpMtksMpnNXcbV7ipR1cbdUZ8X/d+4z18Rtu/I2+vUVlEbsm/oZbOVWpgbsq96637VbN0fl/pOFSmFuRp22eyw/Xuee7MPqwEAAOg9S/54ZZ73/bD9ta/d3IfVAAAAAAAAAAAAAAAAAAAAAAAA4FRBKA0AAKcBk82qormTlDGsMNmlAAD6gGFLlyV7hAxHjkzWVMlil7xu+b0u+V3N8rXXytdWJ19rtSR/sssFAAAAAAAAAAAAAAAAAAAAAAAAAACnGEJpgERITZOu/GRsczweyeuR3G6ptUVqapTqaqTjxyRne0LKBDqzpKVo1KfmxzzP5/HK53LL63LLWd+s9poGNZdVyd3YkoAq0ZlhMmnoJTPlyMtKdim9Zk6xa9R1F8owjMCxinU7Vb/3SFzOP+7zV8TlPCc78Or78rS0xf28ANCZYcuQffgFsg+7QOaMQVHN8Xuc8jYfl7fxqDy1++Sp2SdvU2mCKwUQDdvgOUqf9dWgYw3vfi8u71FL/nhlnvf9sP3OIx+oZdOjvV4HAAAAAAAAAAAAAAAAAAAAAAAAwOmLUBqgv7BYOn7sDik9QxpQ1HHc75fKj0k7tkj1dcmtEQjDZDHLZDHLkuqQPTtDmcUDNWDGeLVW1Kp6yz61VXLtJlLupJGnRSCNJKUPLggKpJGk5tLKJFUDAP2Hbdj5Sp18k0zWtJjmGRa7LNnFsmQXyz7sfPk97ar7x20JqhKJ1F3ISO1rN/dhNYgHa9H0oLa3pYLQqBhknPf/ZM2fELKvbfcratv9ah9XBAAAAAAAAAAAAAAAAAAAAAAAAJxeTMkuAEA3DEMaOFi6+AqpeFSyqwFiklqYq6GXzdaAmeOTXcrpyzCUPXZYsquIm/QhA4La7bWN8rS2J6kaAOgfUibcqPTpS2IOpAHQjxkmWQunBh1yl3+UpGIAAAAAAAAAAAAAAAAAAAAAAAAAoCtLsgsAECWTSZo5V2pvk8qPJbsaIGqGYShnfLEMi0UVa7Ynu5zTjj0nQxaHLWy/u6VNDftL5W5pl/y+wHFnQ0tflBcTw2xSalFe0LHm0sokVQMA/YNt2PlKGXdtsssAEGeWvPEy2YKDplzHNyWpGgAAAAAAAAAAAAAAAAAAAAAAAADoilAa4FRz9kzpzdeTXQUQs+zRQ9ReXa+G/aXJLqXfKf9wm8o/3NajufacjLB9Pq9PR5aulaelvael9am0gfkyWcxBxwilSbyjb69LdgkAwjHblXrWp7sd5vf75Hc2yu91y7A4ZFhTZZjM3c4DkDy2gdOC2j5Xszw1e5JUDQAAAAAAAAAAAAAAAAAAAAAAAAB0RSgNkAyNDdLu7cHHrDYpI0sqHiFZrOHnpmdIuflSbXViawRCcDY0q3b7gaBjhtksa3qqMoYXyZaRGnF+/tQxajx0XH6vN5FlnlHMtvD7hbux5ZQJpJGk9CEDgtrulnY5axuTVA0AJJ99yFyZHNlh+51la+U8uFSe2gOS/+TPVkOm9CJZskfIkjdOtoHTI54HQN+zFk0Parsrtkp+X5KqAQAAAAAAAAAAAAAAAAAAAAAAAICuCKUBksHZLh0pCd23Z4d06SckuyP8/IIBhNIgKbztLjUeOh6yr3rLPhXOmqDsscPCzrek2JU2KE/NRysTVeIZx2Qxh+3zutx9WEnvpQ0pCGq3lPXtdeJubtPB197v0zUBIBJr4dSwfe0ly9S6+YkwvX75mo/L1XxcrtLVat3ypCx5Y2UvvkTWwsmJKRZA1MyZQ2RO6xTGV74pSdUAAAAAAAAAAAAAAAAAAAAAAAAAQGiE0gD9TVurtHeXNHla+DGOCIE1nVksUnqmlJIiOVI62maL5PdJbnfHT3Oj1FAv+Xy9Ll9Wm5SZKaVndPxusUh+SV6P5HRKrS1Sc1NHME9P2R1SerrkSO14Lszmjh+Pp+PH5ex4PM1NvX88iJ7fr4p1O+XIz5IjNyvssLRBBXEJpbHnZMiRmymzwy7DZMjr8qh+z+Go5hpms1Lys2RJtctks8lkNcvn8sjrcsvd1Kr22kbJ7+91jX3CMJJdQVykFGTL4rAHHWs6TcOLTDaLHHnZsqTYZLZZZZjN8rnc8rrccjW2yFl3GuxdhmTPzpAtI00mu1VmW8dXTp/bK3dLm1wNLXI3tya5yMQzO2yypqfKkmqXxWGXYTHJZDbL5/HK5/bI63TL2dAsd2NL4osxJEdetuw5GR3XncmQu6VdjQfLEr82esycGT7ozVmyLKZzeWr2ylOzV4Y1vZdF2WXJGSmTI0eGLV2GxS6/u1V+Z5M8jaXyNR/r3flDMKUWyJw1TIYtXSZrmmSyyO9pl6+9Xr7mcnmbSju+28Z1zXxZckbLlJIrme2S1yXn0ZXyOxuimm/OHCJT+iCZbGkyrOny+9zyu5rka62Rp+6A5OvfwXHmzKEypw/seI2taZLPLZ+7Rf72BnnqDsrvbk54DYY9S5a8sTKnFnS8Bj6PXMfWy9dSnvC1E81aND2o7fe65arYkqRq0K0+2fcMmVLzO35OrGO2SoZFfm+7/O42+Z0N8jQckb+9Lg7rAQAAAAAAAAAAAAAAAAAAAAAAAN0jlAbojxq7udnVHOGtm5YuDR4q5RdI2blSalp0a/p8Um21dOiAVHpY8nqjr9cwpOJR0vARUl6+ZJi6n9PeJtXXSdWVUlWFVFMdfmxevlQ0uOOfWTmS3R5+7Mk8Hqm8rOMxVRyPbg56reHAsYihNLaM1JDHR35yvqzpKSH7jq/eFghOyBw5SHmTR4c8T6RQGsNkKHPkYGWNGiJHXqYMU/jr1OfxqK2yTnV7jqilrCrsuHgpmjdZWaMGh+xrOFCm8g+3BdqWtBSN+tT8qM6bWpircZ+/ImTfnufejL3QBEobMiCo7XN71FZRk6Rq4s+wmJU9ZqgyRw6SPStDhil8mJDX5VZreY3qdh1WW1XibzoeetlspRbmhuyr3rpfNVv3R32u9KEDlD1mqFIKcmSyRv6a6Wl3qrW8Vo2HjoV8n3V3rR949X15WtrC9udNGa38KaND9rVW1Oro2+si1tcTjvxspQ8uUErBv8Nf7Lao5vk8HrWUVat+f6laj0f4POwk3Ptbko68vU5tFbWSIeWMG67ciSNkSQkOtXM3txFK088Z9ozwnT0MT+tRmIjJKvuw82QfdoHM2cUyTOHf3772ermObVD7vn/I19bzfdycPUKOkZfLOmCSTI7siGP9Hqc8tXvlOrZBziMrJJ+ny5i06UtkH3Z+yPnOIx+oZdOjkiRL7hilnPUZWfPHdRnnqT8gT4RQGkvuGNlHLZC1YKJMtvDhP36vS56avWrf/4bclVsjPrasy3/ZEcoShdxPPhu2r233K2rb/WrE+Zac0R31D5gUuX6/T97GMrlKP5Tz0Dvye8LvxSdLGf8ppYy/LmSfu3qXmlb+jyTJlDFIqRM/K2vhVBmd/lzhc9bLMfursmQND3me1u1/Vvv+N6KqJ/XsL8pRfGHIPmfph2rZ8LuoztMTtk6hNJ6a3ZKnF6GdSWTJH6/M874ftr/2tZsjzo/2vSlJtmHnK336kqjqShl/XdjrLZq6+mLfsw2aLUv+OFlyRsucMViGJbo/5/raG+QqWyvn4eXyNh6Nej0AAAAAAAAAAAAAAAAAAAAAAAAgVoTSAP2R3RG5vz3CDYvDR0hnTYl9TZNJyh/Q8TNhkrT+Q6kmijCOtHTp3AulzPAhJCE5UqSiFKloUEcgzivPhx876WypoDC280uSxSINGd7xU3Fc2rBGamuN/TyIibupJWK/2RFdSEMXhqGB50xW5ohBMU9NHzpAhbPP6hLIEI7JYlHaoAKlDSpQe12jjq/aKld9D27iR9TSO4XStByrlt/Xs8CF/iZrzFAVTBsrs80a1XizzaqMYUXKGFak1opaHV+1VZ7W/n2jempRngrnTAwbOhWKxWFXZvFApeRn62DZ+wmsru8UTBsbNuAnEpPFoozhRcoYXqSW49Uq/3B7XF5zk9WsQRdMV9rAvF6fC8lhmMN/ZtqHna/WbSUJr8E2eI5SJ3++22CYE0yObDlGXip78YVq3/+m2nb+VVL0+7kptUBp074ka8FZUc8xLHZZB0yWdcBkuSu3ytcafbjTyewjL1fq5Ju6BKF0x5SSp7QZt8uaPyGq8YbZJuuASbIOmCRP7T41b3hEvtbEh+CFrceRo/TpS2QdMCm68YZJlqyhsmQNlWPs1Wrb9ZKcB9+OSy3WQbOUPuPLEa/99gNvhQ0msQ+/MLpQGsMs26AZYbudh1d0f44eMuxZMueMCDrmOr4pYeshdn2176XP/lqP6jM5suQYdbnsIy+V8+Dbat3xguRz9+hcAAAAAAAAAAAAAAAAAAAAAAAAQCSx3W0HoG8MK47cH01YTG+kZ0jzL+kIqInEYpHmXxp7IE0yFA6ULry8+8Af9FtFcyf1KJCmYPo4DZ4/PepAms4cOZkafsU8ZRQP7NF8dM+anip7VnrQsebSyiRVE0eGoYHnTlHRnIlRB9J0llqYq+FXntOjoJO+kj91jIZcMjOmQBqElzYwX8Mun9PzAK9/MwyDQJrTgN8VPhDNMepypU1fInPW8AStbih1yiKlz/pq1MEMQbNNFqWMvVoZ535XhiUlqjnWQTOVedF/xxRIEy+2YRcobcrNMQfSWAdM7qg5ykCaziy5Y5R54X2y5I/v0fzesuSPV9ZF/x11IE1nJmuq0qYsUtrMOyXD3LtaCiYpfeYdEQNpJMlV+qF8zsaQfeaMQVE9l9YBk2SyZYTs87ZWy1O1o/uCe8hWNK3LdeYu/yhh6yEWfb/v9YZhmOQYtUDps78hyUj4egAAAAAAAAAAAAAAAAAAAAAAADjzEEoD9BcWq5SbJ51zgTSgKPy45iapsjzx9ZjM0rwLJGuEm0LHT5JS0xJfS7ykpUlzzk12Fac9W2bka8LT7or5nFkjBylr1OCY5+VNHqXcs0bEPK8zk8WsgedMVmpR/w0GOZWlDw0OwPL7fGouS3D4Vh8onDOxR0FKnVkcNg2aP122TsE9/UHelNHKmzxKhsGN0PFkTU/RoPOm9uoceZNGEkhzGvA2HYvYbx92vrIu+m9lL/i10mZ9VY7RV8qSN16y9D6EL2XiZ+UYeVmvz2MtOEtps+5Ud4EJHYEkd8pk7fuAK3N6kdKmfiH2edkjlD77GzLZerc/m2wZSp/zTZnSe/+ZEQtzxmBlzPlPmeyZvT6XfchcpZ19S4/nG7YMpc/8igyTpfvBPo+cJe+Fr2X4Rd2ewjZ4btg+15EPJPm7r6OHrAOnB7U99SXytdUkbD1Er6/3vXixFU2VY9wn+2QtAAAAAAAAAAAAAAAAAAAAAAAAnFmiuOMLQNwVFEo33BT7PI9bWrdK8vfgJkm3W/J4JK+nI3DGbpPM3WwBdrs0Zpy0c1vo/qHDI8/3+SRne8c/LVbJapVMccrC8vslt0vyejt+zJaOers7/4AiqWCAVFUZnzrQRebIyOEx7qbWmM+ZWhR7sEJqUZ7ypozudpzP7ZHX5ZbZYZPJbA47zjCZNOj8s3Xo7yvl7UGwDsJLHxIcStNWVS+fy93ndVjTUzTu81fENKds+SY1l3bdT7JGD1H26CHdzvc63fJ5PLI47DLM4fcvs82iwRdOV8nfV8rv88VUY6KkDx2g/CjeY5Lk83jldbpkmE0y26wy4vVZcArw+/3yuTzyeb3ye30yWUwy223dPgepRXlKKcxVW0Vtj9btyb6J/sdduV3WgrO6HWdKyZV98Bxp8BxJkt/nlbfhsNzVu+Q+vlGe2n0xrWsbPEcpY67sdpzf3Sa/p02GPTNimIitcKoc4xaqfc9rYerPU/rsr0UVSOL3eeR3NUt+nwxbhgyztds53bHkjol5jmFNVcacb8qw2COO8/s88jsbZVhSZFhTwo4zWVOVMecuNbz3PcnvjbmemJksSp/zzYg1SR/XL7Ot2/Ad+/D58tQekPPwspjLsWR2/5l5svZD78ox5uqQ14xt0Ey1bkvvuE5CMVm6BMOc4Pf75DzyQUy1xMRsk7VgYtAhd/lHiVsPUevrfS/s+X0e+T1t8nuckt8vw2KXYUuXYUT+3uAYvUDOA2/K72mLaT0AAAAAAAAAAAAAAAAAAAAAAAAgEkJpgFNFS7O0YY1UW9P9WLdbKi/rCF6prZaamzsCbTpLTZMGDpYmTJIcYW5ILR4dOpTGbJbSwtyY2tYqbVwrVZRL/pPCEwxDysiUsrKl/EJpQGFHOxqtrdLxUqm6Uqqv63g+ugQzGFJWljRsREeYjilMwEjxaEJpEsEwVDhrghy5kV/TlmNVvVrG5/WpraJWzoaOa8CSliJHbqZsmWlB4wqmjZVhGGHP01Zdr6qNu9VWVd9xwGQofcgADZgxQdY0R8g5ZrtNuRNHqmrj7l49ht7wtrt0fNWWQDt9aKEyhhWFHOtsaFbt9gN9VVqPmGxWpRRkBx0LFfJyKjFMpm4DkZrLqlT90R456ztumDfMJmWOGKSCaeNktocOWLBlpCprzFDV7zkc95pjZhgqmDYu4hC/16f6/UfVsK9Uzvqmj6eaTXLkZytzeJEyRwxKdKV9zt3SrpaySrVW1slZ1yh3U1vXICFDsmelK3PEIOWMLw4bSJQ9ekiPQ2lO5nN71FpRK1djiyTJkuZQSn5Or8+LxHMeXqaUcQu7DT7pzDCZZckZKUvOSKWMuUre5gq17/unnEfeD/5uFnKyWSlnfTpst8/ZqLa9f5fr6Gr5XY0nJsmSP04p46+TNX9CyHkpo6+U89A7IYNCUiZcL5M1NWJZrvLNat//hjy1eyWfJ7CuOWuYrEXTZB8+X+bU/MiPLUqe+kPy1B6Q390qkyNb5ozBMueMCBrjGH2lTCnh30fOw++r/eDb8jYckdQRKGlKyZN95GVyjFoQMszCnDFQ9uEXyFnycahL67Y/yTDb/90/SCnjFoZds3nDI2H7vI1Hg9r24otlTi8MO97nblHbjr/KeXSV5HX+e/3BSpn4GdmKpoWdlzL+k3IeXSn5ehcu520sk7tmj/yuZhn2TJnTi4LCg/zt9XKVrZN96Dld5hpmm+zDzlf7/jdCnttaODXs9eap3i1fa+++q0ZiHTBZhtkWdMx1fFPC1jvdeKp3B13nKeOulTkjdCCm69h6uY5tiO7ESdj3TvDUl8hduU2emj3yNh2Tr7VaJ/aMAJNVltxRcoy4TLbBs0Oex2RNk23wbDkPvx/pkQIAAAAAAAAAAAAAAAAAAAAAAAAxIZQGOBW0t0sbPowuSOXwIWnPTsnr7X5sa4t0YG/HeS+7siM0prPU1I7wmtaW4ONWW9exJ2zbLJUf63rc75caGzp+jv47VCErWxo8LHKdm9ZJTY2Rx3QsIDXUS9s+6qh32qzQw/IHRHEuhGJ22JQ5YmDQMcNsljU9VRnDi2TLiHxDuafNqZZjUQQrhdFyrFrla7bL09repc9+UhhO6sA8OfKywp6nrbpeR99eJ7/3pBvzfX41H6mQs7ZRwz9xTthgkOwxQ1Wzdb98bk/I/kTze71qPHQ80LZmpCkjzFvI2+4KGtsfpQ8ukGEKDuRoPnpqh9Jkjhwka2roYCNJajpaoWPvfxR0zO/1qWF/qZx1TRq2YE6X5+SE3Ikj+kUoTWbxwC5BUCfzuT0qXbZRbZV1Xfr8/w6WaquoVfWWfcoe281nwCmkYt0OuRpauh/ol5z1zar6aK/cLe0qnH1WyGEpBb0Pjmk8dEwV63fJ5+oaEGHvJkQMyed3Nat1+/NKO3txr85jTi9U2rRbZR95qZrX/Vq+lvD7rG3oOTKnhf6u5GtvUOMHP5avpaJzpfJU71bTyp8q45xvyzpgcpe5hjVF9uJL1L73b0HHTan5sg09N2L9rdv+pPYDb4bo8cvbcFjehsNq3/t3OUYtkN/b8zAUb0uFWjb8Xp66/V36TCl58ntdHQ2zXY6Rl4c9T8vWZ+U8uLTLcV9bjdp2/EXexqNKn/HlkHMdo68KCqVxH98Y+N2SPz5iKI2rdHXYvs5SxlwVts/v86hp9c/lrQsOtvM2lal5zcNKn/MN2QbOCDnXlJIr+7Dzgh5DLHzOJrVs+oPcFVu69Bm2TBnmj/8VRvuBt0KG0kiSffhFYUNpbIPnhl3feWRFjBXHpnOgj7e1Rt6GkoSueTrxtVbJdVJokL34wrChNN7Go1G/J/p63zuh4d3vydtU2n2BPndHIE/1bqXra2GDaSx54wilAQAAAAAAAAAAAAAAAAAAAAAAQFyFvuMZQP/icEjzL5POmS/ZIoTBSFJLc3SBNCdrrJfa28L35+R2PeZ2hR/fXY0na6iXdm6NPCaqQJpOKiIEcaSlxVYjAuxZ6Rp47tSgn6K5k5Q3aWS3gTSSVL1ln/yxXp//1lpRq9JlG0MG0kiSs/bj6yR9cEHEc1Wu3xkcSHMSd3ObanccDDvXZDErtSjEewI9kj4k+AZgZ32z3M2tSaomPtIiXH9+r08V63aG7W+vaVD9vvA3J1tTHbLnZPSqvnjo/Lp1VrFuZ8hAms68Trdqth3odtypIqpAmk5ajleH7bOmp8hkCx2QFY3GkuM6vmpryEAaKXjfRP/lLHlXbbtficu5LFnDlDn/XplSw7+HO4dmnKz94NIQwQwn86v90Hthe62FU0IcO1uGEf6Ppc7D74cJpOm8tFft+/8lv7Oh+7Eh+JxNavzgJyEDaaSOQBm/q6mj5oKzZFhTQo7zNh0PGUhzMtfR1fK5Q+8X5vRCmdIKY6g8duas4TKlhP8u4yxZ3iWQ5mN+tWx5Wn5f+HA+a+HUHtXl97rUtOqBkIE0kuR3NcrXVhtoe+sPylO7L+RYc8ZAWfLHh+iwyVZ0dsg5PnerXGXrYq47eoashcFru8s/Cj0Ufaqv970Togqk6cRduS1snzl7RMznAwAAAAAAAAAAAAAAAAAAAAAAACKxdD8EQL8xaIh04eXSsqWRQ2FOSEuXBg7uCJVJz5RSUiWLpePHFEMmlc3e9ZjXKzU3SekhwhEmT+tYq+K41NQgtUUIvIlVTp5UNEjKyup4THbHvx+TWYpwQ3MXNrvkiuI5RNw0HChTw/7Yb7w8oWLdDsnvj2psamFe2D5nfbPaayIHMTQcLFP+tLEyDCPs+ZuPVkZVCyIwGUodmB90qLn0FH9eDSm1MPyN/i3lNfK2OSOeovFgmXLGDQvbn1qUJ2ddU49LjIdIwUyuxhY1HjrWh9X0T468LKUNypc9O0O2zDSZHVaZLBYZZrMMU+i9JRSzwxY2VCYSn8cbMQAJp5a23a/KU3dQqZNvkjl9YK/OZbJlKH3WHWp8/94QvYYs+RPCzk0960alnnVjj9e25IyUTFbJ9/E1bR0wKex4v9+ntt2v9ni9WLTteln+9u7DtCTJWjAxbJ85Y6ByP/lsr2qx5k+QM2IIRu9YC86K2O888kHEfn97ndyVO2QrCh0+0xEGY0iK7nvbCe0HlsrbeCTGOW8pPXdMyD578cXyVO8OOmYrmibD4gg53lW6JujajDdL7miZHFlBx9zlmxK2HqLV9/teFyaLrAUTZSk4S+b0QTKnF8qwpskw22VYQvxZPNxpbOk9rhMAAAAAAAAAAAAAAAAAAAAAAAAIhVAaIBkaG6Td24OPGUZHwEpeQUeQTLjQmMwsafpsae3K8OfPK5AmnS0VDIhPvTZb6ONHD0sTQtxIbDZL487q+JE+DrBpauh47DVVUnW15PVEX8OwEdJZk0OH4PREuMeEuPP7/arfe0SVG3b1+BxtVXVyNbREPd6amRq2r726vtv53naX3C1tsqWHPo8twvkRvdTCPJltwV9FTvVQGrPDLrPNGrY/muuvvbZRfq9Phjn054AtI7nXn9lhk9kefg9tLqvqw2r6n8wRg5Q3ZXTcXiezzaKeRCQ0H63oUZgN+i93xRY1VG6XbeAM2YfPlyV/ggxz+P0mEkvOKFkHTJa7clvQccOekdBQA8NkkcmRJV9rdeCYOb0o7HhvwxH52moSVs8Jfq9bztJVUY83Rag5HkwpOYk9f1r4+v1et7wNJd2ew1N3IGwojcmaJsOeKb+zIaa6nIffj2m8JLmOrZe3tUbm1K6BhLaBM9VqS5ff1fzxscFzwq9/ZEXM68fCWjQtqO13t8ldRXhYsiVj3wsw2+UYfaUco6+Qydr77w1GHM4BAAAAAAAAAAAAAAAAAAAAAAAAnIxQGiAZnO3SkZIwnbuk7Bzp/Is7QmpCGTq8I9Smob5r36ix0tkzO0Ju4sVsDn18786OWroLijGbpazsjp8TfF7peJm0f49UFSGEwjCkWfM6QmniKdxjQly1VtapZss+tVbU9uo8bVX1UY81LGaZIry+7tb2qM7jaWkPG0oTKZAD0csYGhyc5WlzRhXakiju5jYdfC32G+JPZrZHDohwt0Rx/fn98rQ7ZU1LCbNGcq+/7tZ3NTRH7D9tGYYGnjNZmSMGxfe0Pfy8imXfxCnE75Xr2Dq5jq2TzHZZ88fLkjdOltwxsuSMlGGOfn+wDpzRJZTGZMuMd8VdGLYM6aRwBiPCmt6mYwmvR5K8jUclT3TfDyTJZItTSGIYkZ6TeDDZw9fvc9ZLfl+35+guLMhky5A3hlAan7NRvpbyqMcH+H1yHnpXqRM/3aXLMFtlH3aB2vf/q+OAxSFrYeggHU9jqbx1B2JfPwa2gdOD2u7KbZLfm9A10b1k7HuSZNizlHHO3bJkDYvfOjF8BgAAAAAAAAAAAAAAAAAAAAAAAADRIJQG6I/q66Ttm6UZc8OPGVosNWwOPlY4UJo2K4GFdeJ2SyvelWafK+UXxDbXZJYGD+v4ObhP2rQu9LiJU+MfSIO483m88rk98jrdcjY0qb2mQS2lVXI1tsTl/FEFefyb2Rr5o83vie7mX1+EcaZu1kB00gYHh9I0l1UlqZL46fb688bh+rMl9/ozd7O+z+3po0r6l/ypY+IeSNMb7ta2ZJeARPM65a7YInfFlo62YZYld5SshdNkH3quTCk5EadbckZ1OWZYQ4dhxZNhsUe9pt/TN9dxdwErnRnW0KF18dL5OYr/+SO8zl5XdCfxOiOvEeO15GvreYChs2SZUsYtDPm82YsvDITS2AbOCBva4TryQY/Xj4YpbYDMGYOD1yzflNA1Tx1xDFPtyepJ2PckKX32N+IaSAMAAAAAAAAAAAAAAAAAAAAAAAAkAnfVA/3V8bLI/Tm5XY9NnRF+vM8r7dsjHT0sNTVK3k6hAZ9YKKWlx15na4u0fKlUNEgqHiUVFknWGP+G9pFjpPY2aee24OMpKdLY8eHntbdJu3dI5cc66vD5Pu5LTZOu/GRsdaBbrRW1Ovp2mAChBIol5MLbzVjDYo7qPKYI487U0I14sudmyprmCDrWUlqZpGrip9vrzxyH68+V3OvP2836fRXaZBiRb2Lvy/AoS6pduROKw/Z72pyq2X5QLceq5Glpl/+kzytLWopGfWp+3GvyuaMLQMJpxO+Vp2avPDV71bb7ZaVOWSRH8UVhh5vsmV1P4e77MCO/u02GPSNkX8TwlHjW4Ik+/E6S/O7WBFXSNyKG/YQJbek6LnJwTqzXUqyvQfBazXKWrg55vZvTB8qSP0Ge6l2yDZ4Ter7PI+fRlT1ePxq2gcF/TvP7vHKXb07omv2GYZL8vvDdFkfYvr6QjH3PNniOrHljwvZ76kvUtvfv8tTuk9/ZEPT82Yadr/TpS/qiTAAAAAAAAAAAAAAAAAAAAAAAAIBQGqDfcjoj9zs63aSbmS1lZoUfv36NdLQkfL/FGm1loZUf6/iRIWVnd9STkdkRdJOe3lFbpDXGniXt3SV5Tgo7GDRUMoUJZvB4pGVLpZbm0P29fTw4Zfk9Xvm8XpnChH9YU6O78dWSFn6c1+nqUW34WPqQAUFtn8erluPVSaomfrxOd8T+zkE8IRmGzI7wN/sn+/rrbn1bVg8CznrAMJsi9lvT+ybMQpLShxaGrcfn8erIW2vlbg4dYmGyRhdUBMTE51HrlqdlHTBJ5tSC0GPMXb8r+VxNEU/buOpBeaq2x6PCAL+rUQoTSmPOGBTXteLF5wrz/VOS8+hqtWx8pA+riZ3PGf51Ntmzuw0RkSRTSl7kNbq5lrryxzg+mPPg0rAhTPbii+VtOCzrgMkh+90VW+R3NvZq/e5Yi6YHtT21e+V3tyR0zX7DZJW84f9sa0obELavLyRj3wsXkCRJnoYjalxxv+QL/Z0y2SE+AAAAAAAAAAAAAAAAAAAAAAAAOLNEvpsXQPLYu7nZzDCC2zm54ce63ZEDaVJSJHv4AITY+KX6OunIIWnHFmndKum9t6TXXpQ+eE9qD/M30VssUkFh8LGcCDe7VlWED6SROoJxcMZyN4YOf5AkR36E8KZ/M9ttsqaFD7RwRTg/otM5lKa1vEZ+b+Qb4E8F3nanvK7wwTTRXH+O3AyZIgSuuJqSe/15210Rg2nSBuXHZR2/1xux32yLkK1oSCn52XGpIxqO3PCva2t5TdhAGkly5IQO4wAkSUYvQov8XnnrS8J3u7oGYvidjfJFCMqw5o/reT1heJvLw/aZs4bJlBLhO26S+FrC12zJG9uHlfRMpPoNs1XmrOHdnsOSMzL8+d0tCQ956czbWCp31Y6QfbaBM2QvvliGKfTnhvPwikSWJsOaJkvumKBj7uObErpmX/J7IwfyGdbUCH1pSQ+fSsa+Z84eEbbPdXRV2EAaSTJnDot7PQAAAAAAAAAAAAAAAAAAAAAAAEA4hNIA/dWgIZH7ne3B7UghNn5/5HONGB1dTb3ilyqOS3t3hR+Slh7cjhSU4+8mvKJ4VPSl4bTTWlETts+enSF7bmbE+ZkjB8noHPx08vnLw5//VDP0stka9/krQv4UzZuckDUtqQ45Or0GzaWVCVmrz/ml1orasN2pRfkyp0QOAcscOThif3+4/lrLwz9Ge1a6MooH9noNn9sTsd+amRa2L31IoSzdPM/xZHbYwvb5u/kMzhrVzec9zmjps+5UyoQbZVjTux8cQqQwCJ+zIcRRvzxV4b+r2YddIJl79t6yFk2XYe36vnVXbg87xzBMcoz7ZI/WSyR3ZejwE0kyp+bLOnBGj85rShsgS16EAAxv5H1RJmtU67irdkbstw87L2K/4ciWdcCksP2e6t2SuvnzRwK0H3gr5HHDbFXKhOtD9vnaG+Su2JzAqiRr0dkyTMEBU67y0yiUxh0mdPTfzOnhvxPYh18QNiwoKpECcUzhP5uD9f2+Z7KFD6TzR/hzrmFNlW3wrB7VAgAAAAAAAAAAAAAAAAAAAAAAAPQEoTRAf5STK02cGnlMY2NwO1JIi80m5eaFWStPGjcxtvpOmDRVGjRUihDe0YU1ws2B5k43JEa6kT83X7KEuYFx9DhpQFH0NeG001xWFbG/cNZZMkyhPwKtaSnKmzQy7FyfxxsxdATdSx8yIKjt9/lPn1AaSS0Rrj+T2aTCWRPC9jvyMpU9emjYfndLu5x1Tb2qLx66e70KZ09USkF2t+cxWS3Kmxw6RMzv9cnrdIWdmxkm+MZst2nAjPHdrh1XET6vUvKzZbKaQ/Zljxuu1KIwn8+AJMOSopRx1yr78l8qdfLNMmcNj3quOWeULHnh3wue2v0hj7vKPwo7x5SSq/TpSyQj9DXdhcUh29DzlHnR/yhj7jdlWFO6DHFXbJbf5w17CkfxRbKPvDyKxQw5Rl0hw54VXW294K7eKb/HGbY/bepimSIEcXRmyR2rtBlfUdYlP5O14Kyw4/ye9rB9kmTJGxvVet6Gw/K1RQgXK75Y5uwRYfvTpiyKGCTiLt8SVR3x5i7fLG9zRci+cPU6j67qPuyyl2xF04PansZS+VpOn+89vvbaiEEqtiHzQh43pRXJMXZhr9aO9J6w5I2J+jx9ve9FuubCBz4ZSjv7izKFCLkBAAAAAAAAAAAAAAAAAAAAAAAAEqUXfyU1gB6zO6RhxcHHDKPjeF6BNHCwFCYwI+B4aXC7LfLfUK/Z50qb1kmV5R1ts0UaPkKaPE0yR3mDXWcFRdL4SZLLKVWUS5XHpfp6qalB8niCx9rsHeuNDR/GIGenx9DWGn6sI0Wad4H00Xqp+d8BDQ5HR8DOmD4OI0C/03q8Ru01DXLkhb4xPaUgW0Mvm6XKjXvUXl3fcdAwlD5kgAbMHC+zPXx4Uv3eI/K5PWH70b3OoTTtNQ3ytocPHznVNB48prwpo2VNdYTszxhWpMEXTlfV5r1y1TdLkgyzSRnFAzVg+jgZ5vD7f+3OgwmpOVaNJceVN3mUbJmhb4w22ywaculsNew7oob9pXL++3FKkmEy5MjLUvrQQmWNGiyf26uabQdCnqe9plFpg/JD9qUNzNeAWRNUvXlf4D2ZOjBPhbPOkjU9xA3gCeRpjXBTfIpdgy6Ypor1u+RubJEkmVPsyj1rhHInFPdRhTjVGdYUOUZdLseoy+VpOCp35VZ5avbIU3tAfldwUKEptUC2IXOVMuYaGabw3/HcldtDHncdXSXv+E/KnFoQst82eLYy04vUvvfvclduld990vc1wyRzxmBZckbLUnCWbEXTZFjsER+br7VarqOrZB9+QdgxaVNulrVgotoPvClPzV7J/3GIjTljiKyFUzqCVNIL5Tq+QRFiDePD0672g0uVMvaakN0mR7ayLrxf7fv/JefRD+VrKQ/qNxzZsuSMkiV3rGyDZ4V9rjvztdXI7/fJMEJ/TqTPvEPOw+/L21wu+T7+ruL3OuU+vjFobNu+fyhtyqKQ5zFMFmWc8x217XhBztLVkrcjgMeUMUipZ31atoEzItRYK+fRlVE9nvjzq/3gUqVNuTnqGc4j7yewHkmGWdYBk4MOuSMEoPQl+7DzZR92fszzmtb+Kvh68rTL11Ihc5ggJkfxhfK1Vqn9wJuS1yUZZtkGzlDq5M/LZOtdwIqvrSZsnzVvnNLn/te/96mWoAA5T+0++Vo/DhHs832vvVZm6+DQaxVOVerkz6tt96sddUsyZw5T6qTPdrmWAAAAAAAAAAAAAAAAAAAAAAAAgEQjlAZIhsysjpCYnmqolyqOBx+rqui40c4wQs9Jz5AuuETyeiSnsyPAJcKNyjGx2aWhwzt+TnC7JY9b8nolq7UjcKc7NdXB7coKadTY8OMLB0pXXNsRiuP1dgTVhHv8OONUfbRXQy6ZKSPMNZFSkKPhV8yV1+WRz+WW2WGTyRL5PeF1ulS781Aiyj1jmKxmpRTmBh1rLq1MUjWJ4ff5VLN1v4rmTgo7Jn3IAKUPGSCv0yWfxyuzwy5ThDAaSXI1taph39F4l9szfr+qPtqjwfOnhx1iMpuUM75YOeOL5fN45G13yzCbZLZbZZwUvOZzhw9VazlWFTaURpJyxg1X9uih8rQ7ZbZZZbIm56ttS3mNsscOC9ufNjBfI689X16nW36vV+YUe9i9CeiOJWuoLFlDpTFXSZL8Po/8rmb5fW6ZrOkyrN2HMnkaDstTFTqURn6v2na+pPSZX4lQwzClz7pTkuRzNsrvdcqwpMqwpoQNTImkbfcrsg6aKZM1NewY28Dpsg2c3vF4nR1BPIYtXYY5fJBcIrXv+4fswy6QyRE6AM+wOJQy/jqljL9OfnebfO4WGSZLR82mnu1Vfk+bfM0VMmeEDv8w2TNDBuV4W6vU0CmUxlmyTI6RC2ROLwx9Llua0qbdqtSpi+Rrb5Bhsclky+i2xrbdr0o+dxSPJjGcR1YodcIN0b0Pag/I13QsofVYC87qUov7+KaErpkM7oqtYUNpJCn1rBuVMm6hfM5GmeyZcXvfeuoih/XZiqbKVjS1y/HmTY/KdeTjUJq+3vfcVbtkzggdSiNJjlEL5Bi1QL72eslkjuq9BwAAAAAAAAAAAAAAAAAAAAAAACRC7HcMAkgur0fauDbob3qXJDnbpbIowgrMFik1LTiQpqFeagsfCtAjVquUktoRhhNVIE2V1NQYfOx4qdTa0v1cm71jrZNv8O8c2oMzTmt5jWq27u92nNlmkTU9pdtAGr/Pp2MfbJa33RWvEs9IaQMLuoSvnG6hNJLUsL9U9ftLux1ntttkTUvpNpDG5/aobPkm+X3+iOP6UvPRSlVH8R6TJJOl431mSbEHBdJ0p+HgMfk8nohjDLOp4znsFEjj83ijXqe3Wkqr5G7p/nPUbLfKkuoICqRpOV4dYQbQPcNkkcmRLXNqQVRBHH6fV63b/hxxjKt0tdr3vxnV+iZ7psypBTLZ0noUSCNJvrYaNa/7P/l9kd/v0r8fb0quTCm5SQukkSS/u1VNax+W39v99wLDmiJzar5MjuweB9Kc4Cpb06v5AT6Pmtc+LH+EYDCp4/k2p+ZFFYrhPLxCzsPL41NfT3na5TyyIqqhziPvJ7gYyVoUHN7ma2+Qp+5Awtfta86SZfL7fRHHGGabzKn5Xd630byHwnFVbO72Go76XH247zlL3uv2+ZIkkyO7y3vPXRkmUAwAAAAAAAAAAAAAAAAAAAAAAABIAEJpgFOJyyWtXC7VhrmBfeumjnCamM7plNaulHx9d/N+F263tCHEDbY+n/TRhq4BPN2pq5W2bIxPbTil1Ww7oNqdh3p9Hp/Hq+Ort6m1vDYOVZ3Z0ocOCGq7mlrkamhOUjWJVbFuhxoPHev1ebxOl8qWb+qXz1PN1v2q2X5A/lj36Sj5XG5Vb94X87zmsirV7T6cgIpC8/t8qly/K+bnob22QZUbdieoKiC01i1Py1O9s/tx2/+s9oNv90FFHTxV29W84bfyuVv7bM3e8tYdUPPaX8nnauqzNdsPLJWvLT7fR7xNZWpa+0v5nI3dD+6Gs3SNWjY/EYeqeq/94NJuAz/8HqdcpXEK+InAWjQtqO0q/0hS/wmYixdvU5mcB5fGPK+9ZJlcZWt7vrCnXW17/97z+Z301b7nbTwq54G3Yp7XXrJMztLVCagIAAAAAAAAAAAAAAAAAAAAAAAACI1QGuBU4PFIB/dJb70uVVWEH9faIq16X2qPMpimvV36YJnU2NCzuuIRQtDUKC1f2vHPUI6XSh+t7wioiUZttbTyvY7nDJBUtWmPyt7fJE9bjIFN/9Ze16gjb36oppLjca7sDGQYShuUH3SoubQqScX0AZ9fx1dtVcXaHfK63D06RWtFrUr+tVqtFf03EKl68z6VvrtBrqbEBEnU7T6suj3RB8zU7y/Vsfc3yR/t50acNJdWqmLdzqjXbauuV+m7G+XzJDEUDv2e69g6eRqOxuVc3tZqNa3+mZyHl0U5w6/Wrc+oae2veh2C4murVdu+f3YbfuI+tkGNy34gd/WuXq3Xl9yV2zpqrtzWq/P4vW65jm2Qq3xz5HHuZjV9+At5myP8mSAGnurdalj2A7mrdvRovs/dqpatz6hlw28lf//Yz3wtlXJXbIk4xnV8g/yetoTWYc4aLnNqXtAxd/mmhK6ZTK07XpDreHTBoH6fV217XldrHIKM2vf9XW37/tltEFF0+m7fa93+vJxHPoj6fO0ly9W6+cle1QQAAAAAAAAAAAAAAAAAAAAAAADEypLsAgCcxOuVPG7J7ZZamjvCYqqrpPKyjr5o1FZL7/xLmjRVGjZCMoXInvJ6pSMl0vbNkrNnQR2SpBXvSgUDpMKBUl6+lJUjWaLYVnw+qbqyo4bDB7sPtzm4T6qvlSadLQ0oCj3G2S7t2y3t2SX5fVIq2xs+1ny0Ui1lVcocOVhZo4bIkZcpI9R74998Ho/aKutUt+eIWspO49CUPpYyIEdmuy3oWHNpZZKq6Tv1+46qseSYssYMVeaIQbJnZ8gwjLDjvS63WstrVLf7sNoq6/qw0p5rLa/RoddXKH1IobLHDFVKQbZM1sj7sKfdqdbyWjUeOtbt+SvX71JbVb3yp46RLSM15Ji2qnpVb92v1uPVPXoM8dCw76icdY3KP3us0oryQo7xtLtUt7tEtTsPST6/LJaUPq4SpxJnyTI5S5bJlFog64ApsuSNliVntMzpYb4PdeL3eeSp3S/nkRVyla2VvK6Ya3Af36j68s2yDZkr+7ALZMkdLcNsizjH7/fJ23BE7qodcldslad6l6Towgx9rVVqWvk/MmePlGPUZbIWTJLJkR15PY9Tntq9ch3bKF97fXQPLI58bbVqWv0zmbOGyzFqgawDJndbsyT52us7nqPK7XKXfyS/uyWq9byNR9Tw3ndlGzRb1qJpsmQPl2HPlGFJkWHEnjvrb69T06oHZMkZLfvoK2QtmCiTLT38eL9P3qZjch1dLeehdxIe7tIT7Qfekq1oWth+5+EVCa/BNnBGUNvvccpduT3h6yaNz6Pmtb+WfeSlShl7bcj3gN/nlad6p1p3vihv/aG4Ld224y9yHl4h+7DzZckdI3N6oQxrard7VTh9s+/51bLpUbkrtytl/CdlTh8YcpS36Zjadr0s17F1PXosAAAAAAAAAAAAAAAAAAAAAAAAQG8Yfn93aRBAP/XSn5JdQf9nsXaExqSld/zucUvNTR2BMB5PAhY0pPT0jvVSUiWrVTJbOkJnPG7J5ZKaG6XGRsnbw/VTUqX8AZLDIZnNHedsrJdqqrsPtzmd3XBTTMP3PPdmggo5NRgWs1LysmRJdchst8qwmOVzeeR1ueVublV7TeOZfT0lSMGM8cqdUBxoe50u7X9p2Rn3XJtsFjnysmVJsXdcfyaTfG63vE63XI0tctY1JbvE3jMke06mbBmpMtusMtmskiSf2yN3S5tcDS1yN7f26NT27AzZczNlcdjk9/nkaXWqraZBnpb+FYpgSXUoZUCOLCl2GWaTfC63nHXNaquu79fX/LjPXxHT+NrXbk5QJYjIbJc5rUCm1AKZ7FmSxSHDbJf8Xvk9bfK7W+VtOi5v41HJH2WwYbQMs8zZxTKnFsiwpcmwpkp+n/zudvldTfI2l8vbfFzyueO2pCltgMyZw2Sypcmwpkkmi/yedvnb6+VtKZe3sbQjlLAfMaUWyJw1TIYtXaaTa3a3ytdaLW/TMfldjckuMyxz5lCZMwbJsKbLsKVKPo98rmb5nY3y1B6Q392c7BIjM0zKufqxkEEi3pYqNbz9nwkvIfPCH8uSXRxou45vVPPaXyV83f7BkDlnpCyZQ2TYMuT3uuRrq5Wndp/8zoZkFxe7Ptj3zJlDZM4eKZM9Q5JJPme9vPUlHfs4Tnm5n3w22SUAAAAAAAAAAAAAAAAAAAAAAAD0iCXZBQBIII9bOl7Whwv6O0JvmhMYqNDWKh0tSdz5cUbwe7xqrahNdhlnnPQhBUHt5rKqfh3OkSg+l0etx6uTXUZi+SVnbaOctfEPXHDWN8lZ3/+Dezyt7WoqOZ7sMnC68jrlbSztCGPpa36vvHUH5K070GdL+loq5Wup7LP14sHXWiVfa1Wyy+gxb+PRUzoMwzpwRshAGklyHvkg4eubUnKDAmkkyVX+UcLX7T/8fb5PJFQf7HtJ29MBAAAAAAAAAAAAAAAAAAAAAACACEzJLgAAAJz+bFnpsmWkBR1rLj21AgYAAED/Z1hTlTr++pB9fp9XriPvJ7wGa9H04HX9PrnPqFAaAAAAAAAAAAAAAAAAAAAAAAAAAKcDS7ILAAAAp7/0IQOC2j6vTy3HqpNUDQAAOB2YUgtkyR0jSTIsKTKlFcg2eK7MqXkhx7uPb5SvrTbhddmKpgW1vXUH5Hc2JnxdAAAAAAAAAAAAAAAAAAAAAAAAAIgnQmkAAEDC1e44qNodB5NdBgAAOI1Y8scrffqSqMb6/T617flbgivq0PThz/tkHQAAAAAAAAAAAAAAAAAAAAAAAABIJFOyCwAAAAAAAEgkZ8kyeRuPJLsMAAAAAAAAAAAAAAAAAAAAAAAAADhlEEoDAAAAAABOW576ErVu/3OyywAAAAAAAAAAAAAAAAAAAAAAAACAU4ol2QUAAAAAAAAkgrtym5rX/1byupJdCgAAAAAAAAAAAAAAAAAAAAAAAACcUgilAQAAAAAApwW/xylfe708dQfkOrpK7sqtyS4JAAAAAAAAAAAAAAAAAAAAAAAAAE5JhNIAAAAAAIBTjuvIB6o98kGyywAAAAAAAAAAAAAAAAAAAAAAAACA05Ip2QUAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAPoPQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgglAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEEAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAggFAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEAAoTQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgABCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYTSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQQCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCAUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQAChNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAAEJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIMv9/vT3YRAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAID+wZTsAgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA/QehNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAAEJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIIpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIIJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABBAKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIIBQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABAAKE0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIAAQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGWZBeA+Cv+7j+TXUKPlTxwVeyTPNvjXwhwKrFM6vlc34H41QGcikyjTmocS1oZQP8wqMcz/Qd/Esc6gFOPMfL7gd9dD49OYiVA8tm+uT/w++1j7UmsBEi+P+x1JrsEAAAAAAAAAAAAAAAAAAAAAACAHjMluwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQP9BKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIIBQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABAAKE0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIAAQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgglAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEEAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAggFAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEAAoTQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgABCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYTSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQQCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCAUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQAChNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAAEJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIIpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIIJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABBAKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIIBQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABAAKE0AAAAAAAAAIAz3lNPPSXDMAI/Tz31VLJL6pfuvffeoOdp+fLlyS6pX9uyZYvuvPNOTZkyRbm5uTKbzUHP3+lo+fLlQY/x3nvvTXZJQJDi4uLA9VlcXJzsck457e3teuSRR3TVVVdpyJAhSklJ4T0PnIH4TggAAAAAAAAAAAAAAACcGSzJLgAAAAAAAAAA0L22tjZt2rRJ+/btU11dnVpaWpSSkqLMzEwNGzZMo0aN0siRI2UykUUOJJvP59Pdd9+tX/ziF8kuBQDiZtu2bbrmmmt0+PDhZJcCAAAAAAAAAAAAAAAAAAD6AKE0AAAAAAAAANBP+f1+vf766/r973+vd955Rx6PJ+L4jIwMzZgxQ/Pnz9cnPvEJzZo1i5AaIAkeeOCBPgmkeeqpp3TLLbcE2vPnz9fy5csTvi6AM09NTY0WLFig48ePJ7sUAAAAAAAAAAAAAAAAAADQRwilAQAAAAAAAIB+6PDhw7r11lv13nvvRT2nqalJy5cv1/Lly3Xffffptdde08KFCxNYZf+xePFiPf3004H2oUOHVFxcnLyCcMaqra3Vj3/840DbMAzdfPPNuuKKK5Sfny/DMJJYHQD0zIMPPhgUSDNixAjdcccdmjBhgux2e+D4yJEjk1EeQjj584bQst679957dd999wXay5Yt04UXXpi8ggAAAAAAAAAAAAAAAACgDxBKAwAAAAAAAAD9zMGDB3XBBReorKysS5/NZtOIESOUlZUlp9Op2tpalZWVyefzdRnr9/v7olwAJ/nrX/+q9vb2QPuHP/xh0E3sAHAqevbZZwO/5+Xlad26dcrPz09iRQAAAAAAAAAAAAAAAAAAINEIpQEAAAAAAACAfsTtduuaa64JCqQxDEM33XSTbr/9ds2dO1cWS/C/2m1ubtbGjRv1xhtv6KWXXtKBAwf6umycIe69917de++9yS6jX/vwww+D2rfffnuSKgEQSklJSbJLOOUcOnRI5eXlgfZ1111HIA0AAAAAAAAAAAAAAAAAAGcAQmkAAAAAAAAAoB/5/e9/r507dwbaDodDL7/8sq688sqwc9LT0zV//nzNnz9fDzzwgN5//309/PDDMpvNfVEygJPs3bs38Ht2drYGDRqUxGoAoPdO3tckaeLEiUmqBAAAAAAAAAAAAAAAAAAA9CVCaQAAAAAAAACgH3n66aeD2j/60Y8iBtKEciKgBkDfq6+vD/yemZmZvEIAIE5O3tck9jYAAAAAAAAAAAAAAAAAAM4UpmQXAAAAAAAAAADoUFtbq40bNwbaJpNJt912WxIrAhCr9vb2wO+GYSSxEgCIj5P3NYm9DQAAAAAAAAAAAAAAAACAM4Ul2QUAAAAAAAAAADqUlZUFtfPz85WXl5ekavrGnj17tHbtWh07dkxms1kDBgzQ7NmzNWHChGSXFsTn82ndunXasmWLampqlJaWpoEDB+qCCy5QUVFRXNaorKzUmjVrVF5erpqaGqWnpweejxEjRvTq3LW1tdq0aZP279+vhoYGeTwepaamKj8/XyNGjNDEiROVk5MTl8cRTltbm7Zs2aKdO3eqrq5ObW1tSklJUWZmpoqLizV+/HgNHTo0oTV01traqpUrV+ro0aOqqqqSw+HQgAEDNHHiRE2dOrVH5/T7/XGuEv3JkSNHtHr1ah05ckR+v18FBQU6++yzNW3atFM2qGP79u3atWuXjh8/rubmZhUWFmrRokWyWq0R5yVyz+qsL/bgnnC73Vq9erW2b9+u+vp6ZWZmaujQoZo/f37c9tQjR45ow4YNqqioUF1dnbKyslRUVKRzzz23zx57ovY1r9erNWvWqKSkRMePH5fX69XEiRN19dVXR5xXX1+vVatW6dixY6qurg5ce9OmTdPYsWPjXmdZWZnWr1+v0tJSNTU1qbCwUOeee67GjRvX7dzt27dr/fr1qqiokM1m09ChQ3XxxRefFt/vmpubA69DeXm5HA6H5s+fr+nTpye7tCDbt2/Xhg0bVF5eLqvVqsGDB2vu3LkqLi5OdmmS+v563r9/v9auXRv4c8fgwYM1ffr0uH/3TuT+WFpaqh07dujQoUNqaGiQJOXm5mrw4MGaN29eQr7TtrW1acWKFdq9e7eam5uVk5Oj4uJizZ8/X2lpaXFfDwAAAAAAAAAAAAAAAOgvCKUBAAAAAAAAgH6iqakpqO31euO+RmlpqYqLiwPnHjt2rPbs2RPzeV5//XUtXLgw0P70pz+tF154IWhMSUlJUDDBF77wBT311FOSpKVLl+r73/++NmzYEPL8EyZM0IMPPqhrrrkmbA1PPfWUbrnllpB9kQIRhg8frpKSkrD9J/P5fHrkkUf0wAMPqLS0tEu/YRi6/PLL9dBDD2nSpElRnbPz+Z977jn93//9nzZu3Bj2xv8JEybo7rvv1qJFi2QymaI+/7vvvqsHH3xQ7777rnw+X9hxhmFo3LhxWrhwoe68886w4TD33nuv7rvvvkB72bJluvDCCyPWsH//ft1///165ZVX1NLSEnHsoEGDtGDBAi1ZskRz586NOLY3tmzZoh/96Ed666231N7eHnLM4MGDdeutt+ruu+9Wenp62HN1fk5Odvjw4bBBJYTXBCsvL9eVV16pjz76KHBs1qxZevnllzVlyhTV19dLkrKysnT8+HGlpKTEdP6tW7cGBQ3Nnj1ba9eu7TLu5Ndr/vz5Wr58uSRp/fr1+u53v6v33nsv5PmHDh2q+++/X4sXL46qnuLiYh0+fFhSbHuSFNv7cPny5brooosC7R/96Ee699575fF49Mgjj+gPf/iDduzY0WXe9ddfr+zs7C7HE7Vndd7Pn3zySS1evDghe3Asz32459rpdOpnP/uZfvWrX6m2trbLPLPZrBtuuEEPPvighg8fHlVdJ3O5XHrkkUf06KOPaufOnSHHGIahGTNm6Ic//KGuvfbamNfoTqSQpVtuuSXk5++J6+uEcM9fbW2tfvrTn+q5555TeXl50DmmTp0aNpRmxYoVuu+++7RixQp5PJ6QY0aPHq077rhDd955p2w2W6SHGBDumlizZo3uv/9+LV26NOR3sosuuki//e1vQ4Z5vPzyy7rnnntCvn5ms1k333yzHnroobiH01x44YV6//33uxx///33I76mnV+7UOc68X7fuXOn/vu//1t/+9vf1NraGjTnG9/4hiZMmKBBgwYlfd9+5ZVX9MMf/jDka2AYhs455xz9/Oc/17x588LW0HkPPVm44yd09znf19fz8uXL9b3vfU9r1qwJOW/q1Kn6yU9+oquuuiqqdcJJxP7o8Xj0zjvv6KWXXtI777wTeHyhGIahuXPn6u6779bChQujDoxbvHixnn766UD70KFDKi4uVkNDg+677z49+uijIb/H2mw2ffGLX9T999+v/Pz8qNYCAAAAAAAAAAAAAAAATiXR/5/rAAAAAAAAAICE6hwAUFNTo/3798d1jSFDhgTdbLp3716tWLEi5vM89thjQe0lS5ZEPffuu+/WggULwgbSSNKuXbt07bXX6sc//nHMtcVLY2OjLr/8cn31q18NGYYgddxw/NZbb2nOnDl66623Yjr/vn37NH36dH3hC1/Qhg0bIt68vGvXLt1yyy0699xzVVVV1e25/X6/vva1r+nSSy/V22+/HTGQ5sT43bt368EHH4z5cUTy7LPPatKkSXr22We7DaSRpGPHjunJJ5/Ub37zm7jVcDK/36/vfOc7mj59uv72t7+FDaSRpLKyMv34xz/WmDFjtHLlyoTUgw67d+/WvHnzggJprrzySi1btkxDhw7VokWLAscbGhr04osvxrxGb/as//3f/9W8efPCBtJI0tGjR3XLLbdoyZIl3b7fkq2urk4XXXSRvv71r4cMpAknkXtWKIneg3uqrKxMc+fO1T333BMycEHqCJV74YUXNHPmzKDrOhpr167V+PHjddddd4UNpJE6HvuGDRu0cOFCXXvttVHtsf3B5s2bNXnyZD300ENdAmnCcblcWrRokebPn6/33nsvbICH1BGE9p//+Z+aNGmSdu/e3eM6H3nkEZ133nl64403woYELlu2THPnztXq1asDx3w+n5YsWaIbbrgh7Ovn9Xr11FNP6dxzz1VFRUWPa0yGP/3pT5o2bZqef/75LoE0J6SkpCR13z7xHej6668P+xr4/X6tWrVK5513Xp9/10zG9fzzn/9cF198cdhAGqkjsO/qq6/Wl7/85R4H5yVqf/zsZz+rT3ziE/rjH/8YMZBG6nhtP/zwQ33qU5/SDTfc0Ku9cefOnZo6daoefvjhsOc5ESI2d+7cmALmAAAAAAAAAAAAAAAAgFMFoTQAAAAAAAAA0E+MHDlSDocj6Nh3vvOdHt8YGs6Xv/zloHbnm367U1ZWpjfeeCPQHjlypC6++OKo5n73u9/Vz3/+80A7IyNDEydO1MyZM1VQUNBl/D333KNXXnklpvriwe126+qrr9a7774bODZgwABNnz5dU6ZMUVpaWtD41tZW3XDDDd3eKHvC2rVrdc4552jLli1Bx81ms0aPHq3Zs2frrLPO6nI9rFmzRvPmzes25OGee+4JGeySm5urqVOnau7cuZo8ebIGDhwYVb098fbbb+sLX/iCnE5n0PHU1FSdddZZmjt3rqZNm6bi4mKZTIn/zxV+v1+LFy/Wz372sy6hIfn5+Zo+fbomTJjQ5TkvLy/X5Zdf3meBF2ea1atX69xzzw26kfvWW2/V3/72t8D77Pbbbw+aE+ue1dbWpueeey7QzsjI0Gc/+9mo5v7+97/XN77xjUAoRUpKiiZMmKBZs2Zp0KBBXcY/9thj+tWvfhVTfX3J4/Ho2muvDQpaysnJ0ZQpUzRlyhRlZWWFnJfoPauzRO/BPVVfX69LL71UmzdvDhwbPHiwZs6cqUmTJslutweNr66u1rXXXqvGxsaozv/3v/9dF110kQ4dOhR03Gazady4cZo9e7bGjx8vi8XSZd7FF18cMWirPzh69KgWLFigY8eOBY4NHjxYM2bM0NixY7tcP5LkdDp11VVX6dlnn+3SN3DgQM2cOVNjx46V1WoN6tu3b5/OO++8mEOBJOmvf/2r7rzzzsD7Pj09XZMmTdL06dO7BAg2NjbquuuuCwRw3HHHHUF7VH5+vqZNm6bJkyd3eXx79uzR5z//+ZjrS5Z//etfWrRokVwulyTJZDJp1KhRmjVrloYPHy6z2RwYm8x9+7//+7+DvgOlpqZq4sSJOvvss7u8fj6fT/fcc48eeOCBmOrrqWRcz88++6zuvvvuwJ8n7Ha7xo0bp+nTp4f87v2HP/yhy58VopHI/THU3lZQUKCzzjpLc+bM0dSpU5Wfn99lzCuvvKKFCxf2KCyupKREl1xySdDnyvDhwzVr1ixNmDAh6HqXpAMHDui6666LGDIEAAAAAAAAAAAAAAAAnIoIpQEAAAAAAACAfsLhcOiSSy4JOvbKK6/okksu0apVq+K2zoIFC1RcXBxov/TSS6qrq4t6/hNPPBG4UVuSvvSlL8kwjG7nrVixQg8++KAkac6cOVq6dKlqa2u1fft2rV+/XhUVFVq2bJnGjx8fNO/rX/96yBs8FyxYoLfffltvv/22Lr/88qC+5557LtDX+edPf/pTt7U+8MAD+uCDDyRJN910k7Zu3aqKigpt3LhRW7ZsUU1NjZ544gllZmYG5jQ3N+vuu+/u9tzl5eW69tprVV1dHTg2ZcoUPf/886qvr9e+ffu0du1a7dixQ3V1dXrhhRc0atSowNgDBw5o8eLFYcOKysrKAs/zCbfffrt27typmpoabd68WR9++KG2bt2qY8eOqba2Vv/4xz/05S9/OWwgRU/cddddQTVedNFFWr58uRobG7Vjxw59+OGH2rRpkw4dOqTm5matXr1aP/jBDzRy5Mi41XCy3/zmN3rmmWeCjp1//vlatWqVKisrtXHjRu3cuVNVVVV67LHHlJubGxjX1tam//iP/wgKUjhh0aJFQddXYWFhoK+wsDDsdQjp1Vdf1SWXXBIIc5CkH/zgB/rjH/8YFLpx1lln6fzzzw+0V65cqd27d0e9zksvvaT6+vpA+3Of+1yXUJNQ9u/fr2984xuSpAkTJujll19WbW2tdu7cqXXr1qmsrEwbN27UvHnzgub98Ic/VE1NTdT19aXHH388EEhz6aWXatWqVaqurtaWLVu0ZcsW1dXV6e2331ZKSkpgTqL3rFASuQf3xre+9S3t3r1bFotFX/3qV7V//36VlpZq/fr12rZtm2pqavTQQw8FBUqUlpbqJz/5Sbfn3rFjhz7zmc+ora0tcOz888/XP/7xDzU0NGj37t1au3atdu3apdraWj366KNB+826dev0zW9+M26P9eT96tvf/nZQ37e//e2Q+9qiRYsinvPb3/62KisrZTab9ZWvfEX79u1TaWmpNmzYoD179qi2tlYPPfRQ0Jz/9//+n955552gY5/85Ce1ZcsWHTt2TOvXr9eePXtUXl6un/3sZ0HXbk1NjW688UY1NzdH/bgbGhq0ZMkS+f1+jR07Vq+++qpqamq0bds2bdy4UdXV1XrhhReUk5MTmFNRUaGf/vSn+stf/qI//OEPkqTLL79ca9asUWVlpTZt2qStW7equrpa9957b9B3pnfeeUd///vfo66vO7/4xS9Cfs5MmTIl7OdRNK+d1BEY5vP5lJWVpV/84heqqKjQ/v37tW7dOpWUlKisrEz/8R//ISm5+/b9998vqeMz+JlnnlFNTY22b9+ujz76SNXV1XrzzTc1YcKEoHnf//739eGHH3Y539SpUwPP0c033xzU99BDD0V8TkNJxvX8ta99TVJHsM+vf/1rVVZWavfu3dq4caMqKyu1atWqLp9jjz76qF544YWo15ESuz9KHQFPd9xxh/75z3+qqqpKlZWV2rFjh9asWaPNmzerqqpK+/bt0/e+972gAKh3331Xv/71r2N6LJL0xS9+UeXl5UpJSdE999yjsrIylZSUaN26dYHvi9/5zneC3s8fffRRzAFMAAAAAAAAAAAAAAAAQH9n+OP9V+wi6Yq/+89kl9BjJQ9cFfskz/b4FwKcSiyTej7XdyB+dQCnItOokxpdbyoDzixd/3b3aPkPRnfjAHC6MkZ+P/C76+HRSawESD7bN/cHfr99rD3CSOD094e9zh7PXblyZdBNvCcbPny4Lr/8cs2bN0+zZ8/WhAkTZDL1LHv8f/7nf/T973/8Ofa///u/gZtWI/H7/Ro5cqRKSkokSRaLRUePHlVRUVGXsSUlJRoxYkSX44sXL9bjjz8us9kcco2qqiqdffbZQQEgr732mhYuXBi2rsWLF+vpp58OtA8dOhQUvNOdp556SrfcckvQMcMw9Oijj+pLX/pS2HkffPCBLrzwQvl8PkmS1WpVWVmZCgoKws75xCc+oTfffDPQXrJkiX7zm98E3aTbWX19va666iqtXr06cOyVV17Rpz71qS5jf/e73+nOO+8MtO+55x7dd999Yc99submZtXV1Wno0KEh+++9996gcy1btkwXXnhhl3E7d+7UxIkTA+2LLrpI77zzTlTXq8/n0/79+zV27Nioao5GaWmpxo4dGxT28IUvfEFPPvlk2EClkpISnXfeeSorKwscW7hwoV577bWIaxUXF+vw4cOSOt6zJ94ridD5up0/f76WL1+esPWisXz5cl100UWB9o9+9CPde++9Xcb97ne/09e+9rXAe8dsNut3v/udlixZEvK8f/7zn3XTTTcF2v/5n/+pX/ziF1HVNH/+fK1YsSLQXr9+vWbOnBlybKjrYcGCBXrllVeUmpoack5ra6vOOeccbdmyJXDsV7/6VSDQJpTeXCfRvg+lrq/HCXfddZcefvjhqNZL9J4l9e0eHMtz3/m5liS73a5XXnlFV155Zdh5f/rTn/T5z38+0B4wYIBKS0vDPmcej0fTpk3T9u0f/zev++67Tz/84Q8jhr6VlZXpoosu0r59+wLHNm3apGnTpoWd0xOdX58nn3xSixcv7nZeqOfPYrHo+eef1w033NDt/PXr12vOnDlBgUbdfaZt3LhRF198sRobGwPHvvGNb+hXv/pV2DknXxMnnHvuufrXv/4VFHx0svfffz/ofZednS2Hw6Hy8nLddddd+uUvfxn2tev8vETz2dITJ6/fk8+HCy+8UO+//37QsaKiopABgqEkc98ePny4Vq1apcGDB4ec09raqssvvzwo9HHSpEnaunVr1K9bpL03lGRezzk5OVqxYoUmTQr93xS9Xq8+85nP6OWXXw4cKyws1P79+5Wenh5yTl/tj5L04Ycfatq0aUFhM5Fs3rw5KPRu8ODBKikpCQq866zznyUkKTc3V2+++aZmzZoVdt5PfvIT/eAHPwi0zz77bH300UdR1QkAAAAAAAAAAAAAAACcCnp2twIAAAAAAAAAICHOO+88/fCHPwzZd/jwYT322GO69dZbNWnSJGVlZemSSy7Rj3/8Y23YsCGmdW699dagmz8fe+yxqOa9/fbbQTfwX3311SEDacKZOXOmHn300bCBNJJUUFDQ5Tl44403ol4jXr7+9a9HDEOQpPPPP1833nhjoO12u/Xuu++GHb9mzZqgcIdPfOIT+v3vfx/xRlyp42b3l19+WRkZGYFj4W7s3rt3b1D7jjvuiHjuk6Wnp4cNpIlF5xpuv/32qAOUTCZTXANppI4AlJMDaaZOnarHH388YthDcXGxXnzxxaAxr7/+elD4A3rm//2//6c777wzECSSkpKiV199NWwgjSRdf/31ys/PD7SfeeYZuVyubtfas2dPULDB2WefHTbYIJRhw4bphRdeCBtII0mpqal64IEHgo4lY8+K1jnnnKNf/vKXUY3tiz0rnETswfHwwAMPRAxckKSbbrpJc+bMCbQrKyu1cePGsONfeumloECa22+/Xffcc0/EPUrqCFp4+eWXg/bXWJ/nvvZf//VfUQXSSNLDDz8cFOBx9dVXdxuyNmPGDD366KNBxx5//HE1NDREXWNOTo7++te/hg2kkTpCU6644opAu76+XuXl5TrvvPP0i1/8IuJr993vfldZWVmB9tKlS+X1eqOuL5meeuqpqAJppOTt24Zh6MUXXwwbSCN17NuvvPJK0Ouwfft2vf3221GvE6tkXc9Sx/f8cIE0Ukcw3HPPPRcU6FhRUaE///nPMa2TiP1RkubNmxd1II3Ucc387Gc/C7TLysq0dOnSqOef8Mc//jFiII0kfec739GQIUMC7c2bN6uioiLmtQAAAAAAAAAAAAAAAID+ilAaAAAAAAAAAOhn7r//fv3617/u9ubL5uZmvffee7rnnns0a9YsTZo0SU888UQg6CGSoqIiLVy4MNDetm2b1q5d2+28zuE1t912W7dzTnb//fd3G2YgSZ/+9KeD2ps2bYppnd5KSUkJGw7U2Wc+85mgdqRaf/WrXwW1H3744W5DB04oKioKCmhYtWpVyJteTw5fkRTV8x1v/aGGE/x+v/74xz8GHXvooYdksVi6nTtv3ryg19fv9+vxxx+Pe41nCrfbrUWLFumnP/1p4FheXp7ee+89XXPNNRHn2u12LV68ONCurq7Wq6++2u2anV+vWPeszuER4Vx22WXKyckJtPt6z4rF/fffH/W+0xd7ViiJ2oN7a/DgwbrzzjujGtvTz4bU1NSg90h3Jk+eHPR5/re//a3fBpykpqbqe9/7XlRj6+vr9fLLLwfahmFEHbjzmc98RnPnzg20W1paYgrX+MpXvqJBgwZ1O+7aa6/tcuzee+/tNoTN4XDo8ssvD7Tb2tq0Z8+eqOtLlvPOO08LFiyIenyy9u0bbrih2yARSRowYIC+9a1vBR174oknYlorWsm8nmfNmqXrr7++23EOh0P3339/0LFYno9E7Y899dnPfjYoBHP16tUxzZ89e7Y++clPdjvOYrHouuuuCzrWXcgOAAAAAAAAAAAAAAAAcCohlAYAAAAAAAAA+qGvf/3r2rdvn+64446oAhEkaceOHfriF7+o2bNn6/Dhw92Ov/3224PanQNnOquqqtLrr78eaA8dOlRXXHFFVLVJUlZWVtQ3M+fm5mrYsGGB9tGjR6NeJx4uvfRS5eXlRTX27LPPDmqHq9Xn8+nNN98MtGfPnq1x48bFVNfJN7FL0gcffNBlTOcb6Z977rmY1oiHzjX86U9/6vMaTti9e7cqKysD7WHDhumSSy6Jev6tt94a1F6xYkXcajuTNDU16eqrr9azzz4bODZixAitXr066Gb7SJYsWRIUiNLdnuV2u/XMM88E2qmpqbrpppuirtkwjC4BWeGYzWZNnjw50K6qqpLT6Yx6rb5SWFioiy++OKqxfbVnhZKIPTgerrvuuqhDtqKtq6amRuvWrQu0r7766qCAo2ic/Dw3Nzfro48+iml+X7nqqquUmZkZ1dgPP/xQLpcr0D7vvPM0duzYqNfqzd594403RjVu0qRJQe2cnJyo318n7xeSdOTIkeiKS6LPfe5zMc/p631bkhYtWhTT2JPre//992NaK1rJvJ5jeT6uv/56paenB9obNmxQS0tLVHMTsT/2RlpamgYMGBBox7ovdg7OiaQvP4cAAAAAAAAAAAAAAACAvkYoDQAAAAAAAAD0U0OGDNFvf/tbVVRU6PXXX9c3v/lNzZw5UzabLeK8jRs3avbs2Tpw4EDEcZdcconGjBkTaP/lL39RU1NT2PFPP/100A21t956q0ym6P818/Tp02Maf/KNpA0NDVHPi4eZM2dGPfbkOqXwtW7bti2oL5Y1Tjg5qEeSdu3a1WXMZZddFtT+1re+pR/84AcqLy+Peb2emjNnTlDwwCuvvKJPf/rT2rZtW5/VcMLatWuD2hdddFHQDejdueCCC2SxWALtjz76KOh9gO6Vl5dr/vz5Wrp0aeDYtGnTtHr16phuyh8zZkxQ4MN7772ngwcPhh3/t7/9LSiQ6MYbb4w65EuSiouLow5GkaLfC5Jp5syZUV//fbVnhZKIPTgeElHXypUr5ff7e7TGCT19nvva7Nmzox7bee+ONuzlhM7hY2vWrIlqntVq7RIYE07n/WHatGlRv786z21sbIxqXjLF8vqd0Nf7tmEYmj9/ftTjhw8fruLi4kC7vLw8IQFBybqeJenCCy+MemxqaqpmzZoVaHu9Xm3cuDGquX21b+/YsUP33XefFi5cqDFjxig/P182m02GYXT5OX78eGBedXV11GtI/fdzCAAAAAAAAAAAAAAAAOhrlu6HoCf+/ve/66GHHlJzc3NCzh/t//wFAAAAAAAA4NRnt9t1zTXX6JprrpEkuVwubdu2TatWrdI777yjt956q0tIRmVlpa6//npt3LhRZrM55HkNw9CSJUv07W9/W5LU0tKi559/XkuWLAk5/vHHHw/8bjKZdOutt8b0ODrfsNmdtLS0wO9tbW0xze2tWGo9uU4pfK2dQwJ+97vf6Xe/+13sxZ2ktra2y7FzzjlHl112md5++21Jksfj0U9+8hP99Kc/1TnnnKNLLrlE559/vmbPnq2MjIxerR+Ow+HQd77zHX3/+98PHHvxxRf14osvasKECbr88ss1f/58zZs3T0VFRQmp4YTDhw8HtadMmRLTfLvdrvHjx2v79u2SJKfTqYqKCg0dOjRuNfYn77zzTtRjL7300m7H7N27V/PmzVNJSUng2GWXXaaXX365R9ffl7/8Zb377ruSJL/fr8cff1z/8z//E3LsY489FtS+7bbbYlqrN3uW1Pf7VjRGjBgR9di+2rNCScQeHA998dlw99136+677469uJNE+zz3tViuv97u3SNHjlRGRkYgbO/o0aPy+/3dhsbk5uaG/d7UWWpqalC7oKAg6vo6z+2P+0Vnsbx+J+vLfXvYsGExf7ZMmjRJhw4dCrQPHTrUJeipt5J1PZvNZo0fPz6mtSZNmqRly5YF2ocOHdIFF1zQ7bxE79vbtm3TV7/6Va1YsSLqdU5WX18f0/j++jkEAAAAAAAAAAAAAAAA9DVCaRLkoYce0v79+5NdBgAAAAAAAIDTkM1m04wZMzRjxgx9/etfV01NjR588EH98pe/lNfrDYzbsmWL/vKXv+imm24Ke67FixfrBz/4gZxOp6SO4JlQoTQffPCB9uzZE2gvWLAg5ht2HQ5HTOOTqTe1+v3+kMdramp6fM5wGhoaQh7/85//rGuuuUZr1qwJHPP5fFq5cqVWrlwpSbJYLJo5c6auvvpq3XTTTSouLo5rbd/73vd0+PBhPfroo0HHd+3apV27dunXv/61JGncuHFasGCB/uM//kNz5syJaw2SVFdXF9TOz8+P+Ryd59TV1Z22oTSXXXZZ1GPDXesne/7554Pa1157rV566SVZrdaYa5OkhQsXqqioSOXl5ZKkp556Svfff78sluD/5HX48OGggJ0JEybo3HPPjWmt3u5Z0Tw/fS0zMzPqsX25Z3WWiD04Hk71z4Zki+X6i8fenZeXFwjx8Hq9ampq6raG3rzG/fW6jZdYXr+T9eW+nZeXF3N9nefEGl4SjWRdz1lZWV2e52jWOVm0z0cir/9//OMfuv7667uEcMbixJ91onW6v58BAAAAAAAAAAAAAACAaJmSXcDpqrm5WVLH3xZcVFQU9x8AAAAAAAAAOCEvL08/+9nP9Nprr8lsNgf1PffccxHn5ufn6/rrrw+0169fry1btnQZ99hjjwW1b7vttl5UfGZKxE3OPp8v5PH8/HytWLFCv/vd7zR69OiQYzwej9asWaMf/OAHGjVqlG6++WZVVFTErTbDMPSHP/xBb775ps4777yw4/bs2aP//d//1dy5c3Xeeedpw4YNcatB+vjf15+QlpYW8zk6zzlxUzi6Z7fbg9qrV6/Wjh07enw+q9WqW2+9NdA+fvy4/vnPf3YZ98c//jHo/cGe1SGWMKC+3LPOZGfS8xzL9cfe3f/0NEysL/ft1NTUmOd0vk46X3vxkKzrub8+H7HYu3evbrjhhqBAGsMwNGfOHH3zm9/U//3f/+nFF1/U3//+d7399ttBP4WFhUmsHAAAAAAAAAAAAAAAADg9xPbXYiFmAwYM0MaNG5NdBgAAAAAAAIAzwNVXX60vfOELeuKJJwLHVq5c2e2822+/XX/+858D7ccee0y/+c1vAu36+nq99NJLgXZRUZGuueaaOFV95uh8Y/DnPve5oJu0e2LQoEFh+6xWq77yla/oK1/5ijZs2KB3331Xy5cv1+rVq9XY2Bg01ufz6bnnntM777yj5cuXa9y4cb2q62QLFizQggULdOjQIS1dulTLly/XihUrdOzYsS5jV61apXPPPVfPPfecbrzxxrisn56eHtRuaWmJ+Ryd52RkZPSqpjPJXXfdpa1bt+qNN96QJFVXV+viiy/WW2+9pVmzZvXonLfddpseeOCBQHjBY489poULFwb6vV6vnnzyyUDbbrdr0aJFvXgUZ6a+3rPOVJ2f57vuuktXXXVVr845cuTIXs3vD9i7Ty99tW+3trbGPKfzddL52ouHZF3P/fX5iMV3v/tdOZ3OQHv27Nl6+umnNX78+G7nGoaRyNIAAAAAAAAAAAAAAACAMwKhNAAAAAAAAABwGvn0pz8dFErT3NyshoYGZWVlhZ1zwQUXaMKECdq1a5ck6U9/+pN+/vOfKyUlJdBua2sLjF+8eLEsFv71cqzy8/OD2tnZ2br00kv7ZO2ZM2dq5syZ+s53viOfz6ctW7bozTff1AsvvKAtW7YExpWXl+uGG27Qli1bZDKZ4lrDiBEjdPvtt+v222+XJB08eFDvvvuuXnnlFS1dujRwo7rL5dKiRYs0Z84cDRs2rNfr5uTkBLVrampiPkd1dXXEc55O/H5/XM/ncDj02muv6cYbb9Trr78uSaqrq9Oll16qN954Q+ecc07M5ywuLtaCBQsCQTdvvvmmSktLNWTIkKD2CZ/61KeUl5cXh0cTP725Ub4nIQM9kcw960zS+XkeOHAgz7Pis3efPMdsNhNKk0R9tW93/ryORudrKzs7O+ZzdCdZ13NDQ4PcbresVmuP1pES83xEq7m5Wf/85z8D7cLCQr355ptRfw+rq6tLVGkAAAAAAAAAAAAAAADAGSO+/0c5AAAAAAAAACCpiouLuxyLJsDgRFCIJNXX1+vFF18MtB977LHA74Zh6LbbbutdkWeoESNGBLX379+flDpMJpOmTZum733ve9q8ebNefvnlQACRJG3fvl1vvfVWwusYOXKkbrvtNr3xxhvasmWLRo4cGehrb2/Xb3/727isM3z48KD2ySE80XA6ndqzZ0+gbbfbVVhYGJfazhQ2m00vvfSSbrjhhsCxxsZGLViwQCtWrOjROU/es7xeb1AY18l7liQtWbKkR2skksPhCPx+cuhXNKqqquJdTkj9Zc863fE8h9bbvfvgwYNqamoKtIcNG9arMCj0Xl/s20ePHlVjY2NMc7Zt2xbU7vyejIdkXc9er1e7d++Oaa2+eD6itWnTJrlcrkD7c5/7XNSBNPv375fT6UxUaQAAAAAAAAAAAAAAAMAZg1AaAAAAAAAAADiNtLS0dDmWl5fX7bwvfOELQcEkJ24OXr9+fdCNsxdffHFQeEh/YTIF/+tuv9+fpErCmz17tlJTUwPt1atXq729PYkVdbjuuuv0rW99K+jYypUr+7SGSZMm6dFHH01IDXPnzg1qL1++PKbr44MPPpDb7Q60p0+fLpvNFpfaziRWq1V/+ctfdNNNNwWONTc36xOf+ITefffdmM939dVXa/DgwYH2E088IZ/Pp+PHj+uf//xn4Pjo0aN14YUX9qr2RMjMzAz8XldXF3SNdWf9+vWJKKmL/rpnnW4uuuiioPZ7772XpEr6l857d6zPS+fxnc93Jjg5tKQ/fC/qi33b7/fHFHZ2+PBhlZSUBNpFRUUaNmxYyLG9+a6ZzOv5/fffj3psa2urNmzYEGibzWbNmDEj6vnxVlFREdQeN25c1HPZSwEAAAAAAAAAAAAAAID4IJQGAAAAAAAAAE4jncMKioqKogrQyM7O1qc//elAe+XKldq9e7cef/zxoHG33XZbfAqNs7S0tKB2a2trkioJz2az6eKLLw60W1pa9OSTTyaxoo+de+65Qe3q6urTpoZx48apsLAw0D58+LCWLVsW9fwnnngiqD1//vy41HUmMpvNeuaZZ3TLLbcEjrW2turqq6/Wm2++GfO5vvSlLwXahw8f1tKlS/XUU0/J4/EEjn/pS18KCmboL4YPHx743e12a/v27VHN2759u3bs2JGosoL05z3rdDJ48GBNmjQp0D5w4IDeeOONJFbUP8ydOzfo+8vKlSu1f//+qOezdwd/N+oP34v6at9+5plnejz2ggsuCDu2N981k3k9x/J8vPzyy2pubg60Z8yY0eVx96XOwT8ulyvqeY888kgiSgIAAAAAAAAAAAAAAADOOITSAAAAAAAAAEA/UV1drT//+c/y+Xw9mu9yufR///d/QccWLFgQ9fwvf/nLQe2HH35Yzz//fKCdn5+vT33qUz2qLdFyc3OD2ocOHUpSJZF9+9vfDmr/6Ec/0pEjR5JUzcc6B8Dk5OScNjUYhqEvfvGLQce+/e1vy+v1djt33bp1+stf/hJ0rpNvqEfsTCaT/vjHP+r2228PHGtvb9fChQv1+uuvx3Su2267TWazOdB+9NFHg4K0rFarFi9e3OuaE2H69OlB7b/+9a9Rzfv+97+fiHLC6q971umm8/N81113qaGhIUnV9A/Z2dm64YYbAm2/36//+q//imruSy+9pA8//DDQTk9P1+c+97m419jfnfzdqKSkJHmFnKQv9u2XXnqpS0hjKJWVlfrFL34RdOzWW28NO7433zWTeT2vX79eL7/8crfj2tvb9aMf/SjoWKTnoy8UFRUFtVeuXBnVvEceeUSbN29OQEUAAAAAAAAAAAAAAADAmYdQGgAAAAAAAADoJ5qbm3XTTTdp8uTJeu6559TW1hb13Pb2dn3+85/Xjh07go4vWrQo6nPMnTtXU6f+f/buPCyquv//+GtAxQXcQERJxSXN3MVwTUlLcyFNzazcsgxNs1Vts1zu7ttMv2ZZue95V+aSS5lpkvuGgvsOqCBuIIoCoszvD3+e22GdwYEBfT6ui+vic85nec/MmTNH4byoZ7SnT5+ua9euWcxVqFAhq+fLTbVq1bJo//rrrw6qJHMtW7a0CAq6ePGi2rZtqyNHjlg9R0pKipYvX64RI0aku3/w4MFauXKlzGazVfMlJSXpm2++sdjm6+trdT3pmTx5sr777jvduHHD6jFfffWVXWu416BBg1SkSBGjvWfPHg0cODDT5+j06dPq3r27RZ/OnTuratWqdqvrYWUymTR16lQNHTrU2Hbz5k11797dpveut7e3OnXqZLSXLVumU6dOGe2AgACVLVvWPkXbWYcOHSza3377rY4fP57pmJEjR9oc3HO/cuOcBemVV16x+Bw7duyY2rdvr6ioKKvnSE5O1rx58/Tll1/mRIkO8e6778rJ6X8/zv7tt9/0r3/9K9MxISEhacLDXn/9dRUvXjxHaszL7j2mLl26pKCgIMcV8//lxnnbbDarR48emb5/EhIS1LVrV4vwp8cff1xt27bNcEzqa01rgl7u5cjjecCAATpw4ECG+1NSUtS7d2+LoB1PT0+9/PLLNq1jb76+vhb/9li6dKm2bt2a6ZhVq1bpvffey+nSAAAAAAAAAAAAAAAAgIcGoTQAAAAAAAAAkMccOnRIvXv3lpeXlwYMGKDFixfr3Llz6faNiorS999/r5o1a2rx4sUW+7p27arWrVvbtHZgYGCG+1LfFJuX+Pv7q2DBgkZ73rx56tq1q2bPnq0//vhD69atM762bNniwEql+fPnq0KFCkb76NGj8vX11TvvvKPQ0NB0g1JiY2O1bt06vfvuu/Lx8dHzzz+vHTt2pDv/li1b9Nxzz6ly5cr64IMPFBQUpKtXr6bpl5ycrDVr1qh58+batWuXsd3Ly8vihvHsCAsL05AhQ1S+fHn17dtXy5Yty/AYDgkJUc+ePS2CcZycnNS/f//7quFejzzyiMaPH2+xbebMmWrdurW2b99usf369euaNWuWfH19debMGWN76dKl9d1339mtppxw9zjJzte9oQC5ZfLkyRo2bJjRTk5OVs+ePbVo0SKr58jsnDVgwID7qi8n1a1bV35+fkb7+vXreuqpp/Tbb7/p9u3bxnaz2azt27erffv2RnhBbgcj5fQ5C5Kzs7OWLFmiEiVKGNu2bdum2rVr6/PPP9exY8fSHXf+/HmtWrVKgYGB8vb2Vr9+/XT48OHcKjvHNWrUSO+++67FtpEjR6p79+5pAjZiY2M1YcIENW/e3CJopGrVqlkGfzyoUgesPP/88/rwww+1ePFirV271mGfATl53vb29laBAgUUHh6uhg0bauHChUpMTDT2p6Sk6M8//1SjRo0srgdNJpOmT58uk8mU4dwNGjSQh4eH0Q4KClLr1q01depU/f7772k+V1NzxPFcsmRJFS9eXLGxsWrWrJm+/fbbNNeE27Zt05NPPpkmFO7rr7+Wm5ub1WvlhGLFiqlbt25G+/bt22rfvr2mT59u8bpK0vHjx/Xmm2+qc+fOSkpKkqenp9zd3XO7ZAAAAAAAAAAAAAAAAOCBU8DRBQAAAAAAAAAA0nf16lXNnDlTM2fOlCS5u7vLw8NDJUuWVGJios6dO6cLFy6kO7Zx48aaM2eOzWv26tVLw4cPV3x8vMX2Fi1aqGbNmrY/iFzi6emp3r17a/bs2ca2ZcuWadmyZWn6VqpUSeHh4blYnSVPT0+tXr1aHTt2NEJPbty4ocmTJ2vy5MkqUaKEvL295ebmpvj4eMXExGQY6JKZiIgITZw4URMnTpTJZJK3t7fc3d1VpEgRXb16VadOnUpzQ6+zs7NmzpypIkWK2OWxxsXFaf78+Zo/f74kqUyZMvL09JSbm5sSExMVHh6uK1eupBk3fPhw+fr62qWGuwYPHqydO3dqwYIFxragoCA1bdpUZcqUUYUKFZSYmKiwsDAlJCRYjC1SpIgWLVqk8uXL27Ume9u3b5+eeeaZbI39/PPPNWrUKPsWZIXx48fLxcXFuMn+9u3b6t27t27evKl+/fplOb5du3by8fFJ856uWLFimkCGvObbb79Vs2bNjBCayMhIdenSRSVLllSVKlWUkpKi06dPKyYmxhjz0ksvqXr16ho9enSu1Zlb56yHXY0aNbRs2TJ169ZNsbGxku4EU4wZM0ZjxoyRh4eHvLy8VKxYMV29elWXLl3SxYsXHVx1zvviiy8UGhpqEfKxZMkSLVmyROXLl1f58uV17do1nTp1SsnJyRZj3d3d9csvv6hYsWK5XXae0KdPH33xxRe6dOmSJOnKlSv68ssv0+2bm58BOXnerlatmgYOHKiRI0fq/Pnz6t27twYOHKjKlSurYMGCCg8PN95f9xozZoyaN2+e6dwFCxbU0KFD9dlnnxnbNmzYoA0bNqTbP73Artw+nkuUKKGxY8eqT58+unbtmoYOHaphw4apSpUqKlq0qM6cOZPuvyVee+01vfTSS1avk5PGjh2r1atXG2E6V69eVWBgoN5++21Vr15dLi4uOnfunM6ePWuMcXZ21ty5czVo0CBdvnzZUaUDAAAAAAAAAAAAAAAADwQnRxcAAAAAAAAAALjD1dVV9erVy3D/5cuXdfToUe3YsUOhoaHp3kTq5OSkgQMH6q+//lLx4sVtrsHNzS3dm1AHDBhg81y57euvv852IEduq1OnjoKDg9WuXbs0++Li4nTo0CHt2LFDBw8ezDDcoWLFilavZzabdfbsWYWGhmr79u06dOhQmkCaUqVKacmSJerYsaNtD8YGFy9e1MGDB7V9+3aFhISkCaRxdnbWZ599pv/85z92X9tkMmnevHkaNmyYnJwsfzxy8eJF7dmzR4cOHUoTSOPl5aW1a9em+1rBPsaOHauxY8ca7ZSUFPXv318zZszIcqyTk1O656fXXnstzeuc1/j5+WnmzJlydna22H7lyhXt2bNHISEhaQJp5s6dm8tV3pHb56yH1VNPPaVdu3bpiSeeSLPv0qVLOnDggHbs2KHDhw+nG0hjMplUoUKF3Cg117i4uGj16tXq1atXmn1RUVHavXu3jh49mibA49FHH9XmzZvVsGHD3Co1zyldurSWLFkiT09PR5diIafP259++qmGDh1qtK9fv64DBw5o7969aQJpnJyc9Pnnn+vTTz+1au6PP/443WPRWo44nnv37q2vvvpKJpNJkpSUlKTDhw8rODg4w0Ca6dOn27xOTqlataoWL14sV1dXi+2JiYnat2+fdu3aZRFIU7hwYf34449q3759bpcKAAAAAAAAAAAAAAAAPJDy9m/jAgAAAAAAAMBDxMPDQyEhITp58qT+7//+TwEBASpVqpRVY8uVK6e3335bISEh+uGHH+Tm5pbtOvr372/RLlmypF544YVsz5db3Nzc9Oeff2rNmjXq37+/6tevr9KlS6tgwYKOLi1dZcqU0Zo1a7Rx40YFBASoWLFimfY3mUxq0KCBPvzwQ+3bt0/z589Pt9/KlSs1ZcoUdezYUSVLlsyyjvLly2vYsGE6duyYOnfunJ2HksaYMWP0008/qVevXlYFJLi6uqpXr17au3evRo8ebZca0mMymTR+/Hjt2bNHzz33nFxcXDLsW758eY0cOVLHjx9XixYtcqwm3PHpp5/qq6++Mtpms1mBgYGaMmVKlmNfffVVi7azs3Oa81he1a9fP23cuFFNmzbNsE/VqlW1YMECLVq0SIUKFcrF6izl1DkLlqpWraqdO3dqxYoVat26dZavubOzs5o2baoxY8boxIkTFgFPD4pChQppwYIFCgoKUuvWrVWgQIEM+1atWlUTJ07UgQMH9Nhjj+VilXlTy5YtdeTIEU2ZMkUBAQGqXLmy3NzcHB7aldPn7cmTJ2vJkiWqWbNmhn2aNWumTZs2adSoUVbP6+zsrAULFmjz5s0aPHiw/Pz85OHhken1RGqOOJ4/+OADbdiwQX5+fhn2qVOnjlasWKGZM2c6/PhIrW3bttq1a5cCAgIy7FOgQAF1795doaGhevHFF3OxOgAAAAAAAAAAAAAAAODBZjKbzWZHF/Eg8vX1VXR0tLy8vBQcHJyra/t8uDpX17On8HHZ+Au8tw7YvxAgPylQO/tjU07arw4gP3Kqek8jymFlAHlD+WyPNJ/6wo51APmPqconxvc3J1VzYCWA4xV694TxfWB162/KAx5E044l2W0us9msiIgIHTt2TKdPn1ZcXJwSEhJUtGhRubm5qXz58qpXr54eeeQRu605e/Zsvfbaa0Z78ODBVgVD4P4kJydr586dCgsL06VLl3T9+nUVK1ZMpUqVUvXq1fX444+rRIkSNs1pNpt17NgxHT9+XKdPn9bVq1d1+/Ztubm5ycvLS3Xr1lX16tVz/ObjyMhIHTlyRGFhYYqNjVVSUpKKFi0qd3d31apVS3Xq1LHphm57uXHjhjZv3qzTp0/r0qVLcnFxkaenp2rVqqX69evnej3Inr///ltt2rQx2h07dtSqVascWFH2REREaPPmzYqOjlZiYqI8PT3VsGFDNWzYUCaTydHlpZET5yykdePGDW3fvl1nzpzR5cuXlZCQIFdXV3l4eKhGjRqqWbNmlgFBD5orV65o8+bNioqK0uXLl1WsWDGVLVtW9evXV40aNRxdHqxgz/P2vefHVq1aKSgoyGL//v37tXv3bkVHR6tQoUIqV66cmjZtqsqVK2drPXuz9/Hs4+OjiIgISVKlSpUUHh5usf/EiRPavn27IiMjZTKZVK5cOTVs2FC1atWyx8PJcefOndOmTZt09uxZ3bhxQ8WLF1e1atXUrFkzq8IYAQAAAAAAAAAAAAAAANgm4z+7BQAAAAAAAABwOJPJJB8fH/n4+OTamjNnzrRoDxgwINfWfpgVLFhQzZs3V/Pmze02p8lkUo0aNRx+k763t7e8vb0dWkN6ihYtqrZt2zq6DNynB+WcValSJVWqVMnRZVgtJ85ZSKto0aJq3bq1o8vIU0qWLKlOnTo5ugzch9w8b9epU0d16tTJsfnvV24fz9WqVVO1avk3VLlcuXLq0aOHo8sAAAAAAAAAAAAAAAAAHho5+6dPAQAAAAAAAAD5yr59+7Rt2zaj3bhxY9WrV8+BFQFAxi5duqSlS5cabW9vb3Xs2NGBFQEAMsN5GwAAAAAAAAAAAAAAAADyD0JpAAAAAAAAAACG8ePHW7QHDx7soEoAIGtff/21kpKSjHZgYKAKFCjgwIoAAJnhvA0AAAAAAAAAAAAAAAAA+QehNAAAAAAAAAAASdKGDRu0aNEio+3t7a0ePXo4sCIAyNjhw4f1f//3f0a7aNGiCgwMdGBFAIDMcN4GAAAAAAAAAAAAAAAAgPyFPzkGAAAAAAAAAA+h2NhYBQcHS5JiYmK0ZcsWTZ8+XWaz2ejzySefyMXFxVElAoAhMTFRmzdvliRdvXpVe/bs0ZQpU5SQkGD0GTJkiDw9PR1VIgDgHpy3AQAAAAAAAAAAAAAAACD/I5QGAAAAAAAAAB5CoaGheuaZZzLc37hxYwUGBuZiRQCQsejo6EzPWT4+Pvrss89ysSIAQGY4bwMAAAAAAAAAAAAAAABA/ufk6AIAAAAAAAAAAHlLtWrVtGTJEjk58V/IAPI+T09PrVixQsWKFXN0KQAAK3DeBgAAAAAAAAAAAAAAAID8oYCjCwAAAAAAAAAAOJ6rq6tq1Kihrl27aujQoXJ1dXV0SQCQoaJFi6pKlSrq1KmT3nvvPZUpU8bRJQEAMsF5GwAAAAAAAAAAAAAAAADyH0JpAAAAAAAAAOAh5O/vL7PZ7OgyAMAqPj4+nLMAIB/JrfM2nw2WwsPDHV0CAAAAAAAAAAAAAAAAgAeIk6MLAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADkHYTSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJrPZbHZ0EfnBypUrNWHCBMXHx1vV/8KFC0pJSZGXl5eCg4NzuDoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAsI8Cji4gv5gwYYJOnDjh6DIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIEcRSmOl+Ph4SZKTk5M8PT2z7H/hwgWlpKTkdFkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYFeE0tjI09NTwcHBWfbz9fVVdHR0LlSUls+Hqx2yrj2Ej+to+6CUk/YvBMhPnKpmfyzvHzzsLN4/UQ4rA8gbymd7pHltWzvWAeQ/prZrje+TJtzHtRnwAHD54H//xgis7uLASgDHm3YsydElAAAAAAAAAAAAAAAAAAAAAAAAZJuTowsAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAOQdhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyE0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADITSAAAAAAAAAAAeenPnzpXJZDK+5s6d6+iS8qRRo0ZZPE9BQUGOLilPCw0N1eDBg1W3bl2VLl1azs7OFs/f/fL397frfIA14uLi9NVXX6lNmzYqV66cXFxcOH8CD6F+/fpZvPfDw8MdXRIAAAAAAAAAAAAAAAAAwM4KOLoAAAAAAAAAAEDWEhIStGfPHh0/flyxsbG6fv26ihQpouLFi6tixYqqWrWqqlSpIicnssgBR0tJSdHw4cM1ceJER5cC2FVQUJC6deummJgYR5cCAAAAAAAAAAAAAAAAAACAHEYoDQAAAAAAAADkUWazWStWrNDUqVO1bt063bp1K9P+bm5u8vX1VatWrdS+fXs98cQThNQADjBu3DgCafDAOXHihDp16qTr1687uhQAAAAAAAAAAAAAAAAAAADkAu5GAAAAAAAAAIA8KCIiQk8//bS6dOmiNWvWZBlII0nXrl1TUFCQRo8erSZNmmjlypW5UGne0K9fP5lMJuMrPDzc0SXhIRUTE6OxY8cabZPJpD59+mjRokVau3at/vrrL+MLD45Ro0ZZnIOCgoIcXZLdffzxxxaBNHXr1tWUKVO0evVqi+O6Xbt2DqwSd4WHh1sck/369XN0Sfke1xoAAAAAAAAAAAAAAAAAgIdNAUcXAAAAAAAAAACwdOrUKbVs2VKRkZFp9hUqVEiVK1dWiRIllJSUpJiYGEVGRiolJSVNX7PZnBvlArjHL7/8osTERKM9cuRIjR492oEVAffv6tWr+u2334x2jRo1tGPHDhUuXNiBVQEAAAAAAAAAAAAAAAAAACAnOTm6AAAAAAAAAADA/yQnJysgIMAikMZkMqlXr17atGmTrl+/riNHjmjHjh0KCQnR6dOnFRcXp6CgII0YMUJVq1Z1YPV40I0aNUpms9n48vf3d3RJec62bdss2oGBgQ6qBLCf4OBg3bx502j36dOHQBoAAAAAAAAAAAAAAAAAAIAHXAFHFwAAAAAAAAAA+J+pU6fq0KFDRrtw4cJasmSJOnTokOEYV1dXtWrVSq1atdK4ceP0zz//aNKkSXJ2ds6NkgHc49ixY8b3JUuWVPny5R1YDWAf9x7XklSrVi0HVQIAAAAAAAAAAAAAAAAAAIDcQigNAAAAAAAAAOQh8+bNs2h//vnnmQbSpOduQA2A3HflyhXj++LFizuuEMCO7j2uJY5tAAAAAAAAAAAAAAAAAACAh4GTowsAAAAAAAAAANwRExOj4OBgo+3k5KQBAwY4sCIAtkpMTDS+N5lMDqwEsJ97j2uJYxsAAAAAAAAAAAAAAAAAAOBhUMDRBQAAAAAAAAAA7oiMjLRoe3h4yN3d3UHV5I6jR49qx44dioqKkrOzszw9PeXn56eaNWs6ujQLKSkp2rlzp0JDQ3X58mUVK1ZM5cqVU8uWLeXl5WWXNS5cuKDt27crOjpaly9flqurq/F8VK5c+b7mjomJ0Z49e3TixAnFxcXp1q1bKlq0qDw8PFS5cmXVqlVLpUqVssvjyEhCQoJCQ0N16NAhxcbGKiEhQUWKFFHx4sXl4+Ojxx57TBUqVMjRGlK7ceOGNm/erDNnzujixYsqXLiwPD09VatWLdWrVy9bc5rNZjtX+XBKTk7W1q1bdeDAAV25ckXFixdXhQoV1KpVq/s+Vs+ePauDBw8qLCxMcXFxkqTSpUvL29tbTZs2zfH3gq2OHj2q0NBQXbx4UXFxcSpdurTKly+vFi1aqHTp0rlSQ04d10lJSdqyZYvOnj2rc+fOydnZWU888YRatWqV6bgLFy5o69atio6OVkxMjEqUKKGyZcuqcePGOXIeOXnypPbu3avIyEglJCTokUceUcuWLVWxYsVMx5nNZu3evVshISG6ePGiihUrJh8fH7Vu3Vpubm52rzO3Xb58Wdu3b1dUVJQuXrwoV1dXPfvss6pevbqjSzOYzWYFBwcbr0HRokXl7e2tJ598UmXLlnV0eZJy/3g+cOCAdu/erejoaBUsWFDe3t5q0qSJfHx87LpOQkKCNm7cqCNHjig+Pl6lSpWSj4+PWrVqpWLFit3X3GFhYTp48KBOnz6tuLg4FShQQKVLl1alSpXUpEkTubq62ulR/E9cXJw2btyoEydOKCEhQe7u7qpWrZpatGghFxcXu68HAAAAAAAAAAAAAAAAAHkBoTQAAAAAAAAAkEdcu3bNon379m27r3H27Fn5+PgYc1evXl1Hjx61eZ4VK1aoc+fORrtHjx76+eefLfqEh4dbhKn07dtXc+fOlSStXbtWn3zyiXbv3p3u/DVr1tSXX36pgICADGuYO3euXn311XT3ZRbiUqlSJYWHh2e4/14pKSn64YcfNG7cOJ09ezbNfpPJpLZt22rChAmqXbu2VXOmnn/hwoX69ttvFRwcnGHwQ82aNTV8+HD16dNHTk5OVs+/fv16ffnll1q/fr1SUlIy7GcymVSjRg117txZgwcPzvAm+FGjRmn06NFGe8OGDfL398+0hhMnTmjMmDFaunSprl+/nmnf8uXLq127dnrjjTfUpEmTTPvej9DQUH3++ef6888/lZiYmG4fb29v9e/fX8OHD8/05vbUz8m9IiIiZDKZ0t3nyPCa77//Xm+99ZZxTDg7O2vq1Km6dOmSPvroI6PfF198oY8//tjm+bt27aply5YZ7d9//13t27e36JPRsZSUlKTx48fr66+/VkxMTJq5nZ2d1b17d3355ZeqVKmSVfXcunVL69at06+//qp169YpIiIiw74mk0lNmjTR8OHD1blz5wxfP0kKCgrSU089le6+jLbfldXrHx8fr4kTJ2revHkKCwtLt4+zs7OefPJJjRkzRk8++WSm89kq9fk7tYwe35w5c9SvXz+j3a9fP82bN89oh4WFycfHR2fPntXYsWP1yy+/6MqVKxZzdO7cOcNQmuXLl2vcuHHauXNnhs9hnTp19P7776t3795Wny/vfZ1btWqloKAgSdIff/yhf//739q8eXO6Y7p06aJvvvlGjzzyiMU+s9msGTNm6N///ne6x5uLi4veeustjR49WkWLFrWqRmv5+Piku+a8efMsXovUUr92qee697Nz27Zt+s9//qM1a9YoOTnZYsykSZNUtGhRh19rmM1mzZw5U1988UW6z4ezs7Patm2riRMnZhqEl5PXGrl9PC9dulQjR47UoUOH0h3TrFkzffXVV2ratKlV62QkLi5Oo0eP1vTp09P93C9UqJBee+01jRkzRh4eHlbNmZiYqNWrV2vp0qX6+++/FR0dnWFfZ2dntWnTRh999FGW1yj38vf31z///GO0774m586d0yeffKIff/xRN2/eTDOuWLFieuedd/TRRx/dd9gOAAAAAAAAAAAAAAAAAOQ11v/mOgAAAAAAAAAgR5UsWdKiffnyZZ04ccKuazzyyCPq2LGj0T527Jg2btxo8zwzZsywaL/xxhtWjx0+fLjatWuXYSCNJB0+fFjPPfecxo4da3Nt9nL16lW1bdtWQ4YMSTeQRrpzw/Kff/6pxo0b688//7Rp/uPHj6thw4bq27evdu/enWlIxeHDh/Xqq6+qefPmunjxYpZzm81mvfXWW3r66af1119/ZRpIc7f/kSNH9OWXX9r8ODKzYMEC1a5dWwsWLMgykEaSoqKiNGfOHE2ZMsVuNdzLbDZrxIgRatiwoX777bcMA2kkKTIyUmPHjtWjjz6abiBFfvXxxx9r8ODBxjFRtGhR/fbbb3r99dfVv39/FSxY0Og7a9Ysm8NzoqOjtXLlSqNdsWJFtWvXzqqxkZGRatKkiT777LN0A2mkO2FdP//8sxo1aqS9e/daNW/Pnj3Vvn17zZo1K9NAGunOMbJt2zY9//zz6t69u1XHrb2tWrVKVatW1ahRozIMpJHuPBdBQUFq2bKlAgMDdevWrVysMvvWrVun2rVra/r06WkCaTISFxendu3a6fnnn9eOHTsyPS7379+vfv36qUmTJjp37ly26/z444/VoUOHDN//ZrNZy5Ytk5+fn44dO2ZsT0hIUEBAgAIDAzM83pKSkjRhwgS1a9fOIcfY/Rg/fryaN2+ulStXpgmkucvR1xo3b95U9+7d9cYbb2T4Gty+fVt//PGHGjRooDlz5thc2/3I7eP57jVBt27d0g2kudtny5YtatGixX1dex06dEj16tXTpEmTMjy2b968qR9++EFNmjSxOiSwRYsW6t69uxYtWpRpII1057Vdu3atnnrqKQ0ZMuS+zo2bNm1S7dq1NWfOnHQDaSTp+vXr+uKLL/TUU09l+NkFAAAAAAAAAAAAAAAAAPkVoTQAAAAAAAAAkEdUqVJFhQsXttg2YsQIm0MhsjJw4ECLduqbvrMSGRmpP/74w2hXqVJFrVu3tmrshx9+qK+++spou7m5qVatWmrUqJHKlCmTpv9nn32mpUuX2lSfPSQnJ6tTp05av369sc3T01MNGzZU3bp1VaxYMYv+N27cUPfu3bMMvLhrx44datasmUJDQy22Ozs7q1q1avLz89Pjjz+e5njYvn27mjZtmmUwzWeffZZusEvp0qVVr149NWnSRHXq1FG5cuWsqjc7/vrrL/Xt21dJSUkW24sWLarHH39cTZo0UYMGDeTj4yMnp5z/cYXZbFa/fv00fvz4NCE9Hh4eatiwoWrWrJnmOY+Ojlbbtm3tGtbjCMnJyerTp4/+85//GNvKlCmjDRs2GOERnp6eev755439p06d0t9//23TOnPnzrUIAHjttdesen2vXLmip59+WiEhIcY2b29vNWrUSLVr15aLi4tF/0uXLum5557T1atXs5w7vfChMmXK6PHHH1fjxo1Vr149eXh4pOmzdOlSde7cOctQJ3uaPn26unTpogsXLlhsL1q0qGrWrCk/Pz9Vq1YtzXM6ffp0de/e3e6fF/a2d+9ede7cWXFxcca2SpUqqVGjRqpatapFKNJdMTEx8vf319q1a9Psq1ixoho1aqQqVaqkeU527dql5s2bW31evtdXX31l8V4pWbKk6tWrp3r16qU5/587d07PP/+8kpOTlZKSou7du2v16tXG/nLlysnX11e1atVSgQIFLMZu3rxZ77zzjs31Ocq0adMsrksKFSqk6tWr64knnpC3t7dMJpPR15HXGoMGDbK4dihevLjq1q2r2rVry9XV1aJvUlKSXn/9df3444821Zddjjie//Wvf1lcExQtWlS1atVS/fr10wQipqSk6LPPPtO4ceNsXic8PFxt2rSxqLFSpUp64oknVLNmTTk7O1v0P3nypLp27WpVaEx65/Hy5curdu3axjVNiRIl0vT57rvvbApOvFdwcLDat29vBM04OTmpatWq8vPz06OPPmpxvEt3XqN+/fplay0AAAAAAAAAAAAAAAAAyKsKZN0FAAAAAAAAAJAbChcurDZt2ljczL506VK1adNGY8eOVfPmze2yTrt27eTj46Pw8HBJ0q+//qpvvvlGpUqVsmr87Nmzdfv2baP9+uuvp7kxNz0bN25UWFiYJKlx48YaO3asnnrqKeMmfbPZrH/++UeDBg3SkSNHjHFDhw7Vc889l+Zm/nbt2umvv/6SdCdA4N4bvBcuXKiyZcumW0eRIkWyrHXcuHE6deqUJOmVV17RiBEjVKdOHWN/UlKSFi1apHfeeccIxYiPj9fw4cP1888/Zzp3dHS0nnvuOV26dMnYVrduXX300Ufq1KmTxQ3ziYmJWrFihT7++GOdPHlS0p2buPv166dVq1al+7xHRkbqyy+/tNgWGBiot99+WzVr1kzTPzY2Vlu3btWqVav03//+N6unxmrvvPOORUDGU089pc8//1wtWrRIc2N6QkKCQkJC9Pvvv2vRokV2q+FeU6ZM0fz58y22Pfnkkxo3bpyaNm1qPJfx8fH66aefNGLECONG9ISEBL388svav3+/ypcvbzFHnz591KJFC6Pdq1cvnT9/XpJUtmxZLVy4MEcejy2uXbumbt26Ge8X6U7Aw5o1a/Too49a9A0MDNQvv/xitGfMmKE2bdpYtY7ZbNasWbOMtrOzs/r372/V2Pfff1+nTp1SgQIFNHDgQL3zzjuqWrWqsf/69euaOnWqPvroIyUnJ0uSzp49qy+++CLN8Z4eDw8P9ejRQx07dpSfn1+6ITQnTpzQ7NmzNWnSJCMAYf369Zo8ebLefffdNP3r1atnPKfz58/XggULjH0TJkxQvXr1rHrsd61fv16DBg2yCMEJCAjQ+++/r+bNm1ucA2NiYjRz5kz961//0rVr1yRJv/32m8aPH68RI0bYtG56vLy8LI4Xax9frVq1Mp130KBBunHjhgoXLqwPPvhAAwcOlLe3t7H/ypUr2rdvn8WYN954wyKsSJIGDBigESNGWBwjUVFR+vbbbzVhwgQj5CIsLEwvv/yyNm7cmOa8k5ETJ07ok08+kSQ98cQTGjdunFq1amWMT0pK0uzZs/Xuu+8aoVuHDh3S9OnTde3aNf3++++SpJdeekmffPKJxXMSExOjTz/9VD/88IOxbdasWXrrrbdUt25dq+rLyo8//qiEhASdP39evXr1Mra3bdtWw4YNy3BcVq9dTEyM8T4oV66c/vWvf+mFF16Qm5ub0ScsLEzXr1+X5LhrjaCgICMUpWrVqvq///s/dejQwXj/3Lx5U8uXL9d7772nyMhISXeCWN544w01a9ZMlStXtpjP3tcajjiet2zZIunOZ9JXX32lF154wQhgu337ttatW6d3331Xhw8fNsZ98sknatWqlZo2bWrVOtKdELLo6GgVKVJEw4YNU2BgoMVnZmxsrL788kuNHz/euD7Yu3evZsyYoUGDBmU5f8WKFfXCCy+oQ4cOatSokYoXL26x32w2a//+/Zo6daqmT59uHD9z5sxRQECAReiaNXr06KHr16+rdOnS+vTTT9WnTx+5u7sb+6OiovTJJ59o7ty5xraVK1fqjz/+UPv27W1aCwAAAAAAAAAAAAAAAADyKpM5r//JxDzC19dX0dHR8vLyUnBwsN3725PPh6uz7pRHhY/raPuglJP2LwTIT5yqZt0nI7x/8LCzeP9EOawMIG8on3WXDJjXtrVjHUD+Y2r7vxvSkibcx7UZ8ABw+eB//8YIrO7iwEoAx5t2LCnbYzdv3qwnn3wy3X2VKlVS27Zt1bRpU/n5+almzZpycnLK1jr//ve/jZvuJembb77RW2+9leU4s9msKlWqGDeZFyhQQGfOnJGXl1eavuHh4Wlu7pakfv36aebMmRneUH3x4kXVr19fUVH/+7fa8uXL1blz5wzr6tevn+bNm2e0w8LC5OPjk+XjuWvu3Ll69dVXLbaZTCZNnz5dr7/+eobjNm3aJH9/fyNEomDBgoqMjFSZMmUyHNO+fXutWbPGaL/xxhuaMmWKChYsmOGYK1euqGPHjtq6dauxbenSpeneYP39999r8ODBRvuzzz7T6NGjM5z7XvHx8YqNjVWFChXS3T9q1CiLuTZs2CB/f/80/Q4dOmQRcPDUU09p3bp1Vh2vKSkpOnHihKpXr25VzdY4e/asqlevroSEBGNb3759NWfOnAxDDsLDw9WiRQsjtECSOnfurOXLl2e6lo+PjxGIUKlSJeO9khP8/f31zz//GO30fuQTHR2tDh06aO/evcY2X19frV69OsMwhRo1aujYsWOSpEKFCikyMjLdEJfUNmzYoNatWxvtjh07atWqVen2TX0sSZKLi4uWLl2qDh06ZLjGjz/+aBG04enpqbNnz2b6/tm2bZsaNGhghDBkJSQkRG3atDFCiby9vRUeHp4mGCuzx5PReyMjV65cUc2aNRUdHS1JcnJy0owZM7IM9Tl06JD8/f118eJFSXder4iIiHTPyfcju48v9blZklxdXfXHH39YhDllZMmSJerevbvFtpkzZ+q1117LcMzvv/+uLl26GOFFkjRp0iS98847GY5J7zzQtWtX/fTTTxkeW/Pnz1ffvn2NdqVKlXThwgUlJCRkuV7q5+Xtt9/W119/nWH/7Ej9Gdy3b1+L8Axr3Hs+u6tmzZr6+++/rTrGHHmt0bBhQ23YsCFNcMldFy9eVIsWLYxznSR16NDBIhgwtfu91nDk8VypUiVt2bLFIgTqXjdu3FDbtm2NABtJql27tvbt25fh52R67+/SpUtrzZo1euKJJzKs74svvtCnn35qtOvXr2/xGZWeTZs2qVmzZlaH8fz1118KCAgwgqP8/Py0Y8eOTMek/kyV7rwH1q1bZxEYlNobb7yhGTNmGO0uXbpo2bJlVtUJAAAAAAAAAAAAAAAAAHld9u5WAAAAAAAAAADkiBYtWmjkyJHp7ouIiDBCCmrXrq0SJUqoTZs2Gjt2rHbv3m3TOv3797e40f7em2kz89dff1mEbHTq1Mmm8INGjRpp+vTpmd5UXKZMmTTPwR9//GH1GvYydOjQTANpJOnJJ5/UCy+8YLSTk5O1fv36DPtv377dIpCmffv2mjp1aqaBGpJUsmRJLVmyRG5ubsa2iRMnptv33hvsJenNN9/MdO57ubq6ZhhIY4vUNQQGBlodoOTk5GTXQBrpTlDPvYE09erV08yZMzO80V66cyP64sWLLfqsWLFCx48ft2ttOenIkSNq2rSpxc3+zz77rIKCgjIMpJHu3GB/182bNzV//nyr1kt9HhkwYIBN9Y4bNy7TQBpJeuWVV9S4cWOjfeHChSz/OEDTpk2tDqSR7gQkjB8/3mhHRkZq7dq1mYy4f1OnTjUCaaQ7oQ1ZBdJI0uOPP24RNHLz5k1NmTIlJ0q0mwkTJlgVSCOlPc8NGTIk0wAP6U6wyNixYy22ff3117p9+7bVNVapUkXz58/P9Nzcp08f1axZ02hHREQoISFBL7/8cqaBIdKd1/fec6IjPuOyo2DBgvrll1+s/tx31LVG0aJFtWzZsgwDaaQ71xpLliyxCJv6/fff03x+2ZOjjmeTyaTFixdnGEgj3XnOli5dqhIlShjbDhw4oL/++svqdSRp1qxZmQbSSNKIESP0yCOPGO2QkBCdP38+0zFPPvmk1YE0kvTMM89o2LBhRnvnzp06dOiQ1eMlydnZWYsXL840kEaSvvzyS4vPmLVr1xphhQAAAAAAAAAAAAAAAACQ3xFKAwAAAAAAAAB5zJgxYzR58uQsQxTi4+P1999/67PPPtMTTzyh2rVra/bs2VbdCOvl5aXOnTsb7f3792vHjh1Zjrvf0IkxY8ZkGcAiST169LBo79mzx6Z17leRIkUyDAdK7cUXX7RoZ1br119/bdGeNGlSpsEo9/Ly8rIIydmyZUu6N3HfG74iyarn297yQg13mc1mzZo1y2LbhAkTLIIIMtK0aVOL19dsNmvmzJl2rzEnbN26Vc2bN7cIdujbt69WrlwpV1fXTMf269dPLi4uRtuaxxwTE6OlS5ca7XLlyqljx45W1+vt7a3Bgwdb1deW91x29ezZ0yIAYevWrXZf467bt2/r22+/NdoVK1bU+++/b/X4Dh06qEGDBkZ7yZIldq3PnipWrGj158ahQ4e0bds2o12sWLE04RwZee+99ywCtiIiImwKFvrwww9VrFixLPsFBARYtE0mk0aNGpXlOG9vb/n6+hrtEydOKD4+3ur6HOXFF19U7dq1re7vqGuNwYMHq2LFiln2q127tvr06WOxbfbs2TatZS1HHs/du3fPMihGkjw9PdOce2x5Pvz8/NSlS5cs+xUoUEBdu3a12JZVuFh29OrVy6Jt63m8W7duatSoUZb9SpUqpbZt2xrtGzdu6MiRIzatBQAAAAAAAAAAAAAAAAB5FaE0AAAAAAAAAJAHDR06VMePH9ebb76pEiVKWDXm4MGDeu211+Tn56eIiIgs+wcGBlq0U98EntrFixe1YsUKo12hQgU9++yzVtUmSSVKlFC7du2s6lu6dGmLG8rPnDlj9Tr28PTTT8vd3d2qvvXr17doZ1RrSkqK1qxZY7T9/PxUo0YNm+q696ZnSdq0aVOaPuXLl7doL1y40KY17CF1DT/++GOu13DXkSNHdOHCBaNdsWJFtWnTxurx/fv3t2hv3LjRbrXllOXLl+vpp59WTEyMse3jjz/W3LlzrQrjcXd3V/fu3Y324cOHtXnz5kzHLFiwQElJSUb71VdftWqtu7p27Wp1eJG177n7UaxYMXl6ehrtvXv32n2Nu0JDQxUVFWW0e/bsaXOQ073nhiNHjujSpUt2q8+eXnzxRTk5WffjyX/++cei3bVrV5UsWdKqsQULFlTv3r0ttln73jWZTOrWrZtVfVMHtNSrV0+PPvqoVWPr1KljfJ+SkqKzZ89aNc6RXnrpJZvH5Pa1hqQ0QTOZ6du3r0U7KCjIprWs5ajjWbLt+ejTp49FWF7qujOTOjAsM7lxHq9cubJF29bzeF57PAAAAAAAAAAAAAAAAADgCITSAAAAAAAAAEAe9cgjj+i7777T+fPntWLFCr377rtq1KiRChUqlOm44OBg+fn56eTJk5n2a9OmjcXN8z/99JOuXbuWYf958+bp5s2bRrt///5WhwtIUsOGDW3qf28gRFxcnNXj7KFRo0ZW9723TinjWvfv32+xz5Y17ro3qEe6ExaS2jPPPGPRfv/99/Xpp58qOjra5vWyq3HjxipevLjRXrp0qXr06KH9+/fnWg137dixw6L91FNPWdxwn5WWLVtahKvs3bvX4n2Q1/zwww/q1q2bEhISJElOTk76/vvv9cUXX9g0z8CBAy3aWQVJ3LvfZDLptddes2m9nHjPpefgwYMaPXq0OnfurEcffVQeHh4qVKiQTCZTmq9z584Z43Iy5CV1uFROnRvyAj8/P6v7pn7vtm7d2qa1UodPbd++3apxlStXVunSpa3qmzq8rGHDhtYVl87Yq1evWj3WUWx5/e7K7WsNd3f3NGFBmWnatKnFdVVISIiSk5OtHm8tRx3PJpNJrVq1snqdSpUqycfHx2hHR0fr9OnTVo3NrfP4zp079fHHH6tDhw6qUqWKSpcurYIFC6Y5h7u4uFiMs/U8nluPBwAAAAAAAAAAAAAAAADyMuv/RCWy5cKFC/L19bX7vMHBwXafEwAAAAAAAEDe5OLiooCAAAUEBEiSbt68qf3792vLli1at26d/vzzzzQhGRcuXFC3bt0UHBwsZ2fndOc1mUx64403NGzYMEnS9evX9d///ldvvPFGuv1nzpxpfO/k5KT+/fvb9DhS37CblWLFihnf3w3YyC221HpvnVLGtaYOifj+++/1/fff217cPWJiYtJsa9asmZ555hn99ddfkqRbt27piy++0H/+8x81a9ZMbdq00ZNPPik/Pz+5ubnd1/oZKVy4sEaMGKFPPvnE2LZ48WItXrxYNWvWVNu2bdWqVSs1bdpUXl5eOVLDXRERERbtunXr2jTexcVFjz32mA4cOCBJSkpK0vnz51WhQgW71Wgvn3zyif79738b7SJFimjRokXq0qWLzXO1aNFCtWrV0sGDByXdef2++eYblShRIk3fbdu2Gf2kO+EJVapUsWm9nHjP3Wv//v0aMmSINm7caFNdd125ciVb46yR+tzQo0eP+54zvXNDXlC5cmWr+97ve7devXoWbWuDNcqUKWP1GkWLFrXb2Nz+nLOVq6urPDw8bB6X29catgTSSFLBggVVo0YNIzQtKSlJUVFRqlSpkk3zZMVRx3PFihVt/qyvXbu2wsLCjHZYWFia4Kv05PR5fNOmTRoyZIj27dtn9Tr3svU8ntOPBwAAAAAAAAAAAAAAAADyA+v/rBxs4urqKklKSUlRdHS03b8AAAAAAAAAPLwKFSokX19fDR06VCtWrFBUVJSGDRuWJnwmNDRUP/30U6Zz9evXTy4uLkb73pvB77Vp0yYdPXrUaLdr186qG5TvVbhwYZv6O9L91Go2m9Pdfvny5WzPmZG4uLh0ty9atEhNmjSx2JaSkqLNmzdr9OjRevrpp1W6dGk1bdpUX3zxhcLDw+1e20cffZRu6MDhw4c1efJkde3aVeXKldNjjz2mt99+Wzt27LB7DZIUGxtr0c5OqELqMannzCvuDaQpUKCA/vrrr2wF0twVGBhofJ+QkKAff/wx3X6pzxsDBgywea2ceM/dtWrVKjVq1CjbgTTSnaCKnJKb5wZHK168uNV97/e9W7p0aTk5/e9Hoda+b+/nWMzJ49jRbHntUsvNaw13d3eb60s9JidCqBx1POfm85GTx/+0adPUqlWrbAfSSLafxx/k9zMAAAAAAAAAAAAAAAAAWKuAowt4UA0bNkxfffWV4uPjHV0KAAAAAAAAgAecu7u7xo8fr5YtW6pLly66ffu2sW/hwoV65ZVXMhzr4eGhbt26adGiRZKkXbt2KTQ0VPXq1bPoN2PGDIt2dkInHnY5cZN7SkpKuts9PDy0ceNGzZw5U//3f/+nEydOpOlz69Ytbd++Xdu3b9dnn32ml19+WRMmTFDZsmXtUpvJZNK0adPUtWtX/etf/9LmzZvT7Xf06FEdPXpU33zzjZo3b66vv/5ajRo1sksNktL8P32xYsVsniP1mGvXrt1XTTnFxcXFuOn+1q1bmjdvnpo1ayaTyZSt+fr06aMPP/xQN27ckHTnPPDmm29a9Ll27Zp+/vlno+3h4XFfQTj2duzYMXXv3l03b940tplMJvn5+alZs2aqUqWKvLy8VLhw4TQBBL169dL58+dzvMbcPDc4WsGCBa3ue7/vXZPJpCJFiuj69euS8u77Nr+w5bVLLTevNYoWLWrzmNTHVk78fNdRx3NefT5ssWHDBg0aNMgi6KVAgQJq0aKFGjdurEqVKsnT01OFCxe2CD+SpGeeeSa3ywUAAAAAAAAAAAAAAACABwqhNDmkU6dO6tSpk6PLAAAAAAAAAPAQ6dSpk/r27avZs2cb2zIKArlXYGCgcaO4dOem8ClTphjtK1eu6NdffzXaXl5eCggIsFPVD4/UN4a/9NJL6t+//33NWb58+Qz3FSxYUIMGDdKgQYO0e/durV+/XkFBQdq6dauuXr1q0TclJUULFy7UunXrFBQUpBo1atxXXfdq166d2rVrp7CwMK1du1ZBQUHauHGjoqKi0vTdsmWLmjdvroULF+qFF16wy/qurq4W7bs39Nsi9Rg3N7f7qimnLF++XM8//7wSExMl3Xkv37x5U7Nnz5aTk5PN85UoUUIvvvii5syZI0kKCQnR7t27LUKDFi1aZPH89O3bV4UKFbrPR2I/H374oRHUI0l+fn6aN2+eHnvssSzHZjfMx1apzw3jxo2Tr6/vfc1Zq1at+xqfF6T33nV3d7d6vNlsVkJCgtHOq+/bh0VuXWvcDdGyRepzfOpjzx4cdTzn1efDFu+//75FIE3Hjh01depUPfLII5mOu/fcDwAAAAAAAAAAAAAAAADIHkJpAAAAAAAAAOAB0qNHD4tQmvj4eMXFxalEiRIZjmnZsqVq1qypw4cPS5J+/PFHffXVVypSpIjRvvdG6H79+qlAAf572VYeHh4W7ZIlS+rpp5/OlbUbNWqkRo0aacSIEUpJSVFoaKjWrFmjn3/+WaGhoUa/6Ohode/eXaGhodkKMclM5cqVFRgYqMDAQEnSqVOntH79ei1dulRr165VSkqKJOnmzZvq06ePGjdurIoVK973uqVKlbJoX7582eY5Ll26lOmcecWzzz6r1atXKyAgwAgimDdvnm7evKn58+dn6307cOBAI5RGuhMkcW8ozcyZMy36DxgwIJvV2198fLxWr15ttMuWLas1a9ZY/frFxsbmVGkWUp8bKleunGvnhrwsvfeuLeeEmJgY47yS3nzIXbl1rZH6fG2N1J8LJUuWtHmOrDjqeM6rz4e1jh07pr179xrt2rVra+nSpVaFn8XExORkaQAAAAAAAAAAAAAAAADwULDvb5QDAAAAAAAAABzKx8cnzba74RSZuRsUIklXrlzR4sWLjfaMGTOM700mU54KnchPKleubNE+ceKEQ+pwcnJSgwYN9NFHHykkJERLliwxQgEk6cCBA/rzzz9zvI4qVapowIAB+uOPPxQaGqoqVaoY+xITE/Xdd9/ZZZ1KlSpZtO8N4bFGUlKSjh49arRdXFxUtmxZu9SWE1q3bq01a9bIzc3N2Pbf//5XPXv2VHJyss3z+fn5qX79+hZzXb9+XZIUEhKi3bt3G/tatmypGjVqZL94O9uzZ49u3rxptF966SWrgxxOnDihpKSknCrNQl45N+Q19/veTd0/9XzIfblxrXHgwAGb+icnJ6c5x5cvXz5ba2fGUcfzmTNndPXqVZvW2r9/v0U79TkqN23fvt2i/frrr1sVSCNJBw8ezImSAAAAAAAAAAAAAAAAAOChQigNAAAAAAAAADxA7oZF3Mvd3T3LcX379rUIJrl7c/iuXbssboRu3bq1RXhIXuHkZPnf3Waz2UGVZMzPz09FixY12lu3blViYqIDK7qja9euev/99y22bd68OVdrqF27tqZPn54jNTRp0sSiHRQUZNPxsWnTJoswl4YNG1p9Q7yjPPnkk1q7dq1KlChhbFuyZIm6deuWraCVgQMHGt9fu3ZNP/30kyTLEAlJeS6w6vz58xZtWwJz/v77b5vWup9z0FNPPXVfaz+oUr93bX1eUvdPPd+DLi9+LubGtUZMTIxNwTTbtm2zCK+qX7++ChYsmG7f+3lOHXU8m81mbdy40ep1IiIiFB4ebrS9vLxUsWJFq8fbW26exwEAAAAAAAAAAAAAAAAAaRFKAwAAAAAAAAAPkF27dlm0vby8rArQKFmypHr06GG0N2/erCNHjmjmzJkW/fJa6MRdxYoVs2jfuHHDQZVkrFChQmrdurXRvn79uubMmePAiv6nefPmFu1Lly49MDXUqFFDZcuWNdoRERHasGGD1eNnz55t0W7VqpVd6sppTZo00fr161W6dGlj28qVK9WlSxebw5Befvllubq6Gu0ZM2YoISFBixYtMraVKlVK3bt3v//C7Sh1YMS9wRNZjfvhhx9sWut+zkF+fn4qVaqU0f7777916NAhm9Z/EKV+ry1btkxxcXFWjU1OTtaCBQsyne9Blxc/F3PrWmP+/PlW9503b55FO7Pj5H6eU0cez7Y8H6n7tmzZ0uqxOSG75/GkpKQ0n98AAAAAAAAAAAAAAAAAANsRSgMAAAAAAAAAecSlS5e0aNEipaSkZGv8zZs39e2331psa9eundXjBw4caNGeNGmS/vvf/xptDw8PPf/889mqLafdG7whSWFhYQ6qJHPDhg2zaH/++ec6ffq0g6r5n9QBMPcGZOT3Gkwmk1577TWLbcOGDdPt27ezHLtz50799NNPFnO9/vrrdqkrN/j6+mrDhg0qU6aMsW3NmjXq1KmTTWEKbm5ueuWVV4z2jh07NHLkSF25csXY1qtXLxUuXNgudduLl5eXRXvz5s1Wjfvhhx8UEhJi01r3cw4qWLCg3nnnHaNtNpsVGBio5ORkm2p40NSsWVPNmjUz2vHx8fr888+tGjt58mSLc6uPj4+eeeYZu9eYlxUvXlzOzs5GO698LubGtcZ3332nM2fOZNnv4MGDaUJY+vfvn2H/+3mfO/J4/vXXX9OEFqbnwoULmjhxosW2zJ6P3JDd8/jIkSN1/vz5nCgJAAAAAAAAAAAAAAAAAB4qhNIAAAAAAAAAQB4RHx+vV155RXXq1NHChQuVkJBg9djExET16tVLBw8etNjep08fq+do0qSJ6tWrZ7SnT5+ua9euWcxVqFAhq+fLTbVq1bJo//rrrw6qJHMtW7a0CAq6ePGi2rZtqyNHjlg9R0pKipYvX64RI0aku3/w4MFauXKlzGazVfMlJSXpm2++sdjm6+trdT3pmTx5sr777jubgk+++uoru9Zwr0GDBqlIkSJGe8+ePRo4cGCmz9Hp06fVvXt3iz6dO3dW1apV7VZXbqhbt66CgoIsbuxfv369nn32WcXHx1s9T+ogidTBBQMGDLi/QnOAr6+vxTlr6dKl2rp1a6ZjVq1apffee8/mtVKfg5YsWWLT+Lfffltly5Y12ps3b1b37t0VFxdn9RzXr1/XN998o1mzZtm0dl72/vvvW7S/+eabNCEiqf3555/65JNPLLa98847cnJ6uH4sWrBgQVWvXt1oh4SE6OTJkw6s6I7cuNa4ceOGnn/+eYt5U7t48aK6du2qW7duGdueffZZ1ahRI8Mx93ut4ajj2Ww2q0ePHoqKisqwT0JCgrp27Wpxznn88cfVtm1bq9fJCfcG+UjS1KlTdeLEiUzHTJs2TRMmTMjJsgAAAAAAAAAAAAAAAADgofFw/fYlAAAAAAAAAOQDhw4dUu/eveXl5aUBAwZo8eLFOnfuXLp9o6Ki9P3336tmzZpavHixxb6uXbuqdevWNq0dGBiY4b7XX3/dprlyk7+/vwoWLGi0582bp65du2r27Nn6448/tG7dOuNry5YtDqxUmj9/vipUqGC0jx49Kl9fX73zzjsKDQ1NNyglNjZW69at07vvvisfHx89//zz2rFjR7rzb9myRc8995wqV66sDz74QEFBQbp69WqafsnJyVqzZo2aN2+uXbt2Gdu9vLzUqVOn+3qMYWFhGjJkiMqXL6++fftq2bJlGR7DISEh6tmzp0UwjpOTk/r3739fNdzrkUce0fjx4y22zZw5U61bt9b27dsttl+/fl2zZs2Sr6+vzpw5Y2wvXbq0vvvuO7vVlJsef/xx/fPPP/L29ja2bdq0SW3btrU69KR+/fry8/NLd1/jxo1Vp04du9RqT8WKFVO3bt2M9u3bt9W+fXtNnz5diYmJFn2PHz+uN998U507d1ZSUpI8PT3l7u5u9VoNGjSQh4eH0Q4KClLr1q01depU/f777xbnoHXr1qUZX6JECS1evNjiPLZixQrVqlVLEydO1OnTp9Nd98yZM/r111/Vq1cvlS9fXm+//bbFcZvfde3a1eI1NJvN6tevnwYOHKhTp05Z9D137pw+/vhjderUSTdv3jS2N2vWTEOGDMm1mvOSewNFbt++rZYtW2r06NFatmyZ/vrrL4tjMqNzdE7IyWuNSpUqSZKCg4PVoEEDrVy50iJ45ubNm1q8eLEaNmyoY8eOGduLFCmiKVOmZDr3/V5rOOJ49vb2VoECBRQeHq6GDRtq4cKFFue/lJQU/fnnn2rUqJFFzSaTSdOnT5fJZLJ6rZxQrVo1NW3a1Ghfu3ZNLVu21OLFiy1eV0kKDQ3Viy++aITO1axZM7fLBQAAAAAAAAAAAAAAAIAHTgFHFwAAAAAAAAAASN/Vq1c1c+ZMzZw5U5Lk7u4uDw8PlSxZUomJiTp37pwuXLiQ7tjGjRtrzpw5Nq/Zq1cvDR8+XPHx8RbbW7Rokadv7vX09FTv3r01e/ZsY9uyZcu0bNmyNH0rVaqk8PDwXKzOkqenp1avXq2OHTsa4RE3btzQ5MmTNXnyZJUoUULe3t5yc3NTfHy8YmJishUWEBERoYkTJ2rixIkymUzy9vaWu7u7ihQpoqtXr+rUqVNpgjmcnZ01c+ZMFSlSxC6PNS4uTvPnz9f8+fMlSWXKlJGnp6fc3NyUmJio8PBwXblyJc244cOHy9fX1y413DV48GDt3LlTCxYsMLYFBQWpadOmKlOmjCpUqKDExESFhYUpISHBYmyRIkW0aNEilS9f3q415abq1atr48aNat26tSIiIiRJ27Zt09NPP621a9eqVKlSWc4xcOBA7dy5M832AQMG2L1eexk7dqxWr15tBDNdvXpVgYGBevvtt1W9enW5uLjo3LlzOnv2rDHG2dlZc+fO1aBBg3T58mWr1ilYsKCGDh2qzz77zNi2YcMGbdiwId3+6YVPPfnkk5o/f75effVV470ZGRmpDz74QB988IHKlSsnT09Pubi4KC4uThcuXFBsbKzVz0V+NX36dJ04cUKhoaGS7jx306ZN07Rp0+Tj46MyZcooJiZGYWFhSklJsRhbuXJlLVq0SM7Ozo4o3eHefPNNTZs2zTieoqKiNGrUqHT7zpkzR/369cuVunLyWsPf318FChTQrFmzdPLkST333HMqUaKEfHx8ZDabFRYWpmvXrlmMMZlMmjp1qqpWrZrp3Pa41sjt47latWoaOHCgRo4cqfPnz6t3794aOHCgKleurIIFCyo8PDzd88iYMWPUvHlzq9fJSRMmTJC/v7+Sk5Ml3Qns6dGjh1xdXfXoo4/KyclJZ8+e1fnz540xxYoV048//qiGDRs6qmwAAAAAAAAAAAAAAAAAeCA4OboAAAAAAAAAAMAdrq6uqlevXob7L1++rKNHj2rHjh0KDQ1NN5DGyclJAwcO1F9//aXixYvbXIObm5teeumlNNvzcujEXV9//bWeeeYZR5dhlTp16ig4OFjt2rVLsy8uLk6HDh3Sjh07dPDgwQwDaSpWrGj1emazWWfPnlVoaKi2b9+uQ4cOpQmkKVWqlJYsWaKOHTva9mBscPHiRR08eFDbt29XSEhImkAaZ2dnffbZZ/rPf/5j97VNJpPmzZunYcOGycnJ8scjFy9e1J49e3To0KE0gTReXl5au3Ztuq9VflOlShVt3LjRInhh9+7dat26tS5dupTl+BdffFElS5a02Obm5qaePXvau1S7qVq1qhYvXixXV1eL7YmJidq3b5927dplEUhTuHBh/fjjj2rfvr3Na3388cfq1avXfdXbs2dPbd68WdWrV0+z79y5cwoNDdXOnTt19OjRdIMknJ2d83V4UnpKly6tf/75J93ze3h4uHbt2qWTJ0+mCfB44okntGXLFlWqVCm3Ss1zqlevrgULFqQ5/h0tp681vv/+e3Xr1s1ox8XFKTQ0VPv27UsTSFOoUCFNmzZNffr0sWru+73WcMTx/Omnn2ro0KFG+/r16zpw4ID27t2b5jzi5OSkzz//XJ9++qnN6+SUZs2aacaMGSpYsKDF9vj4eO3du1fBwcEWgTSlSpXSqlWr1KBBg9wuFQAAAAAAAAAAAAAAAAAeOITSAAAAAAAAAEAe4eHhoZCQEJ08eVL/93//p4CAAJUqVcqqseXKldPbb7+tkJAQ/fDDD3Jzc8t2Hf3797dolyxZUi+88EK258stbm5u+vPPP7VmzRr1799f9evXV+nSpdPcxJxXlClTRmvWrNHGjRsVEBCgYsWKZdrfZDKpQYMG+vDDD7Vv3z7Nnz8/3X4rV67UlClT1LFjxzQBIukpX768hg0bpmPHjqlz587ZeShpjBkzRj/99JN69eqlChUqZNnf1dVVvXr10t69ezV69Gi71JAek8mk8ePHa8+ePXruuefk4uKSYd/y5ctr5MiROn78uFq0aJFjNeW2ihUr6p9//lGNGjWMbSEhIfL397e4qT89RYsW1Ysvvmix7aWXXsry2HW0tm3bateuXQoICMiwT4ECBdS9e3eFhoameYzWcnZ21oIFC7R582YNHjxYfn5+8vDwyPQ4S4+vr68OHTqk+fPnq0mTJnJ2ds60v4uLi1q3bq0JEybozJkzeuONN7JVf15WokQJrV27VkuXLpWfn59MJlOGfWvXrq05c+Zo+/btKleuXC5WmTd1795dx44d07hx49SuXTtVqFBBrq6umT6HuSEnrzUKFSqkxYsXa/r06RkGuDk7O6tdu3bau3evTWE49rjWcMTxPHnyZC1ZskQ1a9bMsE+zZs20adMmjRo1Ktvr5JS+fftq48aNatmyZYZ9ChcurP79++vgwYPy9/fPveIAAAAAAAAAAAAAAAAA4AFmMpvNZkcXkR/4+voqOjpaXl5eCg4OdnQ5mfL5cLWjS8i28HHZ+Au8KSftXwiQnzhVzbpPRnj/4GFn8f6JclgZQN6Q/b+ibl7b1o51APmPqe1a4/ukCfdxbQY8AFw++N+/MQKr23bzNfCgmXYsyW5zmc1mRURE6NixYzp9+rTi4uKUkJCgokWLys3NTeXLl1e9evX0yCOP2G3N2bNn67XXXjPagwcP1pQpU+w2P9KXnJysnTt3KiwsTJcuXdL169dVrFgxlSpVStWrV9fjjz+uEiVK2DSn2WzWsWPHdPz4cZ0+fVpXr17V7du35ebmJi8vL9WtW1fVq1eXk1POZthHRkbqyJEjCgsLU2xsrJKSklS0aFG5u7urVq1aqlOnjs3BHfZw48YNbd68WadPn9alS5fk4uIiT09P1apVS/Xr18/1evKDli1batOmTUZ7165datSokQMrss25c+e0adMmnT17Vjdu3FDx4sVVrVo1NWvWzKoQJ0eIi4vT9u3bFRUVpUuXLik5OVlubm7y9PTUY489pho1aqhw4cKOLjNXnT9/Xlu3blV0dLRiY2NVvHhxlS1bVo0bN84whAR5i72uNcLDw1W5cmWj3bdvX82dO9dop6SkKDg4WHv37tWlS5dUpEgReXt7q2XLlvLy8rqvx2Av9j6e7w25adWqlYKCgiz279+/X7t371Z0dLQKFSqkcuXKqWnTphbPY14WHh6uLVu26Ny5c0pKSlLJkiVVo0YNNWvWTEWLFnV0eQAAAAAAAAAAAAAAAADwQCGUxkqE0uQOQmmAbCCUBsg+QmmAexBKA2QXoTTA/xBKA/yPPUNpHKFZs2batm2b0Q4JCVG9evUcWBGAvODo0aN67LHHjHb9+vW1d+9eB1YEIL+y17VGVqE0D6OsQmkAAAAAAAAAAAAAAAAAALBWzv7pUwAAAAAAAABAvrJv3z6Lm8QbN25MIA0ASdL06dMt2oGBgQ6qBEB+xrUGAAAAAAAAAAAAAAAAAAD5A6E0AAAAAAAAAADD+PHjLdqDBw92UCUA8pKYmBjNmDHDaBcvXly9evVyYEUA8iuuNQAAAAAAAAAAAAAAAAAAyB8IpQEAAAAAAAAASJI2bNigRYsWGW1vb2/16NHDgRUByCvee+89Xbt2zWi/8cYbcnV1dWBFAPIjrjUAAAAAAAAAAAAAAAAAAMg/Cji6AAAAAAAAAABA7ouNjVVwcLAkKSYmRlu2bNH06dNlNpuNPp988olcXFwcVSIABzl06JCioqKUkpKiM2fOaOHChQoKCjL2u7q6avjw4Y4rEEC+wLUGAAAAAAAAAAAAAAAAAAD5G6E0AAAAAAAAAPAQCg0N1TPPPJPh/saNGyswMDAXKwKQV4wfP17z5s3LcP+4ceNUpkyZXKwIQH7EtQYAAAAAAAAAAAAAAAAAAPmbk6MLAAAAAAAAAADkLdWqVdOSJUvk5MR/IQOwNGjQIA0ePNjRZQDI57jWAAAAAAAAAAAAAAAAAAAg7yvg6AIAAAAAAAAAAI7n6uqqGjVqqGvXrho6dKhcXV0dXRKAPMDZ2Vnu7u7y8/PTwIED1bFjR0eXBCCf4loDAAAAAAAAAAAAAAAAAID8hVAaAAAAAAAAAHgI+fv7y2w2O7oMAHnQ3LlzNXfuXEeXASCfy41rDR8fH65nUuH5AAAAAAAAAAAAAAAAAADYi5OjCwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA5B0FHF1AdqxcuVITJkxQfHx8rq154cKFXFsLAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABwlX4bSTJgwQSdOnHB0GQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADwwMmXoTTx8fGSJCcnJ3l6eubKmhcuXFBKSkqurAUAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAjpIvQ2nu8vT0VHBwcK6s5evrq+jo6FxZCwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAcxcnRBQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA8g5CaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABkJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAZCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABkJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAZCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABkJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAZCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABkJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAZCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABkJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAZCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABkJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAZCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABkJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACGAo4uAPYXPq6jo0vIXU5VHV0BkH/x/gHuUd7RBQD5lqntWkeXAOQZLh+cdHQJQJ4x7ViSo0sAAAAAAAAAAAAAAAAAAAAAAAAAkE1Oji4AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAJB3EEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAUcHQBsD+fD1c7uoRsCx/XMRujouxeB5C/lM/+UHOE/coA8iNTpf99z/sBD7t73w82Ml9eZMdCgPzH5P6y8f2tb6s7sBLA8Qq8dcz4PrC6iwMrARxv2rEkR5cAAAAAAAAAAAAAAAAAAAAAAACQbU6OLgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAkHcQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBBKAwAAAAAAAAAAAAAAAAAAAAAAAAAAANjAx8dHJpMp3S8fHx9Hlwcgj8nofGEymeTv7+/o8nJEeHj4Q/m4AeBBQigNAAAAAAAAAAAAAAAAAAAAAAAAAAAAHCqzkJd7vzw9PZWUlJStNYYOHWrVGiaTSUFBQfZ9gAAAAEA+QygNAAAAAAAAAAAAAAAAAAAAAAAAAAAA8oWLFy/ql19+sXlcfHy85s2blwMVAQAAAA8mQmkAAAAAAAAAAAAAAAAAAAAAAAAAAACQb0yZMsXmMfPmzdPVq1dzoBoAAADgwUQoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAPKNnTt3avfu3TaN+e6773KoGgAAAODBRCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAA8pVvv/3W6r7r1q3T4cOHc7AaAAAA4MFDKA0AAAAAAAAAAAAAAAAAAAAAAAAAAADylZ9//lmXLl2yqu+UKVPsvn54eLjMZnO6X+Hh4XZfDwAAAMhthNIAAAAAAAAAAB56c+fOlclkMr7mzp3r6JLypFGjRlk8T0FBQY4uKU8LDQ3V4MGDVbduXZUuXVrOzs4Wzx/uOHnypIYNG6YnnnhCHh4eKlCggMXzxC9p5U8+Pj7Ga+jj4+PocvKVoKAgi/fAqFGjHF3SQ8Xf39/qczWvlWPdvn1bP/74o7p166ZKlSqpWLFiFq9Hv379HF0igFzAv+UAAAAAAAAA4OGWlJSkmTNnZtkvIiJCq1atyoWKAAAAgAdLAUcXAAAAAAAAAADIWkJCgvbs2aPjx48rNjZW169fV5EiRVS8eHFVrFhRVatWVZUqVeTkRBY54GgpKSkaPny4Jk6c6OhS8rxJkyZp2LBhun37tqNLAQDkI2fOnFFAQIBCQ0MdXQoAAAAAAAAAAAAcbOrUqRo2bJicnZ0z7PP999/zuwkAAABANhBKAwAAAAAAAAB5lNls1ooVKzR16lStW7dOt27dyrS/m5ubfH191apVK7Vv315PPPEEITWAA4wbNy5XAmnmzp2rV199Nc32WbNmqX///vc119tvv62vv/76fkvM1KJFi/Tee+/l6BoAgAdPUlKSOnbsqP379zu6FAAAAAAAAAAAAOQyk8kks9lssS0iIkIrV65Uly5d0h2TmJioWbNmWT1fXhYVFaVdu3YpMjJScXFxcnd3V9WqVdW8eXMVLlzY0eVZbe/evTp8+LDOnj0rZ2dnVa1aVU8++aTc3d2zHHv69Glt2bJFp0+flslkUpkyZVS7dm35+vre9+/KXbt2TSdPnlRkZKTOnTun+Ph4JSQkKCUlRW5ubipevLg8PT1Vt25dPfLII/e1Vn6WmJioY8eO6dixY7p48aLi4+OVmJioIkWKyNXVVeXKlVPFihX16KOPytXV1W7rXr9+Xbt379a5c+cUGxur2NhYOTs7y83NzVivevXqMplMNs+dkJBgvPZRUVG6evWqEhISdOvWLbm5ucnNzU3u7u6qU6eOKleunK01ctLt27cVGhqqkydPKiYmRrGxsSpYsKA8PDzk5eWlxo0bq2TJknZdc+/evTp48KDOnTsnFxcXeXt7q06dOqpevbpd1wEAOB6hNAAAAAAAAACQB0VERKh///76+++/rR5z7do1BQUFKSgoSKNHj9by5cvVuXPnHKwy7+jXr5/mzZtntMPCwuTj4+O4gvDQiomJ0dixY422yWRS79699eyzz8rDwyNXfiFhzJgx6tWrlwoVKpTja2XXrVu39MEHH1hsCwgIULdu3VSuXDmLXxLy8vLK7fKA+zJq1CiNHj3aaG/YsEH+/v6OKwgPlaCgID311FNG+/PPP9eoUaMcV1AOmDVrlkUgjaenp4YMGaJ69eqpaNGixvby5cs7ojykw8fHRxEREZKkSpUqKTw83LEF5XOpwwTnzJmjfv36Oa4gAAAAAAAAAAByUZs2bbRu3bo026dMmZJhKM1///tfXb58Oc12Z2dntWrVyqbfz0rt3p+DpJbVz0VS/75Tavf+/tPSpUv17bffauPGjUpJSUnTt0iRIurXr58+//xzlS1b1qbHcL9S/4w8tXt/ljF79mxNnDhRhw4dStOvYMGC6tmzp8aNG5fuz/q2bNmiTz/9VEFBQemuU6ZMGQ0fPlxvvfWWXFxcrKp9y5YtCgoK0tatW7V//36dOXPGqnGSVLp0aQUEBKhv377y9/fP8neCZs6cqQEDBmS4v1GjRtq2bZsKFMj4tu+EhAQ1atQo3efvrv/+97/q2bNn1g/ARikpKVq8eLHmz5+vv//+W4mJiVmOMZlMqlKliurXr68WLVqoZcuWatCggU2/P3X+/HlNnTpVv/32m/bt26fbt29n2r9kyZJq0aKFOnfurJ49e2YYihMSEqJ169Zp69atCg0NVXh4eLrvrfS4ubmpbdu26tu3r9q3b5/pa5aTUlJStGzZMs2cOVNbtmzRtWvXMuzr5OSkhg0bqk+fPhowYEC2Q6xu376tKVOm6Ntvv9XJkyfT7ePr66vBgwerX79+eS68BwCQPYTSAAAAAAAAAEAec+rUKbVs2VKRkZFp9hUqVEiVK1dWiRIllJSUpJiYGEVGRqb7A9H89Fd8gAfFL7/8YvFLFyNHjsz0F29yQkREhKZPn64hQ4bk6rq2WLdunc6dO2e0+/Tpk+kvWgEAcNf8+fON7wsWLKiNGzeqRo0aDqwIAAAAAAAAAAAAuWXIkCHphtKsX79eR44c0WOPPZZm35QpU9Kd67nnnlPx4sXtXqM9Xb58WX379tXq1asz7ZeQkKAffvhBy5cv15o1a1S3bt1cqtA6V65cUe/evbVq1aoM+yQnJ2vBggX6+++/tXr1atWrV0/Snd+BGzlypP79739n+vtwFy9e1LBhw7R8+XKtXLlSpUqVyrKuvn37ZhiskZWYmBjNmzdP8+bNU5s2bTRjxgxVrlw5w/6vv/66NmzYoEWLFqW7f/fu3Ro9erTFH8NK7YMPPsg0kGbQoEE5EkgTFhamrl27KiQkxKZxZrNZJ0+e1MmTJ7VkyRJjLmv+2Ny1a9f0wQcfaO7cubp586bVa165ckWrVq3SqlWrVK1atQz/iNAHH3yg9evXWz1v6tqWLFmiJUuWqEGDBpozZ45xvOaWVatW6Z133rH6+E1JSdHu3bu1e/du/fvf/9akSZNsPlYiIiLUtWtX7dmzJ9N+wcHB6t+/vxYuXKiff/7ZpjUAAHmTU9ZdAAAAAAAAAAC5JTk5WQEBARaBNCaTSb169dKmTZt0/fp1HTlyRDt27FBISIhOnz6tuLg4BQUFacSIEapataoDq8eDbtSoUTKbzcZXRj+0f5ht27bNoh0YGOiQOr744gvduHHDIWtbI688TwCQWlBQkMVnHfKWmzdvWvyCm7+/P4E0AAAAAAAAAAAAD5GAgABVqlQp3X3phc9s3bo1wwCFvPzHfiTpzJkzatq0aZaBNPc6d+6cnnnmGV26dCkHK7NNYmKiAgICMg2kuVdkZKRatmyp06dPS5LefPNNffHFF1b//HbLli3q3r17rv68d/369fL19dXhw4cz7Tdt2rRMf775n//8J83v1Ny1evVqff/99xmObdCggSZNmmRdwTaIjo5Ws2bNbA6kuR87duxQ/fr1NX36dJsCaRxh79698vPz08aNG3NlvVu3bumDDz7Qc889l+1ApejoaL300kt67733dPv2bavGREREyN/fP8tAmnv9/fff8vf3z1PnIwBA9hBKAwAAAAAAAAB5yNSpUy3+mknhwoW1atUqLViwQC1atFCBAgXSjHF1dVWrVq00btw4nThxQkFBQercubOcnZ1zs3QAko4dO2Z8X7JkSZUvX94hdURHR+vbb791yNrWuPd5kqRatWo5qBIAQH4SFham5ORko83nBwAAAAAAAAAAwMPFyclJgwYNSnff/Pnzde3aNYtt6QXVSNLjjz+u1q1b270+e+rWrZuOHz9u87gLFy7o/fffz4GKsmfkyJHavHmzTWOuXr2qwYMHa8qUKZo6darNa/7999+aO3euzePuR2xsrDp27Kjr169n2MfV1VW//PKLChcunO7+27dvq3fv3oqPj7fYfuHCBfXv3z/DeYsXL65ffvlFLi4u2Ss+E++9956io6PtPm9GgoOD9fTTT+vUqVO5tub9unnzprp06aKzZ8/m+FqDBw/WxIkT7RK6NGnSJKvOFcnJyercubPCw8NtXuPgwYN65ZVXslEdACAvIZQGAAAAAAAAAPKQefPmWbQ///xzdejQwaY5WrVqpeXLlysgIMCepQGwwpUrV4zvixcvnqtrN23a1KI9fvx4Xb16NVdrsNa9z5OU+88VACB/4vMDAAAAAAAAAAAAr7/+erqhHteuXdP8+fONdnR0tH799dd05xg8eHCO1WcvFy9ezPbYn3766b7G29OlS5eyNW7VqlV65513sr3uN998k+2x2RUWFqYffvgh0z5169bNtLaTJ0+medz9+/fXhQsXMhwzc+ZMVatWzaZarREXF6clS5bYfd6MnD17Vh06dEgTypMfxMbGaty4cTm6xqRJkzR9+nS7zjl58uQsA5z+9a9/KTQ0NNtrpP7jZQCA/IdQGgAAAAAAAADII2JiYhQcHGy0nZycNGDAAAdWBMBWiYmJxvcmkylX1/bz81Pnzp2NdkxMjCZOnJirNVjr3udJyv3nCgCQP/H5AQAAAAAAAAAAAHd3d/Xs2TPdfd99953x/bRp05ScnJymT/HixdWnT58cq8/ePD09NWnSJB04cEBnz57Vn3/+qZYtW2Y65ubNm1q+fHnuFGilKlWqaMGCBTp58qROnTqlKVOmqEiRIpmOuX37tvF9t27dFBQUpLNnzyo4OFgvv/xypmNDQkJ0/PjxLOsqUKCAmjRporfeekvTp0/Xn3/+qQMHDigsLEzR0dGKiorSsWPHtGHDBk2cOFENGzbMdL5JkyZlueaAAQMyrX/WrFn67bffJElTpkzR6tWrM+w7ePBgvfDCC1mumR179uzRzZs3M9zfpUsXLVu2TEePHlV0dLROnz6t/fv3a82aNZo0aZJeffVVVa5c2er1Pvroo0zDd+7q0KGD5syZo3379ikyMlInT57U5s2bNWHCBLVq1crq9ZycnNSgQQO98cYb+v7777V69Wrt27dPJ0+e1Llz53Tu3DmdOHFCmzdv1nfffZfl3LNmzdK1a9esXt8W58+f18iRIzPtU6VKFU2aNEnBwcGKjIzUqVOn9Ntvv6l9+/aZjhs2bFiGQUBXrlzRhAkTMh1vMpnUt29fbdiwQWfOnNHRo0c1b9481axZM/MHBQDINwo4ugAAAAAAAAAAwB2RkZEWbQ8PD7m7uzuomtxx9OhR7dixQ1FRUXJ2dpanp6f8/Pzy3A8kU1JStHPnToWGhury5csqVqyYypUrp5YtW8rLy8sua1y4cEHbt29XdHS0Ll++LFdXV+P5sOWH8+mJiYnRnj17dOLECcXFxenWrVsqWrSoPDw8VLlyZdWqVUulSpWyy+PISEJCgkJDQ3Xo0CHFxsYqISFBRYoUUfHixeXj46PHHntMFSpUyNEaUrtx44Y2b96sM2fO6OLFiypcuLA8PT1Vq1Yt1atXL1tzms1mO1dpm7Fjx2rFihVGHZMmTdLQoUPz3LnEns/T3ffn8ePHdeHCBd2+fVuenp6qXLmymjVrpoIFC9ptLenOLzxt375d4eHhOnfunG7fvq1atWqpU6dOVs9x4MABHTx4UGfPntXt27fl4+Mjf39/eXp6Zjru1q1b2r59u/bv36/Y2FgVL15c1apVk7+/f7p/BS6/Sk5O1tGjR3Xo0CFFR0fr2rVrKlasmEqXLq3HHntMDRs2VIEC9v8x5+nTp7V161adPn1aZrNZZcqUUf369dWgQYM8F3wRGhqq3bt368KFC3JxcZGXl5eaNWsmHx8fu65z+fJlbd++XVFRUbp48aJcXV317LPPqnr16hmOiY+P18GDB3XkyBFdvnxZCQkJKlGihDw8PNSwYcNMx96P3HpO7OnKlSs6cOCAjh49qtjYWN28eVMlS5aUp6ennnjiCVWqVMnRJVrIyWsVa+Xk5+zu3bt14sQJnTt3TomJiapUqVKWv8iaE9cSmbl8+bK2bt2qyMhIxcTEyMPDQ35+fqpfv36WY0+cOGG8n00mk8qXLy9/f395e3vbvc7clpSUpC1btujs2bM6d+6cnJ2d9cQTT9j0C6e54cSJE9qxY4fx7z5vb281bNgwz/zbJ7eP58jISG3dulURERG6deuWypUrp9q1a8vX19eu6+Tkv+UuXLigAwcO6OTJk7py5Ypu3bql0qVLy8vLS40bN7bbvxXvlZycrK1bt+rAgQO6cuWKihcvrgoVKqhVq1Y5/m86AAAAAAAAAMhLhgwZorlz56bZfvjwYa1fv14tW7bU9OnT0x3bt29fubq65nCF9lG9enVt3LhRZcuWNbZ5e3urdevW8vf315YtWzIcu2vXrjzzB9EeffRR7dixw+L/sgcPHqxr167po48+ynL88OHD9eWXXxptb29vLVy4UKdPn9bmzZszHLdr1y49+uijGe7/+uuv1bx5c6v+j/3RRx+Vv7+/3n33XfXu3Vs//vhjuv2ioqJ05MgRPfbYY5nON23aNO3evVvHjh1Ld/+AAQPk5uam4cOHZziHr69vjv7BqvPnz2e4LyAgQMuWLUuzvUKFCqpdu7batWtnbDt9+rSWLl2qX375RU5OTunOt2/fvgyf07vc3d31888/q02bNmn2ValSRc2bN9f777+vQ4cOacSIEZn+rseIESO0cOFCq36eU7VqVTVv3lxvvvmmPv74Y/3nP/+PvfuOjqJ82zh+JQHS6TWhhBo6GDrSu9JEOiL1R1FQkCagVFFAilJUpHcUAaUpVXqvAem9t0BoISSQ5P2Dw77ZZHezm2yyQb+fczgnzzzt3tnJ7ISduZ/RJts9f/5cu3btUr169eIc01ajR49WSEiI2fomTZpo8eLFse4fyp07txo1aqSvv/5aX375pcm+QUFBmjx5sgYPHhyrbvbs2Xr27JnF2ObMmaP27dsbbStQoIBatGihd999V1u2bLHYHwCQ/JGUBgAAAAAAAACSiZirZERf6cVerl+/Lj8/P8PYBQoU0JkzZ2weZ9WqVWrcuLGh3KJFC/36669GbS5fvmz0gHL79u0NN4Ns2LBBX3zxhQ4ePGhy/EKFCmns2LFq2LCh2Rjmzp2rjh07mqyz9GB0rly5dPnyZbP10UVGRuqnn37SmDFjdP369Vj1Tk5OqlOnjsaPH6+iRYtaNWbM8RcuXKgpU6bo0KFDZh+0LlSokAYMGKB27dqZ/WLelM2bN2vs2LHavHmzIiMjzbZzcnKSv7+/GjdurB49ephNDjN8+HCNGDHCUN6yZYuqVatmMYbz589r5MiRWrFihcUvxiXJx8dHdevWVdeuXVW+fHmLbRMiMDBQw4YN0/r16/X8+XOTbXx9fdWpUycNGDDA4o1IMfdJdFeuXDF7c0NiPVRfrFgxtWrVSkuWLJH06rwyevToOFesSWyWfl8lmd1Ply5dMptQ4u7duxo5cqR++eUX3b9/32Qbb29vNWnSRCNHjrQ6uYO54/zBgwcaPXq0Fi5cqNu3bxv1KVGihCEpjaVz38KFCzVhwgQdPXo01rwpU6ZUu3btNG7cuFg3O7148UITJkzQxIkTde/ePZOv84svvlDfvn3NJmsJDQ2Vj4+PHj58KElKkyaNbt26FeeqYzEdO3bM6MH0smXLat++fTaNYUpQUJCWLl2qNWvWaMeOHWZXYJIkT09PtWrVSgMHDlS+fPmsniP6cVa1alVt3bpV0qub0AYOHKi///7bZL8cOXJo5MiR6tChg9mxt27dqurVq5usM7f9NVvOB0uWLNGIESPMfnaXK1dO48ePV6VKlawaz8/PT1euXJFk/Pm4Z88ejR49WuvWrYu1cuB3330XK7HM2bNntWTJEq1bt04HDx7Uy5cvzc6ZNWtWffTRR/rkk0+sfng+5v4dNmyYhg8fLsn++6RatWratm2boWzv8/WRI0f0yy+/aMOGDQoMDLQ4fp48edSrVy916dIlzt9Vc+fRESNGmP2ckuL+LE/saxVrRD9OYzL3+qKfe6XYn0Nz5sxRhw4dFBoaqvHjx2vOnDm6dOmS0Rhp0qQxm5TGntcS0Zk7/k6fPq2hQ4dq5cqVJlcifOutt/TDDz+oQoUKseq2bNmiwYMHa+/evbHqnJyc1LBhQ02ZMkU5c+a0KkZrdejQQfPmzYu13dL1kRT7vTM11uvrg+vXr+urr77S0qVLDZ9vrzVu3FhVq1ZVoUKFdPr0aUmvVjq8cuWKsmfPbtNrCQoKkq+vr2Hf+/j46OrVq3JxcTFqZ+6cunXrVg0aNMjkeyC9uo74+uuvVb9+fbMxxLy+iK5jx44Wr7MsXU9JSX88BwYGasCAAdq4caPJc0revHn15ZdfWvzctUZi/C0XFRWlnTt3aunSpdq4cWOcf0sXK1ZMffv21QcffGB1Qj1z16JhYWH69ttv9f333+vBgwex+rm4uKhZs2YaO3ZssktqBgAAAAAAAACJoVSpUqpQoYL27NkTq27q1KkKCgrSzZs3Y9U5OTmpZ8+eSRFigjk7O2vJkiVGCWleS5EihXr37m0xKU187glLLD/++KPJ74cbNWoUZ1KaPHnyaNSoUbG2Ozk5qX379haT0sS1D2xZgCj6vO3atbOYQGXXrl1xJqXx8vLSb7/9pnLlypn8jubevXuqXbu22Xu+0qRJo6VLl8rV1dW2F2ADS2Pb8n1Ezpw51bt3b/Xu3dtsm8WLF1v8/jxlypRavXq1ye9EYypcuLBWr15t8f7L2rVrxzmOKR06dDCblEZSoiSliYqK0uLFi83W+/j4aOHChRYXtBo8eLB+++03BQYGmqxfsmSJyaQ0y5cvtxhbixYtYiWkec3NzU0LFixQgQIF4kxsAwBI3ux7NxgAAAAAAAAAIN7Spk1rVL5//77Onz9v1zmyZ89u9LDj2bNntX37dpvHmTFjhlG5a9euVvcdMGCA6tatazYhjfRq1aJGjRrpq6++sjk2e3n8+LHq1Kmjnj17mnyIUXr1he/69etVrlw5rV+/3qbxz507p4CAALVv314HDx60+KX6qVOn1LFjR7399tsmE1KYiuuTTz5RrVq1tHHjRosJaV63P336tMaOHWvz67BkwYIFKlq0qBYsWBBnQhrp1UpBc+bM0dSpU+0WQ3RRUVH6/PPPFRAQoJUrV5p96FaSbty4oa+++kr58+e3ePNKcjRy5Eijh15//PFHkzdavcmWLl2qfPny6YcffjCbkEZ6lZRn/vz58vf31w8//BDv+Y4ePapixYpp/PjxsRLSWCMiIkLt2rXThx9+aDIhjfQq8cysWbNi/Z4HBQWpUqVKGjRokNnf/ydPnmjgwIFq3bq12Rtq3N3d1a5dO0P50aNH+u2332x+LQk5/5sTHBysbNmyqUePHvrrr78sJqSRpJCQEM2aNUtFixY1ufKdLSZPnqwKFSqYTUgjSdeuXVPHjh3VtWvXOM+niSU8PFxt27ZVmzZtLN40t2/fPlWrVi1B++Xbb7/V22+/rdWrV8dKSGPKmjVr5O/vr+HDh2vv3r0WE9JI0u3btzVs2DAVK1ZMBw4ciHecSblP7GXq1KkKCAjQt99+q6NHj8aZ8ObixYvq1auXSpcubfdrQmsk5rVKcnDlyhWVLl1aQ4cOjZWQxhxHXEusXLlSAQEB+u2330wmpJFeJTuqWrVqrJUIR4wYoZo1a5pNhhIVFaVVq1apXLlyyeqmZGts2rRJRYsW1fTp02MlpIku+udUZGSkZs+ebfNc8+bNM9r3nTp1ipWQxpxx48apRo0aZt8D6VWSlgYNGqh79+6JlrjQFEccz0uWLFGZMmW0YcMGs6/1woUL6tixoxo1aqSwsLB4zZNYf8v1799fVapU0dSpU636nTl+/Lg6dOig6tWr6+7duza9huhu3Lih8uXLa+jQoSYT0kivrjd//fVXlS5dWkeOHIn3XAAAAAAAAADwJjGXXGb16tVm7zmqXbt2rEU4kqu6desqICDAbH3JkiUt9jf3HcrWrVvl5ORk87/4fueaJ08e1apVy2RdgQIFLC5oIL1K0J8yZUqTdcWKFbPY19L3SDFduXJFP/zwg7p06aIqVaooV65cypgxo9zc3GLti7p161ocy9z3EzEVL15ckyZNMltv6R6FWbNmKU+ePFbNE1+WFj6YO3eufvvtN6vuK7DGX3/9ZbG+c+fOViWkic7a7/Tu3Lmj2bNn66OPPlLNmjWVJ08eZcqUSR4eHrHee39/f4tjWfve2+Lw4cMWv4dv3769PDw8LI7h5OSkmjVrmq3/559/dOvWLaNtYWFhOnTokMVxP/nkE4v1vr6+ev/99y22AQAkfySlAQAAAAAAAIBkIk+ePLFWq/j888/t/mBi9+7djcoxEwzE5caNG0ZfAufJk0c1atSwqu/AgQM1btw4Q9nb21tFihRR6dKllSlTpljthw4dqhUrVtgUnz28ePFCDRo00ObNmw3bMmfOrICAABUvXlyenp5G7Z89e6ZmzZrpypUrVo2/b98+VaxYMdbKIy4uLsqXL5/Kli2rwoULxzoe9u7dqwoVKsT5sPfQoUNNJnZJnz69SpQoofLly6tYsWLKli2bVfHGx8aNG9W+fftYD5J6eHiocOHCKl++vN566y35+fnJ2Tnxv66IiopShw4d9O2338a6YSNjxowKCAhQoUKFYu3z27dvq06dOnZN1pPY8uXLpw4dOhjKoaGhJleMelPNmDFDrVu31pMnT4y2e3l5qUiRIipZsmSsJF9hYWHq2bOnRo4cafN8165dU926dY0S+/j6+qpUqVIqUKCAxVWGXvv000+1YMECQzlTpkwKCAhQsWLFYq0qderUKX3wwQeSXr13derU0f79+w31OXPmVJkyZeTv7x/rxqxly5ZpzJgxZuPo1q2bUdnW839oaKgWLlxoKHt7e6tVq1Y2jWFKRERErEQmLi4uyp07t0qWLKly5cqpYMGCsfZVWFiYOnbsqPnz58dr3mnTpqlXr16GRD7u7u4qVKiQypQpIx8fn1jtZ8yYoe+//z5ecyVU+/btjVZZS5cunYoXL66AgIBYx3tERIT+97//xSvhy88//2x07ZEqVSoVKFBAZcqUka+vr8mbAU0lMXB3d1eBAgVUqlQplS5dWrlz5451rr9x44aqVaumU6dO2RynlHT7xJ5M7Stvb2/5+/urbNmyCggIUI4cOWK1OXnypCpXrpyghAa2SuxrFUd7nbDi5MmThm2ZM2fWW2+9pcKFC8e61pMccy2xa9cutWjRQqGhoZJerSRXqFAhk9fOL168UNu2bXXu3DlJ0pgxYzR8+HDD73OaNGlUvHhxlSxZUl5eXrFifP/99+12w2hiO3LkiBo3bqxHjx4ZtuXKlUulS5dW3rx5jW5Kbt++vdF7Mnv2bJsTjM2cOdPws5OTkzp37mxVvwULFmjAgAGG98DV1VX+/v4KCAgw+bfPzz//HOtvtcTiiON5y5YtateuneE4e30+KV26tMnP3dWrV6tp06ZxJjuLKTH/ljN1Hk+XLp0KFSqkcuXKqWTJkib/xtq5c6dq1Khh+F22xcOHD1WrVi2jxIa+vr4qXbq0ihYtGuv6KCgoSI0aNdLjx49tngsAAAAAAAAA3jTNmjVTlixZYm2PiIjQiRMnTPYxl8gmOWrYsKHF+owZM1qsj3lfhaNUq1bNbF2KFClifb9rS//06dNb7GvNPti8ebMqVKggPz8/9ezZUzNnztSOHTt09epV3b9/P15J9IODg61u27VrV7Vp08am8T/55BM1bdrU1rBsZu67D0l6+vSpWrRooTRp0qhUqVJq0aKFBg0apDlz5mjPnj1G3+XF5dmzZzp27JjFNp06dbIpdmscPHhQ9erVU7Zs2dS5c2dNmzZNf//9ty5duqSgoKB4fbdjy3tvLUsLYEjS6NGjrUosNXHiRIvjxFxk8OTJkxaPf3d3d1WsWDHO+GvXrh1nGwBA8pYi7iYAAAAAAAAAgKTg5uammjVrau3atYZtK1asUM2aNfXVV1/p7bfftss8devWlZ+fny5fvizpVRKDyZMnK126dFb1nz17tuEBfkn63//+F+eKNZK0fft2Xbp0SZJUrlw5ffXVV6pevbpSpHj1X9VRUVHatm2bPvroI50+fdrQ79NPP1WjRo0M7aK/jo0bN0qSxo0bpw0bNhjqFi5caPKmE+nVl6FxGTNmjC5evChJ+uCDD/T5558brawTFhamxYsXq3fv3oaH/Z4+faoBAwbo119/tTj27du31ahRIwUFBRm2FS9eXIMGDVKDBg2MHhJ+/vy5Vq1apcGDB+vChQuSpAsXLqhDhw5as2aNyf1+48YNjR071mhbt27d1KtXLxUqVChW++DgYO3evVtr1qzRkiVL4to1Vuvdu7dRQqXq1atr2LBhqlSpUqxVaEJDQ3X06FH9+eefWrx4sd1iiG7q1KmxEldUrlxZY8aMUYUKFQz78unTp/rll1/0+eef68GDB4b42rRpo+PHj8d6YLZdu3aqVKmSody2bVvduXNHkpQlSxajBB5JaejQoVqwYIHhxoCZM2eqf//+yp07t0Piif77Kkl9+/Y1upkkel10WbNmNSofPnxYPXr0MHp4OmfOnBo/frwaNWpkeCg3IiJCmzZtUt++fY1uNBs2bJjKlCmjd955x+rY+/fvr7t378rFxUVdu3ZVnz59lC9fPkN9aGiodu3aZbb/tm3bDOfbOnXqaNSoUSpTpoyh/unTp5o4caJR4oCNGzdq7dq1Wr16tY4cOSJnZ2d99NFH6tevn9EqVNevX1evXr2Mknd9/fXX6tKlizJnzhwrlsKFC6ty5crasWOHpFcPSJ8+fVoFCxa0al8sW7bMaBWx1q1bm0zcEF+lS5fW+++/r7p166pIkSKxHrJ++fKltm3bpnHjxhk9DN+jRw9Vr17dZDIPc86fP69evXpJkgoVKqRRo0bp3XffNXr4/vDhw+rZs6f27Nlj2DZkyBC1b99eGTJkMBqvRIkShuN4/vz5RkmIxo8frxIlSlgdW0wLFiwwfCbVq1dPw4YNU7ly5QznrYiICK1evVo9evQwJE+KiIhQz549tW/fPqvnefDggT777DNJUrZs2TRq1Cg1b95c3t7ehjaXLl1SSEhIrL4uLi6qV6+eGjVqpBo1aihPnjyxktA8efJEq1at0siRI3X27FlJr24sa9OmjQ4fPmzVtURS75PE4OrqqsaNG6tBgwaqWrWqcubMGavN/fv39dtvv+mrr74yxH/79m116dJFK1euNDnu6+MvMDBQ/fr1M2z/8MMP1a5dO7PxmDo2E/taxVaLFi0y3Oxn7eszleAium+++cbwed2yZUsNHjxYxYsXN9S/ePFCmzZtMuqTWNcSlrRt21bh4eHy8fHRN998o+bNmxtWuIuKitLGjRvVpUsXXb16VdKr36nBgwerd+/e+uKLLyRJZcqU0ejRo1WtWjXDNVhYWJhmz56tzz77zHCtcPLkSU2bNi3OleysNWDAALVt29bwOqy9PrJm/3z00Ud69uyZ3Nzc1K9fP3Xv3l2+vr6G+ocPHxquM9KnT6/mzZsbzstXrlzRhg0bVK9ePatex44dO4z+Nqldu7bFFSFfe/TokWFfent7a9SoUerQoYNSp05taLN7927169fP6HNm+vTpqlGjhlq2bGk0XtasWQ2/5xs2bDBK9Nm/f3/VqVPHbCwxr6ckxxzPHTt21MuXL5UqVSoNHjxYH3/8sVFynmPHjumLL77QmjVrDNvWrl2rCRMm6PPPP7d6nsT8W0569X42a9ZM9evXV8WKFU3eiH3jxg0tXLhQY8aMMVw7nThxQgMHDrS42qkpffv21cWLF5UiRQp1795dvXv3Vt68eQ31ISEhmjZtmgYNGmRI+HP9+nV9/fXXsf4uBAAAAAAAAIB/m1SpUqlr16766quvrGqfO3du1a9fP5Gjsp/o31+ZEtc9ULYm6k8s/v7+FuvjWgwof/78ZuuiL1ZgiqV9EBUVpV69emnKlCkWx4gPW5PH//zzzzp48KDhu3RLSpcurfHjx8c3NJs4OTnpyy+/VI8ePcy2CQ0N1eHDh3X48GGj7c7OzipatKiqVq2q1q1bq0KFCmbHiGuBFldXVwUEBNgWfBzGjRuXKAsGJsbCAUm1gE3MeeJajCZ//vxWLUZn7X1JAIDki6Q0AAAAAAAAAJCMDBw40CgpjfRqRflKlSopV65cqlOnjipUqKCyZcuqUKFCVn2pF5Ozs7O6dOlieFD1+fPnWrhwoVUPoEZFRWn27NmGcooUKdSxY0er5n2dkKZDhw6aOXNmrMQkTk5OqlatmrZv366SJUsaHsK+ceOG1q5dq8aNGxu1z5Ytm+EBwJgPtr799ttWPSxqzsWLF+Xk5KTp06frf//7X6x6V1dXdezYUfny5VO1atUMNzD8/vvvunfvntHDlTF17NjR6Avcrl27aurUqSZvknBzc1OLFi1Up04d1a9fX7t375Yk/fnnn/rjjz/UpEmTWH1WrlxpeBhRepWcZMSIEWbjSZcunerXr6/69etr3Lhxdlmt5eTJkzp58qShXL16dW3atMns8eru7q4KFSqoQoUKGjFihM6fP5/gGKK7fv16rAdZ27dvrzlz5sR6WN7Ly0v/+9//VKtWLVWqVEk3btyQ9CpZw8cff6w//vjDqH2ePHmUJ08eQzn6jTJubm6qVauWXV+LtXLkyKHu3bsbHnp98eKFhg8frnnz5jkknui/r5JiJcGyZj9FRUWpQ4cORsd3iRIltHXr1lirZrm4uKhu3bqqVq2a3n33Xf3999+Gus6dO+vChQtWJaiSpDt37ihFihRasmSJmjVrFqve3d3dYvyvE9J88sknmjRpksljbujQoZJeJc15rW/fvjp79qxcXFz0yy+/mJw7e/bsWrp0qerUqWN4jaGhoYYHrU3p3r27ISmNJM2YMUMTJkwwG390M2fONCp36dLFqn5x8fT01J49e1S+fHmL7VKkSKGaNWsakrW93m9Pnz7V1KlTbXrw+vXvdt26dbVixQpDkofoAgICtGnTJlWsWFGBgYGSXiV8WLhwoSGhzWvp0qUzHAc7d+40qitVqpTFldvi8vrB+iFDhmjkyJGx6l1cXPTee++pcOHCeuutt/Ts2TNJ0v79+xUYGGh1QpzXK8QVKlRIf//9t8kkBqYSW5UqVUoXLlxQrly5LI7v7e2tDz74QE2bNlXTpk31559/SpKOHj2qdevW2ZQsKqn2ib298847ateuncmkUdFlyJBB3bt3V8uWLVW7dm0dOnRIkrRq1SqdPHlShQsXjtXn9fEXM4lfnjx5bP4sSuxrFVtFT8xoj9cnyZAg5fvvv4/1+yy9unk1+jGZmNcSlly+fFn+/v76+++/YyX/cHJygxrJjAABAABJREFUUp06dbRx40YVL17ckFzm999/V2BgoCIjI9WsWTMtXrw41nvn6uqqjz76SO7u7kbX8rNmzbJbUprChQsbjlV7Xx/duXNHXl5e+uuvv4ySA76WNm1aValSxVDu3r27UbKwGTNmWJ2UJr6ffa8TkaRLl07bt29X0aJFY7WpWLGiduzYoZYtW2r58uWG7b169VL9+vWNEkBF32/Xr183Gqdw4cI27VNHHc9XrlyRq6ur/vzzT9WoUSNWffHixbV69Wr16dNH3333nWH7iBEj1Lp1a5NJvExJ7L/lvvnmG6PkQqb4+vrq888/V6tWrVS9enXD38IzZszQsGHD4ly1NebrcXV11YoVK/Tuu+/Gqvf09FTfvn2VNWtWQyIoSZo7d65GjRoV5834AAAAAAAAAPCm6969u0aPHq2XL1/G2fajjz6K171WjhJzsZSY3pT/A06TJo3F+lSpUlmsj3lPiL0MGjQoURLSSLYnBPLy8tLChQtVtmxZi+2cnJy0aNGiOPeZPX300UcKDAzU9OnTbeoXGRmpY8eO6dixY5oyZYpKliypH3/80WRymriSn2TIkCHWvYYJMW3aNA0YMMBu40WXGMmg4to/iTVPXPcRxvW7bWs7AEDy9eZcQQMAAAAAAADAf0ClSpU0ZMgQk3VXrlzRjBkz1KlTJxUtWlRp0qQxPJh/8OBBm+bp1KmT0Y0JM2bMsKrfxo0bDQkWJKlBgwYmH1o3p3Tp0po+fbrFL4kzZcoUax/89ddfVs9hL59++qnJhxijq1y5spo3b24ov3jxQps3bzbbfu/evVq3bp2h/M4772jatGlx3iSSNm1aLV++XN7e3oZt5pJIxFwx5+OPP7Y4dnReXl7KkSOH1e3NiRlDt27drL6px9nZWQUKFEhwDNH9+OOPCg0NNZRLlCihmTNnxnroNjo/Pz/99ttvRm1WrVqlc+fO2TW2xDR48GB5enoaygsXLtSpU6ccGFHCbNy4UcePHzeUPTw8tGrVKos3H71+gDf6eerWrVtavHixTXP369fPZFIYa1WoUEHff/+9xWNuwIABRq/lzJkzioqK0ueff25xbhcXF40aNcpom6VzZtOmTZUxY0ZDef78+QoPD4/zNZw5c0bbt283lEuWLKnSpUvH2c8a7u7ucSakiWnIkCGqXLmyoTx37lyb582ZM6d+/fVXkwlpXvPw8NCYMWOMtjniM6lx48Ymk69EV6BAgVjJJGyNNWXKlFq6dKlNn+25c+eOMyFNdG5ublqwYIHRTUdz5syxKU4p6faJPRUpUiTOhDTRpUuXTkuWLDH6DI3PsW6LpLhWSS5atWplMiGNKY66lnj9OxkzIU10BQoUMEosExERoXPnzilfvnyaN2+exfeuffv2RitTBgYG6tatW1bH50jjx483mZDGlIoVK6pYsWKG8urVqw2JiSx5+PChfvvtN0M5c+bMsRJlxmXGjBkmE9K85uLiooULFxol1Lxz547N1yq2cOS18ddff20yIU10EyZMMLoZOTQ0VNOmTbNpnsT4W056lYgtroQ00eXKlcvob+3Q0FD98ssvVvd/bcyYMSYT0kT3wQcfqFy5coby3bt3DUnNAAAAAAAAAODfzMfHx6qFEtzd3dW5c+ckiMh+oif+N+VNSbDj6urq0P6mnDhxItl9nzl//vw420RFRdn8vUlCOTk56eeff9Zvv/1m8XuvuBw9elTVqlUz+j7aEe7duxdrAQe88vz5c0eHAABIpt6Mq04AAAAAAAAA+A8ZOXKkJk2aFOeNBU+fPtXff/+toUOHqkyZMipatKhmz55t1WobWbNmNXqg8vjx49q3b1+c/WImr+nSpUucfaIbOXKkVav0tGjRwqh8+PBhm+ZJKHd3d7PJgWJq2bKlUdlSrN9//71R+bvvvrP48Gd0WbNmNXqwcteuXSYfpo3+gKnkmFWRkkMMr0VFRWnWrFlG28aPH68UKVLE2bdChQpG729UVJRmzpxp9xgTS+bMmY0eto+MjNTQoUMdGFHCxNz3n332mXLmzBlnvzRp0mj48OFG22xZvcnDw0ODBg2yur0pI0aMiPNmMDc3N9WpU8dom6enp1UrM1WoUMEo0cWRI0fMtnV1dVWHDh0M5aCgIP3+++9xzhFz/9t6/k8MH3zwgeHnu3fv6vz58zb1HzhwoFWrMdWuXVvp0qUzlJP6M0mSvvnmG6va2fKZZK5/Qm4is1b69On1zjvvGMq7d++2eYyk2ieOlj9/fqPV8OKzr2yRFNcqycVXX31lVTtHXks0b95cxYsXj7Ndo0aNYm0bOHCgxaRb0qsbSGP2tfQZklzkzJnT5s+hbt26GX5+8eKFVQmeFi1aZHRd2759e5uua8uUKaOmTZvG2c7NzS1Wkq3Zs2dbPY8tHHk8+/r66tNPP42znZOTk8aOHWu0bc6cOYqKirJqnsT6Wy6+atasqWzZshnKtp7HfX191aNHD6vavumfeQAAAAAAAAAQXz179oyzTevWrZU+ffokiAZvgvnz5+vly5dm611cXNSzZ0/t2LFD9+/f18uXLxUVFWX4t2XLFrvGs3z5ck2dOtWqtt99951WrVpl1/mt0axZMx0/flwHDhzQV199pVq1ailLliw2jREeHq6OHTsqJCTEaHumTJks9rt//74iIiJsjtmUZcuW6fHjxxbbtG3bVhs3btSdO3f04sULo/f+0qVLdonDFnHtn8QS/V4ZUx49emTVOA8fPrRDNAAARyIpDQAAAAAAAAAkQ59++qnOnTunjz/+2KoH5qVXK7h07txZZcuW1ZUrV+JsH/3BTCl2wpmY7t27Z/SFdo4cOVSvXj2rYpNeJYaoW7euVW3Tp09vlGzi2rVrVs9jD7Vq1VKGDBmsaluyZEmjsrlYIyMjjVZ6KVu2rPz9/W2KK2bCih07dsRq4+PjY1ReuHChTXPYQ8wYFi1alOQxvHb69GndvXvXUM6ZM6dq1qxpdf9OnToZlbdv32632JJC//79lTZtWkN5+fLlb8TD5qbE3PcdO3a0um/r1q3l7u5uKB86dEjPnj2zqm/9+vWVOnVqq+eKKW3atKpVq5ZVbWMmA6ldu7bVnwHR+967d8/i6kVdu3Y1SjIR1/n/xYsXRityeXh4GCWEcZTcuXMblW05tp2cnGIlQDPHxcVFxYoVM5Tv3bunsLAwq+dKqGLFiqlw4cJWtS1atKhRYgFbPz9bt25tU/uEiP7+3bhxQ/fu3bO6b1Luk+Qg+r5KzHN4Ul2rJAdlypRRvnz5rGrryGuJ5s2bW9Uu5ueHk5OT3n//fav6Rj+/SdLVq1etC86BWrZsafPKn23btpWnp6ehPHPmzDiTnMRMuBI96ZI12rVrZ3Xbpk2bysvLy1A+ePBgrJtx7cGRx3OrVq2sTupTuXJl5cmTx1C+ffu2zpw5Y1XfxPhbLqH8/PwMP9t6Hn///fet3m9J9XoAAAAAAAAAILmpUqVKnIn+rUlc819RrVo1oyQb1v6LvgDOm27Dhg0W6+fNm6cpU6aoUqVKSp8+vVxcXIzqrU3GYY2LFy+qc+fONvXp0KGDw77XK126tL788ktt3LhRt2/f1sOHD3Xo0CH9/vvvGjt2rJo2bWr0vVxMt2/f1ooVK4y2RV+IyZSwsDC7JeOP670fNWqUFixYoFq1ailz5syxFnew53tvrbj2z08//RSv3+mY/2Iu+hVXMpxz585ZlSzo9OnTcbYBACRvJKUBAAAAAAAAgGQqe/bs+uGHH3Tnzh2tWrVKn332mUqXLq1UqVJZ7Hfo0CGVLVtWFy5csNiuZs2ayp8/v6H8yy+/6MmTJ2bbz5s3T+Hh4YZyp06dbHoYNCAgwKb20b9MTeovc0uXLm1125hf+pqL9fjx40Z1tszxWvREPZJ06tSpWG1q165tVO7bt6++/PJL3b592+b54qtcuXJGSTxWrFihFi1a6Pjx40kWw2v79u0zKlevXt0oGUdcqlSpYnRzwZEjR4x+D5K7tGnTqn///oZyVFSUvvjiCwdGFD+XL1/WnTt3DOVcuXIpb968VvdPnTq10e9cRESEDhw4YFXfsmXLWh+oCQEBAVYfczEfoA4ICLB6nph9La3qlD9/ftWoUcNQ/vvvv3Xx4kWz7VeuXGn0AHvz5s2tTpZjqxcvXmj16tX65JNPVKVKFWXPnl2pU6eWs7OznJycjP7FTHQWFBRk9Tx+fn5WP7AuWX+uTwy2fF6kTJnSKBGVrXEm9Hh/+PChZs2apY4dOyogIEDZsmWTp6dnrPfOyclJo0ePNupry/uXlPsksdy5c0dTpkzRBx98oGLFiilLlixyd3c3ua+WLFli6Pfs2TOFhoYmSkxJda2SHNhyrDvyWqJUqVJWtYt5PsudO3ecq9aZ6xvXqoDJQXzOVWnSpFGrVq0M5fPnz2vr1q1m2x88eFBHjx41lKtWraoCBQrYNGe1atWsbuvh4aEyZcoYyhERETp06JBN81nDkcezLftDerXPo9u/f79V/RLjbzlTLl++rG+//VbNmzdXoUKFlClTJrm6upo8j+/Zs8fQz5bPOynpXg8AAAAAAAAAvOl69Ohhtq5ixYp66623kjAaJHfXr183W5c2bVq1adPGYv/o//efEOHh4WrRooXN/6cfHBysli1b6uXLl3aJIyHSpEmjgIAAvffeexowYICWLVumEydOWExoEnNxEw8Pj1iLacQ0e/Zsu8Rr6b2XpI8//thivb3ee1vE9f3oxo0bE2XewoULy9XV1Wx9aGiodu/eHec4iRUfACDppIi7CaK7e/eu1Td+JabEuPkIAAAAAAAAQPLk6uqqhg0bqmHDhpJefRl9/Phx7dq1S5s2bdL69etjPQh49+5dNW3aVIcOHYq1UstrTk5O6tq1qyFhRUhIiJYsWaKuXbuabD9z5kzDz87OzurUqZNNryOuFTtiir5iSmI9fG2OLbHGXNnFXKwxH8r+8ccf9eOPP9oeXDQPHjyIta1ixYqqXbu24cvcly9f6uuvv9bo0aNVsWJF1axZU5UrV1bZsmXl7e2doPnNcXNz0+eff26U/OS3337Tb7/9pkKFCqlOnTqqWrWqKlSooKxZsyZKDK9duXLFqBzXylgxubq6qmDBgvrnn38kvVp5586dO8qRI4fdYkxsvXr10qRJkwxJRf766y/t2rVLb7/9ts1j3bp1SydOnLCqrY+PjwoXLmzzHKYk9H2UpBIlShjd1GLtilG5c+e2ea7o4lo1KDoPDw+79Y3rvNm9e3dt3rxZ0qtkRTNnztQ333xjsu2MGTOMyl26dLE6LmtFRUVp9uzZGjx4sFECHFs8fPjQ6rYJ+UySkvZzKT6xvn7g3ZY4vby8lDFjRpvmei0kJEQjRozQ5MmTFRYWFq8xEvv9i88+SQxBQUEaMGCA5s+fb9WKYaY8fPhQ7u7udo4s6a5VkgNbzu2OvJaw9nMgKT8/koP4fjZ3795ds2bNMpRnzJih6tWrm2yb0M8+FxcXFSxY0KY+RYsW1ZYtWwzlS5cuqUqVKjaNERdHHs9Fixa1aa6Y7S9dumRVv8T4Wy66K1euqFevXlq1apWioqKsnus1Wz7vpMR/PQAAAAAAAADwb9G2bVsNHDhQwcHBseo++eQTB0SE5MxSEpjw8HBFRkaavefu/v37sb5Liq++fftafFa4cuXKsRK4vLZ3714NGjRI48aNs0ss5oSHh8e5iF5MuXLlUpUqVbR8+XKT9bdu3Yq17Z133rG44Nns2bPVoUMHlStXzuo4Xr58abTgghR3Uv/nz5+brQsLC9P3339v9fz2UqpUKWXMmNHs4gerVq3SkSNH4pV86+XLl1qxYoWyZcumypUrG9W5urqqVKlSFhPPTJ06NVa/6K5fv67ff//d5rgAAMmL9UvS/sd5eXlJkiIjI3X79m2H/wMAAAAAAADw35UqVSqVKlVKn376qVatWqWbN2+qf//+sb4IDwwM1C+//GJxrA4dOhitZhE98Ux0O3bs0JkzZwzlunXrKmfOnDbF7ebmZlN7R0pIrOYeTLx//368xzTH3JfkixcvVvny5Y22RUZGaufOnRoxYoRq1aql9OnTq0KFCvr66691+fJlu8c2aNAgkwmOTp06pUmTJun9999XtmzZVLBgQfXq1Uv79u2zewySYt1sFJ+ECzH7mLqBKTnz9PTUoEGDjLZFTxhki/Xr16t27dpW/fv222/tEb4kx76PqVOntnmu6BJyPkmMc9FrjRs3NkoKNXfuXJMraF25ckWbNm0ylAsVKhSvhEaWREZGql27dvrf//4X74Q0kmxKhpLQz6T4PIQeX4l5HEQX32M9KChIFSpU0Lhx4+KdkEZKuvcvKd+7mC5cuKC33npLc+bMiXdCGsm2fWWLpLxWcTRbjndHfgbF91h/U39HrBXf81Xp0qUVEBBgKK9YscJk4qTXyTpfS5cunZo2bWrTXGnSpIl1g2tcMmTIYFS2NXmJNRx5PMd8fba2t3Z/JObxv3//fpUoUUIrV66M9+9KzISycfm3/z4DAAAAAAAAgL14eHjom2++UcuWLY3+tWvXzub/58e/X7p06czWPXv2TL/++qvJuuDgYL333nt2WZxj+fLlmjp1qtn65s2ba8uWLapYsaLZNhMmTNCaNWsSHIslPj4+6tWrlw4cOKDIyEir+oSEhOjw4cNm6019h9GmTRs5OTmZ7RMeHq6GDRtq69atcc5/6dIlNWvWTLt27YpVZ+m9l6Q5c+aY3P78+XN98MEHRvdQJhVnZ2e1bNnSbP3Lly/VqFEji/s8pmPHjmn48OHKnTu3WrZsqQsXLphsF9f5c+nSpZo3b57JurCwMLVr107Pnj2zOi4AQPJk2x0w/2H9+/fXuHHj9PTpU0eHAgAAAAAAAABGMmTIoG+//VZVqlTRe++9Z/SQ88KFC/XBBx+Y7ZsxY0Y1bdpUixcvliQdOHBAgYGBKlGihFG7mKu7dOnSxY6v4L8hMR4qNfdFf8aMGbV9+3bNnDlTEydO1Pnz52O1efnypfbu3au9e/dq6NChatOmjcaPH68sWbLYJTYnJyf9/PPPev/99zVq1Cjt3LnTZLszZ87ozJkzmjx5st5++219//33Kl26tF1ikBTr//U9PT1tHiNmnydPniQoJkf46KOPNHHiRF27dk2StG3bNm3cuFG1a9d2cGTWceT7mDJlSpvnehOkTJlSnTp10jfffCPp1SpUa9euVePGjY3azZo1y+hckxjn/6+++koLFy402pY6dWpVq1ZNpUqVUo4cOZQ2bVq5ubkZvR+BgYHq16+f3eP5r4rvsd68efNYq5XlyJFD1atXV+HChZU9e3Z5eXnJ3d1dzs7/v2bK/PnztWDBggTF/CYJDw/Xu+++q+vXrxttz58/v6pWrSp/f3/5+vrK09NT7u7uRjfbjRs3Ths2bEj0GJPyWsXRbDneuZZIfhLy2dy9e3dD4sSwsDAtWLBAvXr1Mmrz66+/Gr1HH374oc2JQTw8PGyOLeZxkhj3pzjyeLZ1nyTF/rDF/fv39e6778ZKtlW8eHFVrlxZ+fLlk4+Pj9zd3eXm5mZ0Hu/bt6+OHTuW1CEDAAAAAAAAwH9O9+7d1b17d0eHgTdAoUKFLC6a87///U/Hjx9XkyZNlD17dj18+FCbNm3SuHHjYn3nGx8XL15U586dzdZnz55dP//8s1xcXLR48WKVKFHC5IIgUVFRat++vY4ePaocOXIkOC5T7t+/r8mTJ2vy5MnKkCGDqlevrpIlS6pgwYLKnTu3UqdOLW9vb0VGRur27dvat2+fJk+erEuXLll8fTGVKFFCrVu3NtzHaMq9e/dUo0YN1a9fX82bN1dAQIAyZMig58+f686dOzpw4ID++usvrV+/XpGRkerZs2esMQoVKqT9+/ebnWPo0KG6du2a2rZtq1y5cunZs2fasWOHxo8fr9OnT8extxLPF198oTlz5phN8HL9+nWVLVtWTZo00XvvvafixYsrQ4YMioqKUnBwsO7du6d//vlHhw8f1vbt23Xx4kWr5u3UqZOGDBliMbFMx44dtWXLFnXs2FH58uVTaGio9uzZozFjxujkyZPxer0AgOSFpDRWatCggRo0aODoMAAAAAAAAADArAYNGqh9+/aaPXu2YZu5RCDRdevWzejL3BkzZhitwvLw4UMtW7bMUM6aNasaNmxop6j/O2I+hNm6dWt16tQpQWP6+PiYrUuZMqU++ugjffTRRzp48KA2b96srVu3avfu3Xr8+LFR28jISC1cuFCbNm3S1q1b5e/vn6C4oqtbt67q1q2rS5cuacOGDdq6dau2b9+umzdvxmq7a9cuvf3221q4cKGaN29ul/m9vLyMyiEhITaPEbOPt7d3gmJyBFdXVw0ZMsTwELb06maFNyUpDe9j4ujSpYvGjBljSBoxY8YMo6Q0ERERRitAubq6ql27dnaN4c6dOxo7dqzRtkGDBmnw4MGx3veYoidhg2OsWrXKaBUyb29v/fTTT2rdurVRAhpTNm/enMjRJS/Tpk3T2bNnDeUsWbJo7ty5qlevXpx9Z82alZihGST1tcqbgs+gf5fWrVurb9++hkQqM2bMiJWUxh4JOeOz2l/M4ySuz8H4cOTx/OzZM5uO/aTYH7b4+uuvdf/+fUM5f/78WrhwocqWLRtn3/gkKQIAAAAAAAAAAImnQYMG2rZtm9n60NBQjRkzRmPGjLH73GFhYWrRooXJJDOS5OzsrAULFihdunSSpFy5cmn69Olq2bKlyfYPHjxQq1attG3bNqVIkbiPjN+/f1/Lli0zupcwPurUqWNy++jRo7VhwwYFBQWZ7RsVFaU1a9ZozZo18Zq7QYMGmjdvntn6iIgITZs2TdOmTYvX+IklW7ZsGjZsmD7//HOzbSIiIuzy/kSXNm1a9evXTyNHjjTbJioqSvPmzbO4XwEAbzaS0gAAAAAAAADAv0iLFi2MktI8ffpUjx49Upo0acz2qVKligoVKqRTp05JkhYtWqRx48bJ3d3dUA4NDTW079ChQ6J/gf1vlDFjRqNy2rRpVatWrSSZu3Tp0ipdurQ+//xzRUZGKjAwUOvWrdOvv/6qwMBAQ7vbt2+rWbNmCgwMjDOZgK1y586tbt26qVu3bpJerfizefNmrVixQhs2bDAkxQgPD1e7du1Urlw55cyZM8Hzvr5B47XoD5JaK+aNDjHHfFN07NhR3377rc6fPy9JOnDggP744w+99957Vo/RoUMHdejQIXECtID3MXH4+fmpbt26+uuvvyRJ69at0/Xr1w0rUr0uv9akSRNlyJDBrjGsXLnS6DOma9eu+uabb6zq++DBA7vGAtv98ssvRuWff/5ZrVu3tqrvf+39i7mvfv/9d1WoUMGqvkm1rxx5rZKc8Rn07+Ll5aW2bdvqp59+kiSdOHFCe/bsMfw+njhxQnv37jW0L1++vIoWLWrzPI8ePdKLFy+UMmVKq/vEPLbSpk1r87xxceTxHBQUZFNSmqTYH7b49ddfDT+7ublp3bp1ypMnj1V9/2ufeQAAAAAAAAAAJHddu3bVmDFj4vVdiSR17tw53ouL9O3bV4cOHTJb//nnn6tatWpG21q0aKH169cb3RcY3e7du/XFF1/EWhQoOcqfP7/q169vsi5nzpxau3atatSoEa/FFazRpEkTFSxYUKdPn45X/4S89wk1YMAAnTt3TjNnzkzSeb/88kutXLnS6D5DW+TJk0cXL160c1QAgKRk3zvKAQAAAAAAAAAO5efnF2vbs2fP4uz3OlGIJD18+FC//faboTxjxgzDz05OTurSpUvCgvyPyp07t1H5dWKQpObs7Ky33npLgwYN0tGjR7V8+XJDAiJJ+ueff7R+/fpEjyNPnjzq0qWL/vrrLwUGBho90Pn8+XP98MMPdpknV65cRmVbvxwPCwvTmTNnDGVXV1dlyZLFLrEltRQpUmjEiBFG24YMGWJICJScJfR9NNUn5pj/VdHP/xEREUY3MEU//0uvbsqyt+gP/kvSxx9/bHXfEydO2Dsc2Cj6+5chQwa1aNHC6r7/pfcvMjJSBw4cMJRLlixpdUIaKen2VXK5VkluuJb494n+2ScZf97Z67MvIiLC5htZjx8/blSO+TtpD448nv/55x+b5kqK/WGtq1ev6ubNm4ZyvXr1rE5IExoaqkuXLiVWaAAAAAAAAAAAIB5Sp06tmTNnxmvRrs8++0xt27aN17zLli2zeE9U6dKlY93b89rkyZPl7+9vtu+4ceP0559/xiuupOLt7a1FixbJ1dXVbJuyZctqw4YNiXZfj4uLi+bOnSs3Nzeb+zZt2lSDBw9OhKisN23aNH322WdycnJKsjlTpkypP/74w+S9qXHJnz+/Fi1aZP+gAABJiqQ0AAAAAAAAAPAvYmqFkAwZMsTZr3379kaJSV4/jHngwAGjBxVr1Khh9cN3SSnmDQJRUVEOisS8smXLysPDw1DevXu3nj9/7sCIXnn//ffVt29fo207d+5M0hiKFi2q6dOnJ0oM5cuXNypv3brVpuNjx44devHihaEcEBCgVKlS2SU2R2jVqpWKFStmKP/zzz/65ZdfHBiRdfz8/IweeL5y5YpNK+g8efJEBw8eNJRTpEih0qVL2zXGN1WDBg3k6+trKM+ePVuRkZG6deuW1q5da9ieL1++WCtx2cOdO3eMypZuoIrp77//tnc4dvEmfCbZS/T3L1++fHJxcbGq3+PHjy2u/PZvc//+fb18+dJQtuU4P3v2rG7cuGF1+4Qcf8n1WsXRuJZIuOjHZXI4J5YoUcLofV26dKkeP36ssLAwLViwwLA9derUNiXbimnbtm1Wt3327JnRtYqLi4tKlSplsm1Cfs8deTzbsj8kafv27UblsmXL2tTfnhJyvRJznwEAAAAAAAAAgOThvffe0+zZs61OTuLk5KRBgwZpwoQJ8Zrv4sWL+t///me23tPTU4sXL1bKlCnN1i9ZssTsdzNRUVFq166drl+/Hq/4zIl+T2FCFC9eXLt371aZMmXibFuxYkUFBgaqc+fOZvdHQpQrV05//PGH0qZNa3Wfdu3aafHixfFKZGRPLi4umjhxotavX2/Td1bm5MyZU59//rlq165tsZ2fn5+2bt2qgIAAq8euUKGCtm3bpqxZsyY0TACAg5GUBgAAAAAAAAD+RQ4cOGBUzpo1q1UPCaZNm9bogcudO3fq9OnTmjlzplG7Ll262CdQO/P09DQqP3v2zEGRmJcqVSrVqFHDUA4JCdGcOXMcGNH/e/vtt43KQUFB/5oY/P39YyUz2bJli9X9Z8+ebVSuWrWqXeJyFGdnZ3311VdG24YNG2aULCG5irnv586da3XfJUuWKDQ01FAuXbq0UeKF/zIXFxejm56uXLmiDRs2aO7cuUbHxf/+979EWWUp5oPw4eHhVvU7evSo9uzZY/d47OFN+Eyyl+jvn7XvnfTq3PpfSnYS3+Nckn788Ueb5krI8Zecr1UciWuJhIt+XCaXc2K3bt0MP4eEhGjx4sVasWKFHjx4YNjepk2bWL9Ttpg/f77VbZcvX66nT58ayqVKlTI7d0J+zx15PP/yyy9WJ2fZsWOHUQLCrFmz2uWm2vhKyvM4AAAAAAAAAABIOu3bt9eBAwfUtGlTi8lGqlSpoi1btuibb76J170TYWFhatGihR49emS2zaRJk5Q/f36L47z11lsaM2aM2fr79++rdevWdr0P6MGDB9q4caMGDhyo6tWr25TIxcvLS02aNNGyZct09OhRFS1a1Oq+adKk0cyZM3XlyhUNHTpUJUqUsGqhnNSpU+vdd9/Vzz//bHHBqrp16+ro0aPq1KmTxfsr33rrLa1YsULz5s1LVouP1K5dW6dOndK6devUtGlTZcqUyap+bm5uqlKlioYPH65du3bp8uXLGjNmjNGCVubkypVL+/fv13fffWdxYcNixYrphx9+0I4dO5QtWzarXxMAIPlK4egAAAAAAAAAAACvBAUFacOGDWrVqlW8VtQIDw/XlClTjLbVrVvX6v7du3fXvHnzDOXvvvtOS5YsMZQzZsyoJk2a2BxXUkifPr1R+dKlSypSpIiDojGvf//+WrNmjaE8bNgw1a9fXzlz5nRgVLETwKRLl+5fE4OTk5M6d+6sb775xrCtf//+2r9/f5w3Kuzfv1+//PKL0ViWVix6UzRu3Fhly5bV/v37JUnnz59/I5IO/O9//9PSpUsN5YkTJ6pr167Knj27xX6PHz/W8OHDjbYl1wRbjtKlSxeNGjVKERERkqTp06crMDDQUJ8yZUp16NAhUeaOuRrSzp079e6771rsExERoY8//jhR4rEHU59J/1ZZs2Y1vL4TJ07o4cOHcd6AduPGDY0YMSIJoks+MmTIoBQpUhhu/Nu7d69evnypFCksf11/9OhRm5MZJPT4S67XKo7EtUTCRT8u79+/rydPnsjb29uBEUktW7ZUnz59FBwcLEmaMWOG0qRJY9QmodcLBw4c0PLly9W0aVOL7Z4/f65hw4YZbevUqZPZ9gn5PXfk8Xzjxg1NnjxZffv2tdguKipKn3/+udG2Dh06JEpyPGuZul6xxp9//qmVK1cmRkgAAAAAAAAA8K91+fLlJJ1v7ty5Ni2KE1NC4k3o3FLsxOqOMHz48Fj3ZdgiIfvQz88vwfugaNGiWrZsmYKDg7Vjxw5duXJFjx49koeHh3LlyqXy5cvHSthRrVo1m+Z1dXXVwYMHExTna5999pk+++wzu4xlDTc3N9WqVUu1atUybLt27ZouX76sq1ev6sGDBwoJCdGLFy/k4eEhT09PZcuWTf7+/sqXL1+c34vHJVu2bBoxYoRGjBihp0+f6uDBg7p586aCg4P18OFDubi4yNvbW9mzZ1e+fPlUqFAhq++9zJUrl2bNmqXJkydrx44dunDhgh4+fKhUqVIpe/bsKlu2rPLmzWvUJ6HHnD1/Z52cnFS3bl3DPaLnz5/XP//8owcPHujhw4d69uyZvLy85O3trWzZsqlgwYLy8/OL172pr7m4uKh3797q3bu3Dh8+rBMnTujWrVtKlSqVfHx8VKxYMRUqVMiojz1+TwEAjkVSGgAAAAAAAABIJp4+faoPPvhAX3/9tQYNGqSmTZvK3d3dqr7Pnz9Xu3btdOLECaPt7dq1s3r+8uXLq0SJEoZEBNOnT481VnJa7SO6mAloli1bpgYNGjgoGvOqVKmiunXrav369ZKke/fuqU6dOvrjjz9UsGBBq8aIjIzUqlWrtGfPHo0dOzZWfY8ePVSvXj01aNDAqgc3w8LCNHnyZKNtpUqVsioWcyZNmqQUKVKoY8eO8vDwsKrPuHHj7BpDdB999JG+++47hYaGSpIOHz6s7t27a/r06Wb30dWrV9WsWTOjL8QbN24c60aDN9XXX3+t2rVrG8q7d+92YDTWqVWrlooXL65jx45JkkJCQtS4cWP9/fffsR4ify08PFzNmzfXrVu3DNuyZcumNm3aJEnMbwpfX181aNDA8ND077//blTfsGFDZcmSJVHmrlixolFSpCFDhqh69epmP/8iIiLUqVMn7dmzJ1HisYeYn0nLly9Xz549HRRN4qpYsaIhGUJ4eLgGDRqkn376yWz7e/fuqUGDBnr48GESRZg8uLi4qFy5ctq1a5ck6datW5owYUKspAvRnT9/Xo0bN9aLFy9smitXrlzy8vLS06dPJUmbN29WcHCw1cnekuJa5U3EtUTCFClSRNu3b5f06ibL5cuXJ1qyM2u5u7urXbt2mjRpkqRX72l0AQEBCggISPA8Xbp0kb+/v9lVHyMjI/Xhhx8aJZbJnDmzxWuVmJ8zq1at0ujRo5UyZUqrYnLk8fzFF18oICBA1atXN9umb9++Rp/zbm5u6t69u03z2FvOnDnl6+urGzduSHqVcOjXX39Vy5YtzfbZv3+/2rZtm1QhAgAAAAAAAACABEqXLp0aNWrk6DDeCDly5FCOHDmSfF4vLy9Vq1bN7uN6enqqXr16dh83qeXLl0/58uVLsvns9Z0qACD5i386MwAAAAAAAABAojh58qQ+/PBDZc2aVV26dNFvv/1mlFAhups3b+rHH39UoUKF9NtvvxnVvf/++6pRo4ZNc3fr1s1s3f/+9z+bxkpK1apVM3oIc968eXr//fc1e/Zs/fXXX9q0aZPh3+sHwh1l/vz5Rl/KnzlzRqVKlVLv3r0VGBhoclWQ4OBgbdq0SZ999pn8/PzUpEkT7du3z+T4u3btUqNGjZQ7d27169dPW7du1ePHj2O1e/HihdatW6e3335bBw4cMGzPmjVrghP6XLp0ST179pSPj4/at2+v33//3ewxfPToUbVq1cooMY6zs7M6deqUoBiiy549u7799lujbTNnzlSNGjW0d+9eo+0hISGaNWuWSpUqpWvXrhm2p0+fXj/88IPdYnK0WrVqJcpNGonJyclJc+bMMfpdP3z4sEqWLKnly5crPDzcsD0yMlIbN25U6dKltWHDBqNxZs+eLTc3tySL+01h6fzfpUuXRJu3SZMm8vb2NpQPHz6s6tWrxzrHvXz5UuvXr1e5cuU0f/58SYq1slJy8dZbbyljxoyG8tatW1WjRg1NmzZNf/75p9Fn0qZNmxwYacLFTH43bdo0tW/fXleuXDHa/uTJE82aNUvFixfX0aNHJSXf9y+xxNxXgwYNUr9+/XT37l2j7UFBQZowYYJKly6tq1evysnJSf7+/lbP4+zsrJo1axrKDx8+VPny5TV27FitXLky1vEXHBwca4zEvlZ5E3EtkTB16tQxKn/00Uf65JNPtGTJEq1fv97omDx58mSSxZWYn31p06ZV6tSpFRwcrIoVK2rKlCmxrsn37NmjypUra9myZUbbv//+e6PPxpgyZ86sEiVKGMrnzp1ThQoVNGnSJK1ZsybW7/nz58+N+jvqeM6VK5fCwsJUr149jRgxQvfu3TOqP378uBo1aqTvvvvOaPvQoUOVK1cum+ZKDDHP4+3atdPo0aNjva/Xr1/Xl19+qSpVqig4OFhubm7y8/NLwkgBAAAAAAAAAAAAAADsJ4WjAwAAAAAAAAAAmPb48WPNnDlTM2fOlCRlyJBBGTNmVNq0afX8+XPdunUr1oPMr5UrV05z5syxec62bdtqwIABevr0qdH2SpUqJeuHxzNnzqwPP/xQs2fPNmz7/fff9fvvv8dqmytXLl2+fDkJozOWOXNmrV27VvXr1zc82Pns2TNNmjRJkyZNUpo0aeTr6ytvb289ffpUDx48MJvQxZIrV65owoQJmjBhgpycnOTr66sMGTLI3d1djx8/1sWLF2M9oOri4qKZM2fK3d3dLq/10aNHmj9/viGBRKZMmZQ5c2Z5e3vr+fPnunz5sh4+fBir34ABA1SqVCm7xPBajx49tH//fi1YsMCwbevWrapQoYIyZcqkHDly6Pnz57p06ZJCQ0ON+rq7u2vx4sXy8fGxa0yO9vXXX+vtt992dBg2CQgI0A8//KDu3bsrMjJSknT58mU1a9ZM3t7e8vPzk4uLi65cuWIy0cKIESP+FSsbJYa6devKz88v1vkxZ86csZIJ2FOGDBk0ZMgQDRgwwLBt3759Kl++vLJkyaKcOXPq+fPnunLlitFD3wULFtSYMWPUuHHjRIstvlKmTKlPP/1UQ4cONWzbsmWLtmzZYrK9qQQfb4o6dero3Xff1Z9//mnY9vq8nydPHmXKlEkPHz7UpUuXjBJHtWnTRvnz59eIESMcEbZDdOzYUT/99JMhKU9UVJQmTJig7777Tvnz51fatGl1//59Xbp0SREREYZ+gwYN0o0bN3TmzBmr5/rss8+0atUqw7F19uxZDRw40GTbLVu2xEpSllTXKm8ariXir0GDBvL39zccx8+fP9fUqVM1derUWG3bt2+vuXPnJklchQoVUpUqVbR9+3aj7R4eHmrTpk2Cxk6TJo2++uortWvXTk+ePNGnn36q/v37K0+ePPLw8NC1a9dM/i3XuXNntW7dOs7x+/bta5Qk5dChQzp06JDJtpcuXYqVFMURx/OcOXNUp04dhYeHa/jw4Ro1apRy586tNGnS6NatW7px40asPnXr1lW/fv1smiex9OvXTwsWLND169clSeHh4Ro8eLCGDh0qf39/eXp66t69e7p8+bLRZ/vkyZO1aNEih/4NCgAAAAAAAAAAAAAAEF/Ojg4AAAAAAAAAAPCKl5eX0Yr3Md2/f19nzpzRvn37FBgYaPIhRmdnZ3Xv3l0bN25U6tSpbY7B29vb5EOQXbp0sXmspPb999+rdu3ajg7DKsWKFdOhQ4dUt27dWHWPHj3SyZMntW/fPp04ccLsQ945c+a0er6oqChdv35dgYGB2rt3r06ePBkrIU26dOm0fPly1a9f37YXY4N79+7pxIkT2rt3r44ePRorIY2Li4uGDh2q0aNH231uJycnzZs3T/3795ezs/HXI/fu3dPhw4d18uTJWA/dZs2aVRs2bDD5Xr3pKlasmKjvd2Lp0qWLlixZIm9vb6PtT5480fHjx3X06NFYCWlcXV01depUoyQhMObs7GzyXN+5c+dYvzP21r9/f3Xr1i3W9jt37ujAgQM6fvy4UUKaYsWKxftzLqkMHjxYbdu2dXQYSWLRokUqW7ZsrO0XL17Uvn37dObMGaOENK1atYpX4rw3XcqUKbVy5Urlz5/faHtkZKTh+u78+fNGCWn69OmjUaNG2TxX1apVNWnSJKVMmTLe8Sb1tcqbgGuJ+EuRIoWWL1+uvHnzOjqUWEx9/rRs2dIunzEffvihxo0bJycnJ0lSWFiYTp06pUOHDplNSDN9+nSrxx4wYIBhbFs54niuXr26FixYoFSpUkmSXr58qXPnzungwYMmE9K8++67+v333xN0LrOn9OnTa9WqVcqSJYvR9pcvX+rEiRPav3+/Ll26ZEhI4+zsrIkTJ74Rf0sDAAAAAAAAAAAAAACYQ1IaAAAAAAAAAEgmMmbMqKNHj+rChQuaOHGiGjZsqHTp0lnVN1u2bOrVq5eOHj2qn376KVayBlt06tTJqJw2bVo1b9483uMlFW9vb61fv17r1q1Tp06dVLJkSaVPnz7ZPMQYU6ZMmbRu3Tpt375dDRs2lKenp8X2Tk5OeuuttzRw4EAdO3ZM8+fPN9lu9erVmjp1qurXr6+0adPGGYePj4/69++vs2fPqnHjxvF5KbGMHDlSv/zyi9q2bascOXLE2d7Ly0tt27bVkSNHNGLECLvEYIqTk5O+/fZbHT58WI0aNZKrq6vZtj4+PhoyZIjOnTunSpUqJVpMjjZq1Kh4P8zsSC1atND58+fVo0cPpU+f3mw7b29vtWvXTqdPn1aPHj2SMMI3U8eOHY3KLi4usT4TEsu0adM0f/585cmTx2ybzJkz66uvvtKBAweUPXv2JIkrvlxcXLRgwQLt3LlTPXr0UNmyZZUxY0aL5503Vdq0abV9+3YNGTJEadKkMduuSJEiWrx4sZYsWWJISPBfkzNnTh04cEA9e/aUm5ub2Xbly5fX+vXrNWHChHifoz/55BOdOnVKQ4cOVY0aNeTj4yMPDw+bxkusa5U3GdcS8VekSBEdO3ZMc+bMUbNmzVSgQAGlSZNGLi4uDo2rWbNmsRLQ2DOJSL9+/bRlyxaTybteK1asmFatWqWZM2falAhu7NixOnr0qPr166dKlSopc+bMFs8tMTnieG7VqpX2799vMZlonjx5NHv2bK1du1bu7u7xnisxvPXWWzp06JDatm1r9th1cnJS7dq1tXfvXn322WdJHCEAAAAAAAAAAAAAAIB9OUW9XqLnDVKqVCndvn1bWbNm1aFDhxwdTrLjN3Cto0OIt8tj4rMi6027xwG8WXzi3zXqiv3CAN5ETrn+/2d+H/BfF/33wUZR9xfbMRDgzeOUoY3h55dTCjgwEsDxUnxy1vBztwL/vodtAVv8fDbMbmNFRUXpypUrOnv2rK5evapHjx4pNDRUHh4e8vb2lo+Pj0qUKGHXh/Nnz56tzp07G8o9evTQ1KlT7TY+THvx4oX279+vS5cuKSgoSCEhIfL09FS6dOlUoEABFS5c2OLD/qZERUXp7NmzOnfunK5evarHjx8rIiJC3t7eypo1q4oXL64CBQrY9PBrfNy4cUOnT5/WpUuXFBwcrLCwMHl4eChDhgwqUqSIihUr5pBEDc+ePdPOnTt19epVBQUFydXVVZkzZ1aRIkVUsmTJJI8H8RMREaH9+/fr3Llzunv3riIjI5UpUyblyZNHFStWTLaJqZKjv//+WzVr1jSU69evrzVr1iRpDFFRUTp69KgOHTqkoKAgRUVFKXPmzCpatKhKly7t8OQFsOz58+fas2ePTp06peDgYKVKlUo+Pj4qU6aMChTgb+bonj59qh07duj8+fN69OiR3N3dlSNHDpUvX145c+Z0dHgmJca1ypuOa4k334ULF5Q/f369vnWmSJEi+ueff+I1lp+fn65cefV9U65cuXT58mWj+vPnz2vv3r26ceOGnJyclC1bNgUEBKhIkSIJeg32Yu/juVq1atq2bZuhHPP2pOvXr2vXrl26evWqXr58qWzZshk+798EDx480Pbt23XlyhU9efJEnp6eyp07typWrKjMmTM7OjwAAAAAAAAAAAAAAAC7ICnNvxBJaYD/GpLSAPFGUhrg/5GUBog3ktIA/4+kNMD/s2dSGkeoWLGi9uzZYygfPXpUJUqUcGBEAICk0KZNGy1ZssRQ/uOPP9S4cWMHRgQAQOIaPHiwRo8ebSh///336tWrV7zGiispzX9NXElpAAAAAAAAAAAAAAAAkPwl7tKnAAAAAAAAAIA3yrFjx4wS0pQrV46ENADwHxAUFKQVK1YYyr6+vqpfPz6LCQAA8GZ48eKFZs+ebSi7u7vrww8/dGBEAAAAAAAAAAAAAAAAQPJCUhoAAAAAAAAAgMG3335rVO7Ro4eDIgEAJKXvv/9eYWFhhnK3bt2UIkUKB0YEAEDimjdvnu7cuWMot27dWunTp3dgRAAAAAAAAAAAAAAAAEDyQlIaAAAAAAAAAIAkacuWLVq8eLGh7OvrqxYtWjgwIgBAUjh16pQmTpxoKHt4eKhbt24OjAgAgMR1584dffnll4ayk5OTevfu7biAAAAAAAAAAAAAAAAAgGSI5Q0BAAAAAAAA4D8oODhYhw4dkiQ9ePBAu3bt0vTp0xUVFWVo88UXX8jV1dVRIQIAEsHz58+1c+dOSdLjx491+PBhTZ06VaGhoYY2PXv2VObMmR0VIgAAdrdp0yZJ0rNnz/TPP/9oypQpunPnjqG+efPmKlasmKPCAwAAAAAAAAAAAAAAAJIlktIAAAAAAAAAwH9QYGCgateubba+XLly6tatWxJGBABICrdv37Z4/vfz89PQoUOTMCIAABKfpc++NGnSaOLEiUkYDQAAAAAAAAAAAAAAAPBmcHZ0AAAAAAAAAACA5CVfvnxavny5nJ35L2QA+C/JnDmzVq1aJU9PT0eHAgBAkvDy8tKKFSvk6+vr6FAAAAAAAAAAAAAAAACAZCeFowMAAAAAAAAAADiel5eX/P399f777+vTTz+Vl5eXo0MCACQBDw8P5cmTRw0aNFCfPn2UKVMmR4cEAECicnV1Va5cuVSnTh317dtXfn5+jg4JAAAAAAAAAAAAAAAASJZISgMAAAAAAAAA/0HVqlVTVFSUo8MAACQxPz8/zv8AgP+cpPjsu3z5cqLP8SbZunWro0MAAAAAAAAAAAAAAABAAjk7OgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA/03Dhw+Xk5OT2X//1oT4ly9ftvi6O3ToYLG/n5+f2b5+fn5J8hoAAMC/G0lpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGKRwdAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADbDB8+3Gydn5+fOnTokGSxAAAA4N+HpDQAAAAAAAAAAAAAAAAAAAAAAAAAAADAG2bEiBFm66pWrUpSGgAAACSIs6MDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAkHySlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYpHB0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA612+fNnRIQAAgH85Z0cHAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABIPkhKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwSOHoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABbXLlyRQcPHtTVq1cVHh4uX19fVaxYUXny5Imz78OHD7Vjxw6dO3dOz58/V8aMGZU7d25VrlxZbm5udosxPDxchw8f1pUrV/TgwQMFBwfL3d1dGTNmlK+vr8qVKydPT0+7zZcYbty4oStXrujmzZu6e/eunj17pufPnytVqlTy9vZW6tSplSdPHhUrVkxeXl5JGtehQ4d048YNPXr0SFmyZFHevHlVsWJFpUhhv8enIyIidOzYMZ0/f17BwcF68OCBXFxclD59eqVPn17FihVTvnz57DZfcnH37l0dOnRIQUFBCg4O1uPHj+Xm5qa0adMqd+7cKliwoHx9fZM8rhcvXuj8+fM6c+aMbt++rZCQEIWGhipVqlTy9PRU1qxZlTNnTuXPn19p06a127xhYWE6fPiwrl27puDgYAUHB0uSvLy8lD17duXLl0+FChWSi4tLguYJDQ3VwYMHdf36dT148ECPHj2Sh4eH0qVLp0yZMql06dLKnDmzPV6SVW7evKl9+/bp0qVLevr0qZydnVWqVCm98847Vo9x48YNHTlyREFBQXrw4IHCwsKUPn16ZcyYUcWKFVOBAgUS8RUAABKKpDQAAAAAAAAAAAAAAAAAAAAAAAAAAABwmK1bt6p69epm69u3b6+5c+dKknbs2KHhw4dry5YtioqKitW2SpUqGjdunMqWLRur7urVqxoyZIh++eUXhYeHx6p3c3NThw4dNHz4cGXJkiVeryU8PFwLFy7U/PnztW/fPj1//txs25QpU6p8+fLq0qWL2rRpE2dCCz8/P125csWqOLZt2yYnJyez9VWrVtXWrVuNtoWEhGjt2rXatWuX9u/fr5MnT+rx48dWzefk5KRChQqpdevWateunXLmzGlVP1utXLlSkyZN0rZt2xQZGRmrPmPGjGrfvr2GDh2q1KlTx2uOyMhIrVq1SjNmzNDOnTvj3AdZs2ZVzZo19cknn6hcuXLxmjM+LB0PuXLl0uXLl20a7/jx4/rxxx+1YcMGXbx4Mc72OXLkUPXq1dWkSRO99957Ns1lq3Xr1mnWrFlav369njx5YlWfHDlyqGTJknr77bdVuXJllS1b1qaERY8ePdLs2bO1dOlSHT582OQ5IzpPT0+VL19ejRo1Ups2bZQxY0ar5gkJCdHs2bO1ePFiHTp0SC9evLDYvkCBAmrYsKE+/fRTm37P5s6dq44dO5qtnzNnjjp06CDp1flj2LBh2rZtW6x2jRs3jjMpzalTpzRp0iStX78+zuPQ19dXDRs2VP/+/a1KKgYASFrOjg4AAAAAAAAAAAAAAAAAAAAAAAAAAAAAsCQqKkpffvmlqlWrpr///ttkQhpJ2r59u6pUqaJFixYZbV+xYoWKFi2q+fPnm00u8fz5c02bNk0lS5bUsWPHbI5xzpw5ypUrlzp37qxt27ZZTEgjSS9evNCOHTvUrl07+fv7a/PmzTbPaU9HjhxRy5YtNXnyZO3du9fqhDTSq/fn5MmTGjJkiPLly6eRI0fGmVzDFs+ePVObNm303nvvacuWLSYT0khSUFCQJkyYoEKFCmn79u02z7NhwwYVLFhQTZo00Z9//mnVPrh9+7YWLVqk8uXLq2rVqjp37pzN8zrSuXPnVLt2bRUvXlzTpk2zKiGNJF27dk3z589XkyZNEi22+/fvq1atWnrnnXe0bNkyqxPSvI5v9erVGjhwoN5++23t3LnTqn7h4eEaPHiwsmfPrj59+mjv3r1xJqSRXiWX2bx5s3r16qU1a9bE2T4qKkrfffedcuTIoU8//VR79+616nfm7NmzmjBhgvLmzavOnTvbtE+siWnQoEGqXr26yYQ0cbl165bee+89FSlSRD///LNViZFu3LihadOmyd/fX926ddOzZ8/iETkAILGQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAADJWv/+/fX111+bTUYSXVhYmNq2bavff/9dkrRkyRI1b97c6uQNt2/fVu3atRUUFGRV+5CQELVt21adOnXS7du3reoT04ULF1S3bl2NHz8+Xv2TkxcvXmjYsGFq0qSJIiIiEjxeaGio3nnnHS1ZssTqPjdv3tQ777yjLVu2WNU+KipKX3zxherVq5egpDLbt29XqVKl9Ouvv8Z7jKQ0Z84cvfXWW9q0aZOjQ4nl2bNnqlKlSpImazp79qzKly+v0aNH6+nTp4k2T3BwsBo2bKg+ffooODg4XmO8fPlSs2fPVkBAgAIDA+0S12effaYxY8aYTfplyYYNG1SiRAmtXLkyXv1fvnyp6dOnq2LFirp06ZLN/QEAiYOkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAEi21q5dqwkTJtjcr2fPntqyZYs6dOhgVTKb6O7evau+ffvG2S4iIkItWrTQokWLbI7P1Fj9+/fX999/n+CxkoO1a9dq0KBBCR6nV69e2r59u839nj17piZNmujatWtxth0wYIC++eabeCXTiOnJkydq06aNVq5cmeCxEtPPP/+sTp06KSQkxNGhmDRq1CidPHkyyea7fPmyqlWrpiNHjiTqPGFhYWrUqJHWrl1rl/HOnz+v2rVr6+LFiwkaZ/HixZo0aVK8+m7btk0NGzbUvXv3EhSDJAUGBqp27dp68OBBgscCACQcSWkAAAAAAAAAAAAAAAAAAAAAAAAAAACQbAUFBcWr382bN1W7dm2Fh4fHq/8vv/wSZ5KFPn366M8//4zX+Ob069dPmzdvtuuYjjJ58mTduHEjQWOcO3cu3n0fPXqkzp07W2wzd+5cjR8/Pt5zmBIZGakPPvhA//zzj13HtZe//vpLPXr0cHQYZkVGRmru3LlJNt+TJ09Ur1493bp1K9Hn6tatm3bu3GnXMe/du6cGDRro2bNn8R5j48aN8ep34cIFNWnSJN7nWXNjNmvWzC5JogAACZPC0QEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAcfH09NSXX36pRo0aKU2aNDp06JB69eqly5cvm+0TERFh+Pmtt97Sl19+qVKlSik8PFxLly7VsGHDjNpEFx4erj/++ENdunQxWX/8+HFNmTLFYswlSpTQxx9/rAoVKihjxowKCQnRwYMHNXnyZO3Zs8dszL169dKxY8fk7Oxs2N6+fXvdv3/fUP7hhx/Mzuvj46MmTZqYrc+fP7/ZugwZMqh8+fIqW7as8ufPr1y5ciljxoxyd3eXq6urwsPD9eDBA128eFE7duzQnDlzFBwcbHKssLAw/fjjj/r666/NzmetrFmzauDAgapZs6bSp0+vmzdv6o8//tDEiRMVGhpqtt/GjRu1ZcsWVa9ePVZdSEiIBgwYYHFeJycnffjhh2rXrp3y58+vFy9eKDAwUFOmTNHWrVvN9ns9tr2TFiVUeHi4evbsafa4f83V1VXt2rVT/fr15e/vrzRp0ujx48e6fPmydu3apV9//VVnz55NlBivXLliMUFMtWrV1K1bN5UoUULp0qVTRESEHj16pNu3b+vUqVM6fvy4tm/frlOnTlk137fffqszZ87E2a5SpUr64IMPVK5cOWXKlEkvX77U3bt3deDAAf35559at26dIiMjzfbfv3+/5s2bZ3EOT09PffbZZ2rUqJF8fX316NEj7dixQ2PHjtXFixfN9jt16pSmTp0a5/FsraJFi6pixYrKkCGDgoKCdPnyZe3duzdWu88//9zs77/06jjq3LmzmjRpovz58ytlypS6evWqVq1apUmTJplNpLNlyxYtXbpULVu2tMvrAQDED0lpAAAAAAAAAAAAAAAAAAAAAAAAAAAAkKy5u7try5YtKlOmjGGbr6+vMmfOrAoVKsTZv3Llytq4caNcXV0N27744gs9e/ZM33zzjdl+Bw4cMJuUZsiQIYqKijLbt0ePHpo8ebJRYhlJypcvn1q0aKGPPvpI06dPN9n3xIkT+uWXX9SmTRvDthEjRhi1sZSUJn/+/Jo6darZelPy5MmjLVu2qHLlynJxcbHYNnv27CpevLjee+899enTR2+99Zbu3btnsu3mzZsTnJSmYMGC2r59uzJlymTY5uPjo9KlS+v9999X9erV9fjxY7P9J0+ebDIpzZQpU8zGLUnOzs5atGiRWrVqZbQ9b968ev/999W7d29NmjTJbP+//vpLe/fuVfny5S29vCQ1ffp0i8lNJKlkyZL6/fff5efnZ7Q9W7Zs8vf3V926dTVy5EitW7dO/fr1s3uMd+7csRjb5s2bY/1e+fr6qnDhwqpRo4Zh2927d7Vy5UotXbpUKVKYfqz+zp07mjhxosV43N3dNWvWLLVu3TpWnZ+fn8qWLasePXro6tWrGjJkSKzYXhs6dKjFeTJmzKht27apcOHChm0+Pj4qVKiQPvjgA9WpU0e7d+822//bb7/Vxx9/LC8vL4vzWJIvXz7Nnj1blStXjlUXFhamEydOGMpHjhzRihUrzI6VOXNmbdy4UcWLFzfa7uPjo/Lly6tNmzaqUqWK2aQ2w4cPV/Pmzc3uTwBA4uMMDAAAAAAAAAAAAAAAAAAAAAAAAAAAgGStT58+RglpXitfvrxRohJTXFxcNHPmTKOENK917NjRYt8zZ86Y3P7w4UOtWbPGbL+AgACTCWlec3Z21qRJk5Q5c2azYyxZssRibPbm4+OjatWqxZmQJiZfX1/VqlXLbP3hw4f1/PnzeMfl5OSk+fPnm32fAwICNGrUKItjrF271mQMv/zyi8V+Xbt2jZWQJroJEyaoWLFiFseIa46ktnDhQov1uXLl0qZNm2IlpDGlXr16CgwMtFNk/8/U7+pr2bNntzpJSebMmdWlSxdt3LhRlSpVMtnm999/17NnzyyOs2DBApMJaWLKmTOn5s2bpw8++CBW3YMHD7Rx40aL/adOnWqUkCY6T09PLV68WO7u7mb7379/X5s2bYozTnNy5sypXbt2mUxII716XwICAgzlRYsWWUzM9fPPP8dKSBNd0aJFNWTIELP1p0+f1pEjR6yIHACQWEhKAwAAAAAAAAAAAAAAAAAAAAAAAAAAgGSta9euZuv8/f0t9q1WrZoKFChgsi5fvnwWkzw8fPjQ5PbNmzcrIiLCbL9u3brFmTjDzc3NbKIMSdq6datevHhhcYzEEhERoZ07d2rYsGFq0aKFSpYsKV9fX6VJk0YpUqSQk5OT0T9LCXRevHihO3fuxDuWihUrmkxIFF3nzp3l6elpMYYDBw4Ybbtz546OHTtmcdy+fftarHdxcdGnn35qsU1CkoTY2/3792Pth5jGjh2rDBkyWD2mrUmMrJErVy45OTmZrFu3bp1++umnOBPJWOuvv/6yWF+vXj01bdrUpjFN7ZNNmzYpMjLSbJ8cOXKoRYsWFsfNlSuX3n//fYttEnK8TZ061WKirJjWr19vti5btmx677334hzDUkIrSXEm8gEAJK4Ujg4AAAAAAAAAAAAAAAAAAAAAAAAAAAAAMCdPnjzKmTOn2fpMmTJZ7F+tWjWL9enSpVNoaKjJuidPnpjcvnfvXotjduvWTd26dbPYJi5Pnz7V6dOnVaxYsQSNY4vQ0FBNmTJF48eP17179+w2bnBwsHLlyhWvvrVr146zjYeHhypWrGgxgcWhQ4dUuXJlQ/nAgQOKiooy2z5PnjzKly9fnHPXrVvXYv2JEycUEhJiMWlOUtm3b5/FxCipU6dWs2bNkjAi09KnT6+yZctq3759sepevnypjz/+WL1791bBggWVJ08e5c2bVwUKFFDBggVVuHBhZcyY0eq59uzZY7G+U6dONsdvyv79+y3W165d22winujq1q2rRYsWma03tc+s4efnpwYNGljdPiQkRP/884/Z+lu3bln1euISVxIlAEDiIikNAAAAAAAAAAAAAAAAAAAAAAAAAAAAki1/f3+L9W5ubhbr8+fPb7E+ZcqUZuvMJfC4e/euxTHtJanmkaRr166pbt26OnXqlN3Hfvz4cbz7FipUyKp2BQsWtJiUJmaSnbj2bZEiRayaN0eOHPLy8tLTp08tzp0cktLE9ZrLlCkjFxeXJIrGsqFDh6p+/fpm68PDw3Xs2DEdO3YsVl2BAgVUpUoVtWjRQjVr1pSzs7PJMSIjI3X//n2LcVSoUMG2wM2w1/FWuHBhi/XxTSZVvXp1m5LI/BvPgQCA2Ex/ggIAAAAAAAAAAAAAAAAAAAAAAAAAAADJQJo0aSzWp0qVymJ92rRp7RjNK/FN/JBc5wkJCVG1atUSJSGNZD65jzWsff/iOk4ePHhgVA4KCrLLvNa0Tar3MS5xxZElS5YkiiRu7777rkaNGhWvvmfPntXMmTNVp04d5c+fX2vXrjXZ7v79+3Eem/baJ/Y63hLrWLM2+VNC57FVcvndAYD/KpLSAAAAAAAAAAAAAAAAAAAAAAAAAAAAINlydXV1aH9Hev78eZLMM2rUKF28eDFJ5gKs9cUXX+jvv/9W+fLl4z3GxYsX1aBBA82aNcuOkf37JEbyLntIqnMgAMA0ktIAAAAAAAAAAAAAAAAAAAAAAAAAAAAANsiUKZOjQ7CbqKgozZ0712KbnDlzasqUKTp16pRCQkIUFRVl9K99+/aJFt/Dhw/t0i59+vRG5YwZM9plXmvaJpfjJa447ty5k0SRWK969eras2ePTpw4oXHjxqlBgwbKnj27nJycbBrnk08+0fXr1422ZciQQc7Olh+3t9c+sdfxlljHWsqUKW1qn1yOaQBA4krh6AAAAAAAAAAAAAAAAAAAAAAAAAAAAACAN0nmzJkt1v/111+qV69eEkWTMMeOHdPt27fN1mfLlk0HDx60mITi0aNHiRGaJOnUqVNWtTt9+rTF+pjxx/Uenjx50qp5r1+/rqdPn9o0t6PE9ZoPHDigiIgIubi4JFFE1itcuLAKFy6sfv36SZJCQkJ04cIFXb16VefOndPhw4e1du1aBQcHm+wfGhqqOXPmaMiQIYZtzs7OypAhg+7du2d23j179qh58+YJjt9ex1tcvw9JdazF9XrKlSunvXv3JkksAIDEYzl1GwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAjZcuWtVi/cePGJIok4a5fv26xvnnz5hYTXURGRmrfvn32DsvAmn0ZEhKi3bt3W2xTqlQpo3KZMmXk5ORktv2FCxd04cKFOOdev369xfoiRYrI09MzznGSQtmyZeXsbP7x8sePH2vZsmVJGFH8eXp6qnjx4mrQoIE+++wzLViwQBcuXJC/v7/ZPjt27Ii1rXz58hbnmT17doJjlaw7Z0RFRcU5TlzHW7ly5WyKK748PT1VuHBhs/VHjhxRUFBQksQCAEg8KRwdAOzv8pj6jg4hifk4OgDgzeWUy9ERAMkHvw9AvDllaOPoEIBkI8UnZx0dApBs/Hw2zNEhAAAAAAAAAAAAAAAAAAAAJJratWvLxcVFERERJutnzpypzz77TNmzZ7d57NDQUC1atEgVK1a0mPTBw8NDz549M1l3//59q+d79OiRxfrnz59brJ8/f75u3bpl9Xy22r17tw4cOKAyZcqYbTN79myz+0KSUqZMGat/lixZVLx4cQUGBprtN3HiRP3www9m6yMiIjRlyhQL0Uu1atWyWJ+UMmbMqNKlS2v//v1m2wwaNEh16tRRunTprBrz5cuXSpHC/o+sh4eHK1WqVDb1SZcunRo0aKAzZ86YrDd1nL7zzjtavXq12THXrVun33//XU2aNLE6DlP7pFatWnJ2dlZkZKTJPlevXtWyZcvUvHlzs+Neu3ZNK1assDh3Uh5v9erV08mTJ03WhYeHa/To0ZowYUK8xj527Jh27typjz/+OCEhAgASyHwqOwAAAAAAAAAAAAAAAAAAAAAAAAAAAACxpE2bVvXq1TNb//jxY73zzju6ePGiVeNFRUVp79696tevn3LkyKEuXbro7t27ccZgzokTJ3To0CGr5o4r+ciaNWsUFBRksm7btm3q2bOnVfPEV1RUlNq1a6d79+6ZrD9y5Ii+/PJLi2PUr19fbm5usba3atXKYr9p06Zp2bJlZusHDBhgMamNJLVs2dJifVJr27atxfpLly6pdu3aunr1apxj7dixQ6VLl7ZXaEZKlSqlzp07a9u2bXr58qVVfSIiIrR7926z9VFRUbG2NWnSRB4eHhbH/fDDD7V06dI4579z5466d++uxYsXx6pLnz69ateubbF/z549dfr0aZN1z5490wcffGAx+VKGDBmSNClNXL8/3333ncaNG2c2EU9MQUFBmjNnjmrUqKESJUpYtc8BAInL/mnnAAAAAAAAAAAAAAAAAAAAAAAAAAAAgH+5UaNG6c8//zSZ6EKS/vnnHxUuXFitW7dW/fr1VaRIEaVNm1YvX75UcHCw7ty5o2PHjunw4cPasmWLbt26ZdP8/v7+unnzpsm6qKgovf3226patar8/PyUMmVKo/qxY8fK09NTklSoUCGL89y8eVOVKlXS4MGDVbp0aaVJk0YXLlzQkiVLNHPmTKsThiTE6dOnVaJECQ0cOFC1atVSunTpdPPmTf3xxx+aMGGCQkNDLfb/9NNPTW7/5JNPNHHiRLMJbyIjI9WiRQu1b99eH374ofLnz6+XL18qMDBQU6ZM0d9//21x3nfeeUcVKlSw7kUmkW7dumnixIm6fPmy2TaHDh2Sv7+/2rdvr3fffVcFCxZU6tSp9fjxY127dk179uzR8uXLdfTo0USL88mTJ5o9e7Zmz54tb29vVa1aVaVKlVLBggWVN29epU2bVt7e3pKke/fu6fDhw/rpp5+0b98+s2Nmz5491rasWbOqd+/e+uabb8z2CwkJUcuWLfXDDz/ogw8+UNmyZZUpUya9fPnSMPemTZu0cuVKhYeHq3z58ibHGTlypNavX292nrt376pMmTLq06ePGjZsKB8fHz1+/Fg7duzQ2LFjdeHCBbN9pVdJkry8vCy2sacyZcrovffe0x9//GGyPioqSgMGDNCCBQvUrl07lS9fXjly5JCbm5seP36s4OBgnTt3TkeOHNH+/fu1e/duRUREJFn8AIC4kZTmX8hv4FpHhxBvl8fUj0cv038wAf8dPgnoe8NuUQBvJt9oP/N5gv+6BHyehG6wXxjAm8i9juHHl1MKODAQwPFSfHLW8HO3Aq4OjARwvJ/Phjk6BAAAAAAAAAAAAAAAAABAIitZsqS6d++un376yWybsLAwzZ07V3PnzrX7/BUrVtSWLVsszr1hg+l7/ocPH25ISuPn56ciRYroxIkTZsc6c+aM2rdvn7CAE+jWrVvq1auXzf1q166t6tWrm6zz9PTUt99+q44dO5rtHxUVFa/38PXYyU2qVKk0ZcoUNW7cWJGRkWbbPX/+XD///LN+/vnnJIzOtCdPnmjNmjVas2ZNgsapU6eOye2ff/65li5dqvPnz1vsv337dm3fvj3e85ctW1bt27fXvHnzzLZ5+vSpRo4cqZEjR9o0dqFChdSzZ894xxZfY8eO1ZYtW/To0SOzbY4fP67+/fsnYVQAAHtxdnQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwJto8uTJqlevnkPm7tChg5ycnOwy1qBBg+LdN1u2bHr33XftEocpefLkiXff1KlTa9asWRbbdOjQQf369Yv3HKY4Oztr0aJFKlq0qF3HtZcGDRpo8uTJjg4jSWXIkEEffvihybrUqVNr/fr1ypIlS6LH8fPPP+vtt9+265gZM2bUmjVr5OHhYddxrVGgQAGtWLFCqVKlSvK5AQCJj6Q0AAAAAAAAAAAAAAAAAAAAAAAAAAAAQDykSJFCy5YtU5s2bZJ87nz58ql37952GatNmzZ6//33be7n4eGh5cuXK1OmTHaJw5TJkyerfPnyNvfz8PDQH3/8oRw5csTZduzYsRo0aJBdkvx4eXlp8eLFaty4cYLHSkw9evTQ9OnTHZLIJKmlTJlSc+bMsXic5smTR1u3blXx4sUTNRZXV1etXr3abomc8uXLp02bNiUoeVNC1ahRQ6tWrVLmzJkdFgMAIHGQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAACIJ09PTy1atEiLFi1S9uzZEzyev7+/Ro4cqZIlS8bZ9ttvv1Xfvn0TnEzFyclJCxcutCkxTaZMmbR+/XpVqFAhQXPHxdPTUxs3brQpNh8fH/3111+qXr26Ve2dnZ31zTffaN26dcqXL198Q1WVKlV06NAhtWzZMt5jJKUuXbro8OHDVu+npGSvZDl+fn7atGmTGjZsGGfbggULav/+/RowYIA8PT3tMr8p6dKl05o1azRhwgSlS5cuXmOkSJFCHTt21OHDh1WiRAk7R2i7unXr6vjx42rRooWcnROWwsDNzU3NmzfXkCFD7BQdACC+SEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAJFCbNm106dIlLV26VO+8847SpEljVT9vb2/VqVNHY8aM0ZEjR3T69GkNGTJEadOmjbNvihQpNH78eJ0/f17Dhg1T7dq1lT17dnl5edmcqMbd3V3Lly/X/PnzVbBgQbPtPD099fHHH+vEiROqVKmSTXPEl5eXl5YvX67ffvtNlStXNvvaMmbMqD59+ujUqVOqUqWKzfPUqVNHZ86c0YoVK/TOO+8oderUcfbJkiWL2rRpo71792rbtm0qUKCAzfM6kr+/v/7++28dPXpUXbt2lZ+fn1X9fHx81LZtW61YsSJR4jp27Jh27typ4cOHq27dusqcObPVfV1dXVW3bl3Nnj1bZ8+etelYcHV11dixY3X9+nWNGzdO5cqVU8qUKePs5+7ururVq+u7775TgwYN4mzv5OSkPn366Nq1a5o0aZLKly9v1Tz58+dXnz59dP78ec2ePVve3t5Wva6kkDlzZv3666+6cOGC+vfvr8KFC1t1HnJyclKBAgXUrVs3LV26VHfu3NHSpUtVs2bNJIgaAGCJU1RUVJSjg7BVqVKldPv2bWXNmlWHDh1ydDjJjt/AtY4OId4uj6kfj1437R4H8GbxSUDfG3aLAngz+Ub7mc8T/Ncl4PMkdIP9wgDeRO51DD++nPJmfXkA2FuKT84afu5WwNWBkQCO9/PZMEeHAAAAAAAAAAAAAAAAAABwsKioKJ08eVKnT59WcHCwgoODFR4eLi8vL6VOnVq+vr4qVKiQcuTI4ehQTTp9+rT279+vu3fvKjw8XGnTplWhQoVUoUIFubm5OTS269ev6+DBg7px44aePHmizJkzK2/evHr77beVIkUKu80TERGhY8eO6dy5c4b30NnZWenTp1eGDBlUtGhR5c+f327zJRe3b9/W4cOHFRQUpODgYD158kSurq5KkyaNcuXKpYIFCypXrlwOievSpUu6cuWKgoKC9OzZM4WFhcnd3V0eHh7KkiWLChQooAIFCsjV1X73Mz9//lyHDh3StWvXFBwcrIcPH0p6lSzJx8dHefPmVZEiRaxKKmNJaGioDhw4oOvXr+vBgwd69OiRPDw8lC5dOmXOnFmlS5e2KTlPcvDw4UMdPHhQt2/f1sOHD/Xo0SOlTJlSqVOnVvr06ZU/f375+/vLw8PD0aECAEwgKc2/EElpgP8aktIA8UdSGuD/kZQGiDeS0gAGJKUB/h9JaQAAAAAAAAAAAAAAAAAAAAAAwJvM2dEBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACSD5LSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA5LSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA5LSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA5LSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA5LSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgxSODgAAAAAAAAAAAEebO3euOnbsaCjPmTNHHTp0cFxAydTw4cM1YsQIQ3nLli2qVq2a4wJK5gIDAzV9+nTt2LFD169f16NHjxQZGWmoj4qKcmB0/z4cn47l5ORk+Llq1araunWr2ba8V4716NEjTZ8+XevWrdPJkyf14MEDhYeHG+r5DAT+Gzp06KB58+YZypcuXZKfn5/jAgIAAAAAAAAAAPgXevDggXbt2qXr16/r8ePHypYtm/LkyaOKFSvK2dk5yeJ4/PixTpw4odOnT+v+/fsKDw9XunTplD17dpUvX16ZMmVKsliskVz225smue63e/fuae/evbp48aKePHkiV1dXZcqUSfny5VPJkiXl5eXlsNiS0z6LjIzUyZMndfToUQUFBSkkJEReXl7KkiWLAgIClD9/fqP7cwAASCokpQEAAAAAAACAN0BoaKgOHz6sc+fOKTg4WCEhIXJ3d1fq1KmVM2dO5c2bV3ny5OFLdyAZiIyM1IABAzRhwgRHhwIARrZu3aqmTZvqwYMHjg4FAAAAAAAAAAAAAP61zp07p4EDB2rNmjVGi4S85uPjo65du2rQoEFKlSpVosRw8OBBLV++XJs2bdLhw4eNFlKKqWzZsurdu7datWrl0KQXyWG/WWPixInq27ev0ba4FvBJTMl1v61evVrjx4/Xjh07zC7e5ezsrFKlSunTTz9V27Ztkyy25LTPHj16pHHjxmnWrFm6ffu22XZ+fn7q1q2bevfuLTc3t0SNCQCA6EhKAwAAAAAAAADJVFRUlFatWqVp06Zp06ZNevnypcX23t7eKlWqlKpWrap33nlHZcqUIUkN4ABjxowhIQ2AZOf8+fNq0KCBQkJCHB0KAAAAAAAAAAAAAPxrLVq0SN26dbP43ezNmzc1fPhwrVq1SitWrFCuXLnsGkPt2rW1adMmq9vv379fbdq00axZs7RgwQJly5bNrvFYIznsN2tcvnxZQ4cOTfJ5zUmO++3+/fvq0KGD1qxZE2fbyMhIHThwQGvWrEmypDTJaZ/t2bNHzZs3140bN+Jse/nyZQ0aNEhz587V77//rkKFCiVKTAAAxERSGgAAAAAAAABIhq5cuaJOnTrp77//trrPkydPtHXrVm3dulUjRozQH3/8ocaNGydilMlHhw4dNG/ePEP50qVL8vPzc1xA+M968OCBvvrqK0PZyclJH374oerVq6eMGTM6dDWp5K5atWratm2boWxuhSQgMQwfPlwjRowwlLds2aJq1ao5LqBEMHjwYKMbqooXL66uXbsqd+7cRqt6FSlSxBHhIYbLly8rd+7chnL79u01d+5cxwX0L8D1IgAAAAAAAAAAABLbunXr1L59e0VERBi25c+fXzVq1FD69Ol14cIFrV69WqGhoZKkw4cPq0GDBtq9e7e8vb3tFse9e/dibcuZM6cqVKggHx8feXl56fbt29q6davOnTtnaLN582bVrFlTO3bsUIYMGewWT1ySy36zRlzJTJJSctxvN27cUM2aNXXmzBmj7QEBAQoICFDWrFkVHh6u69ev68CBA0bHX1JITvssMDBQ9erV0+PHjw3bnJycVKlSJZUqVUpp0qRRcHCwDh48qN27dxvanDlzRjVr1tS+ffuUI0cOu8YEAIApJKUBAAAAAAAAgGTm4sWLqlKlisnVL1KlSqXcuXMrTZo0CgsL04MHD3Tjxg1FRkbGaktCByDpLV26VM+fPzeUhwwZYpToAgAc4fHjx1q5cqWh7O/vr3379snNzc2BUQEAAAAAAAAAAADAv8ft27fVqlUrQ7ILJycnjR8/Xr1795azs7Oh3b1799S8eXPDwj3//POPunfvrkWLFtk9pixZsqhjx47q2LGjChQoEKs+KipKy5cvV9euXRUcHCxJOnXqlLp166Zly5bZPR5TkuN+M2fBggXasGGDJClbtmy6detWks0dU3Lcb8+fP1f9+vWNEtK88847mjRpkvLnz2+yz7lz57RgwQI9fPjQ7vHElJz2WVRUlLp162aUkKZo0aJasmSJihYtGqv9kSNH1Lp1a8O+vXXrlnr16qUVK1bYLSYAAMxxjrsJAAAAAAAAACCpvHjxQg0bNjRKSOPk5KS2bdtqx44dCgkJ0enTp7Vv3z4dPXpUV69e1aNHj7R161Z9/vnnyps3rwOjx7/d8OHDFRUVZfhXrVo1R4eU7OzZs8eo3K1bNwdFAiSt6OeGrVu3OjocxHDo0CGFh4cbyu3atSMhDQAAAAAAAAAAAADY0ddff61Hjx4ZyiNGjFCfPn2Mkl1IUqZMmbRu3ToVKlTIsG3JkiUKDAy0WyyZMmXShAkTdOXKFY0ePdpkQhrp1X1pzZo106ZNm+Th4WHYvnz5ch04cMBu8ViSnPabJUFBQerTp4+kV/tt4sSJSTKvOclxv3311VdG4/bu3Vt//vmn2YQ0kpQ/f36NHDlSkydPtns8MSWnfXbgwAHt27fPUE6fPr02btxoMiGNJL311lvavHmz0qZNa9j2xx9/6ObNm3aLCQAAc0hKAwAAAAAAAADJyLRp03Ty5ElD2c3NTWvWrNGCBQtUqVIlpUiRIlYfLy8vVa1aVWPGjNH58+e1detWNW7cWC4uLkkZOgBJZ8+eNfycNm1a+fj4ODAaAHgl+rlJkooUKeKgSAAAAAAAAAAAAADg3+fu3buaMWOGoZw3b14NHDjQbHs3NzdNnTrVUI6KitJXX31lt3j++usv9enTR66urla1DwgI0GeffWa0bdmyZXaLx5zktt8s6d27t4KCgiS9WqSqfPnySTKvKclxv508eVLjxo0zlOvUqaPvvvvOrnMkRHLbZ5s2bTIqd+nSRVmzZrXYx9fXV126dDGKacuWLXaLCQAAc0hKAwAAAAAAAADJyLx584zKw4YN07vvvmvTGFWrVtUff/yhhg0b2jM0AFZ4+PCh4efUqVM7LhAAiCb6uUni/AQAAAAAAAAAAAAA9rRy5UqFhYUZyl27dlXKlCkt9qlRo4b8/f0N5T///FPPnj2zSzymFj6LS6tWrYzK+/fvt0ssliS3/WbOunXrtGjRIklS1qxZNXr06ESdLy7Jcb9NnDhRL168kCQ5Oztr0qRJdhvbHpLbPrtx44ZR2dokRxUqVDAq37x50y7xAABgCUlpAAAAAAAAACCZePDggQ4dOmQoOzs7G61sASD5e/78ueFnJycnB0YCAP8v+rlJ4vwEAAAAAAAAAAAAAPa0atUqo3KzZs2s6te8eXPDz6GhodqwYYNd47JFvnz5jMp37txJ9DnfhP0WEhKijz76yFD+7rvvlDZt2kSbzxrJbb89ffpUv/76q6FcpUoVFSxY0C5j20ty22eRkZFGZQ8PD6v6xWzH/R8AgKRge7pDAAAAAAAAAECiiLn6RcaMGZUhQwYHRZM0zpw5o3379unmzZtycXFR5syZVbZsWRUqVMjRoRmJjIzU/v37FRgYqPv378vT01PZsmVTlSpVlDVrVrvMcffuXe3du1e3b9/W/fv35eXlZdgfuXPnTtDYDx480OHDh3X+/Hk9evRIL1++lIeHhzJmzKjcuXOrSJEiSpcunV1ehzmhoaEKDAzUyZMnFRwcrNDQULm7uyt16tTy8/NTwYIFlSNHjkSNIaZnz55p586dunbtmu7duyc3NzdlzpxZRYoUUYkSJeI1ZlRUlJ2jtN7Tp0914sQJnT59Wvfv31doaKjSpEmjjBkzKiAgQAUKFEiUeQMDA3Xw4EHdvXtXrq6uypo1qypWrCg/P79EmS++Xrx4od27d+uff/7Rw4cPlTp1auXIkUNVq1a1+/F/4cIFHTt2TDdv3tTDhw+VMWNGtWrVSmnSpDHb5+7du/rnn3904cIFPXz4UC9fvlT69OmVNWtWlStXzm7nmuiScp/Y0/Xr13XixAldunRJjx49kiSlT59evr6+qlChQrKL/cyZMwoMDNS9e/f06NEjpU+fXj4+PqpUqZLSp0+fJDEk1rkpLCxMu3bt0vXr13Xr1i25uLioTJkyqlq1qsV+d+/e1e7du3X79m09ePBAadKkUZYsWVSuXLlE+Sy4cOGCjhw5ohs3big0NFTZs2dXlSpVlDNnTov9oqKidPDgQR09elT37t2Tp6en/Pz8VKNGDXl7e9s9zqR2//597d27Vzdv3tS9e/fk5eWlevXqJdrnRXxERUXp0KFDhvfAw8NDvr6+qly5srJkyeLo8CQl/fH8zz//6ODBg7p9+7ZSpkwpX19flS9f3u6fu6Ghodq+fbtOnz6tp0+fKl26dPLz81PVqlXl6emZoLEvXbqkEydO6OrVq3r06JFSpEih9OnTK1euXCpfvry8vLzs9Cr+36NHj7R9+3adP39eoaGhypAhg/Lly6dKlSrJ1dXV7vMBAAAAAAAAAID/lh07dhh+zpIli/LkyWNVvwoVKhiVt2/frvfee8+eoVntyZMnRuWUKVMm+pxvwn778ssvdfnyZUlSnTp11KpVq0SZxxbJbb+tXr1aT58+NZSbNm2a4DHtLbnts5j3A165csWqfq+Pxdfy5s2b4Fjwf+zdd3gUVdvH8d+mAiH0EnoogqE3Q4dQHpoUqQIiTSBgVwREkKYoUsSCSu8gioCiIFUC0ot0pBchtNBJQk32/YM3YyZ1N9lkA34/18X15J49Z869Z9o+7s49AIDEUJQGAAAAAAAAANKImF/sR0REOHyM8+fPy9fX11h38eLFdfToUbvXs2zZMrVs2dKI27dvb3raifT4C9DoX5527dpVs2bNkiStXr1agwcP1q5du+Jcv5+fnz777DM1b9483hxmzZql7t27x/laQkVcChUqFOvL2fhERkbqu+++0+jRo3X+/PlYr1ssFjVs2FDjxo1T6dKlbVpnzPXPmzdPX3/9tXbv3h1v0QA/Pz8NGDBAXbp0kYuLi83rX7dunT777DOtW7cu1tNVYr6PEiVKqGXLlnrttdfivYF6+PDhGjFihBGvX79eAQEBCeZw4sQJjRw5UkuWLFFYWFiCbfPmzatGjRqpd+/eqlq1aoJtk2Pfvn0aNmyYVq1apXv37sXZJl++fOrRo4cGDBiQ4I3RMeckurNnz8b7NBpHFYg4duyYvv/+e61cuVK7du3So0eP4m3r4+Ojvn376o033rC5aEdQUJDq1q1rxMOGDdPw4cMlSd9//71GjBgR7zmkSpUqGjdunGrWrBnv+hOav4Se5FOnTh0FBQUl/gb0uGjGmDFj9MUXX+j69euxXnd1dVXbtm312WefqVChQomuL6FzW9TxvGPHjlj9qlSpovLlyxux1WrVpk2b9OOPP2rNmjWJnovLlCmjfv366aWXXpKbm21f8cV3zDp6TiTz9rJn+9ji0aNHWrt2rX766SetXbs2wR/iWCwWVa1aVQMGDFDLli0T3I9i7t/Rxbc8SmLHcGhoqMaPH6/Zs2fr9OnTcbZxdXVVrVq1NHLkSNWqVSvB9dkr5n4aU3zvb+bMmerWrZsRd+vWTbNnzzbi06dPy9fXV+fPn9dHH32kH3/8UTdv3jSto2XLlvEWpfn55581evRo7dixI945jNrXX375ZZuvefHtf7///rs++eQTbdq0Kc4+L7zwgr766ivlz5/f9JrVatXUqVP1ySefxLm/eXp66o033tCIESNsflqZrXx9feMcc/bs2aZtEVPMbRdzXdE//2zdulWffvqpVq5cqYcPH5r6TJgwQRkyZHD650Wr1app06Zp1KhRcc6Hq6urGjZsqPHjxydYzDAlPy+m9v68ZMkSffjhhzp8+HCcfapXr66xY8fG+lGkvW7duqURI0ZoypQpcX528/Dw0CuvvKKRI0cqR44cNq3z3r17Wr58uZYsWaI//vhDly5diretq6ur6tevr0GDBiX6OTO6gIAAbdiwwYijtsnFixc1ePBgzZ8/Xw8ePIjVz8vLS2+//bYGDRqU7GI7AAAAAAAAAADgv+nixYvGA1UkqUKFCjb3rVixoin++++/HZaXvfbv32+KY36P6mhPwrzt3LlTX331lSQpXbp0+vbbb1NkHHukxXnbtm2bKa5UqZJD1usoaXHOGjdurIEDBxrxDz/8oF69eiXab+HChcbfGTJkUL169RySDwAACbH9l+sAAAAAAAAAgBSVJUsWU3zt2jWdOHHCoWPkz59fzz//vBEfO3ZMGzdutHs9U6dONcW9e/e2ue+AAQPUqFGjeAvSSI+/vG3RooU++ugju3NzlNu3b6thw4Z6/fXX4yxIIz2+2XXVqlWqUqWKVq1aZdf6jx8/rooVK6pr167atWtXggUO/v77b3Xv3l01atRQSEhIouu2Wq1644031KBBA61ZsybBgjRR7Y8cOaLPPvvM7veRkLlz56p06dKaO3duogVpJOnChQuaOXOmJk6c6LAcorNarRo4cKAqVqyoX375Jd6CNJIUHBysjz76SM8880ycxQzSgt9++00lSpTQ8OHDtW3btgQL0kjSpUuXNGzYMJUpU0Y7d+5M8rgPHjxQ586d1alTpwSLFGzfvl0BAQFGwRZnCA4OVtWqVTV06NA4i69IjwuA/fDDD6pcubL27NmTpHHu37+vNm3a6OWXX46zIE1c+vfvr9q1a2vixIk2FXs4cOCAunXrprp16+rKlStJylNKvTlxpA4dOqhJkyaaPn16ok+Gslqt2rp1q1q1aqW2bdvadO5xtN9++01FixbV8OHD4y1IIz2e56CgINWuXVuBgYGJHsNpxdq1a1W6dGlNmTIlVkGa+Ny6dUuNGjVSq1attH379gSveVH7etWqVXXx4sUk5/nBBx+oadOm8Z7DrVarli5dKn9/fx07dsxYfvfuXTVv3lyBgYHx7m/379/XuHHj1KhRI6fsY8kxZswY1ahRQ7/++musgjRRnP158cGDB2rbtq169+4d7zaIiIjQ77//rgoVKmjmzJl255Ycqb0/R32ua9OmTZwFaaLabN68WTVr1kzW5+fDhw+rXLlymjBhQrz79oMHD/Tdd9+patWqNhd6rFmzptq2basFCxYkWJBGerxtV69erbp16+r1119P1rnxzz//VOnSpTVz5sw4C9JIUlhYmEaNGqW6devGe10EAAAAAAAAAABIyJEjR0xxwYIFbe6bO3dueXh4xLuu1DRv3jxTnNLFLtL6vD169Eg9e/Y0fnc1ZMgQFS1a1OHj2CstzlvM3wGWKlVKknTjxg198803qlu3rgoWLCgvLy8VKFBANWvW1LBhw5L0cJSkSItzVrZsWdP38uvWrUv0N3Pjx483PSTqnXfeUebMmR2SDwAACaEoDQAAAAAAAACkEUWKFFG6dOlMywYOHJjgja5J0adPH1Mc84bhxAQHB+v333834iJFitj8I4T3339fY8eONWJvb2+VKlVKlStXVs6cOWO1Hzp0qJYsWWJXfo7w8OFDNWvWTOvWrTOW5cqVSxUrVlTZsmXl5eVlah8eHq62bdsmWiwhyvbt21W9enXt27fPtNzV1VXFihWTv7+/SpYsGWt/2LZtm6pVq5ZoYZqhQ4fG+SV1tmzZVK5cOVWtWlVlypRRnjx5bMo3KdasWaOuXbvq/v37puUZMmRQyZIlVbVqVVWoUEG+vr5ycUn5ryusVqu6deumMWPGxCrSkyNHDlWsWFF+fn6x5vzSpUtq2LChQ4v1OEpcRXXSp0+v4sWLq1KlSqpcubIKFy4ca36Dg4MVEBCQ5Cf3dO3aVfPnzzfirFmzqmzZsqpYsWKs4loRERHq2bNnsorgJNXNmzfVoEED7d2711iWL18+Va5cWaVLl5anp6ep/dWrV9WiRQvdvn3b7rG6detmOldFndsqVKigHDlyxNknru2XNWtW+fn5qUqVKipfvnycx+imTZtUr1493b171+48U3NOHCmuucqZM6dKliypKlWqqFy5cnHO85IlS9SyZctEC3M50pQpU/TCCy/EKhyUIUMG+fn5yd/fX8WKFYt1XE6ZMkVt27Z1+DXf0fbs2aOWLVuaniBWqFAhVa5cWUWLFpW7u3usPtevX1dAQIBWr14d67WCBQuqcuXKKlKkSKw52blzp2rUqGHztTW6sWPH6tNPPzXiLFmyqFy5cipXrlysa/jFixfVqlUrPXz4UJGRkWrbtq2WL19uvJ4nTx5VqlRJpUqVkpubm6nvpk2b9Pbbb9udn7NMnjzZ9NnSw8NDxYsX13PPPad8+fLJYrEYbZ35ebFv376mc2qmTJlUtmxZlS5dWhkzZjS1vX//vnr27Gm6LqUkZ+zPH3/8selzXYYMGVSqVCmVL18+1nU3MjJSQ4cO1ejRo+0e58yZM6pfv74px0KFCum5556Tn5+fXF1dTe1Pnjyp1q1b21Q0Jq7zeN68eVW6dGnjc2lcP5b85ptv7Cp+Gd3u3bvVpEkTo9CMi4uLihYtKn9/fz3zzDOm/V16vI26deuWpLEAAAAAAAAAAMB/W3BwsCnOnz+/zX0tFovy5ctnxPE9OCulHT9+XAsWLDBiV1dXtWnTJkXHTOvzNmbMGO3fv1+S5Ofnp/79+zt8jKRIi/MW/eEa6dKlU6ZMmbRs2TI9++yzev311xUUFKRz584pPDxc58+f1+bNmzVy5EiVKlVKvXv3TtJvYOyRFudMevw9fJEiRYz4jTfe0Isvvqj169fr9u3bslqtunXrltauXatWrVrpvffeM9o2bdpUw4YNc1guAAAkxC3xJgAAAAAAAACA1JAuXTrVr1/fdCP0kiVLVL9+fX300UeqUaOGQ8Zp1KiRfH19debMGUnSTz/9pK+++kpZs2a1qf+MGTMUERFhxD179ox1U2dcNm7cqNOnT0uSqlSpoo8++kh169Y1bvC2Wq3asGGD+vbta3qiyJtvvqkWLVrEuhG8UaNGWrNmjaTHN59Hvzl43rx5yp07d5x5pE+fPtFcR48erVOnTkmSXnrpJQ0cOFBlypQxXr9//74WLFigt99+2yjWEBoaqgEDBuiHH35IcN2XLl1SixYtdPXqVWNZ2bJlNWjQIDVr1sx0s/W9e/e0bNkyffDBBzp58qSkxzcAd+vWTb/99luc8x4cHKzPPvvMtCwwMFBvvfWW/Pz8YrW/ceOGtmzZot9++03ff/99YlNjs7fffttUXKFu3boaNmyYatasGeum5rt372rv3r1asWKF6QcujjRx4kTNmTPHtKxWrVoaPXq0qlWrZsxlaGioFi5cqIEDBxo3Md+9e1edOnXSgQMHlDdvXtM6unTpopo1axpx586ddfnyZUmPn5QT8ylSjubq6qrGjRurRYsWqlevXpw3wt+5c0fLli3TyJEjdezYMUmPCyl16tRJf/31l03Hb5S5c+cax0bjxo01bNgwValSxVhHRESEfv31V7322mu6cOGCsez111/X9u3bY60v+vz169fP+DGPJOP4jost56t+/frp1KlTcnNzU58+ffT222+bnlgVFhamSZMmadCgQXr48KGkxz8cGTVqVKxjKCErV640tnnlypU1atQo1atXz3TO2rZtm+kHKlG8vb3Vtm1bPf/886pevXqcRWiCg4M1b948jR49Wjdv3pQkHTp0SO+//76+/PJLm/OUUm9OUkKOHDnUvn17Pf/88/L394+zCM2JEyc0Y8YMTZgwwSiAsG7dOn355Zd65513YrUvV66csZ/NmTNHc+fONV4bN26cypUrZ1eO69atU9++fU1FcJo3b65+/fqpRo0apn3i+vXrmjZtmj7++GPduXNHkvTLL79ozJgxGjhwoF3jxsXHx8d0DNn6/qKeVhafvn37Kjw8XOnSpdN7772nPn36mPbtmzdvmo5jSerdu7epEJIk9erVSwMHDjTtfxcuXNDXX3+tcePGGUUuTp8+rU6dOmnjxo2xrh3xOXHihAYPHixJeu655zR69GjVqVPH6H///n3NmDFD77zzjlE47fDhw5oyZYru3LmjFStWSJI6duyowYMHm+bk+vXrGjJkiL777jtj2fTp0/XGG2+obNmyNuWXmPnz5+vu3bu6fPmyOnfubCxv2LBhgj9yTGzbXb9+3TgO8uTJo48//ljt2rWTt7e30eb06dMKCwuT5LzPi0FBQUZRlKJFi+rzzz9X06ZNjePnwYMH+vnnn/Xuu+8aPxyMjIxU7969Vb16dRUuXNi0Pkd/XnTG/rx582ZJjz9XjB07Vu3atTOK6EVERGjt2rV65513TMXmBg8erDp16qhatWo2jSNJr7zyii5duqT06dOrf//+CgwMNH3uuXHjhj777DONGTPG+Iy3Z88eTZ06VX379k10/QULFlS7du3UtGlTVa5cWZkyZTK9brVadeDAAU2aNElTpkwx9p+ZM2eqefPmatWqlc3vRZLat2+vsLAwZcuWTUOGDFGXLl2UPXt24/ULFy5o8ODBmjVrlrHs119/1e+//64mTZrYNRYAAAAAAAAAAPhvi/rON0r07+BsEb39o0ePdP/+/VgPlUlJkZGR6tmzpx48eGAs69q1q3x9fVN03LQ8b8ePH9dHH30k6XFRksmTJ8vDw8Mh606utDZvkZGRpgfbeHt7a/78+Xr55ZcTfTBPRESEpk6dqn379mnVqlWxHsrhKGltzqLkyZNHW7duNT245ccff9SPP/4Yb59MmTKpf//+GjRokM3fOwMAkFwUpQEAAAAAAACANOT99983FaWRpPXr16tmzZoqVKiQGjZsqGrVqsnf319+fn6xik/YwsXFRb169TJu2L53757mzZunN954I9G+VqtVM2bMMGI3Nzd1797dpnGjCtJ069ZN06ZNi/WlqMViUUBAgDZu3Kjy5csbBS2Cg4O1fPlytWzZ0tQ+T548RgGHmIU/atSokawfRpw6dUoWi0VTpkxRz549Y73u6emp7t27q1ixYgoICDAKECxdulQhISHKmTNnvOvu3r27rly5YsS9e/fWxIkT5e7uHqttunTp1L59ezVs2FDPP/+8tmzZIklasWKFfv755zhvzv3ll1+MYhKSNHToUI0YMSLefLJmzarnn39ezz//vMaOHasbN27E29ZWhw8fNj0Bp27dulq7dm28+2v69OlVrVo1VatWTSNGjNCJEyeSnUN058+fj1XkoWvXrpo5c2asG+QzZsyonj17qkGDBqpZs6Zxw/v169f16quv6ueffza1L1KkiOmJNVE3iUf93aBBA4e+l+gqVaqkkydPqlChQgm28/b21ksvvaQ2bdqoTZs2RsGFvXv3auXKlXbdeB1VkObDDz/UyJEjY73u6uqqF154QSVLllSFChUUHh4uSdqxY4f27dsXqwhG9PmLWegguXN36tQpeXp6asmSJWratGms1728vNSvXz/5+PiYCj/MmjVLH3/8cZzHZFyiCtK0a9dOCxYsiFVAS5KqVq0aa1n37t31ySefxLopP6Z8+fJp4MCB6tChg+rWrWucS6dOnaphw4YpW7ZsNuUppd6cONrgwYNVoUIF0/EVl2LFiumTTz5R+/btVb9+faOw1Pjx4/XGG2/E2jZZs2Y19rNNmzaZXqtUqZICAgJszvHmzZvq3LmzcT1wcXHR1KlT1aNHjzjbZ8uWTQMGDFCzZs0UEBCgkJAQSY/P2V27dpWPj4/NY8cl5vknue8vyuXLl5UxY0b9/vvvpoJcUbJkyaLatWsb8eLFi7V48WJTm2nTpumVV16J1Tdv3rz69NNPVatWLb3wwgvGtWzLli36+uuv9fbbb9uUY9R5u3Xr1lq4cGGs/dbT01N9+/aVl5eXunbtaiwfO3ascX2eMGFCnONly5ZN3377rcLDwzV79mxJ/342+uKLL2zKLzFRRQijisFEyZMnT7LOi1E/svPz89Mff/wR5z4WvaCLsz4vRhWkqVixotavXx/rHOnh4aH27durbt26qlmzpqnY2uuvvx7rc7QjPy86c38uVKiQNm/eHKvAmaurqxo1aqRdu3apYcOGRgGbqEI9+/fvt7n43KlTp5QtWzatXLlSzz33XKzXs2bNqtGjR8vb21tDhgwxlk+ZMiXRojTfffedqlevnuCPIi0Wi8qWLatvv/1WrVq1UvPmzY3CUaNHj7a7KM2pU6fk6+urtWvXmgoGRcmbN69mzpwpd3d3TZ061fR+KEoDAAAAAAAAAADsEfXghyiJfbcdU8z2oaGhqVqUZvjw4dq4caMR58yZU6NHj07xcdPqvFmtVvXu3dt4GE737t1Vq1atZK/XUdLavN2+fdtUfCY0NFQ9evSQ1Wo1vnfu0aOH8VvHo0ePavbs2fr222+NB3zs2LFD3bt319KlS5OcR0LS2pxFlytXLi1evFgrVqxQYGCgzp8/H2/bokWLauLEiWrcuLFDxgYAwFb2360AAAAAAAAAAEgxNWvW1Icffhjna2fPnjVucC9durQyZ86s+vXr66OPPtKuXbvsGqdHjx6mm7Sj34iZkDVr1phukm7WrJldN85XrlxZU6ZMSfCG1Jw5c8aag99//93mMRzlzTffjLMgTXS1atVSu3btjPjhw4dat25dvO23bdumlStXGnGTJk00adKkRAs9ZMmSRYsXLzY9dWX8+PFxto26OTvKq6++muC6o8uYMaMKFChgc/v4xMwhMDDQ5gJKLi4uKl68eLJziO7bb7/V3bt3jbhcuXKaNm1agjdp+/r6atGiRaY2y5Yt0/Hjxx2aW3IULlw40YI00aVLl05z585V5syZjWUzZ860e9yWLVvGWZAmuuLFi8cqXOCM43j06NFxFl+J7qWXXlKVKlWM+MqVK9q9e7dd4xQpUkSzZ8+OsyBNfCpVqpRoQZroChUqZDpX3717VwsXLrQrTyn15sSRqlWrZtcPgsqXL68xY8YYcXBwsFavXp0SqRkmTZqkS5cuGfGoUaPiLUgTXcmSJTVr1iwjfvDggSZOnJgSKTrMuHHj4ixIE5eY16rXX389zgIe0TVt2tR44lyUL774QhERETbnWKRIEc2ZMyfB62uXLl3k5+dnxGfPntXdu3fVqVOnRAuGjBo1ynRdc8b5LSnc3d31448/2vzZzVmfFzNkyKClS5cmeI7MmTOnFi9ebDrvrlixItZnEEdy1v5ssVi0aNGiWAVposuQIYOWLFliusYfPHhQa9assXkcSZo+fXqcBWmiGzhwoPLnz2/Ee/fuNQq0xadWrVp2PaXvf//7n/r372/EO3bsMBU8tIWrq6sWLVoUZ0Ga6D777DPTNWb16tVGgTEAAAAAAAAAAABbRP9NiiS7i1XEbB9zfSlp8eLF+vjjj43YYrFo+vTpCT6Qy1HS6rxNnz5dQUFBkqQcOXKYfn+QFqS1eQsNDY21vgcPHsjd3V0///yzJk2aJH9/f3l7e8vLy0sVK1bUl19+qVWrVil9+vRGv59//jnWw8IcJa3NWXQXLlxQ+/bt1axZswQL0kjSyZMn1aRJE1WvXt3u708BAEgOitIAAAAAAAAAQBozcuRIffnll4negB8aGqo//vhDQ4cO1XPPPafSpUtrxowZNt1E6ePjo5YtWxrxgQMHtH379kT7xbwZuVevXon2iW7kyJGJFmCRpPbt25viv/76y65xkit9+vTxFgeK6cUXXzTFCeX6xRdfmOIJEyYkWBglOh8fH1ORnM2bN8d5A3DML71tmW9HSws5RLFarZo+fbpp2bhx42wqHlKtWjXT9rVarZo2bZrDc0xN2bJlU5MmTYx4y5Ytdq/jk08+samdPcdGSsiXL59ee+01m9omN9cPPvjA9EOZlFK/fn3lyZPHiO3dfqk5J87WoUMHUwGEpOzrtoqIiNDXX39txAULFlS/fv1s7t+0aVNVqFDBiBcvXuzQ/BypYMGCNl/7Dx8+rK1btxqxl5dXrOIc8Xn33XdNRdLOnj1rV2Gh999/X15eXom2a968uSm2WCwaPnx4ov3y5cunSpUqGfGJEydi/dAuLXrxxRdVunRpm9s76/Pia6+9poIFCybarnTp0urSpYtp2YwZM+way1bO3J/btm2baKEY6fET9GKee+yZD39/f73wwguJtnNzc1Pr1q1Ny1KicFnnzp1Nsb3n8TZt2qhy5cqJtsuaNasaNmxoxOHh4Tpy5IhdYwEAAAAAAAAAgP+2mL/xevDggV3979+/n+D6UsqmTZvUuXNnWa1WY9nw4cNjfY+aUtLivF26dMn08IRx48Ype/bsyV6vI6W1eYuvwMvgwYMT3Jfq1aunTz/91LRs7NixycolPmltzqLs27dP5cqV06JFi2S1WmWxWPTSSy9pzZo1CgkJ0YMHDxQSEqJVq1apU6dOxu8Mt27dqueee04bN250SB4AACSGojQAAAAAAAAAkAa9+eabOn78uF599VVlzpzZpj6HDh3SK6+8In9/f509ezbR9oGBgaY45g3EMYWEhGjZsmVGXKBAATVu3Nim3CQpc+bMatSokU1ts2XLZroZ+dy5czaP4wgNGjSw+QcF5cuXN8Xx5RoZGamVK1casb+/v0qUKGFXXtFvmJWkP//8M1abvHnzmuJ58+bZNYYjxMxh/vz5qZ5DlCNHjujKlStGXLBgQdWvX9/m/j169DDFT8OX+YULFzb+Dg4OVkhIiM19y5Qpo5IlS9rUtnTp0qbiP6l9HLdu3drmgki2HsdxcXV1jVVIKyX5+voaf+/Zs8euvqk1J2mBl5eXcuXKZcT2zpU99u3bpwsXLhhxhw4d7C7GFf38fuTIEV29etVh+TnSiy++KBcX275i3rBhgylu3bq1smTJYlNfd3d3vfzyy6Zltp5/LRaL2rRpY1PbmAVaypUrp2eeecamvmXKlDH+joyMTPSJZWlBx44d7e6T2p8XJcUqNJOQrl27muKoJxY6mrP2Z8m++ejSpYup4GHMvBMSsxhZQlLjGhH984pk/3k8rb0fAAAAAAAAAADw9MqYMaMpjvkwp8Tcu3cvwfWlhP3796t58+amsfv27auhQ4em+NhR0uK8vf7667p586YkKSAgINb3kWlBWpu3uPp7eXnpnXfeSbRv3759lTNnTiPeunWrrl27lqx84pLW5kySbty4oaZNmxq/D3F3d9eyZcs0b948NWjQQDly5JC7u7ty5Mihhg0bav78+frll1+M36OEh4erVatWunTpUrJzAQAgMRSlAQAAAAAAAIA0Kn/+/Prmm290+fJlLVu2TO+8844qV64sDw+PBPvt3r1b/v7+OnnyZILt6tevb7rxeuHChbpz50687WfPnm16SkiPHj1svjFdkipWrGhX++jFBG7dumVzP0eoXLmyzW2j5ynFn+uBAwdMr9kzRpTohXok6e+//47V5n//+58p7tevn4YMGZKqX0BXqVJFmTJlMuIlS5aoffv2OnDgQKrlEGX79u2muG7duqabtRNTu3ZtU2GVPXv22P20nNRw8+ZNTZ8+Xd27d1fFihWVJ08eeXl5yWKxxPoX8ylD9hS/sGe/dXd3N92w/zQcx3F59tln5e3tbXP7uJw5c0ZjxoxRu3bt5Ofnp5w5c8rT0zPO7bd161ajn72FS1JrTlLSoUOHNGLECLVs2VLPPPOMcuTIIQ8Pjzjn6uLFi0a/lCzyErNAWEqd39MCf39/m9vGPP/Wq1fPrrFiFhDbtm2bTf0KFy6sbNmy2dQ2ZgG6ihUr2pZcHH1v375tc19nsWf7RUntz4vZs2ePVSwoIdWqVTN9Nt67d68ePnxoc39bOWt/tlgsqlOnjs3jFCpUyFS87NKlS/rnn39s6pta14gdO3bogw8+UNOmTVWkSBFly5ZN7u7usc7hMZ+o+F+85gEAAAAAAAAAgCdDzAIVoaGhdvWP/v2bm5ub0qVL55C84nPy5Ek1atTIKL4iPS74P3HixBQdN6a0Nm+//PKLFi9eLEny8PDQpEmTkrW+lJLW5i19+vRydXU1LatTp47pt2Px8fDwMD3kzmq1mn4X4yhpbc4kadSoUaYHII0aNUrNmjVLsE/z5s318ccfG/H169dNMQAAKcUt8SZwtF9//VXjxo2z+4NLdLt373ZgRgAAAAAAAADSMk9PTzVv3lzNmzeXJD148EAHDhzQ5s2btXbtWq1atSpWkYwrV66oTZs22r17d6wvfaNYLBb17t1b/fv3lySFhYXp+++/V+/eveNsP23aNONvFxcX9ejRw673EfNmz8R4eXkZf9v7dJLksifX6HlK8ecas8DAt99+q2+//db+5KK5fv16rGXVq1fX//73P61Zs0aS9OjRI40aNUqffvqpqlevrvr166tWrVry9/dPdiGN+KRLl04DBw7U4MGDjWWLFi3SokWL5Ofnp4YNG6pOnTqqVq2afHx8UiSHKGfPnjXFZcuWtau/p6ennn32WR08eFCSdP/+fV2+fFkFChRwWI7JERYWphEjRuirr77S/fv3k7SO6D8ySkxSjuOom8ifhuM4LoULF7a5bUxnz57VW2+9pWXLlslqtdrd355tJ6XenKSEAwcO6PXXX9fGjRuT1N/eubJHzPN7+/btk73OuM7vaYE9+3tyz7/lypUzxbYW1oj+FLXEZMiQwWF9nX2MJCZjxozKkSOH3f1S+/OiPQVppMcF0EqUKGEUvrt//74uXLigQoUK2bWexDhrfy5YsKDdn9dKly6t06dPG/Hp06djFb6KS0pfI/7880+9/vrr2r9/v83jRPdfuuYBAAAAAAAAAIAnS758+UzxuXPnbO5rtVoVHBwc77ocLTg4WA0aNDA93Kpx48aaO3euXQ+bcIS0Nm/9+vUz/n7//fdVokSJZK8zJaS1eZMeP3wv+neq9nzvW6ZMGVMcPT9HSWtzZrVaNWfOHCP28vLS66+/blPfN954QyNHjlRYWJgkaf78+frqq69S/fgFAPy3cJVxgnHjxunEiRO6dOlSkv8BAAAAAAAA+O/y8PBQpUqV9Oabb2rZsmW6cOGC+vfvH6v4zL59+7Rw4cIE19WtWzd5enoacfQbiaP7888/dfToUSNu1KiRTTe3RpfST/FxpOTkGl9hiWvXriV5nfG5detWnMsXLFigqlWrmpZFRkZq06ZNGjFihBo0aKBs2bKpWrVqGjVqlM6cOePw3AYNGhTnDet///23vvzyS7Vu3Vp58uTRs88+q7feekvbt293eA6SdOPGDVOclBvyY/aJuU5nuXr1qqpVq6axY8cmuSCNJLv6psSxkVJSK1dbnuwUlx07dqhcuXL65Zdfkjw3MQuSJeZJ2n7R/fbbb6pcuXKSC9JI9u3n9krN87uz2bO/J/f8my1bNtOPpmw99yZnP39SjxFbJPVcJaXu58Xs2bPbnV/MPilRhMpZ+3NqzkdK7v+TJ09WnTp1klyQRrL/PP40H88AAAAAAAAAACBtefbZZ02xrQ8okKTLly+bvvuPuS5Hunr1qv73v/+ZfidUq1YtLVmyRO7u7ik2bnzS2rxFPXRJkkaNGiU3N7cE/xUrVszUf8OGDabX69evn+yc4pLW5k2SSpYsaYqzZs1qc9+YbVPiIT5pbc5OnjypkJAQI65SpYrSp09vU9/06dPL39/fiG/evKkTJ04kOycAABLi5uwE/otCQ0MlPX4ynL1PFAUAAAAAAACAmLJnz64xY8aodu3aeuGFFxQREWG8Nm/ePL300kvx9s2RI4fatGmjBQsWSJJ27typffv2qVy5cqZ2U6dONcW9evVy4Dv4b0iJG6QjIyPjXJ4jRw5t3LhR06ZN0+effx7nF8+PHj3Stm3btG3bNg0dOlSdOnXSuHHjlDt3bofkZrFYNHnyZLVu3Voff/yxNm3aFGe7o0eP6ujRo/rqq69Uo0YNffHFF6pcubJDcpD+/W/yUby8vOxeR8w+d+7cSVZOjtKuXTsdOHDAtKxAgQKqW7euSpYsqfz58ytjxoxKnz696Ub4OXPmaO7cuamd7lMrKT/Munbtmpo2bRqr8EjZsmVVq1YtFStWTHnz5lX69OmVLl06WSwWo02/fv2SdVP/k+bYsWNq27at6Qc+FotF/v7+ql69uooUKSIfHx+lS5cuVgGCzp076/LlyymeY2qe353Nnv09uedfi8Wi9OnTG0/3Sivn3idVcn5EmpqfFzNkyGB3n5j7Vsx9zxGctT+n1fmwx/r169W3b19ToRc3NzfVrFlTVapUUaFChZQrVy6lS5fOVPxIkv73v/+ldroAAAAAAAAAAAB2y5s3rzJnzmz8BmDPnj029/3rr79MsZ+fn0Nzi3L79m01btxYf//9t7GsUqVK+u2332wuhuFoaXneov/+Lqn9krqOxKTFeStVqpR+//13I7bngRMx26bEg+/S2pxduXLFFPv4+NjVP2b7q1evqnjx4snOCwCA+FCUxoly5cql3bt3OzsNAAAAAAAAAE+JZs2aqWvXrpoxY4axLL5CINEFBgYaNxlLj28onjhxohHfvHlTP/30kxH7+PioefPmDsr6vyPmTcUdO3ZUjx49krXOvHnzxvuau7u7+vbtq759+2rXrl1at26dgoKCtGXLFt2+fdvUNjIyUvPmzdPatWsVFBSkEiVKJCuv6Bo1aqRGjRrp9OnTWr16tYKCgrRx40ZduHAhVtvNmzerRo0amjdvntq1a+eQ8TNmzGiKo24Gt0fMPt7e3snKyRGWLVumoKAgI/b29tZ3332njh07mgrQxGXdunUpnB0SM2rUKF27ds2In3nmGc2bN8/0JKP4JKVAwZPs/fffN/0Ayd/fX7Nnz7bpyVPRi/mkpJjbZPTo0apUqVKy1lmqVKlk9U8L4jr/Zs+e3eb+VqtVd+/eNeK0cO79L0utz4vh4eF294l5nY657zmCs/bntDof9ujXr5+pIM3zzz+vSZMmKX/+/An2s+eHqgAAAAAAAAAAAM5Ws2ZNLV++XJJ0+fJlnTp1SkWKFEm035YtW0xx7dq1HZ7b3bt31axZM9O9rKVKldKqVauUKVMmh49nj7Q8b2lZWpu3OnXqaNy4cUYcHBxsc9/z58+b4pw5czokp5jS0pzFLLwT/btkW8T8HjkpD2kDAMAeFKUBAAAAAAAAgKdI+/btTUVpQkNDdevWLWXOnDnePrVr15afn5/xJJz58+dr7NixxlNw5s+fb/ris1u3bnJz4z8v2ytHjhymOEuWLGrQoEGqjF25cmVVrlxZAwcOVGRkpPbt26eVK1fqhx9+0L59+4x2ly5dUtu2bbVv375EC5vYq3DhwgoMDFRgYKAk6dSpU1q3bp2WLFmi1atXKzIyUpL04MEDdenSRVWqVFHBggWTPW7WrFlNcfRCILa6evVqgut0hoULF5riyZMnq2PHjjb1vX79ekqkBDv88MMPxt/p0qXTypUrbfqhi/Tf2n6hoaHGD4IkKXfu3Fq5cqXNx+CNGzdSKjWTmOf3woULp9r5PS2L6/xrz3n9+vXrxrUhrvUhdaXW58WY11xbxLy2Z8mSxe51JMZZ+3NanQ9bHTt2zPSUwdKlS2vJkiXy8PBItO9/6XoHAAAAAAAAAACefC1atDB9v71o0SINHDgw0X7RH/yQLl06NWzY0KF5PXz4UG3bttWff/5pLCtWrJjWrFlj10MYUkpamrebN2/a1f7MmTMqXLiwEdepU8f0gKmUlJbmTZIaNGigjBkzKjQ0VNLjB5PZKmbRlwoVKjgkp5jS0pzlypXLFEd9D2+rw4cPJ7g+AAAczbG/KAcAAAAAAAAAOJWvr2+sZTGfjBGXqEIh0uMv2BctWmTEU6dONf62WCzq1atX8pL8j4r+IwRJOnHihFPycHFxUYUKFTRo0CDt3btXixcvNm4ol6SDBw9q1apVKZ5HkSJF1KtXL/3+++/at2+fqSDHvXv39M033zhknEKFCpni6EV4bHH//n0dPXrUiD09PZU7d26H5JYc27ZtM/7Onj272rdvb3PfQ4cOpURKsNE///yjCxcuGHHjxo1tLkhz9+5dnT59OqVSS3P++usvPXjwwIg7duxocyGHEydO6P79+ymVmklaOb+nNck9/8ZsH3N9SH2p8Xnx4MGDdrV/+PBhrOt03rx5kzR2Qpy1P587d063b9+2a6wDBw6Y4pjnqNQU/fOKJPXs2dOmgjQSn1cAAAAAAAAAAMCTpWXLlqbvQaZOnaqHDx8m2OePP/4wfdfVtGlTeXl5OSynyMhIdenSRStWrDCWFShQQGvXrlWePHkcNk5ypMV5exKktXlLly6dXnjhBSM+fPhwrO8K43LkyBFt2rTJiPPmzatSpUo5JKeY0tKc5c+f3/S99pEjR2z+DnrXrl06fvy4ERcqVCjNHM8AgKcXRWkAAAAAAAAA4CkSFhYWa5ktT7Xp2rWrqTBJ1I3FO3fuNH3hWa9ePZuLJ6QmFxfzf+62Wq1OyiR+/v7+ypAhgxFv2bJF9+7dc2JGj7Vu3Vr9+vUzLYv+ZX9qKF26tKZMmZIiOVStWtUUBwUF2bV//Pnnn6YfIFSsWNHmm6lT0uXLl42/ixUrJldXV5v63b59W7t3706ptJLlSTiOHSH6tpOkEiVK2Nw35v74tEvOXP3xxx92jZWc/a9u3brJGvtpFfP8a++8xGwfc31Pu7R4TkyNz4vXr1+3qzDN1q1bTcWrypcvL3d39zjbJmdOnbU/W61Wbdy40eZxzp49qzNnzhixj4+PChYsaHN/R0vN8zgAAAAAAAAAAIAz5c6dWz179jTikydPavTo0fG2v3fvnt544w0jtlgsGjJkSLztz5w5I4vFYvyL68FlMb366qtauHChKcd169alqQeCpMV5exKkxXn78MMP5ebmZsSvvfZagr+Ne/Tokfr06WP63jZ6jo6W1uasefPmpvjVV181ffcdX06vvvqqaVmLFi0S7AMAgCNQlAYAAAAAAAAAniI7d+40xT4+PjYV0MiSJYvat29vxJs2bdKRI0c0bdo0U7tevXo5JlEHi/kEkvDwcCdlEj8PDw/Vq1fPiMPCwjRz5kwnZvSvGjVqmOKrV68+NTmUKFFCuXPnNuKzZ89q/fr1NvefMWOGKa5Tp45D8kqu6D/ISOwHCdHNmDEjTRRDisuTcBw7QswiCPZsv2+//dbR6aRpSZ0rq9Wq7777zq6xkrP/+fv7K2vWrEb8xx9/6PDhw3aN/zSKeb5cunSpbt26ZVPfhw8fau7cuQmu72mXFs+JqfV5cc6cOTa3nT17tilOaD9Jzpw6c3+2Zz5itq1du7bNfVNCUs/j9+/fj/UZDAAAAAAAAAAAIK0bPHiwvL29jXjYsGGaMGGCIiMjTe1CQkLUpEkT0/fKL774oipUqOCwXD744ANNnjzZiLNnz661a9fqmWeecdgYjpKW5u1JktbmrXjx4goMDDTiv/76S02bNtX58+djtb18+bJeeOEFbdiwwVjm6+ur1157zaE5xZSW5uz99983/a5zy5YtatiwoU6ePBln+6NHj6pevXqm34emS5dOAwYMcFhOAADEh6I0AAAAAAAAAJBGXL16VQsWLIj1JaetHjx4oK+//tq0rFGjRjb379OnjymeMGGCvv/+eyPOkSOHWrVqlaTcUlq2bNlM8enTp52UScL69+9viocNG6Z//vnHSdn8K2YBmOjFFZ70HCwWi1555RXTsv79+ysiIiLRvjt27DA9McpisZiemONMPj4+xt+HDh3SzZs3E+0THBysESNGpGBWyfOkHMfJFX3bSY+LOthixYoV+uWXX1IipTQrqXP13Xffae/evXaNlZz9z93dXW+//bYRW61WBQYG6uHDh3bl8LTx8/NT9erVjTg0NFTDhg2zqe+XX35puj76+vrqf//7n8NzTMsyZcokV1dXI04r58TU+Lz4zTff6Ny5c4m2O3ToUKwiLD169Ii3fXKOc2fuzz/99FOswpNxuXLlisaPH29altB8pIaknsc//PBDXb58OSVSAgAAAAAAAAAASDF58+bV999/LxeXx7ftWq1Wvfvuu3r22WfVt29fDR48WC+++KIKFSqkoKAgo1/JkiVNBWQc4dNPPzXF169fV/ny5eXm5mbXv9SQlubtSZIW5238+PGqWbOmEa9fv17PPPOMWrRooffff1+DBg1S69atVbRoUS1fvtxolyFDBi1evNhUMCYlpKU58/X1jfVbzw0bNqhEiRIKCAjQu+++q+HDh+udd95R7dq1VbJkSW3dutXUfvLkycqfP79D8wIAIC4UpQEAAAAAAACANCI0NFQvvfSSypQpo3nz5unu3bs297137546d+6sQ4cOmZZ36dLF5nVUrVpV5cqVM+IpU6bozp07pnVFfzpHWlKqVClT/NNPPzkpk4TVrl3bVCgoJCREDRs21JEjR2xeR2RkpH7++WcNHDgwztdfe+01/frrr7JarTat7/79+/rqq69MyypVqmRzPnH58ssv9c033yg8PNzmPmPHjnVoDtH17dtX6dOnN+K//vpLffr0SXCO/vnnH7Vt29bUpmXLlipatKjD8kqO6DfGP3jwQIMGDUqwfUhIiJo1a2ZT8RpneVKO4+QqWLCg8uXLZ8Q7d+7UDz/8kGCfHTt2qHPnzimdWppTqVIl03VnyZIl2rJlS4J9fvvtN7377rt2jxVz/1u8eLFd/d966y3lzp3biDdt2qS2bdvq1q1bNq8jLCxMX331laZPn27X2GlZv379TPFXX30Vq4hITKtWrdLgwYNNy95++23jR2H/Fe7u7ipevLgR7927N94noqWm1Pi8GB4erlatWpnWG1NISIhat26tR48eGcsaN26sEiVKxNsnudcZZ+3PVqtV7du314ULF+Jtc/fuXbVu3dp0zilZsqQaNmxo8zgpIfrnFUmaNGmSTpw4kWCfyZMna9y4cSmZFgAAAAAAAAAAQIp5/vnnNXPmTGXIkMFYdvz4cU2aNEmffPKJfvzxR9NvwsqXL6/ly5crU6ZMKZqX1WpVRESE3f9SS1qdt7Qurc2bp6enfvnlF9NDOu7du6dff/1Vn332mUaPHq2lS5cqLCzMeD1PnjzasGGDKlasmCI5xZSW5qx3796aPHmy6XdtERER2rBhgyZMmKARI0boiy++0J9//ml60KGXl5dmzpxp129DAQBIjv/WL/cAAAAAAAAA4Alw+PBhvfzyy/Lx8VGvXr20aNEiXbx4Mc62Fy5c0Lfffis/Pz8tWrTI9Frr1q1Vr149u8YODAyM97WePXvata7UFBAQIHd3dyOePXu2WrdurRkzZuj333/X2rVrjX+bN292YqbSnDlzVKBAASM+evSoKlWqpLffflv79u2Ls1DKjRs3tHbtWr3zzjvy9fVVq1attH379jjXv3nzZrVo0UKFCxfWe++9p6CgIN2+fTtWu4cPH2rlypWqUaOGdu7caSz38fFRs2bNkvUeT58+rddff1158+ZV165dtXTp0nj34b1796pDhw6mwjguLi7q0aNHsnKILn/+/BozZoxp2bRp01SvXj1t27bNtDwsLEzTp09XpUqVdO7cOWN5tmzZ9M033zgsp+SK+aOCSZMmqWvXrjp79qxp+Z07dzR9+nSVLVtWe/fulST5+fmlVpp2if6DFEn66KOP1L17d82dO1crV640Hce7d+92UpaOEXP7denSRZ9++mmsY/X8+fMaMmSIateurRs3bihdunTy9fVNxUydy8vLS23atDHiiIgINWnSRFOmTNG9e/dMbY8fP65XX31VLVu21P3795UrVy5lz57d5rEqVKigHDlyGHFQUJDq1aunSZMmacWKFab9b+3atbH6Z86cWYsWLTJdi5YtW6ZSpUpp/Pjx+ueff+Ic99y5c/rpp5/UuXNn5c2bV2+99Zbp3POka926tWkbWq1WdevWTX369NGpU6dMbS9evKgPPvhAzZo104MHD4zl1atX1+uvv55qOacl0QuKREREqHbt2hoxYoSWLl2qNWvWmPbJ+K6zKSElPy8WKlRIkrR7925VqFBBv/76q6nwzIMHD7Ro0SJVrFhRx44dM5anT59eEydOTHDdyf286Iz9OV++fHJzc9OZM2dUsWJFzZs3z3T+i4yM1KpVq1S5cmVTzhaLRVOmTJHFYrF5rJRQrFgxVatWzYjv3Lmj2rVra9GiRabtKkn79u3Tiy++aBQOTKufVwAAAAAAAAAAABLTpUsX/fXXX3rhhRdM309FlydPHg0dOlTbt2//T/0OICHMW9KktXnLli2bVq1apUmTJqlkyZIJths0aJAOHTqkypUrp2hOMaWlOevdu7f279+vvn37ytvbO8G2mTJl0muvvaYDBw6oW7duKZYTAAAxuTk7AQAAAAAAAABA3G7fvq1p06Zp2rRpkqTs2bMrR44cypIli+7du6eLFy/qypUrcfatUqWKZs6cafeYnTt31oABAxQaGmpaXrNmzTR9Y2iuXLn08ssva8aMGcaypUuXaunSpbHaFipUSGfOnEnF7Mxy5cql5cuX6/nnnzcKD4SHh+vLL7/Ul19+qcyZMytfvnzy9vZWaGiorl+/nqQbzc+ePavx48dr/Pjxslgsypcvn7Jnz6706dPr9u3bOnXqVKyiDq6urpo2bZrp6SvJcevWLc2ZM0dz5syRJOXMmVO5cuWSt7e37t27pzNnzujmzZux+g0YMECVKlVySA5RXnvtNe3YsUNz5841lgUFBalatWrKmTOnChQooHv37un06dOmp91Ij290X7BggfLmzevQnJKjYcOGatq0qVasWGEsi5rrIkWKKGfOnLp586ZOnz5tuhm+U6dOeuaZZzRixAhnpJ2gChUqqF69evrjjz8kPb7RftasWZo1a1astnXq1FFQUFDqJuhA7733nubOnavz589Lelxk4YMPPtDQoUNVokQJeXl5KSQkRGfOnDEVqvrqq680f/58p57DUttHH32k5cuXGwV7bt++rcDAQL311lsqXry4PD09dfHiRWMupcfnslmzZqlv3766du2aTeO4u7vrzTff1NChQ41l69ev1/r16+NsH1cBsVq1amnOnDnq3r27cX4NDg7We++9p/fee0958uRRrly55OnpqVu3bunKlSu6ceOGzXPxpJoyZYpOnDihffv2SXo8d5MnT9bkyZPl6+urnDlz6vr16zp9+rTpyV6SVLhwYS1YsECurq7OSN3pXn31VU2ePNnYny5cuKDhw4fH2XbmzJmp9qOzlPy8GBAQIDc3N02fPl0nT55UixYtlDlzZvn6+spqter06dO6c+eOqY/FYtGkSZNUtGjRBNftiM+Lqb0/FytWTH369NGHH36oy5cv6+WXX1afPn1UuHBhubu768yZM3GeR0aOHKkaNWrYPE5KGjdunAICAvTw4UNJjwv2tG/fXhkzZtQzzzwjFxcXnT9/XpcvXzb6eHl5af78+an2NEQAAAAAAAAAAABHK1GihJYuXapr165p8+bNOn/+vG7fvi0fHx8VKVJENWrUsOt7o6jvy2xlT9u0xNnzZo+UXLe90tq8WSwWBQYGKjAwUPv379fhw4cVHBysiIgI5ciRQ6VKldJzzz0nFxeXJI+RXGlpzooVK6Zvv/1WX3/9tfbv368DBw7o+vXrCg0NVcaMGZUtWzaVLVtWZcqU+c/+fgIA4FwUpQEAAAAAAACANCJjxowqV66ccZNrTNeuXUv05noXFxf17t1bY8aMSfTJGXHx9vZWx44dNXXqVNPyXr162b2u1PbFF1/o3LlzWrNmjbNTSVSZMmW0e/duvfzyy1q1apXptVu3bunWrVuJrqNgwYI2j2e1WnX+/HlT0YaYsmbNqpkzZ+r555+3eb32CgkJUUhISLyvu7q6avDgwSlSMMVisWj27Nny8fHR+PHjTTeKJ5SXj4+PFi1apJo1azo8p+SaP3++GjVqpB07dpiWnzp1SqdOnYrVvkOHDpo5c6Y++eST1ErRbnPnzlXz5s31119/OTuVFJUtWzYtW7ZMTZo0Md2E/+jRIx06dChWexcXF40bN069evXS/PnzUzNVpytatKgWLVqkNm3amApg3Lt3T/v374/VPl26dJo1a5aaNGli91gffPCBjh07pnnz5iU53w4dOuiZZ55Rp06ddOzYMdNrFy9eTLTImKura5oqgOUI2bJl04YNG9SuXbtY1+gzZ87EW2Tpueee0y+//KI8efKkQpZpU/HixTV37lx17949VgEYZ0rpz4vffvutbt68qcWLF0t6/Nkovs/HHh4emjhxorp06WLTupP7edEZ+/OQIUMUEhKir776SpIUFhamgwcPxtnWxcVFH374oYYMGWL3OCmlevXqmjp1qnr16mUUppGk0NBQ7dmzJ1b7rFmzasmSJapQoUJqpgkAAAAAAAAAAJAismfPrhYtWjg7jScO85Y0aXHeypYtq7Jlyzo7jXilpTlzdXVVhQoV+K4UAJDmOK+MHAAAAAAAAADAJEeOHNq7d69Onjypzz//XM2bN1fWrFlt6psnTx699dZb2rt3r7777rskFaSJ0qNHD1OcJUsWtWvXLsnrSy3e3t5atWqVVq5cqR49eqh8+fLKli2b3N3dnZ1anHLmzKmVK1dq48aNat68uby8vBJsb7FYVKFCBb3//vvav3+/5syZE2e7X3/9VRMnTtTzzz+vLFmyJJpH3rx51b9/fx07dkwtW7ZMyluJZeTIkVq4cKE6d+6sAgUKJNo+Y8aM6ty5s/bs2ZMiBWmiWCwWjRkzRn/99ZdatGghT0/PeNvmzZtXH374oY4fP54mC9JIj4/NjRs36sMPP1TmzJnjbVeqVCktWLBA33//vTw8PFIxQ/vlzZtX27Zt06JFi9SpUyeVKlVKWbJkkZvb0/echQoVKmj37t3q3LlzvE8xslgs+t///qdt27bpnXfeSeUM046GDRtq586dat68ebxt3Nzc1LZtW+3bt08vvvhiksZxdXXV3LlztWnTJr322mvy9/dXjhw5EjxXxKVSpUo6fPiw5syZo6pVqyb6lCpPT0/Vq1dP48aN07lz59S7d+8k5Z+WZc6cWatXr9aSJUvk7+8vi8USb9vSpUtr5syZ2rZt23+6IE2Utm3b6tixYxo9erQaNWqkAgUKKGPGjAnOYWpIyc+LHh4eWrRokaZMmRJvET5XV1c1atRIe/bssasYjiM+Lzpjf/7yyy+1ePFi+fn5xdumevXq+vPPPzV8+PAkj5NSunbtqo0bN6p27drxtkmXLp169OihQ4cOKSAgIPWSAwAAAAAAAAAAAAAAABAvi9VqtTo7CXtVqlRJly5dko+Pj3bv3u3sdOyW0vn7vr/c4etMLWdGJ+UJvBccngfwZEnOE1ODHZYF8GTKF+1vrif4r0vG9eTuaselATyJ0jc0/nz0dXEnJgI4n9sbx4y/A4vbd+Mu8LSZfOy+w9ZltVp19uxZHTt2TP/8849u3bqlu3fvKkOGDPL29lbevHlVrlw55c+f32FjzpgxQ6+88ooRv/baa5o4caLD1o+4PXz4UDt27NDp06d19epVhYWFycvLS1mzZlXx4sVVsmTJBAuPxMVqterYsWM6fvy4/vnnH92+fVsRERHy9vaWj4+PypYtq+LFi8vFJWVr2AcHB+vIkSM6ffq0bty4ofv37ytDhgzKnj27SpUqpTJlythd9MERwsPDtWnTJv3zzz+6evWqPD09lStXLpUqVUrly5dP9XyS4969e9q6dav+/vtv3bhxQx4eHsqbN6+ee+45FS/O59S07vr169q4caPOnj2rO3fuyMvLS4ULF1b16tWVK1cuZ6eXply8eFF//vmnzp8/r/DwcGXKlEnFihVT9erVbSrE5Qy3bt3Stm3bdOHCBV29elUPHz6Ut7e3cuXKpWeffVYlSpRQunTpnJ1mqrp8+bK2bNmiS5cu6caNG8qUKZNy586tKlWqxFuEBGmLoz4vnjlzRoULFzbirl27atasWUYcGRmp3bt3a8+ePbp69arSp0+vfPnyqXbt2vLx8UnWe3AUR+/P0Yvc1KlTR0FBQabXDxw4oF27dunSpUvy8PBQnjx5VK1aNdM8pmVnzpzR5s2bdfHiRd2/f19ZsmRRiRIlVL16dWXIkMHZ6QEAAAAAAAAAAAAAAACIhqI0TkBRmvhRlAZICorSAElHURrgXxSlAZKMojSAgaI0wL8cWZTGGapXr66tW7ca8d69e1WuXDknZgQAAIC0xFGfFxMrSvNflFhRGgAAAAAAAAAAAAAAAABILSn76FMAAAAAAAAAwBNl//79phuMq1SpQkEaAAAAGPi8CAAAAAAAAAAAAAAAAAD/DRSlAQAAAAAAAAAYxowZY4pfe+01J2UCAACAtIjPiwAAAAAAAAAAAAAAAADw30BRGgAAAAAAAACAJGn9+vVasGCBEefLl0/t27d3YkYAAABIS/i8CAAAAAAAAAAAAAAAAAD/HW7OTgAAAAAAAAAAkPpu3Lih3bt3S5KuX7+uzZs3a8qUKbJarUabwYMHy9PT01kpAgAAwIn4vAgAAAAAAAAAAAAAAAAA/20UpQEAAAAAAACA/6B9+/bpf//7X7yvV6lSRYGBgamYEQAAANISPi8CAAAAAAAAAAAAAAAAwH+bi7MTAAAAAAAAAACkLcWKFdPixYvl4sJ/QgYAAEBsfF4EAAAAAAAAAAAAAAAAgKefm7MTAAAAAAAAAAA4X8aMGVWiRAm1bt1ab775pjJmzOjslAAAAJCG8HkRAAAAAAAAAAAAAAAAAP5bKEoDAAAAAAAAAP9BAQEBslqtzk4DAAAAaVRqfF709fXlM2kMzAcAAAAAAAAAAAAAAACAtMLF2QkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANIOitIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwUpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGChKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwUJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGCgKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEBRGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgaI0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADRWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAaK0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFEaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIDBzdkJAAAAAAAAAAAAAAAAAAAAAAAAAAAA4L/owv//r1WyRv77t6yJLP//ZdYYbeNcHvnv3wkuj2OceJdH6xszR2v0NjGWKTLa6zHyjauNrLJao7WxeErpnpHuHpMiw2OPH3Od0fO1Rp+nmMsio63DUX9HJG89cS6LZ92RcbynyP+fU4+sUt4m0rnl0oPr5rmOjLG9IqNtG1Ob6HMas5/VPL4t64injTXaOv9t/m+bx5vXGm151LAx2kS9HhltWdTrkdH6WaONGalo40vyyikP/5f1YPscRd6+kug6raZ1xl7/47cR1d6+tnEuj9X337mwN5foy/9/KkybLjLatETfdJFWS6z2bll9lP/53jr32xTdu34pRvvY64m+K8Ue1xLnuMbrSmTd8b2n6OuTxba+imfcaH/HXnfc7yGhvCRp8rH7AgCkHS7OTgAAAAAAAAAAAAAAAAAAAAAAAAAAAABAIlzSy5KhrOSS3tmZPDk8sslS+CXJM5uzM3miWLxzK139frJ453Z2Kk8Uz2x5VKzrUHlmz+PsVAAAcAiK0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFEaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBojQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANFaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABorSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMbs5OAI53ZvTzzk4hleV1dgLAEyyfsxMA0hCuJ0CSpW/o7AyANMPtjWPOTgFIMyYfu+/sFAAAAAAAAAAAAAAAAAAAAAAAAAAkkYuzE/gvCgsLM/0vAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAKQVFKVxAorSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEir3JydABzP9/3lzk4hyc6Mft7+ThFHHZ8I8CRxLZH0vtZ/HJcH8CSyFPz374gjzssDSAtcn01G5wsOSwN4MuU1/rr9YT4n5gE4X6aPgo2/A4t7OjETwPkmH7vv7BQAAAAAAAAAAAAAAAAAAAAAAACSzMXZCQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA0g6K0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAABI6yLvyhq+X4q86+xMnhwPrst6er50/7qzM3miWO9c1r1142W9c9nZqTxR7l+/qBOzR+r+tYvOTgUAAIdwc3YCAAAAAAAAAAAAAAAAAAAAAAAAAAAAABJhvSfdPSBZI52dyZPjwQ3p9Hwpkjmzh/XOFd1fN16yWp2dyhPlwfVLOjlnpLPTAADAYVycnQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIO2gKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEBRGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgaI0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADRWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAaK0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFEaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBojQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANFaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABorSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMFKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgoSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMFCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgoCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMDgltSOv/76q8aNG6fQ0FBH5mOTK1eupPqYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAPBfkOSiNOPGjdOJEyccmQsAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwMmSXJQmNDRUkuTi4qJcuXI5LCFbXLlyRZGRkak6JgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAD8FyS5KE2UXLlyaffu3Y7IxWaVKlXSpUuXUnVMAAAAAAAAAHhanDlzRoULFzbirl27atasWc5LCCYWi8X4u06dOgoKCnJeMv8vLeaUWrp166bZs2cb8enTp+Xr6+u8hPCfwH6Xdg0fPlwjRoww4vXr1ysgIMB5CSHVpObnp7R4DggICNCGDRuM2Gq1OjEb50mL2wYAAAAAAAAAAAAAAAAAUoqLsxMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAKQdbs5OAAAAAAAAAABg5uvrq7NnzybYxsXFRZkzZ1bWrFnl5+en5557Tu3atVPJkiVTKUsAAAAAAAAAAAAAAAAAAAAAAPC0cnF2AgAAAAAAAAAA+0VGRurGjRs6deqUli9fruHDh6tUqVJq0KCBjh8/7uz0APyH+fr6ymKxyGKxyNfX19npAHgCRJ0zLBaLAgICnJ0OAAAAAAAAAAAAAAAAAAAQRWkAAAAAAAAA4Kmybt06lS9fXr///ruzUwEAAAAAAAAAAAAAAAAAAAAAAE8oN2cnAAAAAAAAAABI2Lhx41SuXDnTsoiICF27dk1//fWXFi5cqODgYOO18PBwtWvXTlu2bFHZsmVTO13gqTJr1izNmjXL2WkAAAAAAAAAAAAAAAAAAAAAQKqiKA0AAAAAAAAApHGVKlVSQEBAnK916tRJn3zyiQYMGKAvv/zSWB4WFqZ+/fppzZo1qZQlAAAAAAAAAAAAAAAAAAAAAAB4Wrg4OwEAAAAAAAAAQPJ4eHjoiy++0EsvvWRavnbtWh08eNBJWQEAAAAAAAAAAAAAAAAAAAAAgCcVRWkAAAAAAAAA4CkxevRoubiY/7Pv6tWrnZQNAAAAAAAAAAAAAAAAAAAAAAB4Urk5OwEAAAAAAAAAgGPkz59f5cqV0549e4xlBw4cSNK6jh49qu3bt+vChQtydXVVrly55O/vLz8/P4fkGh4ers2bNys4OFhXrlwxxihZsqQqVqwoi8XikHGipPT7SSvu3bunw4cP6++//1ZISIjCwsLk7e2t7Nmzq0yZMipdunSswkVPa05pdZvv27dPu3bt0pUrV+Tp6SkfHx9Vr15dvr6+Dln/zZs3tWXLFl28eFEhISFKly6dcubMqQoVKqhkyZIOGcNZjh49qj179ujKlSsKCwtTjhw5lDdvXtWsWVOZM2d2dnqSpIMHD2rXrl26dOmS3N3dlS9fPlWtWtVh2zfK3bt3tXHjRh05ckShoaHKmjWrfH19VadOHXl5eTlkjKNHj2rfvn0KCQnRrVu3lC1bNmO+s2XL5pAxoqTG+0krzp8/r0OHDun06dO6deuWJClbtmzKly+fqlWrpqxZszpsrIiICO3atUsnT57U1atXdefOHWXMmFH58uVTyZIl5efnl+TrbWpfx+0VGhqqQ4cO6ciRI7p27Zru3r2rzJkzK0eOHKpYsaKKFy/ulLwuX76sjRs3Kjg4WHfv3lXOnDlVvnx5VapUyeFzZrVatX//fv3999+m82b+/PlVq1YtZcyY0aHj3bp1Sxs3btSJEyd09+5dZc+eXcWKFVPNmjXl6enp0LFSwtO8bQAAAAAAAAAAAAAAAAAgqShKAwAAAAAAAABPkSJFipiK0ly9etWu/qtXr9bgwYO1a9euOF/38/PTZ599pubNmycpv82bN2vUqFH6448/dP/+/Tjb5MqVS4GBgerfv7+8vb0TXeeZM2dUuHBhI+7atatmzZolyTHv5/z58/L19VVERIQkqXjx4jp69GiiecW0bNkytWzZ0ojbt2+vH374we71xJXfwoULtXz5cm3dujXeeZWkrFmzqnv37urXr5/y5s2b7LGdlVNKb/PounXrptmzZxvx6dOn4y0wEhQUpLp16xrxsGHDNHz4cEnS999/rxEjRsS771SpUkXjxo1TzZo1bcorpl9//VXjxo3Tli1b9OjRozjbFCxYUG+//bZeffVVhxcIiDlPUc6ePZvgzfzRt11c7t+/r6+//lqTJk3SyZMn42zj5uamOnXqaPjw4UmeP1tFfy916tRRUFCQJGnJkiX68MMPdfjw4Tj7VK9eXWPHjlW1atWSNf6tW7c0YsQITZkyRWFhYbFe9/Dw0CuvvKKRI0cqR44cdq8/NDRU48eP1+zZs3X69Ok427i6uqpWrVoaOXKkatWqZdN64zuOHPV++vTpo8mTJxvx/Pnz1alTJ5tyi65ixYqma+jhw4eTXcjq0aNHWrt2rX766SetXbtWZ8+ejbetxWJR1apVNWDAALVs2TLJhTAOHTqkTz75RMuXLzcK38TFx8dHLVu2VN++fVWuXDmb1u3I63hAQIA2bNgQa/mGDRsSfO/Rz63RHTt2TN9//71WrlypXbt2xXsulB6/9759++qNN95waCGg+Pz999969913tWbNGuPzRHSFChXS4MGD1bNnz2QXQAkJCdGnn36qhQsX6uLFi3G28fDwUOPGjfXRRx+pbNmyyRrv4sWLGjx4sObPn68HDx7Eet3Ly0tvv/22Bg0alGCRqcaNG2vVqlVGvHnzZlWvXt2uXB4+fKj8+fPrypUrkqR06dLpwoULCW7jp3nbAAAAAAAAAAAAAAAAAEBype7jUAEAAAAAAAAAKcrd3d0UJ3RDdkwDBgxQo0aN4i3mIT2+cbdFixb66KOP7MorLCxM7du3V82aNfX7778nWKTkypUr+uijj1S8eHHt3LnTrnGic9T7yZ8/v55//nkjPnbsmDZu3Gh3PlOnTjXFvXv3tnsdMe3fv18FCxZU//79FRQUlOC8StKNGzf0+eefq2TJkvr999+TPX5ayykl9+GkevDggTp37qxOnTolWMxo+/btCggISLBAS1yuXLmiunXrqkWLFtq4cWOCx/w///yjd999V2XLlo23wEtacujQIZUsWVL9+/dPMN9Hjx5p3bp1qlWrlnr06KGHDx+mWo5Wq1VvvPGG2rRpE2dBmqg2mzdvVs2aNZO13x0+fFjlypXThAkT4izgIj3e37777jtVrVpVZ86csWv9v/32m4oWLarhw4fHW5BGkiIiIhQUFKTatWsrMDDQrutMdI58P4GBgaZ42rRpdueze/duU0GamjVrJrsgjSR16NBBTZo00fTp0xMsSCM93le2bt2qVq1aqW3btvHOS3wePnyoV199VWXLltWCBQsSLEgjSZcuXdLkyZNNBdPi44zruD1+++03lShRQsOHD9e2bdsS3S8vXbqkYcOGqUyZMime4/Tp01WhQgWtXLkyzqIn0uMCXr1791bbtm3jLOxiz1hFixbVhAkT4i16Ij0+tpYtW6YKFSpo5MiRSR7vzz//VOnSpTVz5sx48w4LC9OoUaNUt25dXb9+Pd51OeI4XrZsmVGQRpLatm2bYEGap3nbAAAAAAAAAAAAAAAAAIAjUJQGAAAAAAAAAJ4iFy5cMMXZs2e3qd/777+vsWPHGrG3t7dKlSqlypUrK2fOnLHaDx06VEuWLLFp3VeuXFGdOnW0aNGiWK/lz59flSpVUvny5WPdNHzp0iUFBARo06ZNNo0TnaPfT58+fUxxzAIziQkODjYVXClSpIjq1atn1zri8uDBA1mtVtMyDw8PFS1aVBUqVJC/v7+eeeYZubm5mdrcunVLzZo10/r165OdQ1rJKSX34eTo2rWr5s+fb8RZs2ZV2bJlVbFiRWXJksXUNiIiQj179rS5QMLx48dVtWpVBQUFmZZbLBb5+vqqcuXKKlOmjDJmzGh6/dixY6pWrZqOHTuWpPeUGnbt2qVatWrp1KlTpuXu7u565plnVLlyZeXNmzdWv5kzZ6pFixbJKhxgj48//lgTJ0404gwZMqhUqVIqX758rO0bGRmpoUOHavTo0XaPc+bMGdWvX99U1KRQoUJ67rnn5OfnJ1dXV1P7kydPqnXr1jYXjJkyZYpeeOEFUzGHqPfj5+cnf39/FStWTC4uLrH6tW3bNtYxn9rvJ+rcEiUoKEgnTpywK6eY5/VevXrZ1T8+9+7di7UsZ86cKlmypKpUqaJy5copR44csdosWbJELVu2VGRkpE3j3Lp1Sw0aNNB3330Xq0/69OlVvHhxValSRX5+frHOCYlxxnXcXnHNc9T7rlSpkipXrqzChQvH2oeDg4MVEBCgv//+O0Xymj9/vnr16hWriE+mTJlUtmxZlSlTRt7e3sbyJUuW6NVXX03SWB9++KF69uypO3fuxBqrVKlS8vf3l6+vr+m1yMhIDRs2TG+99Zbd4+3evVtNmjQxCs24uLioaNGixnXeYrGY2u/cuVPdunWLd33Nmzc3ndd//PFH3b59266c7DmOn+ZtAwAAAAAAAAAAAAAAAACOQlEaAAAAAAAAAHhKhIeHxypmUbhw4UT7bdy4UZ999pkkqUqVKlq9erWuX7+ugwcPaufOnbp8+bLWr1+vZ5991tTvzTffTLTgQWRkpDp06KDdu3cby3LmzKmxY8fq4sWLOnfunHbt2qU9e/bo6tWr2rRpk6lYS3h4uDp27Khr164l+j5S8v00atTIdLPwTz/9pBs3btic04wZMxQREWHEPXv2jHWzdnLUqVNHEyZM0MGDBxUWFqYTJ07or7/+0vbt23Xs2DHduXNHP//8s6loQ2RkpDp37qzQ0FCH5eGsnFJyH06OuXPnauHChZKkxo0ba+vWrbp27Zr27dun3bt36+rVq1q6dKnpJvyIiAi9/vrria47PDxcLVu21OnTp41lhQsX1uTJk3Xt2jWdPn1aO3fu1P79+3Xjxg39/vvvqlixotE2JCREL774Yqyb8ZNqwIABWrNmjdasWaPcuXMby3Pnzm0sj+vfgAEDYq3rzp07ateunekYy5Ahg8aMGaNLly7p2LFj2rlzp4KDg7Vv3z698MILpv4rV67U0KFDHfK+EnLixAmNHDlS0uP3OWfOHF27dk0HDx40zmkrV66Un5+fqd/gwYO1detWu8Z65ZVXdOnSJaVPn15Dhw5VcHCwzpw5ox07dujw4cMKCQnRwIEDTeeVPXv22FRAa926derbt6/pHNW8eXMFBQXp1q1bOnz4sLZv367jx48rJCREn332malQwy+//KIxY8Y4/f0EBgYaf1utVk2bNs3mfMLCwvT9998bcZYsWdSuXTu73lNCcuTIoVdffVXLly9XSEiIrly5okOHDmnbtm3au3evQkJCdPz4cQ0aNEjp0qUz+q1bt05ffvmlTWN0795dGzduNC2rU6eOVq5cqRs3bujo0aPatm2bDh8+rFu3bungwYMaNWpUrP0zppS8jo8fP944F0RXtmzZBM8bXbp0iTNXV1dXPf/885o8ebKOHz+u0NBQHT16VLt27dLOnTt16tQp3bx5U/PmzVPx4sVNOXbq1Mnu4kqJOXXqlHr37m1ab9GiRfXLL78Y14L9+/fr6tWrWrx4sQoVKiRJmj59ujZs2GDXWDNnztTHH39sxBaLRV26dNHOnTt148YNHTx4UNu3b9fp06cVHBysQYMGyd3d3Wj/1Vdf6ccff7RrzPbt2yssLEzZsmXT559/ritXrujEiRPGdf78+fOxitD8+uuvpiJ90bm5uemVV14x4rCwMC1YsMDmfM6ePWval0qUKKHatWvH2fZp3zYAAAAAAAAAAAAAAAAA4CgWaxJ/XVepUiVdunRJPj4+ph8hpgZnju0IBQoUUGRkpFxcXHTu3DmHr9/3/eUOX2dqOTP6efs7RRx1fCLAk8S1RNL7Wv9xXB7Ak8hS8N+/I444Lw8gLXB9NvE28brgsDSAJ9O/NxDf/jCfE/MAnC/TR8HG34HFPZ2YCeB8k48lr8iDr6+vzp49a8Tr169XQEBAov0+++wzvf/++6Zlf/zxh+rWrWtadubMmTiL1XTr1k3Tpk2Tq6trnOsPCQlR+fLldeHCv58Bf/75Z7Vs2dLmnKpUqaJly5YpV65c8faJjIzUW2+9pYkTJxrL3nzzzXhvzE+t9/PJJ59o8ODBRvzVV1/pjTfeiPd9RLFarSpSpIjOnDkj6fFN1+fOnZOPj0+c7aMXYahTp46CgoLiXfeVK1cUEhKiUqVKJZqH9HhuAwMDTcUavv32W/Xt2zfBfmktp9Tch7t166bZs2cb8enTp00FiqILCgqKdbxJ0ocffmgUL4nLsWPHVKFCBYWHhxvL9u7dq3LlysXbp2/fvpo0aZIRN2/eXAsWLFDGjBnj7XP//n117NhRS5cuNZZ9/vnneuedd+LtkxTRz2GFChUy9n1bvfHGG6bjP3PmzFq/fr0qVKgQb58hQ4Zo1KhRRuzi4qKdO3eaCvE4QlzFpAoVKqTNmzcrX764//9AeHi4GjZsqM2bNxvLSpcurf3798dbnCrmfidJ2bJl08qVK/Xcc8/Fm9+oUaM0ZMgQIy5fvrz27NkTb/ubN2/Kz89Ply5dkvR43qZOnaoePXrE20eSDh8+rICAAIWEhEiSPDw8dPbs2XjPa6nxfsLDw5U3b17dunVL0uNCQefOnTMVd4jPzJkzTe/5tddeM+2D0Q0fPlwjRoww4sSu0Vu3blWFChVMxWYSsnfvXtWvX1/Xr1+XJOXLl09nzpyRm5tbvH0mT56sPn36GLHFYtGYMWP03nvvJTqe1WqN99wlpc51PCrnKIldZ+Jy+vRpubi4GMVDEnPv3j21adNGK1asMJatWLFCTZo0ibdPzGtP165dNWvWrHjbN23a1FSApWLFilq/fr0yZcoUZ/sbN26odu3aOnjwYKzXErr2nDp1SmXKlDGuIenTp9fixYsTfC/S44JujRs31t27dyVJuXLl0tmzZ+PdVwMCAmIVZPH19dXatWtVtGjReMfp3bu3qaDUCy+8YLoORXfu3DkVLlzYKJJVsWJFm38PMGzYMNO1duzYsfEeA0/btgEAAAAAAAAAAHjyRf2OwypZI//9W9ZElv//MmuMtnEuj/z37wSXxzFOvMuj9Y2ZozV6mxjLFBnt9Rj5xtVGVlmtcbwnq7mNTG1saB9rWWS0dTjq74jkrSfOZfGsOzKO9xQZfVtYpcg45joyxvaKtMbTJvqcxrVuO9cRTxtrtHX+2/zfNo83rzXa8qhhY7SJej0y2rKo1yOj9bNGGzNS0cb//3ZRudiwTqtpnbHX//htRLW3r22cy2P1/Xcu7M0l+vL/nwrTpov8d1pMmy7SaomzffT1mNvHXk/M9uZxLXGOa7yuRNYd33uKvj5ZbOureMaN9nfsdcf9HhLKS0r+b3ABAI7l4uwEAAAAAAAAAADJ9+uvv+rDDz80LStcuLBq1qxpU//KlStrypQp8RbzkKScOXPGGiP6Db0xhYeHa8yYMUacJ08erVixIsEb2aXHRRG++OILVa1a1Vg2Y8YM3bx5M5F38a+UeD89evQwFTeIfoN1QtasWWMqytGsWbN4CzfYK1euXDYXf5Eez+0333xjunl85syZDsnF2TmlxDZ3hJYtWyZYkEaSihcvHqvAUUJ5nTt3zlTEp2zZslq0aFGCBWkkydPTU/PmzTPdPP/ll18aN/+nBTdv3tSMGTNMy6ZPn55gQRpJ+vjjj003+UdGRmrChAkpkmN0FotFixYtircgjSRlyJBBS5YsUebMmY1lBw8e1Jo1a+waa/r06QkWcJGkgQMHKn/+/Ea8d+9eXb58Od72kyZNMgrSSI+LwCRWkEaSSpYsaSrG8eDBg3iLuMTH0e8nQ4YMevnll4348uXL+vXXX23KJeb5vFevXjb1s0W1atXsKiRRvnx507UzODhYq1evjrf9o0eP9Mknn5iWDRo0yKaCNNLjfTi+gjTOvI7bq3DhwjYXpJGkdOnSae7cuabj0pHXwyNHjpjO4xkyZNDSpUvjLXoiSVmzZtXPP/8sT0/7CnyOGTPGVNRsxowZiRY9kaTatWtr3LhxRnzlyhXNmzfP5nFdXV21aNGiBAvSSI8LG0U/BlavXq3IqB96xlCgQAFT7n/99Zf++uuvRHOJiIgwXTs8PDzUtWvXONv+F7YNAAAAAAAAAAAAAAAAADgKRWkAAAAAAAAA4AkUERGhq1evatWqVerUqZNatmyphw8fmtp8/PHHpiIqCRk5cqRNbdu3b2+KE7pReM6cObp+/boRDx8+XNmyZbMpH1dXVw0aNMiIQ0NDtWrVKpv6Sinzfnx8fNSyZUsjPnDggLZv357oGClZ7CApPDw81K5dOyPes2eP7t6968SMHJNTSmxzR4hZLCI+L774oilOKK9vvvlGjx49MuKxY8fafKN8hgwZ9M477xjx2bNntWvXLpv6poYFCxaYbuCvUaOG2rRpY1Pfzz//3BT/+OOPunXrlkPzi6lt27aJFlaRHhdr6tevn2lZzOI7CfH399cLL7yQaDs3Nze1bt3atGz37t1xto2IiNDXX39txAULFoyVY0KaNm1qKha0ePFim/umxPuRpD59+phiW4qHHTp0SFu3bjXi5557TuXKlUu0X0rq0KGDqcDWli1b4m27ZMkS/fPPP0ZcpEgRDR8+3CF5OPM6nhqyZctmKhCS0DzbK2aBm9dee00FCxZMtF/RokXVt29fm8e5fv265syZY8TVqlVThw4dbO7fq1cvU5Ehe47jNm3aqHLlyom2y5o1qxo2bGjE4eHhOnLkSLztk3Icr1y5UufPnzfili1bKmfOnHG2/S9sGwAAAAAAAAAAADzlLOmk9GUkS3pnZ/Lk8MgqFX5J8rDtO288ZvHOJc/6/WTxTvjhNTDzyOajol2GyiObYx5cBwCAs1GUBgAAAAAAAADSuLp168pisZj+ubm5KWfOnGrcuLG+//57Wa1WU593331XnTp1smn9mTNnVqNGjWxqmy1bNtONu+fOnYu37YoVK4y/3dzc7LoRV5Lq168vF5d//zP2n3/+aVO/lHo/khQYGGiKE7tROiQkRMuWLTPiAgUKqHHjxjbllpIKFy5s/P3o0SMdPHjQidk8lpycUnKbJ0eZMmVUsmRJm9qWLl1abm5uRmzrseXj46MGDRrYlVf04gCS7cdWatiwYYMp7tGjh819n332WVWvXt2IHzx4oG3btjkst7h06dLFrrYWi8WIY77XhMQsWpSQ8uXLm+L49qV9+/bpwoULRtyhQwebC5lFib4vHTlyRFevXrWpX0q8H0kqVaqUatSoYcSrV682FWyJy7Rp00yxswuHSZKXl5epGMWePXvibbt69WpT3LdvX7u3Y3ycdR1PTdGvPcHBwQoJCXHIeoOCgkyxPeeKbt262TVO9CJuL7/8ss19Jcnd3V1169Y14i1btigyMtKmvil1HDdp0sR0nY5ZrCwu9hzH/4VtAwAAAAAAAAAAgKecS3pZMpSVXChKYzOPbLIUfknypCiNPSzeuZWufj9ZvHM7O5Unime2PCrWdag8s+dxdioAADgERWkAAAAAAAAA4Cni5eWlb7/9VuPHj7e5T8WKFU03jScm+o3yt27dirON1WrV5s2bjbh48eLKlCmTzWNIj99L9uzZjfjvv/+2qV9KvJ8o9evX1zPPPGPECxcu1J07d+JtP3v2bD148MCIe/ToYVdu9ggPD9fChQsVGBioqlWrKm/evPL29paLi0usokYxi+vYWkgireaUkts8OSpXrmxzW3d3d2XJksWI48vrxo0bpoI99r53Saab/SXbj63UsH37dlNcr149u/rXr1/fFKdkURqLxaI6derY3L5QoULy9fU14kuXLiVaMCWKPftS9P1bin9filkgxJ4xoiR1X0qJ9xOlT58+xt+RkZGaPn16vG3v37+vuXPnGnHGjBnVsWNHm3Oz16FDhzRixAi1bNlSzzzzjHLkyCEPD49Y50OLxaKLFy8a/RI6H8bcjk2bNnVIrs68jifXzZs3NX36dHXv3l0VK1ZUnjx55OXlFec8f/rpp6a+jrgePnjwQHv37jXi7Nmzq3Tp0jb3L1eunOl6kBBHH8e3b99WcHCwTf1S6jh2cXExFZW5ffu2fvjhh3jbX7p0Sb/99psRFy5cON5ibf+VbQMAAAAAAAAAAAAAAAAAjuKWeBOklMjISFWqVClJfXfv3u3gbAAAAAAAAAA8qdKlS6cKFSqodevW6tatm3LkyGFX/5g3CifGy8vL+Pvu3btxtrl8+bKuX79uxIcPH5bFYrFrnJiiry8hKfF+olgsFvXu3Vv9+/eXJIWFhen7779X796942w/bdo0428XFxf16NHDrtxs8fDhQ33++ecaNWpUggVyEnLz5s0nOqeU3ObJkZS8ogoixJfX0aNHZbVajXjFihWpdmylNKvVqnPnzhlxpkyZTEVcbFGuXDlTbGvRl6QoWLCgvL297epTunRpnT592ohPnz4dq7BLXOzZl6Lv31L8+1LMAiHt27e3eYz4pMR52tb3E6Vdu3Z6++23de3aNUnSjBkzNGzYsDiLNy1ZssRoJ0kdOnRQxowZbc7NVgcOHNDrr7+ujRs3Jql/QufDU6dOGX97eXnJz88vSWPE5MzreFKFhYVpxIgR+uqrr3T//v0krcMR18MLFy6YCtLZU/QkSpkyZWIVNYlLzOPY39/f7rFiun79ugoUKJBou5Q8jl955RWNGDFCjx49kiRNnTpV3bt3j7PtzJkzjXZRfePbV/8r2wYAAAAAAAAAAAAAAAAAHIWiNE4Q/Udwly5dcmImAAAAAAAAAJ4E48aNi1VowdXVVd7e3sqSJYt8fX3l5pb0/9ybLl265KYYS/Sb/B3l1q1bNrVLifcTXbdu3TRkyBDjhvdp06bFWZTmzz//1NGjR424UaNGNhWfsMfdu3fVrFkz/fHHH8laT1Jv3o+LM3JK6W2eVMnJK3rhmeiceWyltFu3bikyMtKIs2fPbvc6YhblunHjRrLzik9S8ovZx9YCGE/bvpQS7yeKp6enunbtqs8//1ySdP78ea1cuVJNmzaN1TZ64TBJ6tWrV5Lzis9vv/2mNm3amAph2Cu+8+Ht27dNxThy5MiR7MIxUZ60c83Vq1dVr149HThwIFnrccT1MOZx7YhzRXye1uM4T548atGihZYsWSJJ2rp1qw4fPqySJUvGWs/06dON2NXVNd7iNdJ/Z9sAAAAAAAAAAAAAAAAAgKNQlMYJvL29dfPmTbm4uNj9lFQAAAAAAAAA/z2VKlVSQECAs9Owi62FFuwRvViFM+XIkUNt2rTRggULJEk7d+7Uvn37YhUOmjp1qilOiWIHr776aqziLzlz5lRAQIDKlSunAgUKKFOmTEqfPr1cXV2NNqtXr9bYsWMdnk9azelp8jQfW6GhoabYy8vL7nXE7HPnzp1k5ZSQDBky2N0nZn4x33Nqepr3pcDAQKMojfT4fByzKM3Jkye1fv16Iy5btqz8/f0dmsexY8fUtm1bU0Eai8Uif39/Va9eXUWKFJGPj4/SpUsXq8BH586ddfny5QTXH3P/zpgxo8Nyf9L2j3bt2sUqSFOgQAHVrVtXJUuWVP78+ZUxY0alT59eLi4uRps5c+Zo7ty5Ds0l5nHtiHNFfJ607WSPPn36GEVppMfH8YQJE0xt1q9fr5MnTxrx888/r7x588a7TrYNAAAAAAAAAAAAAAAAANiHojROEPWj0ly5cmn37t1OzgYAAAAAAAAAHC/mTb4lS5bUl19+max1pk+fPln9HSkwMNAoSiM9vlF64sSJRnzz5k399NNPRuzj46PmzZs7NIe9e/dq9uzZRuzu7q4xY8bo1VdflYeHR4J9o9/A/bTn9LSJeWzVrVtXH3zwQbLWmTVr1mT1d5SYBTXCwsLsXkfMPt7e3snKKSHh4eF294mZnyOLiNgr5r40evRoVapUKVnrLFWqVLL6O0rx4sVVt25do+jMb7/9pkuXLsnHx8doM23aNFmtViNOicJh77//vu7fv2/E/v7+mj17tp599tlE+1oslkTbxNy/HVnk6Em6ji9btkxBQUFG7O3tre+++04dO3Y0FaCJy7p16xyeT8yiJY44V8Qn5naaOXOm8ufPb/d40cUssucsDRo0UNGiRY3PB3PnztXo0aPl6elptLG3ACDbBgAAAAAAAAAAAAAAAADsQ1EaAAAAAAAAAIDD5ciRwxRbrVY1aNDASdk4Xu3ateXn56e///5bkjR//nyNHTvWuOF+/vz5unv3rtG+W7ducnNz7H+S//HHH00FFUaMGKG3337bpr7Xr193aC5pOaenTcxjK126dE/NsZU5c2a5uLgoMjJSknTt2jW713H16lVTnJIFd2KOZYuY7ylLliwOysZ+MfelwoULPzX7kiT16dPHKErz6NEjzZw5U4MGDTLiWbNmGW3Tp0+vzp07O3T80NBQLV++3Ihz586tlStX2rxP3rhxI9E2mTJlkpubmx49eiTp8T5ptVptKmiTmCfpOr5w4UJTPHnyZHXs2NGmvilx7Yl5XDviXBGfmNupZMmS8vf3t3u8tMhisah3794aOHCgpMdzsmTJEmPbXrt2TUuXLjXa58+fX02aNElwnWwbAAAAAAAAAAAAAAAAALBPwo+GAwAAAAAAAAAgCXx8fIwCLZJ09uxZPXz40IkZOV5gYKDx982bN7Vo0SIjnjp1qvG3xWJRr169HD7+tm3bjL9dXFzUp08fm/seOnTI4flIaTOnp03hwoVN8YkTJ5yUieNZLBYVKFDAiG/fvq0zZ87YtY59+/aZ4kKFCjkitTidO3dOt2/ftqvPgQMHTHHM7ZmanuZ9SZJatWqlXLlyGfG0adOMolm//fabLl26ZLzWrl07hxcI+uuvv/TgwQMj7tixo80FaU6cOKH79+/b1LZYsWLG32FhYUaxtOR6kq7j0a892bNnV/v27W3umxLXnnz58snDw8OIDx48aPc6Yp4r4vO0H8fdu3c3zWX0z1dz5841HSc9evSQq6trgutj2wAAAAAAAAAAAAAAAACAfShKAwAAAAAAAABwOHd3d9WoUcOIw8PDtX37didm5Hhdu3Y13bAfdaP0zp07TYUx6tWrpyJFijh8/MuXLxt/58yZ0+ZiB5GRkdqwYYPD80mrOT1t8ufPbypCcfz4cZ07d86JGcXm4vLv109RRUBsVbVqVVP8xx9/2NU/ZvuY63Mkq9WqjRs32tz+7NmzpiI7Pj4+KliwYApkZpu6deuaYnvnOq1zd3dXjx49jPjUqVPGe4xe2EJSihQOi34+lKQSJUrY3NeebVGrVi1TvGLFCpv7JiS1r+MWi8X4297zRvS5LlasWKKFSaLcvn1bu3fvtmssW3h4eKhcuXJGfP36dbuKn+zbt083b960qe3TfhznzJlTrVu3NuKgoCCjuMu0adOM5S4uLqbjPT5sGwAAAAAAAAAAAAAAAACwD0VpAAAAAAAAAAAponHjxqb466+/dlImKSNLlixq3769EW/atElHjhwx3SQtpUyxA8l80/6DBw9s7rds2TKdP38+JVJKkzk9jWIeWxMnTnRSJnHz8vIy/g4PD7erb506dUzxrFmzbO579OhRbd682Yg9PT1VpUoVu8a315w5c5Lctnbt2o5Oxy7+/v6mwlF//PGHDh8+7MSMHK93796mYidTp07V+fPntWrVKmPZs88+q5o1azp87JiFVWw9J1qtVn333Xc2jxPzfPDdd9/p4cOHNve3Z90peR1PznkjqdeeGTNm6N69e3aNZauAgABTbM+5wp7zXoMGDeTm5mbECxcu1LVr12zu/yQIDAw0/rZarZo2bZq2bt2qQ4cOGcsbNmyoQoUK2bQ+tg0AAAAAAAAAAAAAAAAA2I6iNAAAAAAAAACAFNGzZ09lyZLFiH/66SctX77ceQmlgD59+pjiCRMm6PvvvzfiHDlyqFWrVikyto+Pj/H3jRs3bComERoaqn79+qVIPmk1p6fRO++8Y7rR/euvv9Zff/3lxIzMsmXLZvx97do13blzx+a+HTt2NBWn+PPPP/Xzzz/b1DfmftS+fXtlzpzZ5rGT4qefftLOnTsTbXflyhWNHz/etKxHjx4plZZN3N3d9fbbbxux1WpVYGCgwwqapAWFCxdWw4YNjXjp0qUaM2aMIiIijGUpVTgs+vlQely4zBbfffed9u7da/M4LVu2lK+vrxGfOnVKw4cPt7l/QlLzOh79vHHmzBm7+kaf60OHDunmzZuJ9gkODtaIESPsGsce3bt3N8XffPONzp07l2i/kydP2lWUKHfu3Hr55ZeNOCwsTK+99prtiT4BAgIC9OyzzxrxrFmz9O2335ra2HMcs20AAAAAAAAAAAAAAAAAwHYUpQEAAAAAAAAApIjMmTNr4MCBRhwZGamOHTtq2bJldq1n9+7devHFFx2dnkNUrVpV5cqVM+IpU6aYCnB06dJFHh4eKTJ29erVTfGAAQMUGRkZb/vw8HC1bt1ap06dSpF80mpOT6MiRYrolVdeMeK7d++qWbNm2rp1q13r+eOPP9S7d29Hp6dSpUoZf1utVi1evNjmvlmyZIlVrKVHjx7av39/gv2GDRtmKpbh4uKid955x+Zxk8pqtap9+/a6cOFCvG3u3r2r1q1b69atW8aykiVLmoqlOMtbb72l3LlzG/GmTZvUtm1bU66JCQsL01dffaXp06enRIrJFr142IMHD/T1118bsYeHh7p06ZIi41aqVMl0/l+yZIm2bNmSYJ/ffvtN7777rl3juLq6asiQIaZln376aawiSPGxWq1av359nK+l5nU8+nnj6tWrCgoKsnn90a89Dx480KBBgxJsHxISombNmtlUvCap/Pz81KhRIyMODw9Xq1atEizSdePGDbVq1Ur379+3a6whQ4YoQ4YMRvzDDz8oMDBQDx48sHkd169f18cff6xff/3VrrFTS2BgoPH35cuXNW/ePCPOnTu3mjdvbvO62DYAAAAAAAAAAAAAAAAAYDuK0gAAAAAAAAAAUsyAAQPUtGlTI75z545eeOEFtW7dWn/88UecN/feu3dPO3bs0CeffKJKlSqpcuXK+vHHH1MzbbtEv1E6pp49e6bYuJ07d5aLy7//mX/58uVq3ry5Dh8+bGp37949/fTTTypXrpzWrFkj6fEN2f+VnJ5WEyZMUIUKFYz44sWLql27trp3765t27bp0aNHsfqEhoZq06ZNGjx4sJ599lnVr19fq1evdnhuMYut9O3bV2+88Ya+//57rVq1SmvXrjX+xdw3JGnUqFHy9fU14hs3bqh69eoaP368bty4YWp78OBBtWnTRiNHjjQt79+/v2l+UkK+fPnk5uamM2fOqGLFipo3b57u3btnvB4ZGalVq1apcuXK2rx5s7HcYrFoypQpslgsKZqfLTJnzqxFixbJ3d3dWLZs2TKVKlVK48eP1z///BNnv3Pnzumnn35S586dlTdvXr311ls6d+5caqVtl2bNmilv3rxxvtaqVSvlyJEjRcb18vJSmzZtjDgiIkJNmjTRlClTTPuJJB0/flyvvvqqWrZsqfv37ytXrlzKnj27zWO98soratu2rRFbrVa99957qlevnlavXh3rWhsZGalDhw7pk08+kZ+fn7p37x7vulPrOh7zvNGqVSu9//77WrRokVavXm06b8QsZBazsNCkSZPUtWtXnT171rT8zp07mj59usqWLau9e/dKStlrz8SJE5UuXToj3r17typUqKBff/3VdI5++PChlixZogoVKujAgQOSZDoHJqZIkSKxikJNmTJFZcqU0dSpU3X58uVYfaxWq06ePKm5c+eqdevWyp8/vz788ENdu3bNzneZOrp27Wqay+i6detmOofZgm0DAAAAAAAAAAAAAAAAALZxc3YCAAAAAAAAAICnl4uLixYsWKAXXnhBQUFBkh7fbLt06VItXbpUnp6eKlSokLJmzap79+7p5s2bOn/+vCIiIpybuB06d+6sAQMGKDQ01LS8Zs2aKXqz+7PPPqs+ffro22+/NZatWLFCK1asUIECBZQnTx6FhobqzJkzCg8PN9rUrl1bL7/8snr16vWfyOlplT59ei1btkxNmzY1bpR/9OiRZs2apVmzZsnLy0sFChRQ5syZFR4erhs3big4OFhWqzXFc2vWrJlKlCiho0ePSnpcoGLixImaOHFirLZdu3bVrFmzTMu8vb21aNEiNWzY0ChCExYWpvfee0+DBg1S4cKFlSlTJl28eFHBwcGx1tm4ceNYRWpSQrFixdSnTx99+OGHunz5sl5++WX16dNHhQsXlru7u86cOROriI4kjRw5UjVq1Ejx/GxVq1YtzZkzR927dzeKpQQHB+u9997Te++9pzx58ihXrlzy9PTUrVu3dOXKlTjfV1rl5uamnj17xrlPpPQ556OPPtLy5ct1+/ZtSdLt27cVGBiot956S8WLF5enp6cuXryo8+fPG31cXV01a9Ys9e3b164iFNOnT1dISIg2bNhgLFu/fr3Wr1+vDBkyqECBAsqSJYvu3Lmjf/75x3TNKlSoULzrTa3reJcuXTRq1ChdvXpVknTz5k199tlncbYdNmyYhg8fbsQNGzZU06ZNtWLFCmPZnDlzNGfOHBUpUkQ5c+bUzZs3dfr0aT148MBo06lTJz3zzDMaMWKEXbnaqlixYpo0aZK6d+9unHtPnjypFi1aKHPmzPL19ZXVatXp06d1584do1/Pnj318OFDnTlzxuaxOnTooAsXLqh///6KjIyUJB07dky9e/dW7969VaBAAeXIkUNubm66efOmLl26ZBozrcuaNavat2+vOXPmmJZbLJYkFQBk2wAAAAAAAAAAAAAAAACAbVwSbwIAAAAAAAAAQNJlzpxZa9as0bvvvis3N3Ot9Pv37+vYsWPavn279u3bp7Nnz8Z5I3uBAgVSK127eXt7q2PHjrGWp0aBlQkTJqhZs2axlp87d047duzQ4cOHTcVf6tatq19++SXWdnjac3pa5c+fX1u3blXnzp1lsVhMr4WFhenIkSPavn27Dhw4oPPnz8dZkKZgwYIOz8vNzU2LFy9W0aJFk7yOypUra+PGjSpSpIhp+cOHD3Xs2DHt2rUrzoI03bp107Jly+Th4ZHkse0xZMgQvfnmm0YcFhamgwcPas+ePbEKt7i4uGjYsGEaMmRIquRmjw4dOmjTpk0qXrx4rNcuXryoffv2aceOHTp69GicBWlcXV2VN2/e1Eg1SXr27ClXV1fTsiJFiqhevXopOm7RokW1aNEiZcyY0bT83r172r9/v3bu3GkqSJMuXTrNnz9fTZo0sXusTJkyafXq1erZs2es80F4eLiOHj2q7du36/Dhw7GKqCUmNa7j2bJl0+LFi5UrVy67cosyf/58+fv7x1p+6tQpbd++XUePHjUVpOnQoYNmzpyZpLHs0bVrV02ePDnWOenWrVvat2+f9u/fbypA0qZNG33zzTdJGuvdd9/VihUrlCdPnlivnTt3Tnv27NHOnTt1/PjxOIueeHp6Jnn+U0NgYGCsZQEBASpWrFiS1se2AQAAAAAAAAAAAAAAAIDEUZQGAAAAAAAAAJDi3NzcNH78eB09elS9e/e26cZaX19f9e7dW6tXr9aZM2dSPslk6NGjhynOkiWL2rVrl+Ljenh46JdfftGECRPk4+MTbztfX19NnDhRa9euVZYsWf5zOT3NvLy8NHfuXO3du1cdO3a0aS6fffZZvfXWW9qyZYs2btyYInmVKlVK+/fv18yZM9W2bVsVL15cmTNnjlUYJCGlS5fW4cOHNXbs2FjFaaJzc3NT/fr19eeff2rmzJlyd3d3xFuw2ZdffqnFixfLz88v3jbVq1fXn3/+qeHDh6deYnaqVKmSDh8+rDlz5qhq1aqJbitPT0/Vq1dP48aN07lz59S7d+9UytR+BQoUUIMGDUzL4irekhIaNmyo/2PvvqOjKN+/j39SCYSQEFrovVfpnVAURZSuoDQRAVGKInxFRcWKooCKBQQp0hQQRREEFRCQllBD7x1CIIUkJCHl+YMn+8sk2ZZssgHfr3M4Z++Zu1yzMzszZGau2b17tx577DGzddzd3dW7d2/t379fTz75ZJbH8vT01Lfffqs9e/aoZ8+e8vb2tli/fPnyGjNmjNatW2e179w4jrdt21ZHjx7VzJkz9dhjj6lixYry8fGRq6v1S9p+fn76559/NGnSJPn6+pqtV7t2bS1ZskRLly7NteRVzz33nPbs2aOHHnrI7LKUK1dOs2fP1vLly7MVV+fOnXX69Gl9/vnnqlevntVtvGDBgnr00Uf19ddf68qVK+rSpUuWx85pLVu2VPXq1Q3TspsAkHUDAAAAAAAAAAAAAAAAAJa5pGT2WkwbNGrUSFevXlVAQICCg4MdHVeeHdsRcjr+Cq+ucXifueXslEftb5R0zPGBAPcSt+rW65iTct5xcQD3Ipc0bwJPOuq8OIC8wK1GNhpfdlgYwL2plOlT1KTSTowDcL5C714yfR5eLZ8TIwGcb9bxeGeHkOelpKTo0KFDOnTokMLCwhQREaF8+fLJ19dXFStWVK1atVSqVCnrHeUR3333nZ599llT+YUXXtDMmTNzNYbExETt3r1bBw4c0I0bN+Tm5qaAgAA1aNBA9evXz9VY8nJM97vk5GTt2bNHx48fV1hYmKKiolSgQAH5+fmpcuXKqlWrlooVK+bsMLPk6NGj2rt3r0JDQxUbG6siRYqodOnSat26tcUkFI6UNolAu3bttGnTJsP8gwcPKigoSFevXpWnp6dKliypFi1aqGLFirkSnyNFRkZqx44dunz5ssLCwnTnzh35+PioePHiqlGjhqpXry4vLy9nh2mTlJQUVaxYUefOnZN0N8HKhQsXLCbOyglXrlzRli1bdPHiRcXGxqpQoUKqUqWKWrZsmSPJueLj47V9+3adO3dO169fV0JCgnx8fFSuXDnVqVNHlStXznLfefk4HhcXp+3bt+vIkSMKDw+Xp6enSpUqpSZNmqhatWpOiSlV6jZw6dIl3b59W8WKFVODBg3UuHHjHEmSdP36de3cuVNXr17VjRs3lJycrEKFCikgIEA1a9ZU1apVcz2RV1bdunVLJUuWVExMjCSpSJEiunTpkvLlc8z/PVk3AAAAAAAAAAAAzpR6P3qKlJL8f5+VYmX6/5+Wkq5uptOT/++zxemZjGN2epq26WNMSVsn3TQlp5mfLt7M6ihFKSlp6rj5ycWvi1LC10iJNzKOn77PtPGmpP2e0k9LTtOHoz4nZa+fTKeZ6Ts5k2VK/v/facHKcmnyuVJ2vijdOmn8rpPTra/kNOvGUCftd5q+XYpxfFv6MFMnJU2f/1f9/+rcXb0paaanDpuuTur85DTTUucnp2mXkmbMZKUZX3IJqCOfF//QrS8eUtKlg1b7TDH0mbH/u4uRWt++uplOz9D2/74Le2NJO/3/fxWGVZf8f1+LYdUlp7hkqO9d+QG1+Ga3/h3RRJEn9qarn7GftJtSxnFdMh3XNF9W+ja3TGn7k4ttbWVm3DSfM/ad+TJYikviHlwAyGvcnR0AAAAAAAAAAOC/x8XFRXXq1FGdOnWcHYpDzJkzx1B+7rnncj0Gd3d3tWjRQi1atMj1sc3JizHd71xdXdW4cWM1btzY2aE4XI0aNVSjRnaSqua8unXrqm7dus4OwyF8fX3VuXNnZ4fhEBs2bDAlpJGkrl275npCGkkqWbKknnjiiVwbL1++fAoMDMyRvvPycdzLy0vt27dX+/btnR1KBrm9DRQrVkxdu3bNtfFy0tKlS00JaSRpwIABDktII7FuAAAAAAAAAAAAAAAAACAzrs4OAAAAAAAAAACAe9mBAwe0fft2U7lZs2aqX7++EyMCAKQ1a9YsQ3n48OFOigRAVqX/HQ8bNsxJkQAAAAAAAAAAAABOlnxbKbEHpOTbzo7k3pFwUylnFkvxN50dyT0l5dY1xf31qVJuXXN2KPeU+JtXdHLBO4q/ccXZoQAA4BDuzg4AAAAAAAAAAIB72ccff2wov/DCC06KBACQ3vHjx/Xzzz+bylWqVFHnzp2dFxAAu/3555/as2ePqdyhQwfVrFnTiREBAAAAAAAAAADAsUr930cXM1XMTf+PyHTxC9TL7TDufRWfdnYEVuXFTd2r4zhnh3BPqjLoTWeHAACAQ7g6OwAAAAAAAAAAAO5VGzdu1JIlS0zl0qVL64knnnBiRACAVElJSRo5cqSSk5NN08aOHSsXl7x4CxeAzMTExGjs2LGGaS+//LJzggEAAAAAAAAAAAAAAACA/xh3ZwcAAAAAAAAAAMC9IDw8XMHBwZKkmzdvatu2bZo9e7ZSUlJMdV5//XXly5fPWSECwH9acHCwwsPDlZiYqDNnzmj27Nnat2+faX65cuU0dOhQ5wUIwKpt27bp9u3bio+P1/Hjx/Xll1/q1KlTpvnNmjXTo48+6sQIAQAAAAAAAAAAAAAAAOC/g6Q0AAAAAAAAAADYYP/+/XrwwQfNzm/WrJmGDx+eixEBANIaN26cNm/ebHb+V199ReIwII97+umnde7cuUzneXh46JtvvsnliAAAAAAAAAAAAAAAAADgv8vV2QEAAAAAAAAAAHCvq1KlilauXClXV/7sDgB5jYuLi6ZMmaJHH33U2aEAyCIPDw999913atCggbNDAQAAAAAAAAAAAAAAAID/DHdnBwAAAAAAAAAAwL2oYMGCql69unr27KnRo0erYMGCzg4JAPD/ubu7q0SJEmrVqpVGjx6tVq1aOTskAHby9PRUqVKl1L59e7300kuqW7eus0MCAAAAAAAAAAAAAAAAgP8UktIAAAAAAAAAAGCDwMBApaSkODsMAE7EPiBv27Rpk7NDAJBNZ8+edXYIAAAAAAAAAAAAAAAAAID/z9XZAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA8g53Zwdwv/n111/1ySefKDo62myd0NDQXIwIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGxHUhoH++STT3Ty5ElnhwEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAWUJSGgeLjo6WJLm6uqp48eKZ1gkNDVVycnJuhgUAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANiEpTQ4pXry4goODM53XqFEjXb16NZcjAgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADrXJ0dAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAg7yApDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAhKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAHJVYGCgXFxczP6bMWOGs0PMEVu3brW43FWqVLHY3t3d3Wxbd3f3XFoK5EWWtis/Pz9nhwcAuAeRlAYAAAAAAAAAAAAAAAAAAAAAAAAAAABOZSnRRtp/rq6uioqKytIY9evXt2mM+zkhCgAAAGAr0t0BAAAAAAAAAAAAAAAAAAAAAAAAAADgnpCSkqIJEybom2++savd1atXdeDAgRyKKncFBgaanVelShXNmTMn94IBcgjbOQAAzkdSGgAAAAAAAAAAAAAAAAAAAAAAAAAAANwzFi1aZHdSmtGjR+dQNLlv8+bNZuft27ePZB24L7CdAwDgfK7ODgAAAAAAAAAAAAAAAAAAAAAAAAAAAACwVUxMjBYuXGhXm19++SWHogEAAADuTySlAQAAAAAAAAAAAAAAAAAAAAAAAAAAwD1l8uTJNtf9+OOPlZCQkIPRAAAAAPcfktIAAAAAAAAAAAAAAAAAAAAAAAAAAADgnnL69GkdO3bMprrTp0/P4WiA3JOYmKiUlJRM/yUmJjo7PAAAcB8hKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAADuOWPGjLFaZ9u2bbp69WouRAMAAADcX0hKAwAAAAAAAAAAAAAAAAAAAAAAAAAAgHvOn3/+qYSEBIt1Xn755VyKBgAAALi/uDs7AAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBeSUlJeuutt/Thhx9mOj8iIkK7d+/O5aik6OhoLVu2TAcPHlRoaKhu3LihAgUKKCAgQFWrVtWAAQNUvHjxXI/LHkFBQQoODtbx48d16dIlRUdHKy4uTp6envL19VXhwoXVoEEDPf744woICMjVuFatWqXTp08rPDxcpUqV0gMPPKDnnntOXl5eDhsnISFBK1eu1O7duxUaGqqwsDC5ubmpWLFiKlGihB566CF17NjRYePlFYcOHdLKlSt1+fJlXb9+XZGRkfLy8lKRIkVUp04dtW/fXo0bN3Z2mA7jrO1869at2rJli44ePaqoqChFR0fLzc1N3t7eCggIUO3atdW0adP76ru2R2xsrL755hsdPHhQV65ckb+/vypXrqy+ffuqdu3aTo0tKipK3333nXbu3KkbN27ozp078vPz0/fff6+CBQva1MfFixe1dOlSnTlzRqGhoYqMjFShQoVUvHhxlS9fXv369VP58uVzeEnyjlWrVmnLli06c+aM7ty5o/Lly6t9+/bq3bu3s0MDIJLSAAAAAAAAAAAAAAAAAAAAAAAAAAAAII/z9/fXzZs3M0yfNWuW2aQ0L7/8slJSUjKdV7hwYYWHhzssvujoaI0aNUqrVq1SZGSkxbqvvPKKChUqpH79+unzzz+Xp6enxfru7u5KSkqyKY7IyEi5uLiYne/r66uIiAjDtNDQUH344YfauHGjTp06pejoaJvGkqThw4crX758at26td599121aNHC5rb2eO211/T1119niF2S5s2bpzFjxqhhw4b6+eefVaZMmSyNkZiYqEmTJmnu3Lm6fv26xboff/yxXF1dVa5cOb3xxht69tlnszRmVljaHtzc3JSYmGhXfytXrtSbb76p48eP29TWzc1N5cuX1xNPPGH2t5cV9/t2vnr1ar366qs6duyYkpOTbWrj4uIiX19f1ahRQ4888oiGDh2qUqVK2T22La5evaoaNWpY3X998sknGjduXI7EcObMGfXo0UMHDhzIdN/93nvvqVixYnr99dc1ZswYSdLQoUM1d+5cs322a9dOmzZtMjvf1u3oyJEjevLJJ3Xw4MFM6964ccNiUppTp05pxIgR2rZtm27fvm22niRNnDhRXl5eat68ub766ivVrFnTYv20/Pz8LK5Dc8fEtOz9bWVl/NjYWD399NNau3at4uPjM9T76quv5OHhoZ49e2rRokVydyctBuAsrs4OAAAAAAAAAAAAAAAAAAAAAAAAAAAAALBk+PDhmU4PDw/X77//num8ZcuWZTq9ZCF1LOYAAQAASURBVMmS8vf3d1hsQ4YMka+vr+bPn281oUOqqKgozZo1S97e3po6darDYsmKFStWaMaMGdq/f79diTpSxcfH66+//lLLli3VoUMHxcbGOiy2sLAwVahQQR9++KHFRAgpKSkKDg5W+fLl9cUXX9g9zocffqgCBQpoypQpVhPSpEpOTtbZs2c1dOhQ+fn5acOGDXaP60wbNmxQkSJF1Lt3bx0+fNjmZDZJSUk6ffq0pkyZksMROpaztvOEhAQ1bNhQ3bp105EjR2xOSCPd3a4jIiK0Y8cOvfXWW3rqqafsjtsWly9fVrVq1Szuv1xcXDR//vwcS0gzZcoUVa5cWfv377eYOOX69esaO3asHnjgASUkJORILOnNnDlTtWvXNpuQxpLY2Fi1bNlSVapU0Z9//mk1IU2quLg4bdq0SbVq1VLTpk2ztM3mVZs2bVLRokX1888/Z5qQJtWdO3f0ww8/yM/PT3v27MnFCAGkRVIaAAAAAAAAAAAAAAAAAAAAAAAAAAAA5GnvvPOO3NzcMp336quvZpg2a9Yssw//OyqpQmhoqCpWrKh58+bZlWgircTERE2YMEFdu3Z1SEzOtnHjRlWsWNEhySKioqJUpUoVnTt3zuY2ycnJGj16tKZNm2Zz/VatWum1117TnTt3shqqIiMj9dBDD2nMmDFZ7iM3DRkyRA899JBu3rzp7FDuSfZs57Vq1dLevXtzIaqsOX/+vKpXr65bt26ZrePm5qbVq1dr0KBBORLDm2++qYkTJ1pMRpPevn37VK1atSzve201a9YsjRo1yq7YUu3cuVPFixfX9u3bsxXD7t27FRAQoG3btmWrn7zgjz/+UMeOHW1OziNJMTExat68uYKCgnIwMgDmkJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAeZq7u7sefPDBTOcdPHhQly9fNkz78MMPM62bL18+hySlSUhIUJ06dXT27Nls9yVJa9asUY8ePRzSl7OFhoaqdevW2e7nvffeU2RkZJbajhs3Tjt37rRar1mzZvr333+zNEZmPv/8c7322msO6y8nPP3005o3b56zw7jn2bKdf/jhhzp16lQuRWS/c+fOqVatWoqOjjZbx8PDQ1u2bMmxxFl//PGH3n333Sy1PXfunBYvXuzgiP5PXFycRo4cmaW2p06dUps2bRQTE+OQWGJiYtS+ffs8vT3Z4rHHHstSIqE7d+4oMDDQIQnPANiHpDQAAAAAAAAAAAAAAAAAAAAAAAAAAADI8z7//HOz88aOHWv6fODAAZ07dy7Tet27d3dILE2bNtX169cd0leqn3/+WVOnTnVon86ye/duBQUFZauPO3fuZKt9ly5dLM4fOnRotmPMzIcffqhVq1Y5vF9HeOedd7RkyRJnh3HfsLadf/3117kYjX3OnDmjWrVqWUyakj9/fu3du1ctWrTIkRgSExPVq1evbPWRk0lK4uPjs5RAJTk5WY0bN872Piy9O3fuqHHjxlmKKa/IzncSExOjRx991IHRALCFu7MDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAKypWrWqqlSpopMnT2aY98svv5g+jxkzxmwfM2bMyHYcK1eu1P79+y3W8fLyUq9evdSjRw9VrlxZN27c0C+//KJ58+YpOjrabLtJkybppZdekrv7/z0C3KpVK4WHh5vKBw8eNNve1dVVtWvXNju/SpUqZue5uLioWLFiql27tmrXrq06deqoQoUK8vPzU8GCBRUbG6tz584pODhYGzZs0J49e5SSkmK2vzFjxmjbtm1m59vK1dVV3bp104ABA1S+fHmFhIRo9uzZVvu+efOmpk2bppdffjnDvNDQUH333XdWx65UqZJGjBih1q1b6/bt21q3bp2+/fZbRUREWGw3fPhw9ejRw2r/uSk6OlrvvvuuTXVr1KihHj16qHXr1ipVqpSuXLmiPXv26I8//tCOHTscnmxDuj+388uXL5tt5+7urh49eqhbt26qXr26PD09deXKFZ07d047duzQwYMHdfToUYv7i6w6ceKE6tevr9u3b5ut4+vrq5CQEJUpU8bh46d68803LSbFSdW6dWsNHz5ctWrV0unTpzVnzhytX7/e4nrJCS4uLipTpoxKlCihpKQkXbt2TaGhoUpMTDTUGzdunNV9hIuLix5++GENHjxYVapU0ZkzZ7Rw4UL9+uuvFpcrIiJC48aN0/Tp0x2xSE5ToEABDRkyRN27d1fBggW1detWzZw5U2fPnrXY7s8//9TFixdzdLsEYOSSksW9baNGjXT16lUFBAQoODjY0XHl2bGtsSW2nI6/wqtrHN5nbjk7JQvZyZKOOT4Q4F7iVj3rbVPOOy4O4F7kUu7/PicddV4cQF7gViMbjc3/gQz4byhl+hQ1qbQT4wCcr9C7l0yfh1fL58RIAOebdTze2SEAAAAAAAAAAAAAAAAAwD3F3d1dSUlJZuenPg67aNEiDRgwINM6H3/8sV544QX5+PgoOTk5w/x69eqZkslUqVJFp06dMjve9OnTNXbs2EznlSxZUlevXjXbtm7dutqzZ48hsUyqxMRE1a1bV0ePmn+WZeTIkfryyy/NzndxcTE7z9fX12oyhPT27Nmjf/75RyNHjpSnp6fN7YKCgtS0aVOzCRS8vb0tJtQIDAzU5s2bLY7h6empffv2qWbNmhnmLV68WP3797fYvmTJkpkmBnn44Yf1xx9/WGw7atQoff7555nOe+CBB7Rv3z6L7b/99lsNHTo0w/StW7eqTZs2ZttVrlw508RLqSz9Vtzc3DIkyEjVp08frVixwmLM+fPn1/r169W6dWuL9d577z299957iouLs1gvO+6H7dzSMuzfv1/16tWzOn5YWJimTZumJUuWqG7duvr111/N1rXlOzty5IgaNmxocd2VKFFCR48elZ+fn9X4sqNw4cJW1+OSJUvUr1+/DNP//vtvPfjgg5nu69Nq166dNm3aZHa+pe8srUceeUQ//vijChYsmGHe4sWL1a1bNxUsWFDJycnKnz+/EhISzPbl4eGhXbt2qUGDBhnmhYSEqGHDhhYTP3l6eur27dtydXXNMM/Pz0+RkZFm29qSWiI7vz1r40tS2bJldfLkyUx/h4MGDdLChQsttu/Ro4d++ukni3UAOE7GPQ0AAAAAAAAAAAAAAAAAAAAAAAAAAACQB/Xv3z/TpADS3UQyEydONJuk4OOPP872+OfOnbOYkCZ//vxmE9JIdxOKbN++3eJD/9YShzhaw4YNNXbsWLsSdUhS48aNVa5cObPzY2Ji7E4ckt7s2bMzTUgjSU8//bR69eplsf2VK1cyjcFaMpxatWqZTUgjSTt37pSXl5fFPiwlFnKGtWvXWpzv5uam/fv3W01II0lvvPGGoqKiHBVarshr23mtWrVsqle0aFF98MEHOnv2rMWENLY4dOiQHnjgAYsJaSpVqqSzZ8/meEKa8+fPW/3e+vbtm2lCGknq0KGDJkyYkAORZTRq1Cj9/vvvZo89Tz/9tGne8uXLLSakkaSvvvoq04Q0klSnTh19/fXXFtsnJCRo+fLl1gPPg1xdXbVr1y6zv8MFCxaoVKlSmc5L9ddff+VEaADMICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAA7hkDBw7MdPqVK1c0a9asTOcVKVJEnTt3zvbYM2fOtDi/Z8+eZhPSpPLz81PJkiXNzg8NDVVsbGyW4suuhIQEffXVV2rXrp3Kli2r/Pnzy83NTS4uLpn+O3funMX+Dh06lOVYfHx8NGjQIIt1vvnmG6v9LFq0yFAOCQmxmJRDksWENJLk6empJ554wmKdI0eOWI0tt5w4cUIxMTEW67zwwguqWrWqzX3am9wlL8mt7dxS4qJ69epl6/dhr7i4ODVs2FDx8fFm69SvX18nTpywmnDJEebNm2e1jrXkLO+//77V/W12lSpVyur+IK0FCxZYnO/j46OhQ4darPPss8/Kx8fHYp3vv//e5pjykhYtWiggIMBiHWvJhqKiohQdHe3IsABYkLN7WTjF2SmPOjuE3OVW3dkRAPcuF/PZOYH/HLcazo4AuIdZzj4L/JcUeveSs0MA8oxZx81frAAAAAAAAAAAAAAAAAAAAMiOjz76SN98842Sk5MzzDOXcOH55593yNibNm2yOH/x4sVavHhxtsdZu3atevXqle1+bHXz5k31799f69atU0pKisP6vXjxYpbbNmrUyGqdokWLyt/fXzdv3jRb56+//tKLL75oKv/4448W+3R3d1fHjh2tjj1y5EgtXLjQ7Pz4+HiFhoaqePHiVvvKaUuWLLFaZ+rUqbkQiXPl9nZev3597dy5M9N5R44cUZ06deTh4SFfX18FBASoSpUqatSokdq3b69WrVo5LD7J/L4xVfv27fX33387dExLduzYYXF+sWLF5OfnZ7GOq6urKleurGPHjjkwMqPx48fbVf/AgQMW5zdp0sSmfpo0aWJxfezbt8+esPKMPn36WK3z/PPPa+zYsRbrrFmzRk8++aSDogJgiauzAwAAAAAAAAAAAAAAAAAAAAAAAAAAAABsVbBgQbsSNri7u+utt95yyNiWkp840unTp3NlHEnauXOnSpYsqbVr1zo0UYckhYaGZrltgwYNbKpXpkwZu2K4cOGCxfpFihSxadxmzZpZrXP8+HGb+spp1pa5cOHC8vT0zKVonMMZ2/ns2bOttr1z547CwsIUEhKin3/+WZMmTVLr1q3l5uamMmXKaMSIEdn6Hdmid+/euZqQRpJu3LhhcX7ZsmVt6qdq1aqOCMesYcOG2VX/1q1bFufbkmxLkho2bGhxfnR0tM0x5SWdOnWyWsfT01NeXl4W65w5c8ZRIQGwgqQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAuKfMmDHD5roPPfSQ3N3dHTKutYQDjmItiYijhIaGqnXr1kpISMiR/pOSkrLctnjx4jbVK1SokMX5ERERhvK1a9cs1vf29rZpXElyc3OzOP/UqVM295WTrly5YnG+te/wXues7bxevXr68MMPs9RncnKyLl26pFmzZqlEiRLq2rVrjsUfEBCQI/1aYm1f6uPjY1M/hQsXdkQ4mcqXL58KFChgV5u4uDiL80uUKGFTP9b2f9bGyavKly9vUz1rSbKs7dMAOI5jzqCRp1R4dY2zQ8iys1Metb9R0lHHBwLcS9xqZKPxZYeFAdybSv3fx+STzgsDyAtcq2S97e31josDuBflf8j08fqYok4MBHC+Yp+FmT4Pr5bPiZEAzjfreLyzQwAAAAAAAAAAAAAAAACA+1rDhg1VpkwZXbx40Wrdzz//PBcicqzY2NhcGadbt25KTEzMlbEAZ3Hmdv7qq6+qefPm6t+/vy5dupTlftasWaOaNWvmSKKjmTNnyt3dXdOnT3d43+Z4eHhYnH/nzh2b+snJ5CzWEqPcixITEy0mabt582YuRgPgXuDq7AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAe02YMMFqnapVq6py5coOG9PHx8dhfTlbcnKydu3aZbGOm5ubevfurTVr1uj69etKSUkx/HPkd5teaGioTfUiIyMtzvfz8zOUS5QoYbF+TEyMTeNKUlJSksX5Ofn92KNkyZIW50dFReVSJLkvL2zngYGBunjxog4ePKiBAweqQoUKVpOyZOb06dMaP358tmIxZ8aMGTnWd2YKFy5scb6tyVGuXLniiHAy5epqfyoGLy8vi/OvXbtmUz/W9n/WxjHHWsKznEh6lNa5c+dsqhcfb/kFodb2aQAch6Q0AAAAAAAAAAAAAAAAAAAAAAAAAAAAuOeMGjXK6oP5kydPduiY/v7+Fue/++67GRJaZOXfnDlzHBp3ZlasWKHk5GSz811dXXXw4EEtX75cXbp0UdGiRTPUsZbgIDv27dtnU71Lly5ZnF+8eHFDuWzZshbr37hxw6Zxd+/ebbVOtWrVbOorp1lb5vDwcCUkJORSNLkrL23nderU0YIFC3TmzBklJCQoJiZG69at00cffaSnnnpKdevWlbu7u8U+5s6dm6Wxvb29MyRoSu+TTz7RxIkTs9S/vSpUqGBx/oULF2zq5+jRow6IxnGsJS7bs2ePTf3s3bvX4vyCBQtmOt3FxcViO2tJcdauXWs5sGz6888/rdZJSEiwmpSmYsWKjgoJgBUkpQEAAAAAAAAAAAAAAAAAAAAAAAAAAMA9qVevXmbn+fj4qF+/fg4dr02bNhbn//zzzw4dLycdPnzY4vy6deuqZs2aZucnJiZaTXCQHcHBwVbrhIaG6ubNmxbrdOzY0VB+4oknLNZPTEzU33//bXXsL7/80uL8fPnyZUiI4yx9+/a1Wmf8+PG5EEnuy8vbeYECBdS5c2dNmDBBixcv1oEDB3T79m2VK1fObJvw8HAlJibaPZa7u7uOHDliNplJqilTpuj111+3u397Wdp3S1JMTIx27txpsc6hQ4cUFhbmyLCyrV69ehbn25LMypZ6DRo0yHR6vnz5LLaz9nv44YcfLM7PruXLl1ut89VXX1mt8+ijjzoiHAA2ICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAA7kkzZsyQi4tLpvMGDx7s8PHGjBljcX5wcLDNSQfSu3nzpgYNGqTVq1dnqb0kxcXF2Vz3+vXrFufHx8dbnD9ixAglJyfbPJ69bt26pQULFlisM3LkSKv99O/f31CuU6eOvLy8LLaxtp4TEhKsJm+wlOgkt1WvXl3e3t4W63z11Vc6c+aMzX3as6052r2ynUdHR9vdxt3dXYMGDbJYx571lFZAQIBCQkKUP39+i/U++OADvf3221kaw1Zdu3aVq6vlVAfWkoo9/vjjjgzJIaytu6ioKM2bN89inYULFyoqKspinQEDBmQ63drvfM2aNWbnBQUFWU1ak13bt2/X1atXLdaZOnWqxfmFChWymlwJgOOQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAD3pKJFi6pv374qW7as4V+lSpU0ZcoUh49Xvnx5FS9e3GKdNm3aaNOmTTb1l5ycrDlz5qhx48YqWrSoFi5cqNOnT1tsYymRQ3x8vBYtWmTT2CVKlLA4//jx4zp27Fim8z777DPNnTvXpnGyY9iwYTpy5Eim85YuXaqVK1dabF+yZEn5+fllmN6uXTuL7UJCQjRu3Diz81u2bGk1Mcrzzz9vcX5ue/jhhy3OT0xMVJ06dbR9+3arfc2cOVOFCxd2VGiZuh+280aNGql48eIaN26cLl++bHO7devWWZyfnWRQ5cuXV3BwsDw9PS3Wmzx5st57770sj2ONq6urWrZsabHOmTNn1KBBA928edMw/ebNm6pXr57VfaUz9OnTx+p3O3z4cB04cCDTeYcOHdJzzz1nsb2np6f69OmT6bxKlSpZbLt48eJME96EhoaqQ4cOFts6QnJyspo2baqEhIRM5w8ePNjqb6Vjx445ERoAM9ydHQAAAAAAAAAAAAAAAAAAAAAAAAAAAACQVUuWLMnV8T7//HP17dvX7Pz4+Hi1b99eVapU0eOPP6727durTJkyio+P14ULF3Ty5Elt27ZNBw8e1IULF+xOMFGoUCFFRESYnT9gwACNHTtWxYoVk4eHh2Hen3/+aUqq06FDB7311ltm+0lOTla9evU0aNAgde3aVaVKldL27dv1zTff6PDhw3bFnFUJCQmqU6eOunfvrkGDBqlcuXIKCQnRrFmztHXrVqvtX3nllUynL1y4UAEBAUpJSTHbdtq0aVq9erVGjBih1q1bKz4+XmvXrtWsWbMUHh5ucdxixYpp2LBhVuPLTfPnz9cvv/yixMREs3ViY2PVsmVL1axZUz169FDbtm1VsmRJXblyRQcOHNAff/yhf//9V7dv387xeO+H7TwpKUnXr1/XtGnTNG3aNPn4+KhGjRqqXbu2mjZtqrJlyyogIEBeXl66dOmSNm/erMWLF+v8+fMW+61evXq24qpZs6a2bdumFi1aWNweJk2aJHd3d7366qvZGs+cL774Qg888IDFOvv371fRokVVokQJ+fn5KTIyUlevXrX423UmV1dXjRw5UjNmzDBb586dO2rQoIG6dOmiwYMHq0qVKjpz5owWLFigX3/91eoxYeTIkWaTNnXs2FF//vmn2baxsbEqXbq0xo8fry5duuj69etaunSpli5danFbcKQLFy6ocOHCevbZZ9W9e3f5+Phoy5Ytmjlzps6cOWO1/eeff54LUQJI5ZKSxT1uo0aNdPXqVQUEBCg4ONjRceXZsa2xJbacjr/Cq2sc3mduOTvlUfsbJR11fCDAvcStRjYa255ZE7g/lfq/j8knnRcGkBe4Vsl629vrHRcHcC/K/5Dp4/UxRZ0YCOB8xT4LM30eXi2fEyMBnG/W8XhnhwAAAAAAAAAAAAAAAAAA9xR3d3clJSWZne/oBARVqlTRqVOnzM6fPn26xo4da3Z+nTp1dOjQIYfGZOvYrVu31rZt27LU99GjRw0JLby8vBQfn3P3vFlalsDAQG3evDnHxvb399eNGzfMzh86dKjmzp2bI2P/9NNP6tGjR6bztm7dqjZt2phtW7lyZZ08af5ZJ0u/FTc3N4uJJd588029++67ZufbKycTg9wP27m1/UxWlChRQlevXs10nouLi9l2vr6+GZL8/PPPP2rfvr3VJCgff/yxxo8fb3estujevbt++eWXHOlbktq1a6dNmzaZnW/vd2aL5ORkFSlSJEttrfHz89ONGzfMJqWJjo5WoUKFcuy3ae07SU0clFM6deqkDRs25Fj/ADLKfG8DAAAAAAAAAAAAAAAAAAAAAAAAAAAAIFNBQUEqWtQ5L7WcPHmyw/oaMmRIltu6urqqWLFiDoslPXd392y1//333y3OnzNnjho3bpytMTIzceJEswlpnO2dd95Rnz59nB2GTf4r27m9HJkcpm3btvrll18sJmaRpAkTJmjatGkOGzetn376KVvf7wMPPODAaBzD1dVVQUFB8vDwcGi/Hh4eCgoKMpuQRpIKFixoMemVNda2hezKznfi7e2tNWvWODAaALYgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAABgBy8vLx06dEjly5fP9bE7duyohg0bOqSvmTNnqlSpUllq+/XXX6tQoUIOiSMzr7/+ugoWLJiltp9++qmaNWtmtd727dvVokWLLI2RmdGjR+uDDz5wWH854ccff9SAAQOcHYZV/5Xt3B7NmzfXuHHjHNpn165dtXjxYqvJSMaNG6cvvvjCoWNLdxO4HD9+PEuJab744gvVrVvXYp18+fJlNbRsqVy5sjZv3ixvb2+H9FegQAFt3LhRlStXtlr3l19+ydJyu7i4aPHixVkJz2a//vqrxaQ65nh4eGjTpk3y9PTMgagAWEJSGgAAAAAAAAAAAAAAAAAAAAAAAAAAAMBOxYsX19mzZzVy5Ei5ublluz9PT0916NBBPXr0sFp327ZtatSoUbbHdHV11cGDB+1K2OHi4qIvv/xSw4YNy/b4lvj5+enEiRN2xebq6qrPP/9cL7/8sk313d3d9e+//+qDDz6Qh4dHVkOVr6+v1q1bp88++yzLfeSmhQsX6vfff5efn5+zQ7HoXt/Ovby8stw2vd69e2v79u0O6y+tfv366ZtvvrFab/To0frqq68cPr6fn5/Onj2rBx980Kb6hQoV0p9//qkXX3xRoaGhFusGBAQ4IsQsadGihUJDQ9W8efNs9dOkSRNdu3ZNrVq1sqm+n5+fNm7caFcCl/z58+vPP/9Uv379shqmTTp37qwNGzbY9dvw9vbWjh071Lhx4xyMDIA5JKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAsujLL79UdHS0XnrpJRUrVsyutkWKFNHDDz+sZcuWKT4+Xn/99ZfKly9vtZ2Xl5eCgoK0ceNGtWvXTv7+/llOjOPv769Lly5p+PDhVpMY1KlTR4cOHdLIkSOzNJa9AgICdOnSJb388ssqVKiQ2XouLi5q1KiRzp07p1GjRtk9zsSJExUbG6sJEybYvA5dXV1Vvnx5zZkzRxEREercubPd4zrTI488ovDwcP3www+qUaOG3N3dbWrn6uqqihUrasKECTkc4b2/nYeEhOiXX35Rjx49VLp0aZu/41Te3t7q1KmTTp48qeXLl2crFmuGDRumTz75xGq9F154QbNmzXL4+AUKFND69esVHBysrl27ytfX17SuXVxclD9/ftWtW1ezZs1SZGSkOnbsKEk6ePCgxX7LlSvn8FjtUaBAAW3fvl3Hjx9Xx44dlT9/fpvaeXl5qV27djp8+LB27dqlggUL2jVuixYtdObMGbVu3VouLi4Wx+nTp49u3rypDh062DVGVnXo0EHXrl1Tly5dLP4WPTw81KdPH0VERKhhw4a5EhuAjFxSUlJSstKwUaNGunr1qgICAhQcHOzouPLs2NbYEltOx1/h1TUO7zO3nJ3yqP2Nko46PhDgXuJWIxuNLzssDODelCazbfJJ54UB5AWuVbLe9vZ6x8UB3IvyP2T6eH1MUScGAjhfsc/CTJ+HV8vnxEgA55t1PN7ZIQAAAAAAAAAAAAAAAAAAnCQ5OVm//vqrtm7dqtDQUIWFhSkhIUE+Pj7y8/NT1apV1aFDBzVr1szZoWbq999/188//6zLly8rISFB/v7+atq0qYYMGSI/Pz+nxrZ792799NNPOnXqlKKiolSyZEk1aNBAw4cPl5eXl8PGSUhI0MqVK7Vr1y6Fhobqxo0bcnV1VbFixRQQEKBOnTrpwQcfdNh4ecWBAwe0atUqXbp0SWFhYYqKilK+fPlUuHBh1axZU4GBgWrVqpWzw3QIZ23n58+f15YtWxQSEqJz584pKipKt2/flqenpwoWLKhixYqpSZMm6tixo9MTquR1hw4dUp06dSzW2b17txo3bpxLEdnm4sWLWrp0qU6fPq3Q0FBFRUXJx8dHxYsXV4UKFdSvXz+bkpPZKi4uTgsWLNC///6r0NBQ5cuXTxUrVtRDDz2kRx55xGHjSJKfn58iIyPNzs8stcWKFSu0ZcsWnTt3TomJiSpXrpwCAwP1xBNPODQ2AFlDUhoHIylN9pCUBsgCktIA2UBSGsCEpDRA1pGUBjAhKQ3wf0hKAwAAAAAAAAAAAAAAAAAAAFi3YcMGtWnTxq7EUnFxcSpfvrxCQ0PN1smfP79iY2MdESJslJWkNADyNldnBwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAP57nn/+eXl7e6tx48aaNm2axUQyCQkJev/99+Xv728xIY0kPfTQQxbnAwCsc3d2AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA4L8pOTlZwcHBCg4O1rhx4+Tl5aXChQurYMGC8vDw0O3btxUeHq7IyEilpKRY7c/Dw0OLFi3KhcgB4P5GUhoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAJAnxMXF6cqVK1lu/+mnn6pgwYIOjAgA/ptcnR0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABAdr399tsaNWqUs8MAgPuCu7MDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAyKoCBQrou+++05NPPunsUADgvuHq7AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMB/T+fOnZUvX74sty9YsKAGDx6s8PBwEtIAgIORlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAOS6L7/8UnFxcdq4caOGDh2q2rVrq3DhwnJ3d5eLi4upnouLi9zd3VWwYEHVrFlTTz/9tNatW6dbt25p3rx58vT0dOJSAMD9yd3ZAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgP+uwMBABQYGOjsMZENERISzQwDgYK7ODgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAkHeQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYEJSGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACACUlpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAmJKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAJiQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYEJSGgAAAAAAAAD4jzl79qxcXFxM/wYPHuzskJBG2nUTGBjo7HAk5c2YcsvgwYMNy3/27Flnh4T/ALa7vOvtt982rJtNmzY5OyQgxyxevNi0rZctW1ZxcXFOjee/fD6SFeyv8q758+cb1s38+fOdHRJyUW7ty/LiPoBz3Lvy4rp5+OGHTfG8/PLLzg4HAAAAAAAAAAAAAJCHkJQGAAAAAAAAAAAAAACYREdHa8KECaby5MmT5eXl5cSIAABATvnggw/k4uIiSZo5c6aOHTvm5IgAAAAAAAAAAAAAAHkFSWkAAAAAAAAAII+pUKGC4a3Zmf1zc3OTv7+/KleurK5du2ry5Mk6fPiws0MHAADAfeDDDz/U5cuXJUlVq1bVoEGDnBwRAADIKQ0bNlTPnj0lSXfu3NG4ceOcHBEAAAAAAAAAAAAAIK8gKQ0AAAAAAAAA3IOSk5MVHh6u06dPa82aNXr77bdVu3ZtderUSSdOnHB2eAD+w9Im1qpQoYKzwwFwD0ibeC8wMNDZ4WTq7NmzhjgHDx7s7JByzPXr1/XZZ5+Zyq+88orc3NycGBEAWMc5KJA9//vf/0yf16xZox07djgxGgAAAAAAAAAAAABAXkFSGgAAAAAAAAC4j/z1119q0KCB1q5d6+xQAAAAcA+aMmWKYmJiJEklSpTQoEGDnBwRAADIaU2aNDEkB3zzzTedFwwAAAAAAAAAAAAAIM9wd3YAAAAAAAAAAADLPvnkE9WvX98wLSkpSTdu3NCePXu0bNkyXbp0yTQvNjZWffr00b///qt69erldrjAfWX+/PmaP3++s8MAACBXREREaPbs2abyM888o3z58jkxov+TkpLi7BAAALivjRgxQps2bZIkbdiwQfv27VODBg2cGhMAAAAAAAAAAAAAwLlISgMAAAAAAAAAeVyjRo0Mb6tO66mnntIHH3ygCRMm6LPPPjNNj4mJ0bhx47Rhw4ZcihIAAAD3urlz5yo6OlqS5OLioqFDhzo5IgAAkFt69OihIkWK6MaNG5Kkzz77TPPmzXNyVAAAAAAAAAAAAAAAZyIpDQAAAAAAAADc4zw9PTVjxgyFhYVp8eLFpul//vmnQkJCVKdOHSdGBwAAgHvF119/bfrcokULVa5c2YnRAACA3OTp6aknn3xSX331lSRp6dKlmj59uvz8/JwbGAAAAAAAAO5rwefCdeFmrLPDyKCsfwE1Kl/Y2WEAAAAATkdSGgAAAAAAAAC4T0yZMkVLly5VcnKyadr69etJSgMAAACrtm/frlOnTpnKPXv2dGI0AADAGXr06GFKShMfH68VK1Zo6NChTo4KAAAAAAAA96vgc+Hq9fW/zg7DrJXPtyQxDQAAAP7zSEoDAAAAAAAAAPeJMmXKqH79+tq7d69p2sGDB7PU17Fjx7Rz505dvnxZbm5uKl68uJo2baqaNWs6JNbY2Fht27ZNly5dUmhoqGmMWrVqqWHDhnJxcXHIOKlyennyiri4OB0+fFhHjhzR9evXFRMTIx8fHxUpUkR169ZVnTp15Orq+p+IKa+u8/379ysoKEihoaHKly+fAgIC1LJlS1WoUMEh/UdEROjff//VlStXdP36dXl5ealYsWJ64IEHVKtWLYeM4SzHjh3T3r17FRoaqpiYGBUtWlSlSpVS69at5evr6+zwJEkhISEKCgrS1atX5eHhodKlS6t58+YOW7+pbt++rX/++UdHjx5VdHS0ChcurAoVKqhdu3by9vZ2yBjHjh3T/v37df36dUVGRsrf39/0ffv7+ztkjFS5sTx5xcWLF3Xo0CGdOXNGkZGRkiR/f3+VLl1aLVq0UOHCjruhMSkpSUFBQTp16pTCwsJ069YtFSxYUKVLl1atWrVUs2bNLB9vc/s4bq/o6GgdOnRIR48e1Y0bN3T79m35+vqqaNGiatiwoapVq+bU+NLLK/vuJUuWGMrdunXLtbFzWl48RzLn5MmT2rlzpy5duiRJKl26tBo2bOjwc5g7d+7o33//VUhIiCIiIlSoUCGVLVtW7dq1c9i+6Pz58woKCtK1a9cUHh4uX19fBQQEqFWrVgoICHDIGKlyY3nyitDQUIWEhOjUqVOKiIhQYmKi/P39FRAQoGbNmjn0u01JSdGBAwd09OhR0zlBgQIFFBAQoJo1a6pu3bpyc3PLUt8JCQnasWOHzp49q+vXrys5OVnFihVT1apV1bx58yz36yh5db8RERGhTZs26eLFi4qKipK/v7/q1Kmj5s2by93d8beBcU5ou/tl3QQGBsrX19d0rrpkyRKS0gAAAAAAACDHXLgZ6+wQLLpwM5akNAAAAPjPIykNAAAAAAAAANxHKlWqZEhKExYWZlf79evX6/XXX1dQUFCm82vWrKmPPvpIjz32WJbi27Ztm95//339/fffio+Pz7RO8eLFNXz4cI0fP14+Pj5W+zx79qwqVqxoKg8aNEjz58+X5JjluXjxoipUqKCkpCRJUrVq1XTs2DGrcaW3evVqw8PdTzzxhH744Qe7+8ksvmXLlmnNmjXavn272e9VkgoXLqxnnnlG48aNU6lSpbI9trNiyul1ntbgwYO1YMECU/nMmTNmE4xs2rRJ7du3N5Xfeustvf3225KkpUuXavLkyWa3nWbNmumTTz5R69atbYorvV9//VWffPKJ/v33XyUmJmZap1y5cho7dqxGjhypfPnyZWkcc9J/T6nOnTtnMTlF2nWXmfj4eH3xxRf65ptvdOrUqUzruLu7q127dnr77bez/P3ZKu2ytGvXTps2bZIk/fTTT5o0aZIOHz6caZuWLVtq6tSpatGiRbbGj4yM1OTJkzV79mzFxMRkmO/p6alnn31W77zzjooWLWp3/9HR0fr000+1YMECnTlzJtM6bm5uatOmjd555x21adPGpn7N/Y4ctTwjRozQrFmzTOXFixfrqaeesim2tBo2bGg4hh4+fDjbSSASExP1559/asWKFfrzzz917tw5s3VdXFzUvHlzTZgwQd26dctyYpdDhw7pgw8+0Jo1a0wPE2cmICBA3bp10/PPP6/69evb1Lcjj+OBgYHavHlzhumbN2+2uOxp961pHT9+XEuXLtW6desUFBRkdl8o3V32559/XqNGjbKarKJChQqZrrcFCxZkut9LNW/ePA0ePNhi387ed6e3Zs0a0+eyZcuqSpUqFutfvXpVZcqUMZ0jlStXTmfPnrV72928ebMCAwNN5UcffVS//fZbhnrm9sHm5MVzJMm4TZUvX15nz56VdPc8YuLEidqxY0em7erXr6/3339fjz76aLbGj4+P18cff6wZM2bo5s2bGea7ubmpd+/e+uijj1S+fHm7+09ISNDXX3+t2bNnZ3pclO6uy0aNGmnSpEl6/PHHber37bff1uTJk03ljRs3KjAw0GHLM2XKFE2cONFUfv/99/Xaa6/ZFFtaPXv21KpVq0zl33//XY888ojd/aSVkpKirVu36scff9SGDRus/l+kbt26GjdunJ5++uksJ8I4f/68PvjgA61atUqhoaFm6xUuXFhdunTR8OHDbT4vCAkJ0TvvvKO1a9cqOjo60zp+fn7q37+/Jk2apOLFi5vty9HnoHl1v5Ea2yuvvKJVq1YpISEhw/xixYpp3LhxGjduXLYToOTUOaE59/o54f22btzd3dW6dWvTecGWLVsUFRWlQoUKZSt2AAAAAAAAAAAAAMC9KW+86gsAAAAAAAAA4BAeHh6GsqUHstObMGGCOnfubDaZhyQdOXJEjz/+uN5991274oqJidETTzyh1q1ba+3atRYf7gsNDdW7776ratWqaffu3XaNk5ajlqdMmTKGh3+PHz+uf/75x+54vv32W0N52LBhdveR3oEDB1SuXDmNHz9emzZtsvi9SlJ4eLimTZumWrVqae3atdkeP6/FlJPbcFYlJCSof//+euqppyw+QLxz504FBgZaTNCSmdDQULVv316PP/64/vnnH4u/+fPnz+vll19WvXr1zCZ4yUsOHTqkWrVqafz48RbjTUxM1F9//aU2bdpoyJAhunPnTq7FmJKSolGjRqlXr15mH7xPSUnRtm3b1Lp162xtd4cPH1b9+vU1ffr0TB/Wlf4vEUDz5s1NSQ5s9dtvv6ly5cp6++23zT7gKklJSUnatGmT2rZtq+HDh9t1nEnLkcszfPhwQ3nOnDl2xxMcHGx4+Lh169bZTkgjSX379tUjjzyiuXPnWkxII93dVrZv364ePXqod+/eZr8Xc+7cuaORI0eqXr16WrJkicWENNLdhCKzZs0yJEwzxxnHcXv89ttvql69ut5++23t2LHD6nZ59epVvfXWW6pbt26uxZhWXtx3Hz9+3PDbb9u2rdU2AQEB6ty5s6l8/vx5q4liMpM+ocSgQYPs7iO9vHiOZMnUqVPVoUMHswlpJGn//v3q2rWrRowYoZSUlCyNc+nSJTVv3lxvvvlmpglcpLv7+R9++EGNGzc27BdtsXPnTtWoUUNjx441e1yU7u7vgoKC1K1bNz3++ON27+9SOXJ5hgwZYvi/1Ny5c+3+nq9evapff/3VVC5XrpzhN5JV48ePV9u2bTVz5kybkmMePHhQgwcPVvv27S0mlMlMSkqKJk+erKpVq2rWrFlW24eHh2vx4sU27TMSExM1atQo1a9fX8uXLzebkEaSIiIiNHPmTFWpUsWQMCsn5eX9xu+//67atWvrhx9+yDTpiSRdv35dr776qtq3b2/1HMASzgntOye8X9dNu3btTJ9TkywCAAAAAAAAAAAAAP6bSEoDAAAAAAAAAPeRy5cvG8pFihSxqd2rr76qqVOnmso+Pj6qXbu2GjdurGLFimWo/+abb+qnn36yqe/Q0FC1a9dOy5cvzzCvTJkyatSokRo0aKDChQsb5l29elWBgYHaunWrTeOk5ejlGTFihKGcPsGMNZcuXTI8qFipUiV16NDBrj4yk5CQkOFhWU9PT1WuXFkPPPCAmjZtqqpVq2Z4G3tkZKS6du2qjRs3ZjuGvBJTTm7D2TFo0CAtXrzYVC5cuLDq1aunhg0bys/Pz1A3KSlJQ4cOtTlBwokTJ9S8efMMCQBcXFxUoUIFNW7cWHXr1lXBggUN848fP64WLVro+PHjWVqm3BAUFKQ2bdro9OnThukeHh6qWrWqGjdurFKlSmVoN2/ePD3++ONmHwh1tPfee08zZ840lQsUKKDatWurQYMGGdZvcnKy3nzzTU2ZMsXucc6ePauOHTsakpqUL19eTZo0Uc2aNeXm5maof+rUKfXs2dPmB1Bnz56t7t27Z3jwvECBAqpZs6aaNm2qKlWqyNXVNUO73r172/3QvqOXJ3XfkmrTpk06efKkXTGl368/99xzdrU3Jy4uLsO0YsWKqVatWmrWrJnq16+vokWLZqjz008/qVu3bkpOTrZpnMjISHXq1Elff/11hjb58+dXtWrV1KxZM9WsWTPDPsEaZxzH7ZXZ95y63I0aNVLjxo1VsWLFDNvwpUuXFBgYqCNHjuR4jKny6r57y5YthnLjxo1tapc+gczChQvtGjc2NlYrVqwwlQsXLqzHH3/crj4ykxfPkcz5/vvvNWHCBFO8+fLlU/Xq1dWwYcNMz2FmzZqV4dzUFhEREerUqZP27dtnmla6dGk1btxYderUUb58+Qz1w8LC9PjjjysqKsqm/n/99Ve1b98+Q6IET09PVa9eXU2bNlWNGjUyfOe//vqrOnTokOnvODeXp3jx4urRo4epfPr0af399992xTR//nzDserZZ5/NsN/Jisy+m8KFC6tmzZpq1qyZGjRooJIlS2aos3XrVnXo0EG3b9+2aZw7d+7oiSee0Ntvv53hXCr199O0aVPVrl07w3mONbGxseratatmzpyZ4TgVEBCgBg0aqGHDhipevLhh3q1bt9StW7dMj0GOllf3G3///bd69eqVYdtNe96Z9hi8detW9enTJ0vJqzgntO+c8H5eN40aNTKUs5KYFwAAAAAAAAAAAABwfyApDQAAAAAAAADcJ2JjYzMks6hYsaLVdv/8848++ugjSVKzZs20fv163bx5UyEhIdq9e7euXbumjRs3qkaNGoZ2o0ePtprwIDk5WX379lVwcLBpWrFixTR16lRduXJFFy5cUFBQkPbu3auwsDDTg5Npl6lfv366ceOG1eXIyeXp3LmzKlSoYCqvWLFC4eHhNsf03XffKSkpyVQeOnSoXFxcbG5vTbt27TR9+nSFhIQoJiZGJ0+e1J49e7Rz504dP35ct27d0s8//2x4QC85OVn9+/dXdHS0w+JwVkw5uQ1nx/fff69ly5ZJkh5++GFt375dN27c0P79+xUcHKywsDCtWrXKkFwlKSlJL774otW+Y2Nj1a1bN8OD3xUrVtSsWbN048YNnTlzRrt379aBAwcUHh6utWvXqmHDhqa6169f15NPPqn4+HiHLOuECRO0YcMGbdiwQSVKlDBNL1GihGl6Zv8mTJiQoa9bt26pT58+ht9YgQIF9PHHH+vq1as6fvy4du/erUuXLmn//v3q3r27of26dev05ptvOmS5LDl58qTeeecdSXeXc+HChbpx44ZCQkJM+7R169apZs2ahnavv/66tm/fbtdYzz77rK5evar8+fPrzTff1KVLl3T27Fnt2rVLhw8f1vXr1/W///3PsF/Zu3evTQm0/vrrLz3//POGfdRjjz2mTZs2KTIyUocPH9bOnTt14sQJXb9+XR999JF8fHxMdX/55Rd9/PHHTl+e4cOHmz6npKRozpw5NscTExOjpUuXmsp+fn7q06ePXctkSdGiRTVy5EitWbNG169fV2hoqA4dOqQdO3Zo3759un79uk6cOKGJEyfKy8vL1O6vv/7SZ599ZtMYzzzzTIaHhdu1a6d169YpPDxcx44d044dO3T48GFFRkYqJCRE77//fobtM72cPI5/+umnpn1BWvXq1bO43xg4cGCmsbq5uenRRx/VrFmzdOLECUVHR+vYsWMKCgrS7t27dfr0aUVERGjRokWqVq2aIcannnrK7MPaixcv1oYNG7Ro0SLD9IceeshinJ07d87QV17bd6eVdh1LUp06dWxq9/jjjxuSU6xcuVKxsbE2j7tq1SrdunXLVH7yySczJBPJjrx4jpRWZGSkRo0aJeluQr3PPvtMoaGhOnr0qIKDgxUaGqpt27apRYsWhnazZ8/WDz/8YNdY48aN09GjR+Xu7q4XX3xRJ0+e1MWLF7V7924dPHhQN27c0CeffCIPDw9Tm4sXL+r999+32vehQ4f05JNPGpKftGnTRr/99psiIyN19OhR7dy5U0eOHNHNmzc1e/Zsw/nCrl279NJLLzl9edIeSyT7ElGmpKRo7ty5prKbm5uGDBlixxJZ5uPjo2eeeUYrVqzQ5cuXdfPmTR0+fFg7duzQ3r17dfnyZV28eFFTpkwx/CYPHTqkV1991aYxJkyYYEgSJUkNGjTQypUrdfPmTZ08eVI7d+5USEiIwsPDdeLECc2YMUNNmjSx2vfzzz+vP/74w1QuWLCgJk2apNOnT+vKlSvau3evgoODde3aNe3bt0+9e/c21U1KStKzzz6baXIRR56DpspL+42IiAg9/fTThsRExYsX14IFCxQWFmY479ywYYPq1q0rSdqwYYPdScI4J7TvnPB+Xzep8aYKCgqyqz0AAAAAAAAAWOPi4mL6FxgY6OxwkE2p98Kl/suJl9dUqVLF1H/6lwgA1vj5+Zm2H3tfwJEXzJgxw/AbmzFjhtm6gYGBhroA4AgkpQEAAAAAAACA+8QXX3xheBBUkuHBcHNSH4wePHiwtm3bpgcffNBw4Tb14v8///xjSJ5x6dIlrVmzxmLfU6dONbwxvlmzZgoJCdErr7yigIAAQ11XV1e1atVKGzZsMCTluHjxoinxgy1yYnlcXV0Nb0iPi4vL8HC6OSkpKfruu+9MZXd3dz3zzDM2L48l5cqVU0hIiDZt2qSxY8eqdu3amV509/LyUrdu3bR9+3YNHTrUNP3y5cv6/vvvHRKLM2PKyW04O06fPi1JmjRpktauXavmzZsbLvS6ubmpe/fu2rhxowoUKGCavmvXLu3fv99i3+PGjdORI0dM5ccee0wHDhzQsGHDVLhwYUNdd3d3Pfzww/r333/Vo0cP0/R9+/bpq6++ytYypqpVq5Y6deqkTp06GZJqeHl5maZn9q9WrVoZ+nrttdd09uxZU9nX11dbt27V+PHj5e/vb6hbr149rVq1Sq+//rph+tSpU7Vnzx6HLJs5ly5dUmJiosqXL6/g4GANGDDAsOxubm7q3LmzgoKC1KpVK9P05ORkDRs2zGwCjMycPn1a/v7+2rx5syZPnmzYjiWpcOHCmjJlit59913D9NmzZ1vsNyIiQv3791dycrKku/u6uXPnavXq1WrXrl2G366/v78mTJigHTt2qFixYqbpb775pq5everU5enbt698fX1N5fnz5+vOnTs2xfPjjz8qKirKVH766aeVP39+m9pa8/rrr+vChQv68ssv1aVLFxUtWjTTelWqVNEHH3yg7du3G7bzTz/91GryrFmzZmnVqlWmsouLi6ZOnapNmzapc+fOGRJ8uLq6qnbt2nrttdd06NAhzZs3z2zfOXkcb9SokWlfkFbhwoUt7jcqVaqUaV+nTp3Sb7/9pmHDhqlKlSpydc14KdzHx0dPP/209u/fry5dupim79u3T+vWrcv0O2jVqpU6depk+B1LUsmSJS3GWbJkyQx95bV9d1oHDhwwlKtXr25TOy8vLz355JOm8q1bt/TTTz/ZPO6CBQsM5UGDBtnc1pK8eI6UmYiICEVGRqpw4cL6999/NXr0aBUqVMhQp2XLltqyZYt69eplmD5mzBi7EmCcPn1a+fLl0y+//KIvvvhClStXNsz39vbWuHHjMuwTrO1PExMT1bdvX8P/QyZPnqzNmzfr0UcfNRwbpbu/w+eee07BwcGqWrWqafo333yjvXv3OnV5OnToYEhatWrVKoWFhdkUz6ZNmwxJUx5++GGVKVPG1sWx6JlnntHFixf13XffqVevXpnuXySpdOnS+t///qd9+/YZkpN+++23unnzpsUx/vjjjww3Lo4ePVrBwcHq2bOnvL29M7SpUqWKxowZo127dunPP/802/cPP/xgSMJRuXJl7du3T++8806mSVTr16+v5cuXG5Jo3Lp1S+PGjctQ15HnoHlxv/Haa68ZzrHKly+vPXv2aODAgYZzFVdXV3Xq1Em7d+82/Q0g7fm0NZwT3mXPOeH9vm6KFStmOCe19v9TAAAAAAAAAI5z5MgRQzIBFxcXlS9f3tlhAQAA4D+MpDQAAAAAAAAAcB/49ddfNWnSJMO0ihUrqnXr1ja1b9y4sWbPni03NzezdYoVK5ZhjLVr15qtHxsba3iQsGTJkvr9999VvHhxi7G4urpqxowZat68uWnad999p4iICCtL8X9yYnmGDBkiDw8PU9nS29nT2rBhg+Ghs65du2Z4kD+rihcvrtq1a9tc39XVVV9++aXhoV1LyQjupZhyYp07Qrdu3awmVapWrZpGjRplmGYprgsXLmjOnDmmcr169bR8+XIVLFjQ4jj58uXTokWLVKFCBdO0zz77TElJSRbb5aaIiAhDEidJmjt3rh544AGL7d577z098sgjpnJycrKmT5+eIzGm5eLiouXLl6t06dJm6xQoUEA//fST4eHYkJAQbdiwwa6x5s6dqyZNmlis87///c/wAPy+fft07do1s/W/+eYbw8Op77//voYMGWI1llq1amn+/PmmckJCgmbOnGm1XVqOXp4CBQpowIABpvK1a9f066+/2hRL+v152iRk2dWiRYsMCRksadCggeHYeenSJa1fv95s/cTERH3wwQeGaRMnTtQrr7xi03guLi5q3759pvOceRy3V8WKFe26EdPLy0vff/+94Xfp6ONhenl9352a4E26uw7NJb3ITPpEMmmTT1hy6dIl/fXXX6ZytWrVDNtNduTFcyRLvv32W9WpU8fsfDc3twzbwbVr17RkyRK7xpkyZYohIVNmnn76aTVr1sxUDg0NVXBwsNn6K1asUEhIiKk8fPhwvfnmm1bfuFa6dGmtXLnSkEDq008/tbYIBjmxPMOGDTN9TkhIsHl7zsljSaNGjTIkK7KkfPnyhnhu376tZcuWWWwzefJkQ/mpp57SZ599lmmCr8x07Ngx0+kpKSl6++23TeUCBQrojz/+yJBEKDPjx49Xnz59TOVff/1Vx48ftymerMhr+42IiAjD+ZYt55358uXTypUrDclIbME54V22/o7/K+smbZKg6Oho3bhxw672AAAAAAAAALIm/Ut5JOn8+fM6f/68E6IBANtNnDhRgYGBCgwMNLx8BwBw7yMpDQAAAAAAAADcg5KSkhQWFqY//vhDTz31lLp165bh7d/vvfeeIYmKJe+8845NdZ944glDec+ePWbrLly4UDdv3jSV3377bcObti1xc3PTxIkTTeXo6Gj98ccfNrWVcmZ5AgIC1K1bN1P54MGD2rlzp9UxcvIB1azw9PQ0PNy5d+9e3b5924kROSamnFjnjpA+WYQ5Tz75pKFsKa4vv/xSiYmJpvLUqVOVL18+m8YpUKCAXnrpJVP53LlzCgoKsqltbliyZIliY2NN5VatWqlXr142tZ02bZqh/OOPPyoyMtKh8aXXu3dvqw/RSncfch43bpxhWvrkO5Y0bdpU3bt3t1rP3d1dPXv2NEwz9+B9UlKSvvjiC1O5XLlyGWK0pEuXLoZkQStXrrS5bU4sjySNGDHCULYledihQ4e0fft2U7lJkyaqX7++1XY5qW/fvoYEW//++6/Zuj/99JPh5sNKlSoZHv7PDmcex3ODv7+/IZmVpe/ZEfLyvjsxMdHwwHuxYsXk7u5uc/sWLVqoWrVqpvJff/2lS5cuWW23aNEiJScnm8rpk9vkNmedIzVp0sSmY52Xl1eGRHf2HEtKly6tF154waa69pyXzJgxw/S5QIEC+vDDD22OqW7duobz619++cXmhEs5tTyDBw82/DbTJpMy5+bNm/rpp59M5ZIlS+rRRx+1Kbac0rFjR0NyKUv7uF27dhmOhT4+Pvryyy8dEscff/yho0ePmspjxoyxKSFNqjfeeMP0OSUlRatWrXJIXI6Sk/uNpUuXGvqy9bzTz88v0wcWzOGc8C57zgn/C+tGUoYEdRcuXLCrPQAAAAAAAICsWbduXabT0/7NHADyouXLl2vz5s3avHmzzS8NAADcG0hKAwAAAAAAAAB5XPv27eXi4mL45+7urmLFiunhhx/W0qVLlZKSYmjz8ssv66mnnrKpf19fX3Xu3Nmmuv7+/ipXrpypbOmhpN9//9302d3dXX379rVpjFQdO3aUq+v//Rl7y5YtNrXLqeWRpOHDhxvK1h5uu379ulavXm0qly1bVg8//LBNseWkihUrmj4nJiYqJCTEidHclZ2YcnKdZ0fdunVVq1Ytm+rWqVPHkADA1t9WQECAOnXqZFdcDz30kKFs628rN2zevNlQHjJkiM1ta9SooZYtW5rKCQkJ2rFjh8Niy8zAgQPtquvi4mIqp19WS9I/TG9JgwYNDGVz29L+/ft1+fJlU7lv3742JzJLlXZbOnr0qMLCwmxqlxPLI0m1a9dWq1atTOX169dbfVtc+mQDzk4cJkne3t4qXry4qbx3716zddevX28oP//883avR3OcdRzPTWmPPZcuXdL169dzbKy8vO++deuWITlMwYIF7e4j7f4wOTlZixYtstpm4cKFps+urq4aMGCA3eM6mjPOkew5lvTq1cuwfoKCghQTE2NT2549e9q8f7B133vjxg3t2rXLVO7atasKFy5s0xip0m7b0dHRFvd5aeXE8khSkSJF1Lt3b1P5yJEj2rp1q8X+v//+e8XHx5vKzzzzjF2JnXJKhQoVTJ/tOZYMHDhQfn5+Dokh7b5Pkt2/83r16ikgIMBUzuvHEkfuNzZt2mQo27OvePrpp23eBjknvMuec8L/wrqRMp4P5HTCTwAAAAAAAADSihUrzCY///nnn3M3mByWkpJi+pf+766498yZM8ewTlu3bu3skID/rE2bNhl+jwDgCCSlAQAAAAAAAID7iLe3t7766it9+umnNrdp2LCh4aFxa9I+KG/uoaSUlBRt27bNVK5WrZoKFSpk8xjS3WUpUqSIqXzkyBGb2uXE8qTq2LGjqlataiovW7ZMt27dMlt/wYIFSkhIMJWHDBliV2z2iI2N1bJlyzR8+HA1b95cpUqVko+Pj1xdXTMkNUqfXMeeB9PyYkw5uc6zo3HjxjbX9fDwMDz8ay6u8PBww4Ou9i67JENSHsn231Zu2Llzp6HcoUMHu9p37NjRUM7JpDQuLi5q166dzfXLly9veDj86tWrVh+OTWXPtpR2+5bMb0vpH+q2Z4xUWd2WcmJ5Uo0YMcL0OTk5WXPnzjVbNz4+Xt9//72pXLBgQfXr18/m2Ox16NAhTZ48Wd26dVPVqlVVtGhReXp6Ztgfuri46MqVK6Z2lvaH6ddjly5dHBKrM4/j2RUREaG5c+fqmWeeUcOGDVWyZEl5e3tn+j1/+OGHhrY5dTzM6/vu2NhYQzl//vx29zFw4EDDMqX9bWUmKChIhw8fNpXbt2+vsmXL2j2uLfLiOVJagYGBNtctUKCAmjRpYionJSUpODjYprY5se/dunWr4Sa2+/FYIllPRJl2vouLi5599lmbY7PX2bNn9fHHH6tPnz6qWbOmihUrpnz58mW6j9u+fbupnTOOJen79vb2Vo0aNezuI+2+IbeOJXlhv5E24ZO9551FixZVnTp1bKrLOaH954T/hXUj3T3mpGVrEjQAAAAAAAAAWZf++mVat27d0oYNG3IxGgAAAOAu57+a6T8sNDRUjRo1ylJbW28sAwAAAAAAAHD/8/Ly0gMPPKCePXtq8ODBKlq0qF3t0z/cZY23t7fps7m381y7dk03b940lQ8fPiwXFxe7xkkvbX+W5MTypHJxcdGwYcM0fvx4SXcfylq6dKmGDRuWaf20b1t3dXXVkCFD7IrNFnfu3NG0adP0/vvvW0yQY0lERMQ9HVNOrvPsyEpcqQ+xmovr2LFjhoe/f//991z7beW0lJQUXbhwwVQuVKiQIYmLLerXr28o25r0JSvKlSsnHx8fu9rUqVNHZ86cMZXPnDmT4UHRzNizLaXdviXz21L6B1KfeOIJm8cwJyf207YuT6o+ffpo7NixunHjhiTpu+++01tvvZVpApCffvrJVE+S+vbtq4IFC9ocm60OHjyoF198Uf/880+W2lvaH54+fdr02dvbWzVr1szSGOk58zieVTExMZo8ebI+//xzxcfHZ6kPRx8PU91r++6svCmrbNmyat++vf766y9Jd5MwBQcHm70evnDhQkN50KBB9gdqRV48R0rPzc3N7iQdderU0caNG03lM2fOqG3btlbb5caxZMKECZowYYLN42QmLxxLWrdurdq1a+vQoUOSpOXLl+vzzz+Xr69vhrrbt2831ZPuJsirVKmSzbHZ6ty5cxozZoxWr16dpd+opW351KlThnJWEl+Yk3YbiYmJyXaCzpw+luSV/UZycrLOnTtnKmflvLNu3brat2+f1XqcE9p3TvhfWTdS1s4HAAAAAAAAAGRdXFyc9u7da5jWrl07bd682VR+55139OCDD+Z2aAAAAPiPIymNE6TeuJCcnKyrV686ORoAAAAAAAAAed0nn3ySIdGCm5ubfHx85OfnpwoVKsjdPet/7vXy8spuiBmkfaDLUay9CT1VTixPWoMHD9Ybb7xheuB9zpw5mSal2bJli44dO2Yqd+7c2abkE/a4ffu2unbtqr///jtb/WT14f3MOCOmnF7nWZWduMw9AOjM31ZOi4yMVHJysqlcpEgRu/tIn5QrPDw823GZk5X40rex9aHl+21byonlSZUvXz4NGjRI06ZNkyRdvHhR69atU5cuXTLUTZs4TJKee+65LMdlzm+//aZevXopISEhy32Y2x9GRUUpMTHRVC5atGi2E52kutf2NWFhYerQoYMOHjyYrX4ceTxMK69/nwUKFDCU4+ListTPoEGDTElpJGnBggWZJqW5c+eOli5daioXLFhQPXv2zNKY5uTFc6TM+Pr62n0ez7Hkrpw8lkjS8OHDNXr0aEl3t6fFixdr5MiRGerlxrFk165deuihh7L1u7d0HEqbpMLFxcXuJKfmxMTEOPw3lJPHkry034iKijJsp4447zTnfv0d59Q54X9l3UgZE/+kTwwEAAAAAAAAwLHef/99w98fy5cvr88//9xwz9j27duVnJyc7STwAAAAgD1ISuME48eP19SpUxUdHe3sUAAAAAAAAADcAxo1aqTAwEBnh2EXR7wdPr20ySqcqWjRourVq5eWLFkiSdq9e7f279+fIXHQt99+ayjnxAOqI0eOzPDQZLFixRQYGKj69eurbNmyKlSokPLnzy83NzdTnfXr12vq1KkOjyevxnQ/uZ9/W+mvm2Tlocf0bW7dupWtmCxJn8TBFunjc+a1ovt5Wxo+fLjpAWTp7v44/QPIp06d0saNG03levXqqWnTpg6N4/jx4+rdu7chEYCLi4uaNm2qli1bqlKlSgoICJCXl1eGh7L79++va9euWew//fad+mIMR7jXto8+ffpkSEhTtmxZtW/fXrVq1VKZMmVUsGBB5c+f33CD5sKFC/X999/nWFyp8vr36ePjI1dXV1OfWd139uzZUyNHjjTt25YtW6ZPP/1UHh4ehnq///67wsLCTOXevXs7/EH3e+V8hGNJRnnlWDJw4EC9+uqrio2NlXT3WJI+Kc2tW7f0ww8/mMpFixZV9+7dHRrHjRs31KVLlwyJJOrVq6c2bdqoSpUqKlWqlPLnzy8vLy9DcrJx48bpwIEDVsdI+5svUKCAw25kz4ntw5aEQlmVl/Yb6X/XjthXmHM//45z4pzwv7Ru0i+rr6+vo8MBAAAAAAAAkMa8efMM5aFDh6pevXoqWrSo6dpaUlKSpk+frnHjxjlkzKioKH355Zc6cuSIrl69Kn9/f3Xp0kUDBw602C4kJETz5s3TsWPHlJiYqLJly2rw4MFq1aqVQ+Kyx7Zt27Ry5UqdOnVKSUlJKlOmjJ566im1bds2W/0eOXJE8+fP17lz53Tz5k35+/urYsWKGjx4sKpXr+6g6G3zzz//6O+//9bhw4cVGRkpd3d3FS1aVPXr19fAgQMd9sKB3377TX/88YeuXLmiyMhIeXt7q1KlSurUqVOmSd8dJTk5Wd999522bt2qq1evqmjRoqpRo4ZGjhwpf3//bPe/fft2rV69WidOnFBUVJRcXV1VsGBBVapUSU2bNtXjjz8uT09PByyJ7X788Udt2rRJZ8+elaurqx544AGNGjVKxYsXN9smNjZWs2fP1rZt2xQZGakiRYqoTZs2GjZsWLZeaChJa9eu1dq1a3X58mXdunVLxYoVU82aNfXcc89ZjCknBQUFacOGDTp48KDpJRup28bAgQMd+nLAuLg4ffXVVzp48KCuXLmiokWL6oEHHtALL7yQ7Zfl/fHHH/rtt9906dIlxcbGqkSJEqpZs6aGDRvmkO3bkc6cOaPly5dr7969Cg8PV1JSkry9vVW6dGk98MAD6t69e7b2N6m/9V27dun8+fPy8vJSs2bNNGrUKIv3PIWFhWnWrFkKDg5WdHS0ihUrpkceeUT9+/fPciwA7EdSGifo2rWrunbt6uwwAAAAAAAAACDHpH8wq1atWvrss8+y1Wf+/Pmz1d6Rhg8fbkpKI919uG3mzJmmckREhFasWGEqBwQE6LHHHnNoDPv27dOCBQtMZQ8PD3388ccaOXKk1Qvlp06dcmgseTmm+03631b79u312muvZavPwoULZ6u9o6S/uBwTE2N3H+nb+Pj4ZCsmS1IfUrdH+vgcmUTEXum3pSlTpqhRo0bZ6rN27drZau8o1apVU/v27U0PGP/222+6evWqAgICTHXmzJljeLA9JxKHvfrqq4qPjzeVmzZtqgULFqhGjRpW26ZNKmBO+u3bkYkp7qXj+OrVq7Vp0yZT2cfHR19//bX69etnNbHCX3/9lSMxpZfX993u7u4qWbKkLl26JOnuDT2JiYl237jm7e2t3r17a/78+ZKk69eva+3atXr88ccN9dKeK0jSoEGDsh58Ju6l85H77VgyduxYPfroo9nqs1KlStlq7yi+vr568sknTTeA79u3T0FBQWrcuLGpzpIlSwzrY9CgQQ6/YfX999/XjRs3TOWqVatq0aJFNiVSszVZho+Pj8LDwyXd3SYd9YbV9OP7+/sbkvjkJXltv5E+aYkj9hXmcE5o3znhf2ndXL582VB25M3dAAAAAAAAAIxOnTplulYnSa6urpowYYIkqW/fvoZ7kr766iu7ktK4u7srKSlJklS5cmWdPHlSly9f1uOPP649e/ZkSAj/ww8/aPjw4frss880bNgww7xt27apb9++unjxYoZx5syZo+LFi2v16tVq1qyZ1bjSXhNv166d4ZpvWkOHDtXcuXNN5S1btqh169ZauXKlhg0bZkpWkdasWbNUqFAhffPNN+rXr5/VWNL6+OOP9d5775l9kceUKVNUqFAhvfPOOxozZoxdfdsqNjZWU6dO1aJFi3T69GmLCcfHjRunMmXK6NNPP9UTTzxh91inTp3SoEGDtH37drPjTJ8+Xa6urqpdu7amTp2qzp07Z6hjbj1Z89xzz2nevHmmbTStSZMmqUGDBtqwYUOWEmG89NJLmj17tk1/yy9WrJjGjx+v8ePH2z1OZgIDA7V582ZTOfV3Nnr0aH377beKi4sz1F+zZo3ee+89Pfzww1qzZo3hWl1cXJx69uypdevWZfi9Llu2TKNHj9aECRP0wQcf2BXjzZs31b9/f23YsEGJiYmZ1nnjjTdUtmxZzZo1S4888kimdWbMmKGXXnopw/SkpCSL976kX5bExER9+eWX+vbbb00Jr8yZNGmSihQporfeekujRo0yW8+auLg4denSRZs2bcoQz+LFizV+/Hg9+OCD+uWXX+xKTpOQkKBnnnlGP/74o9nlmDhxosqXL6/58+c7/IWV5rY/cxYvXqyXX35ZoaGhFus999xz8vb21uOPP264dzatKlWqmK6furm5KTExUYmJierfv79WrlyZ4fv45Zdf9Prrr2vw4MH67rvvDPPCwsLUtWtX7dy5M8M4S5Ys0dChQzVt2rQML5cBkDNISgMAAAAAAAAAcLj0F4JTUlLUqVMnJ0XjeG3btlXNmjV15MgRSXcvzE2dOtX0wP3ixYt1+/ZtU/3Bgwdn+40k6f3444+GC4aTJ0/W2LFjbWqb2Q0Z92tM95v0vy0vL6/75rfl6+srV1dX000uaR/AtlXq28FS5WTCnfRj2SL9Mvn5+TkoGvul35YqVqx432xLkjRixAjTA8iJiYmaN2+eJk6caCqnJs2Q7iZLcfTbc6Kjo7VmzRpTuUSJElq3bp3N22RqcgBLChUqJHd3d9MNG2FhYUpJSbEpoY0199JxfNmyZYbyrFmzbL6xMLeOPffCvrtChQqmG12Tk5N1+fLlLD18PmjQIMPva8GCBYakNDdv3jT8NsqXL6927dplPfBM3EvnI5GRkbpz5448PDxsbpOXjyUlS5bMc9t2dowYMcLwVtJvv/3WkJRmzpw5hvo5keAsbRIXLy8vrVu3zubEPbZuz/7+/qbjTkpKisLCwhzyxkM/Pz/Dcer27dt5dvvIa/sNX19fubi4mGJyxHmnOZwTzjfVteWc8L+0btImpfHx8clzb+sEAAAAAAAA7iepf7dM1aBBA1PS9MmTJxuS0pw+fTpDAm57BAUFqVWrVkpISDBbJy4uTsOHD9f169f1+uuvS5Jmzpyp0aNHW0xuEBoaqlatWmnTpk02JSTJqhEjRmjWrFkW60RFRempp57SuXPn9Oqrr1rtMy4uTnXr1tXJkyet1o2KitLYsWM1e/Zs7d271+EvLXj33Xc1ZcoUm+tfvHhRTz75pH7++WeziSIy8/rrr9ucyCQ5OVkHDx7Uq6++mmlSGnvFxcWpWrVqunDhgsV6+/btU5kyZQwJNmzpu1atWjpz5ozNba5fv6558+Y5LClNZurXr68DBw5YrLNu3TpVr15dJ06ckHT3b/W1atVSZGSk2TZJSUn68MMPdfnyZcN1D0sWLVqkwYMHZ5oMKL0LFy6oS5cuGjp0qL799lub+s+KlStX2nx9Trp7vWX06NH68ccftWXLFrvHu3jxomrXrq2oqCizdVJSUrR+/XoVLVpUhw8ftukeipCQEDVr1symZEjnzp1T+/bt9eyzz2a49pxb+vbta9eLRWJiYrRy5Uqb68fGxqpq1aoZXoaQVkpKiubNm6czZ86Yruvt27dPzZs3N7wMLL34+Hi98MILCgsL05tvvmlzTACyJvuvNgIAAAAAAAAAIJ2AgABTghbp7gW0O3fuODEixxs+fLjpc0REhJYvX24qp70A6+LikiMPqO7YscP02dXVVSNGjLC57aFDhxwej5Q3Y7rfVKxY0VC25WaYe4WLi4vKli1rKkdFRens2bN29bF//35DuXz58o4ILVMXLlyweGNCZg4ePGgop1+fuel+3pYkqUePHoYH6ufMmWO6Oe63337T1atXTfP69Onj8KQOe/bsMdzE169fP5sT0pw8edLiTRVpValSxfQ5JibGlCwtu+6l43jaY0+RIkXsevtbbh177oXfW7169QzlY8eOZamfdu3aGfa9v/32myHJ0tKlSw2/jYEDBzokkVJa99L5SFJSko4ePWpXG44luadp06Zq0KCBqbx06VLFxMRIunsTXFBQkGle27ZtVb16dYeOf/78ecPNeQ8//LDNCWlu375t8422VatWNZTTLld2uLi4GPYHt2/ftnizoTPltf2Gq6ur4bvLynmntZuaU93vv2NHnxP+V9ZNaGio4fid/jwBAAAAAAAAgGP9/vvvhvIrr7xi+uzv75/hb/lvvPFGlsa5c+eO2rRpY7pelz9/ftWrV09t2rRRjRo15OpqfNx40qRJOnHihH777TeNGjXK9PdVf39/NWrUSK1bt86QpCEpKUmPPfZYluKzxbfffmtISFO8eHE1btxYrVu3zvQekddee83qdfTk5GRVqlQpw99hPTw8VKtWLbVp00a1atXK8KKJw4cPG67Z5xRvb29VrlxZTZo0UWBgoJo1a6Zy5crJzc3NUG/p0qX63//+Z1OfTz31VKYJaXx9fVW3bl21bdtWjRo1UunSpTOM4wi1a9fOkJDG09NTtWvXVuvWrVWjRg3TuPHx8erUqZNNCVQkqXPnzhmukxUoUEA1atRQq1at1LZtWzVs2FAlS5Z0+EvmzHnkkUdM1wdcXFxUqVIltWrVSo0bN5aPj4+h7smTJzVkyBAlJiaqbt26poQ0np6eqlmzptq0aaO6detm2B4XLFigtWvXWo3l008/1YABAwzfp4uLi0qWLKnGjRurbdu2ql27try8vAzt5syZoyFDhmToz9HX+1Plz59fFSpUUKNGjdSuXTs1b95clSpVyrDcW7duVffu3e3uv0WLFqZrPK6urqpcubJat26tevXqqUCBAoa6MTExqlWrlqKjoy32eeLECT3wwAMZEtL4+PioQYMGatWqlSpVqpThO5s7d67NL19ypGnTpmVISOPu7q6KFSuqefPmCgwMVOPGjVWhQoUM24OtmjRpYrpG7ObmpmrVqql169Z64IEHMvS5adMmTZs2TWFhYWrRooXp3qn8+fOb9ktp9w2p3n777Szf4wLAdiSlAQAAAAAAAAA4nIeHh1q1amUqx8bGaufOnU6MyPEGDRpkeGA/NRHN7t27DYkxOnToYPPDo/a4du2a6XOxYsVsTnaQnJxs19tj7vWY7jdlypQx3NBy4sQJq28Nym1pb1Ky9IaszDRv3txQ/vvvv+1qn75++v4cKSUlRf/884/N9c+dO2dIshMQEGDTG3RySvv27Q1le7/rvM7Dw8NwM8zp06dNy5j+zU05kTgs7f5Qkl2JCuxZF23atDGU09+smFW5fRxPe8ONvfuNtN91lSpVbL4hLioqSsHBwTaPk/4GTHvivBf23Y0aNTKU0yc+sZWLi4sGDhxoKickJGjZsmWm8sKFCw3109Z1lHvtfMSeMWNjYw0JQ9zc3DKsu9x0vx9LJBmSk9y6dcu0Pef1Y8mWLVtsTiaWU8cSKXe3keycg+bF/UaTJk1Mn+097wwLC7M5Wc79/jvOiXPC/8K6SX8e0Lhx41wbGwAAAAAAAPivWb16tSkpviTly5cvQ3KCF154wVBeuXJllsY6f/684uLi5ObmphkzZig2Nlb79+/XP//8oyNHjujGjRuGhNkpKSkaOHCg6cUgvr6+2rhxo27cuKGgoCBt2bJF586dU3BwsCG5QEREhD7++OMsxWhN6vXGihUr6vDhw7p27Zp2796tLVu26OzZszp+/Ljh7/wpKSlW//7bu3dvXblyxTBt9OjRSkhI0KFDh/TPP//o0KFDSkhI0MiRIw31Lly4oKeeespBS3dXwYIFValSJX344YcKDw9XdHS0Tp48qV27dmnjxo3asWOHzp07p4SEBL3xxhuGayRTp05VWFiYxf5nzpyppUuXGqZVq1ZN+/fvV0REhA4cOKDNmzcrKChIFy9eVGJiopYsWaK6desa7pXLqpdfflmnT582THvxxRcVHx+vkJAQbdmyRUeOHFFCQoKefPJJSVJ0dLRNL7a6efOm4e/2Hh4eWrZsmeklP1u3btXmzZsVHBysy5cv686dO1q/fr0CAwNVokSJbC+bOevWrZN09z7CqKgonTp1Slu3btXu3bsVFRWl8ePHG+ovXLhQ3bp1082bNyXdTVQVHx+vw4cP659//tGBAwcUHR2tZs2aGdql3z7TCwoKyjDWgAEDFBUVpcuXL2v37t3avHmzQkJCdPv2bX3yySeGezDmzZuXIfHNmDFjlJKSopSUFFWuXNk03c3NzTQ9s3/peXp6qmTJkpo4caKuXLmi2NhYnTlzRkFBQdq0aZO2b9+uU6dOKSEhQV988YU8PT1NbX/55Rft3r3b4rKnFRkZqYsXL0qSAgMDdevWLZ08eVJbtmzR/v37FRMTo08//dTw24qJidGjjz5qsd927dopMTHRVM6XL58WLVqkqKgo7d27V1u3btWpU6cUERGR4Z62ZcuW6ccff7R5GRzh3XffNZRfeukl3blzR6dPn9b27du1ceNG7d69W2fOnNHt27d1+PBhDRgwwPBCCEuSkpJ0+PBhSXcTYcXFxenYsWPasmWL9uzZo9u3b6t3796GNm+//bY6duxoOlZNnz5dsbGxpv3SkSNHFBYWluFY9eyzz2bz2wBgDUlpAAAAAAAAAAA54uGHHzaUv/jiCydFkjP8/PxMN11Id9+6cfToUc2ZM8dQLyceUJWMD1qmvsHIFqtXrzZdVHW0vBjT/Sj9b2vmzJlOiiRz3t7eps/p3/xiTbt27Qzl+fPn29z22LFj2rZtm6mcL1++DDdfOFr65Ar21G3btq2jw7FL06ZNDTdi/f3336YbAe4Xw4YNMyQ7+fbbb3Xx4kX98ccfpmk1atRQ69atHT52+htobN0npqSk6Ouvv7Z5nPT7g6+//trmJAT29p2Tx/Hs7Deyeuz57rvvFBcXZ3P9tDFK9seZ1/fd6ZNS2JOwJ71BgwYZygsWLJAkHT16VLt27TJNb9WqVY68OfBeOx+x51iycuVKw9vXGjVqlGHbzE2lS5dWnTp1TOVTp07Z9Pa9e8lTTz2lggULmsrffvutbt++rSVLlpimFS5cOMPNco6Q1WOJJH311Vc2102/f/r+++8VERFhc3t7+s7JfZ8zjiU5ud8IDAw0lO3ZVyxevNhww6slnBPeZc854X9h3aQ/D3D2/10AAAAAAACA+9n7779vKHfo0CFDnVGjRsnd3d1UjoiI0NatW7M0nouLi/7++2+NGTMmwzw/Pz/t27fPMNaOHTt0+/ZteXt76/z58xn+RipJDRs21PLlyw3TZs+enaX4bFGjRg2dPn1aNWvWzDCvatWq2rt3r+Hvwjt27DDb18WLF7Vq1SrDtG+++UafffZZpvW//PJLffrpp4ZpS5cu1dWrV+1ZBItef/11nTp1Sq+++qr8/PzM1nN1ddW7776rv/76yzQtJSUl03WbKjExUS+//LJhWteuXXXs2DHVq1fPbLt+/frpwIEDWd7uUiUkJOjzzz83TPvwww8zvR/B1dVVy5Yt0/PPP29z//PmzTOUZ8yYYUpsY86DDz6ojRs3auPGjTaPkxU9evTQX3/9Zbj2mOrjjz9WixYtTOWkpCTTiyTmz5+vqVOnZmjj6empHTt2GPo7e/asxaREjz/+uOm6mIuLi1asWKGFCxdmGpMkjRs3Tps3bzb8nl588UUrS5o1PXr00OXLl/XBBx8oICDAYt0XX3xRhw8fNiTMGT16tN1jPvzww9q4caMKFCiQYd7LL7+sX375xTAtNSFQZqZNm2ZIbuXu7q7g4GA9/fTTGeoWKlRI27dvz/CCgrQvTMlpsbGxhmvCHTt21LRp0yy2qVmzphYuXGj3S5heffVVLV682HBsSbV8+XKVL1/eVL5165YOHDhgOlaNHTs2QxtzxyoAOYukNAAAAAAAAACAHDF06FDDhfEVK1ZozZo1zgsoB6S/EDh9+nTDm2SKFi2qHj165MjYaS++hoeH2/RwWnR0tMaNG5cj8eTVmO5HL730kuGi6hdffKE9e/Y4MSIjf39/0+cbN27o1q1bNrft16+f4YHiLVu26Oeff7apbfrt6IknnpCvr6/NY2fFihUrbHrTTmhoaIabkoYMGZJTYdnEw8PDcOE+JSVFw4cPd1hCk7ygYsWKeuihh0zlVatW6eOPP1ZSUpJpWk4lDkt/g4ytN2Z9/fXX2rdvn83jdOvWTRUqVDCVT58+rbffftvm9pbk5nE87X7DlrebpZX2uz506JBNiRQuXbqkyZMn2zVOoUKFDDc0nTlzxq72eX3fXa1aNcO2lPbtcfaqXLmy4cH+nTt36vjx4xke2k+fvMZR7rXzkd27d9v0Nsu4uDi99dZbhmnOPpZIyvAmvbFjxyoyMtJJ0Tiej4+P4UbBnTt3atKkSYZ9Tf/+/Q1v/3SUrB5Lfv/99ww3SFrSsGFDw282KioqwxtXs6p79+6G5FM7d+60K/maPbJzDpoX9xv9+vUzbFe2nndGRERkeIDBEs4J77LnnPC/sG42b95s+uzu7q6OHTvm+JgAAAAAAADAf1FCQoKCgoIM0955550M9VxdXdW8eXPDtDfffDNLYz7xxBMWE1EXKlQo0yTeS5YsUaFChcy269q1q+EejfPnz2cpPmtcXV21fft2i3XKly+vxo0bm8pJSUlmr82mT2TRrFkzDR8+3GL/L7/8surXr2+YNmrUKIttclJgYKDq1q1rKv/5559m644fP97wd+ZSpUrp119/tXksV9fsPZL+7rvvGv4+X7VqVb366qsW23z11VcqXry4Tf2nv9/gqaeesjvGnODt7a2ffvrJYp3MEs80bdrU6nX19El3zI3z+++/G5KmDBw4UL169bLYt3T3ZTNpxzh9+rTOnTtntV1Oq1y5suEFGXv37rWrff78+a1u+127dlXXrl0N08xdH/zkk08M5bfeeku1a9e22P+6deuUP39+Uzk8PDxDkqyccvz4cUM5s4RjjlCmTBl9+OGHFuu89NJLGaY9+eSTVo9VrVq1MpWTkpLsut8KgP1ISgMAAAAAAAAAyBG+vr763//+ZyonJyerX79+Wr16tV39BAcHW31jibM0b97ccJPB7NmzDQ8/Dhw4UJ6enjkydsuWLQ3lCRMmKDk52Wz92NhY9ezZU6dPn86RePJqTPejSpUq6dlnnzWVb9++ra5du1q96Sa9v//+W8OGDXN0eIYL6ikpKTY9aJ/Kz88vwwP2Q4YMMfuWmVRvvfWWIVmGq6trphesHS0lJUVPPPGELl++bLbO7du31bNnT8MD+rVq1TI8GOssY8aMUYkSJUzlrVu3qnfv3nYlE4iJidHnn3+uuXPn5kSI2ZY2eVhCQoLh7Vqenp4aOHBgjozbqFEjw/7/p59+0r///muxzW+//ZbhjWTWuLm56Y033jBM+/DDDzMkQTInJSXF7Nu+cvM4nna/ERYWpk2bNtncf9pjT0JCgiZOnGix/vXr19W1a1ebktek5eHhoWrVqpnK+/bt06lTp2xun9f33ZL06KOPmj5fvHhRJ0+ezHJf6W+Mmz9/vhYtWmQqe3l56Yknnshy/5bci+cjzz33nEJCQszOT05O1oABAwzJkIoXL54nbqJ8+umnDb/h48eP65FHHrF4bEzvzp07WrBggT766KOcCDHb0ieiTL+PzakEZ+XKlVPp0qVN5d27d+uHH36w2GbXrl3q37+/3WO99dZbhjccLlmyRGPHjjW9KdGatG/gTMvNzU3vvvuuYdqYMWP07bff2hXf8ePHNWzYMF26dMlsneycg+bF/UbhwoUN+1Jbzjvj4+PVu3dvXb9+3a6xOCe075zwfl83iYmJ2rZtm6ncqlWrHE/2CQAAAAAAAPxXffzxx4a/RxcuXNiQTCWt9ElotmzZkqUxZ86cabVO+kTVvr6+evzxx622q1GjhunznTt3FBsba3+AVrRo0cLwchVz0iarkMxfy0h/vfybb76xKY70Sfj//vtvm9rllLRJaSz9LTr9taavvvoqx2KyZfz33nvPpna2vigg/bbx448/2tQup/Xp08dqnVatWmVI+pNZopr0evbsaSibuzclbeJ8FxcXm/YFqdIny8or14NatGhh+hwfH6+rV6/a3LZv376GFwuZM2vWLEM57TWUVNHR0YaEP56enhnu5cmMp6dnhnsrcus3WbRoUUM5q8cUa9K+gMGctC+JSZX2Wp45nTp1MpQ3bNhgc1wA7EdSGgAAAAAAAABAjpkwYYK6dOliKt+6dUvdu3dXz5499ffffys+Pj5Dm7i4OO3atUsffPCBGjVqpMaNG+eZC8SZsfR2nKFDh+bYuP379zdciF6zZo0ee+wxHT582FAvLi5OK1asUP369U0X3mrWrPmfiel+NX36dD3wwAOm8pUrV9S2bVs988wz2rFjhxITEzO0iY6O1tatW/X666+rRo0a6tixo9avX+/w2NInW3n++ec1atQoLV26VH/88Yf+/PNP07/024Z09yaIChUqmMrh4eFq2bKlPv30U4WHhxvqhoSEqFevXhlufhg/frzh+8kJpUuXlru7u86ePauGDRtq0aJFiouLM81PTk7WH3/8ocaNGxtuSHBxcdHs2bMND347i6+vr5YvXy4PDw/TtNWrV6t27dr69NNPzb657MKFC1qxYoX69++vUqVKacyYMbpw4UJuhW2Xrl27qlSpUpnO69GjR4abLBzF29vb8EappKQkPfLII5o9e7ZhO5GkEydOaOTIkerWrZvi4+NVvHhxFSlSxOaxnn32WfXu3dtUTklJ0SuvvKIOHTpo/fr1GY61ycnJOnTokD744APVrFlTzzzzjNm+c+s4nn6/0aNHD7366qtavny51q9fb9hvpE8AkP4h8m+++UaDBg3K8GauW7duae7cuapXr57p7Uj2HnvSxpmUlKS2bdtq8uTJWrVqlTZs2GCIM+0NR6ny8r5byviWuJ9//jnLfT3xxBOGN3pNnz7dsJ/o3r17jj3gfi+dj/j5+alQoUKmY90XX3yhqKgoQ53t27erTZs2WrFihWH6jBkz5OPjk5vhZsrNzU0rV640rM/t27erTp06euuttzK8YS3VtWvX9Ntvv2n48OEqXbq0Bg8erCNHjuRW2HZp0KCBmjZtmum8Zs2aGW7ydbT0+7iBAwfqww8/zLCdXLx4UW+88Ybatm2r8PBweXl5Gc6nrOnUqVOGxGifffaZGjVqpJ9++kkxMTEZ2pw8eVKfffaZGjdunOGGv7T69u1r+D/TnTt3NGzYMHXs2FG//fZbpn3fuXNH+/fv14wZM9SmTRvVqFFD3377reHNnell5xw0r+43PvjgA8ObP1PPO7///vsM551//fWXmjZtarqp3p71zzmh/eeE9/O62bRpkyHpTV5IgAYAAAAAAADcr2bPnm0op73GnN6DDz6oggULmsqJiYl2Jy/w8fGx6e+haRPBZ1Y2p2zZsoaypWTzWZX2RRuWpL9nJDQ0NEOdhIQEw8tMChYsqAYNGtjUf4sWLeTt7W0q37x5M9Prvdn1119/6aGHHlKpUqXk6ekpV1dXubi4ZPi3ZMkSU5uUlBRFR0dn6Cs5OdmQtMPLy0vdunVzeMyWpL2O7+bmZvOLTGx9wU/639ALL7xgeHmKs9j6t3YvLy/TZ1dXV7Vt29Zqm4YNGxrKN27cyLTewYMHTZ+LFCli2J9YU7VqVcN9TtZeypRde/bsUffu3VWuXDl5eXmZ3e7TJ37Zv3+/zWOkfUmTJaVKlVKxYsVM5du3b+vmzZuGOumTLaVfJ5ZMmjTJULZnGbKjTJkyhpdtrV+/Xi+99JLD92OW7klKVbRoUcP2ldVjVWb7eQCOYz2NFwAAAAAAAAAAWeTq6qolS5aoe/fu2rRpk6S7F75XrVqlVatWKV++fCpfvrwKFy6suLg4RURE6OLFi0pKSnJu4Hbo37+/JkyYkOFifuvWrXP0AcUaNWpoxIgRhhtMfv/9d/3+++8qW7asSpYsqejoaJ09e9bw5qG2bdtqwIABeu655/4TMd2v8ufPr9WrV6tLly6mmwYSExM1f/58zZ8/X97e3ipbtqx8fX0VGxur8PBwXbp0SSkpKTkeW9euXVW9enUdO3ZM0t2Hd2fOnJnpG3YGDRqk+fPnG6b5+Pho+fLleuihh0xJaGJiYvTKK69o4sSJqlixogoVKqQrV65kegPTww8/nCFJTU6oUqWKRowYoUmTJunatWsaMGCARowYoYoVK8rDw0Nnz57NkERHuvv2oFatWuV4fLZq06aNFi5cqGeeecb0AO2lS5f0yiuv6JVXXlHJkiVVvHhx5cuXT5GRkQoNDc10ufIqd3d3DR06NNNtIqf3Oe+++67WrFljShwQFRWl4cOHa8yYMapWrZry5cunK1eu6OLFi6Y2bm5umj9/vp5//nmzNwplZu7cubp+/bo2b95smrZx40Zt3LhRBQoUUNmyZeXn56dbt27p/PnzhmNW+fLlzfabW8fxgQMH6v3331dYWJgkKSIiQh999FGmdd966y29/fbbpvJDDz2kLl266PfffzdNW7hwoRYuXKhKlSqpWLFiioiI0JkzZ5SQkGCq89RTT6lq1aqaPHmyzXGOHDlSs2bNMv1WLl++bIglrXnz5mnw4MGGaXl53y1JLVu2VKVKlUyJf3766Se98sorWeqrUKFC6t69u5YuXSpJGZIxpX/blyPdS+cjvr6+evfddzVw4EDdunVLo0eP1vjx41WpUiUVKFBAFy5cyPTGqWeffVb9+vXLtTitqV69ulatWqVevXqZjhHh4eF655139M4776ho0aIKCAiQt7e3oqKiFBYWZvENjXnRiBEjtGvXrgzTc3p7eeWVV/T999+bjhUJCQl67bXX9Oabb6p69ery9vbW9evXdfbsWcO+4vPPP9fixYt19uxZm8eaMmWKLly4YEgktnfvXvXq1Uuenp4qX768/P39FRMTo4sXLxpu0rbmiy++UHh4uKHvv//+W3///bfc3d1NfScmJioiIkKXLl0y7LNtkZ1z0Ly63/D399fixYvVtWtXUyK4a9euaeDAgRoxYoQqVaokDw8PnTt3znDz60MPPaTmzZvbdU7MOaF97ud1s2rVKtPnfPny2fT2VgAAAAAAAAD2O3/+fIZk0u+++67FNl27dtWyZctM5c8++0wjR460eUxbXxqR/iUq6ZPNmJP+hQqhoaGqWrWqbcHZqFatWjbVS78MmV3XCA4ONpQtXTvPTLly5QwvXQgODlazZs3s6sOcnTt3qkePHpm+DMUWZ8+eVZ06dQzT9uzZY7ieZE8SdUdITk42XP+x52U97u7uKliwYKbJdtJq0KCBypcvb0p+k5iYqAEDBmjYsGFq1KiRunTpoqefflrlypXL2kJkka0v1kqbpD7tS2AsSZtEX5LZ7+jWrVumz2FhYdl6mVb6pCyOcurUKT366KOm6332Snv/jSWurq6qXr26zf1WqVLFcH3777//Nrw8aufOnYb6zZs3t7nv1IRTqb+N3Lz+1qdPHy1evNhUnjFjhr744gvVqlVLnTp10lNPPaXGjRtnuX8XFxf5+/vbVNfV1dV0r1FWj1X2XL8GYD9X61UAAAAAAAAAAMg6X19fbdiwQS+//LLc3Y250uPj43X8+HHt3LlT+/fv17lz5zJ9kN3WmxucwcfHJ9OHgnPjgebp06era9euGaZfuHBBu3bt0uHDhw0PTbZv316//PJLhvVwv8d0vypTpoy2b9+u/v37Z7hRICYmRkePHtXOnTt18OBBXbx4MdOkBjlxk4W7u7tWrlypypUrZ7mPxo0b659//lGlSpUM0+/cuaPjx48rKCgo04Q0gwcP1urVqw1vcslJb7zxhkaPHm0qx8TEKCQkRHv37s1wk4Crq6veeuutDG/pyQv69u2rrVu3qlq1ahnmXblyRfv379euXbt07NixTG9+cHNzU6lSpXIj1CwZOnSo3NzcDNMqVaqkDh065Oi4lStX1vLlyzO8XSouLk4HDhzQ7t27DTfEeHl5afHixXrkkUfsHqtQoUJav369hg4dmmF/EBsbq2PHjmnnzp06fPiw1Zu00suN47i/v79WrlyZ4WYpWy1evFhNmzbNMP306dPauXOnjh07Zri5rW/fvpo3b57d41SrVk3ff/+9XW8MSy+v7rtTPf/886bPO3bs0MmTJ7Pcl7nEMyVLltSDDz6Y5X5tcS+djwwYMEBTp041bQ/x8fE6cuSIgoODzSakSf/WzLygffv22r17t5o0aZJhXlhYmEJCQrRz504dOXIk04Q0Li4uefqc/8knn5Sfn59hmo+Pj/r27Zuj4/r7+2v16tUqUaKEYXpiYqIOHTqkXbt26cyZM6Z9haurq6ZNm5al/4u4u7tr2bJleuONNww3vEp3k+GcOHFCO3fuVEhIiN039Hl4eOiHH37QRx99lOEG2sTERJ06dUq7d+/W3r17MyQRS1W0aFGLN99m9xw0r+43OnXqpBUrVmS4kT82NtZ03pn2xt/WrVvrxx9/zNKNxJwT2ud+XDcJCQmG5FF9+/ZV4cKF7Y4XAAAAAAAAgHUTJ040lCtUqKCAgACLbT744AND+fjx43Ylh8iXL59N9VxdjY8d23p9NH27nHj5hq2JTGyJJX1SIHuvV6evn76/rFq7dq1atmyZ5YQ0khQZGZlhWvqXGaS//pTT0n8/tiaeSJX+7/Hm/PPPPxnq3r59W1u3btVrr72m8uXLq0CBAmrcuLHmzJljVwxZVbRoUbvb2HoNypZtPfUFQY5i730ntggJCVGtWrWynJBGsj0uW/eFqdLvm9Pfs5b++01/v5s1aa+B5ubLHBctWqS6desapiUlJengwYOaPn26mjRpIg8PD1WtWlWvvfaa3es9/bZpq6weq3LrhU/AfxVJaWz066+/ql27dmrUqJHFf5ndFAYAAAAAAAAA/3Xu7u769NNPdezYMQ0bNsymi/gVKlTQsGHDtH79+gwXxfOaIUOGGMp+fn658jZxT09P/fLLL5o+fbrFG1MqVKigmTNn6s8//8zwMO1/Iab7mbe3t77//nvt27dP/fr1s+m7rFGjhsaMGaN///1X//zzT47EVbt2bR04cEDz5s1T7969Va1aNfn6+mZ4CNSSOnXq6PDhw5o6darFi/Xu7u7q2LGjtmzZonnz5mV4iDqnffbZZ1q5cqVq1qxptk7Lli21ZcsWvf3227kXmJ0aNWqk/8fefcdVXff/H38yRAREwIUD3Hvl3jPNkQ39ambOK7cNy8yrMsuWeWmm2TBLr9TKkVpZplaauPfeKCqgiAqIgAjI+P3hz8/Fh3kOHj2oj/vtxu12Xp/zHq/PGR8s4HmOHTumhQsXqlmzZrk+VwULFlSHDh30ySefKDQ0VMOHD79HnVrPz89PHTt2NB3LKrzlbnjssce0e/duPfHEE9mOcXZ2Vq9evXTw4EH16dMnz3u5uLjo22+/1b59+9SzZ0+5u7vnOL5cuXIaM2aM1q5dm+va9+L7eJs2bXTixAl98cUXeuKJJ1ShQgUVLlzYol9S8fLy0qZNmzRx4sQcf3GtVq1aWrRokRYvXpzn8KpevXopMDBQU6ZMUefOneXn5ycPDw+rXk/59dot3Xpv3H7tpKWlad68eXleq2PHjln+AXy/fv2s+n6QF/fbv0fGjRunDRs2ZBmudFudOnX022+/ae7cuXn+5a27rVKlStq1a5d+++03dejQIdf3mZOTk5o3b673339fp0+fzvXTP+3Jzc0t0zW6b9++uV5rbaF+/frau3ev+vfvn+17x8HBQZ06ddKOHTv06quv5nkvBwcHffDBBzp+/LgGDx6c6/uiZMmSGjp0qHbt2mXR+uPHj9fZs2c1btw4iwK2fH191b9/f/38888KCwvL9Zej7+TfoPn5utG9e3cdPXpUvXv3zvZ9VaxYMX388cfasGGD1b/EnR7/JrTOg/bc/Prrr6ZfnB4zZkye+wUAAAAAAACQs99++81UDx06NNc5FSpUyPTzt4kTJ9q0r4dJxiAJa3/ukzGsJ6sPZrBWUlKSevbsqdTUVOOYk5OT2rRpo8mTJ2vdunUKDQ1VbGys0tLSjK8hQ4aY1skqnCFjf3fyYSx5kfFvr11dXa2ab2lQhb+/vy5fvqxnn302298hunHjhvbu3athw4bJ3d1dn332mVW93G9CQkJsul7616etdOjQwfTBFQ4ODmrcuLEmTpyoP/74Q8HBwbp27ZrpdT9jxgzTGpaGklj7u2UZQ44yfkBATEyMqbY28D/9a/teB6scOnRIH374YbY/w0pOTtbp06f18ccfq0iRIho4cOA97Q9A/uGQlscrVMOGDRUeHi5fX1/t3bvX1n3lu73btm1r1afA2eNxua38G3/YZV9bODflcesnpZywfSPA/cSp+h1MDrNZG8D9Kd3/jEvN+6e9Ag8Ex8p5n3vjL9v1AdyPCj1m3LwyxvoUc+BBUvyz//2AbkRV61LkgQfNnMBEe7eQ76Wlpeno0aM6evSoIiIiFB0drYIFC6pIkSKqUKGCatasma8/YT2j//73v6Yf7r/wwgv64osv7mkPycnJ2r17tw4dOqTIyEg5OTnJ19dXjzzyiOrVq3dPe8nPPT3oUlNTtW/fPgUGBioiIkIxMTFyc3OTl5eXKlWqpJo1a6p48eL2bjNPTpw4of379+vy5cuKj49X0aJFVaZMGbVq1eqO/rjTGun/YLVt27YKCAgw3X/48GHt2bNH4eHhcnFxUalSpdS8eXNVqFDhnvRnS9euXdOOHTsUFhamiIgI3bx5U4ULF1aJEiVUvXp1VatWzepfELKXtLQ0VahQQcHBwZJuBayEhobm+ilztnbx4kVt3rxZ58+fV3x8vDw9PVW5cmW1aNHirvxRe2JiorZv367g4GBduXJFSUlJKly4sPz9/VW7dm1VqlQpz2vn5+/jCQkJ2r59u44fP66rV6/KxcVFpUuXVuPGjVW1alW79JSb/HbtHjdunKZPny7p1qf7hYSEWP1JYflJfvv3SPny5Y3rUbly5TKFNp0+fVo7duzQhQsX5ODgoFKlSqlBgwaqVavWPe/1TsXHx2vHjh0KDQ1VZGSkbty4IQ8PDxUrVkzVqlVTjRo17kmoi620adNGmzdvNurdu3erUaNG97SHqKgobdq0ScHBwYqNjZW7u7sqVKigFi1aWP3pnZZISUnRrl27FBQUpCtXrig+Pl4eHh4qU6aMatWqperVq99RoMfp06d14MABXblyRVevXpWzs7OKFCkif39/1ahRQ+XLl7fdyVghv1030rt69aoCAgKMXzj38fFR7dq11bx5c4s/MdMa/JvQcg/Cc9OhQwdt2LBBkvToo49q3bp1Nu8bAAAAAAAASO/X/Rf0ytID9m4jWzP7PKKn65ex+bp//vmnunTpYpO1fHx8FBkZme39zs7OSklJkXTrAwYs+RvZLVu2qHXr1kY9ZMgQzZ07N9d5Q4cONX3oxebNm9WqVassx+b2+xd5WTM9S87hp59+Mn0oQfv27fXPP//kuvZt7dq108aNG4162bJl6tWrl8Xzs/Lqq69q5syZRu3n56djx47lGiDTu3dvLV++3KizepxWrFhh6i+nx91aljxPwcHBpp/9VKlSRYGBgRbvUbp0aV28eFHSraCe5ORki+YtX75cixYt0p49e3ThwoVsA1UGDx6s7777zuJ+cpLxtWHpn/F7eXnp2rVrkqQiRYooOjraonm5vZ8yPvZ+fn42D6qRpMqVKysoKEiSdc/Rd999Z/pQwCJFiujIkSMqW7ZsjvPeeecd0wefzJgxQ6+88kqWY9M/toUKFVJ8fLxFvUlSz5499csvvxj1zJkzTcH+Ge+fNWuWXnrpJYvXT9+bg4NDlq/RmTNnmj6cJKdzzevrb+fOnfr222+1efNmBQcHKzEx69+Dzum9m9fXwL38XgUgb2z/E+8HVFxcnCTJ0dExx1/kuXz58l1JeQMAAAAAAACAB4mDg4Nq166t2rVr27sVm8j4w6xhw4bd8x6cnZ3VvHlzNW/e/J7vnZ382NODztHRUY0aNbrnfyB9L1SvXl3Vq99JSPfdV6dOHdWpU8febdhEkSJF1LlzZ3u3YRN///238cfHktS9e/d7HkgjSaVKldIzzzxzz/YrWLCg2rVrd1fWzs/fx11dXdW+fXu1b9/e3q1YLL9du8ePH6+vv/5a169f1+XLl7VgwQINHz7c3m3l2f3275HKlSurcuU7CBHPR9zc3NShQwd7t2ETJ0+eNAXSPPLII3Z5z/r4+Ojpp5++Z/s5OTnd1fdPfn295+frhre3t3r06HHP9uPfhJa735+bPXv2GIE0kky/xA0AAAAAAADAtt5//32brRUVFaU9e/bkm5813k/8/PxM9eXLl62an3F8xvXyYuXKlaZ6165duQbSSFJ4eHiuYzJ+oJIlc2wp4+NzO4TDUrGxsXnat1evXqYwnvXr1+vzzz/X2rVrTaEb8+fP16BBg+7a71nYU7ly5Uz17b+Xzy/++9//mupVq1blGkgjKc/BOtmFrWQn43ulTBlzWFmxYuYPNj5z5oxV69+4ccO47eTkZNVcW2ratKmaNm1q1CdPntSMGTO0bNkyRUVFGcdPnTqlUaNGafbs2fZoE4CdONq7gftNiRIltHfv3my/7sYnTwEAAAAAAAAA8q9Dhw5p+/btRt20aVPVq1fPjh0BANKbM2eOqR4xYoSdOgHuDyVKlDB9qtcnn3xifCIV8LD65ptvTDXfS4D7D/8mzNl//vMf43a3bt3yZSgTAAAAAAAA8CBITk7Wzp07bbrm22+/bdP1HhYNGzY01emDzS2RMRAj43p5kT7opnjx4haHq584cSLXMY888ogcHByM2trzvVOOjo5ycXEx6sjISIvnJicn2yxI5dFHH9Wvv/6quLg4ValSxXTfu+++a5M98iNXV1fjdnR0tFJTU+3Yjdm5c+eM2y4uLmrVqpVF83bv3p2n/VJTU3Xy5EmLx58+fdpUZ/xglvRBLpK0Y8cOi9cODw9XUlKSUXt7e1s8926rVq2avv76a0VGRmrIkCGm+xYvXmynrgDYC6E0AAAAAAAAAADcgalTp5rqF154wU6dAAAyCgwM1K+//mrUlStXVufOne3XEHCfePPNN1W6dGlJtz7lasGCBXbuCLCfqKgoffvtt0bt6emp/v3727EjANbi34Q5279/v1asWCFJKlCggKZPn27njgAAAAAAAIAH16effmr6QAhvb2+lpaVZ9bVt2zbTmhs2bLjXp/FAcHFxMQVAxMXF6cCBAxbN3blzp65fv27UPj4+cnZ2vuOe0odTuLm5WTTn5MmTioiIyHWco6OjSpUqZdQJCQn65ZdfrG/yDpQrV864nZKSouXLl1s0b8aMGTbvxdnZWQEBAaZjgYGBNt8nv6hRo4ZxOy0tTZ9//rnN98jreyA+Pt64XbBgQYvnWBLGlJ1p06ZZNC4sLExXrlwx6kKFCsnHx8c0pm/fvqZ63759Fvfx/vvvm+r8+mGIc+fONV2Trl27ZsduANgDoTQAAAAAAAAAAOTRhg0btGjRIqMuU6aMnnnmGTt2BAC4LSUlRaNHjzZ9utMrr7xi+uQvAFnz8PAwBe9NmjRJCQkJduwIsJ+xY8cqNjbWqIcPHy4PDw87dgTAGvybMHdvvvmm0tLSJEkvvviiqlevbueOAAAAAAAAgAfX119/bap79epl9RrNmzeXl5eXUSclJWnevHl32tpDqX379qZ65MiRFs0bPXq0qe7UqZNN+kkf6hEVFWXRnIEDB1q8fr9+/Uz1iy++aPFcW+jdu7epfvvtty2ad7fC1EuXLm36eUH6wKgHzZgxY0z1pEmTlJycbNM93N3djdvWPJaurq7G7fRhTzkZMmSI6Wc/1lq8eLFF80eNGmWqW7ZsmWmMm5ubKfApKSlJH374Ya5rJycnZ/qAoHv9nrRG0aJF7d0CADsilAYAAAAAAAAAAAtcvXpV69at07p16/TTTz9pzJgx6tatm/GHW5I0YcIEiz+tAwBgW3v37tW6deu0du1azZ49W40aNdL69euN+/39/TV06FA7dgjcX/r162d82mJISIjpF8GAB9WxY8e0bt06/fXXX5o3b57at29v+kVADw8PjR8/3o4dAsgN/ya03tq1a43v+Z9++qm92wEAAAAAAAAeWGFhYTp79qzpmCXBBVnp0aOHqZ4xY0ae+3qYffbZZ6Z6586dmjNnTq5z9u3bZzo2a9Ysm/STPtgiNjZWK1asyHH8Rx99pF27dlm8/uTJk+Xi4mLUYWFheuKJJyyefychIJL07rvvysnJyahPnjyZa+DMyy+/rEuXLlm0/qpVqxQeHm5xP6tXrzb97l+JEiUsnnu/GTRokIoXL27U0dHRatasmVXPaUJCgmbOnJnt/eXLlzfVq1atsmjdSpUqGbdTU1P13nvv5Th+6dKlWrJkiUVrZyc+Pl5PPvlkjmPWrFmj3377zXQsu9drxp8hv/feezp+/HiO63ft2lXx8fFG7ePjo6eeeirHObZy6NAh7dy50+LxcXFxCgsLM+r01xEADwdCaQAAAAAAAAAAsMDBgwfVqVMnderUSX369NGsWbOUkJBg3N+0aVONGDHCjh0CwMPttddeU6dOndS1a1eNHj1aBw4cMN3/1VdfERwGAMjR1KlT1alTJ3Xu3FlDhw5VQECA6f4pU6aYflkTQP7DvwkBAAAAAAAA5FcTJkww1RUrVsxzCEbGMJtjx44pJiYmz709rMqWLZsp4GfUqFEaO3ZsluNffvllvfLKK6Zjffv2tVmYSa9evUz1s88+q19++SXTuKSkJPXu3Vtvv/22Ves7OztnChVZtWqVatSooUOHDmU7b/ny5apbt65atWpl1X4Zubi46KWXXjIdGzdunMaMGZNpbGpqqvr166fPP//c4vU/+eQTlS5dWg0aNNA333yTY+DKihUrMj33AwcOtHiv+9Hy5cvl4OBg1Hv37pWvr2+u4Ud///23HnvsMRUuXFhvvvlmtuMyhrz069dPP/74o5KSknJcP+PvXL733nv64osvshz78ssvq2/fvjmuZ6k//vhDHTt2NP0O6G2ff/55psCmNm3aqG7dulmu9corr5hCpZKTk1W/fn0tXbo009i4uDi1atVK69atMx2fPXt2Xk4jT/755x81a9ZM/v7+mjBhgqKjo7Mde/z4cVWtWlUpKSnGscaNG9+DLgHkJ872bgAAAAAAAAAAgPtd5cqVtWLFCjk6kgUPAPmNg4ODPv74Yz3++OP2bgUAcB8bNWqUXnjhBXu3ASCP+DchAAAAAAAAAHv7+eefTfXIkSPzvFbp0qVVvnx5nTt3TpKUlpamSZMm6dNPP72TFh9Ky5cvV5kyZRQeHi7p1mM5Y8YMffnll6pSpYp8fHwUFRWlU6dOZQrX8PPz06JFi2zWy8cff6wvvvhC8fHxkm4FW/Ts2VNFixZV5cqV5ezsrPDwcJ09e9YIXHF0dNSjjz6qv//+26I9Ro0apc2bN2vx4sXGsRMnTqhevXry8vKSv7+/vL29FR8fr/DwcF28eFHJycmSpEceeeSOz3HGjBlauXKlzp49axybNWuWvv76a1WtWlXe3t66cuWKTp8+bezr4eGhYsWKGa/3nKSlpWn//v0aMWKERo4cKW9vb5UuXVpFihSRk5OTIiMjde7cOV2/ft00z9fXV+PHj7/j88vP2rRpoxkzZpiCla5cuaJevXqpYMGCKleunIoWLSpnZ2ddu3ZNly9f1pUrV0xhJM7O2ccSDBo0SKNGjdKNGzckSTExMerfv7/69++faWxaWppxu1+/fnr99dd18eJF476XXnpJEyZMUJUqVeTm5pbpNSFJPXv2zHRdtUSRIkXk4eGhCxcuaP369XJ3d1elSpVUsmRJxcbG6vTp05leH+7u7vrjjz9yXHfjxo2qWbOm0WNiYqKeffZZDR8+XJUqVZK7u7vCwsJ09uxZ0/lLtwKonnnmGavP5U6FhoZq8uTJmjx5stzd3VWmTBl5e3urUKFCiomJUXBwsCIjI01znJycbHrdA3B/IJQGAAAAAAAAAIA88PDwULVq1dSzZ0+9/PLL8vDwsHdLAID/z9nZWSVLllTLli318ssvq2XLlvZuCQBwn3FyclLRokXVpEkTjRw5kiAL4D7EvwkBAAAAAAAA5BcBAQGKiYkxaicnJ7366qt3tObw4cP11ltvGfWPP/5IKE0eODo66uzZs6pdu7aCgoKM40lJSTp69Gi286pXr66DBw/avJc///xT7dq1MwWBREZGZgqGuD1+0aJFFgfS3LZo0SKVLVtW06ZNMx2Pjo5WdHR0nnq3xrFjx1SlShWdP3/eOJaUlKQjR45kGuvi4qJ169apX79+Vu+TlpamqKgoRUVF5TiuZMmSOnz4sNXr34/GjBmjSpUqqVevXkpMTDSOJyYmKjAwMNf5Tk5OOd6/ePFi9ezZ0whNstT27dtVvXp1JSQkGMdiYmK0d+/eLMd//PHHcnV1zVMojSTt2LFDtWrVUkxMjFJTU3Xq1CmdOnUqy7Hu7u46evRorr8fWqVKFe3du1fNmjUzgnlun8f+/fuznTd48GB99913eToPW7p+/Xqur4ECBQpo7dq18vf3v0ddAcgv+MhWAAAAAAAAAAAs0K5dO6WlpRlfsbGx2rNnj9566y0CaYCHRPprQEBAgL3bQQYBAQHG83Pz5k2dP39eS5cu5Y+PAeQr586dM65VlnySH+6t+fPnG89PcnKyLl26pN9//51AGuA+wr8JAQAAAAAAAORHkyZNMtWNGzeWs7PzHa35+uuvy9Hxf38ifPnyZR06dOiO1nxYubq66vTp0/r4449VuHDhHMd6enpqxowZOn78uFxcXGzeS6tWrXT48GFVrFgx2zEODg6qVauWDh06pD59+uRpn6lTp+rYsWNq2LChHBwcchzr5OSkBg0aaMaMGXnaKyNXV1eFhoZqyJAhOYac1KlTR6GhoWratKlF637yySd66qmn5O3tbdH4woULa+zYsQoPD1exYsUsmvMg6N69u6KjozVixAi5ubnlOt7BwUGlS5fW6NGjFRISkuPYp556SmfOnFGPHj1UrFixXENsbitXrpyCg4P1yCOP5Dpuw4YNeuONNyxaNztly5bVpUuX1Lp162xf/w4ODurUqZMuX76scuXKWbRu3bp1FRUVpb59++Z6jff399f69evtEkjz3HPPadiwYSpbtmyu73/pVhhN165ddfnyZXXo0OEedAggv3FIS0tLy8vEhg0bKjw8XL6+vtkmjd0t9tjb0j3t+bjcVv6NP+yyry2cm5KHX+RKOWH7RoD7iVP1O5gcZrM2gPtT6f/dTD1tvzaA/MCxct7n3vjLdn0A96NCjxk3r4x5eP5nNJCV4p9FGLdHVC1ox04A+5sTmJj7IAAAAAAAAAAAAAAAAAB4SP26/4JeWXrA3m1ka2afR/R0/TL2bgN2cPToUc2fP1/BwcG6evWqvL29Vb58ef3rX/9SjRo17lkfJ0+e1Lx58xQYGKiEhASVLl1aNWrU0IgRI+Tp6WmzfVJTU7V48WJt3rxZFy9eVHx8vAoXLqzKlSvr0UcfVefOnW22V1Z7z507V1u2bNGlS5dUrFgxVatWTaNHj76joJiEhAStWrVKO3fu1JkzZxQbGysHBwd5e3urZs2a6tmzp2rXrm3DM7l/BQUF6fvvv9eZM2d0+fJlpaWlydPTUxUrVlTr1q3VpUuXOw7Rssb58+c1d+5cHTp0SHFxcSpVqpQqVaqk4cOHy9fX1+b7xcfH68svv9TRo0d18eJFFS1aVPXr19dLL70kV1fXO1p7zZo1WrVqlcLCwhQfH6+SJUsa72EfHx8bncGdCwgI0MaNG3XixAldvXpVKSkp8vT0VPny5dWlSxd16tTJ3i0CsDNCaWy8J6E0d4ZQGiAPCKUB7gChNICBUBog7wilAQyE0gD/QygNAAAAAAAAAAAAAAAAAGSPUBoAAAAg/3O0dwMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgPyDUBoAAAAAAAAAAAAAAAAAAAAAAAAAAADcM34+bvZuIUf5vT8AAADgXnC2dwMAAAAAAAAAAAAAAAAAAAAAAAAAAAB4eDQs560Vo1ooNCre3q1k4ufjpoblvO3dBgAAAGB3hNIAAAAAAAAAAAAAAAAAAAAAAAAAAADgnmpYzpvwFwAAACAfc7R3AwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACA/INQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgVAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIFQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgVAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIFQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgVAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIFQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgVAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIFQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgVAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgMHZ3g0AAAAAAAAAAAAAAAAAAAAAAAAAAADgIROyS4o+Z+8uMvMqL/k3sXcXAAAAgN0RSgMAAAAAAAAAAAAAAAAAAAAAAAAAAIB7J2SX9N9O9u4ie8//TTANAAAAHnqO9m4AAAAAAAAAAAAAAAAAAAAAAAAAAAAAD5Hoc/buIGf5vT8AAADgHiCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgcLZ3A7C9c1Met3cL95ZTdXt3ANzHStu7ASD/cKxs7w6A+1ehx+zdAZBvFP8swt4tAPnGnMBEe7cAAAAAAAAAAAAAAAAAAAAAAAAAII8c7d0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACD/IJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAyAe2bNmi9957T0OHDtXjjz+url27qn///nrzzTe1fft2e7dnM+3atZODg4Pxdb/ZsmWLqf+hQ4fau6V74n5/3oD7VeXKlY33nbOzs73bsZo118yhQ4eaxm7ZsuUedgogo/vvioNcnX9js71byLOyU1rnYdYFm/cB3F/K5H1qynHbtQHcj5xq/O920jb79QHkBy4t8jx1RNWCNmwEuP/MCUxMV4XZrQ8gfyj9v5spR+3XBpAfONWydwcAAAAAAAAAAAAAAAAAgPvEpk2bNGbMGB07dkxJSUnZjpsyZYocHR1VrVo1vfTSSxo1atQ97BIAAAAPG0d7NwAAAAAAAAAAAAAAAAAAAAAAAAAAAAA8bIKCglS3bl21bdtWBw4cyDGQ5rbU1FQdP35co0ePlqenp+bMmXMPOgUePO3atTO+Zs6cae92kA/MnDnT9LoAAEjO9m4AAAAAAAAAAAAAAAAAAAAAAAAAAAAAeJj8888/6tKli27evJnpPjc3N/n6+srHx0cFCxbUlStXFBkZqcjISNO42NhYjRw5UiNGjLhXbQMPjI0bN5rqV155xT6NIN/49ddfM70uAOBh52jvBgAAAAAAAAAAAAAAAAAAAAAAAAAAAICHxS+//KKOHTtmCqR57LHHdOTIEV2/fl1BQUHavXu3tmzZopMnTyoiIkKRkZF6++23VaRIETt1DgDA3TV37lylpaUZX61atbJ3S8BDjVAaAAAAAAAAAAAAAAAAAAAAAAAAAAAA4B64fPmynn32WaWlpRnH3N3ddfjwYf3555+qVatWtnN9fHz0wQcfKDo6Wm+//bacnJzuRcsAAAB4SBFKAwAAAAAAAAAAAAAAAAAAAAAAAAAAANwDrVq1UlJSklG7u7srMDBQtWvXtmqdDz74QPv27ZO7u7utWwQAAAAkEUoDAAAAAAAAAAAAAAAAAAAAAAAAAAAA3HV//vmnTp06ZTq2cuVKlS5dOk/r1a1bV2FhYbZoDQAAAMjE2d4NAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA+6V155xVS3bt1ajz766B2t6enpadG4kJAQrVq1Srt379aVK1eUlJQkHx8f+fn56ZlnnlHjxo3vqI+M/vzzT/35558KDg5WbGysqlSpoi+//NLi+T/++KO2bt2qc+fOyc3NTVWrVtXo0aNVtmzZO+rr+PHjmj9/voKDgxUVFSUfHx9VqFBBgwcPVrVq1e5o7fTOnz+vOXPm6NSpU4qOjlbJkiXVqVMnPffcc3J0dLTZPrb0008/6e+//1ZoaKi8vb1VuXJlDRs2TP7+/jbfKzo6Wl9//bVOnjypsLAwubm5aeTIkercuXOW45OSkvT7778br4nY2Fi5u7urePHiateunXr37i0XFxeb92mNpKQkLVy4UHv27NGFCxeUkpIiX19fNWvWTIMHD85zf0eOHNGKFSt09OhRXbt2TWlpafLw8FC5cuXUsGFDPf300/Lw8LDx2eTszz//1OrVq3XmzBmlpKSoRo0aGj16tCpVqpTtnOTkZM2fP1///POPIiIiVKRIETVs2FAvvvjiHfe/fft2LV++XKGhoYqOjlbRokVVsWJFDR06VBUqVLijtfPq+PHjWrNmjQ4cOKCIiAilpKSoaNGiqlChgvr3768aNWrYbK/U1FR9++232rNnj0JDQ+Xl5aWaNWvq5ZdflpeX1x2tvXPnTi1ZskTnz59XTEyMihUrpipVqmjo0KF3fD22tfDwcC1btkw7d+5UVFSUkpKS5O7urpIlS6pu3bp68skn7/h69tNPPykgIEDnzp2To6Oj6tevr5deekklSpTIdk58fLy++eYbbd26VdeuXVPRokXVunVrDR8+XM7OxH0g/3NIS0tLy8vEhg0bKjw8XL6+vtq7d6+t+8p3e1u6pz0fl9vOv7HZLvvaQtkprfMw64LN+wDuL2XyPjXluO3aAO5HTun+wy1pm/36APIDlxZ5njqiakEbNgLcf+YEJqar+JQBPOzSfUpHylH7tQHkB0617N0BAAAAAAAAAAAAAAAAAORfh36Sfh5m7y6y1/Nbqe4zNl0yJiZGRYoUMR3bsWOHmjZtatN90lu1apWmTZumPXv2KD4+PsexBQsW1IABAzR79myL/ki+Xbt22rhxo1Hf/nPlt956S5999lmW+6X/k+bs5r/66quaPXu2EhMTM82XpCpVquj333+3OkBm6tSp+vDDDxUbG5vtGE9PT73//vsaM2ZMrutt2bJFrVv/729ihwwZorlz5yooKEhdu3bVqVOnspzn7Oysl156SZ9++mm2axcvXlwRERFGfeXKFRUrVizXnm7btGmT2rZta9R16tTRoUOHsh0/adIkffzxx0pKSsry/ooVK2r16tWqVq1ats9bVhwcHIzbbdu2VUBAgM6ePavHH39cx49n/tvGp59+Wr/88otRBwcH64MPPtCqVat06dKlbPe5rWHDhpo/f75q166d7ZjKlSsrKCgo17XSq1Spkk6fPp3t/fv27dPAgQN17NixbB8PBwcHNWnSRIsXL7Y4JGXatGn66KOPdO3atVzHenl56V//+leOrytrDB06VPPmzTPqzZs3q1WrVrm+jxo2bKiAgIBMITODBg3Sjz/+qJSUlExzHBwcNGDAAC1YsMCqHhMSEjR8+HD99NNP2V4vJKlo0aKaPn26Bg0alOX9Gd/Llrr9mKS3cOFCff755zp8+HCOPUmSh4eHXn75ZX300UcW7Zf+tevk5KTk5GRJ0jPPPKOff/45y8dWuvWcrF69OsfQlKy8+uqr+vrrr5WQkJDtmBIlSmjWrFnq06dPrutld83MSnavv+ysX79ezz//vEJCQnLtw9XVVS1bttS6deuyvD+7a9zLL7+sb7/9NtvHo0uXLvrjjz9MoWMJCQnq2bOn1q5dm+W1wcnJSePHj9fkyZNz7Ruwp/wZpQcAAAAAAAAAAAAAAAAAAAAAAAAAAAA8IBYuXGiqixQpclcDaSTpiSee0KZNm3INpJGkxMREzZ07V2XKlNH58+fztF/dunX18ccfW7RfVurXr6+ZM2fmGOZw6tQp1ahRQ8uXL7dozYSEBFWpUkX//ve/cwykkW4FB73yyiuqVatWtgEtOVm8eLGqVauWbSCNJCUnJ2vGjBnq1KlTtmNGjRplqseNG2dVH+PHjzfVH3zwQbZjGzVqpPfeey/H8z1z5oxq1apl8WOenaVLl6pKlSpZBtJk5emnn9a8efMsCqSRpL1796pevXqaPXv2nbRplWHDhqlhw4Y6evRojgE9aWlp2rlzpypXrqwffvgh13WbNWum8ePHWxRII0nR0dGZrjG21r1791zfR3v37lW5cuWMa0B8fLz8/f21cOHCbENT0tLStHDhQrVp08biXgICAuTl5aXvv/8+1/CXyMhIDR48WI899pjF6+dFcHCwBg0apD179uTakyTFxcVp8uTJqlq1quLi4qzeLyEhQWXLltWyZcuyfWylW89J2bJltXPnTovWDQ8PV/HixTVz5swcA2kk6fLly3r22WfVsWNHq3q3pTfffFMdO3a0KJBGuvW4rV+/3qo96tWrp88//zzHx2Pt2rWmsLSwsDD5+vpqzZo12V4bUlJS9PHHH2vw4MFW9QPca7lHFQIAAAAAAAAAAAAAAAAAAAAAAAAAAADIs99++81UV61a9Z7uX6BAARUrVky+vr7y8PBQWlqaIiMjFRISouvXrxvjLl++rMaNG+vixYtWrd+tWzcdPnzYqEuVKqWyZcvKyclJYWFhunDhQo7zn3nmGR04cMA0v1y5ckpOTta5c+cUERFh3JeWlqZnnnlG69atU4cOHbJdMzU1VRUrVsx0LgUKFFCVKlVUtGhRRUZG6tSpU7p586Zx/7Fjx1S5cmWLQw4kKSQkRP3791dqaqokqXDhwqpUqZI8PT11+fJlnTp1yhQcsW7dOn366acaO3ZsprXeeecdffzxx0pOTpYkLVu2TPPnz7eoj7i4OO3atcuoPT099dRTT2U5tk2bNtq7d6/pmJOTkypXrqzixYsrOjpagYGBSkpKUkpKivr27au6deta1EdGUVFR6tevn/EYuLq6qlKlSvLx8dHVq1d19uxZOTg4ZDvfwcFBRYoUUalSpVSkSBG5uroqNjZW58+fN4XWpKamavTo0apVq1aWISeOjo5W957dnC5duujPP/80HXN2dpa/v79KlCghJycnXbx4UcHBwcZ5p6amasCAAXJ3d1ePHj2yXPf555/PFCDi4uJirOvi4qLY2FhdvnxZly5dylOAkrU+/PBD07mWLVtWfn5+SklJUVBQkCIjI437oqKi1LVrV23cuFH169dXaGiopFuvrYoVK6pkyZK6fv26Tpw4oRs3bhjzNm/erJkzZ+qVV17JsZcVK1aod+/emYI+ihcvLn9/f7m7uys6OlpBQUGma9vff/+ttm3bauPGjaZ5Ob3ucpLbvIIFC6pEiRIqXry4PDw8lJycrIiICIWEhJjCTU6dOqVWrVqZrn+WaN26tXFddXBwkL+/v8qUKaMbN27ozJkzpkCjmzdvqlWrVjpx4oQqVaqU7ZoxMTGqXLmy6XGTpEKFCqly5coqUqSILl++rKCgINP1bP369WrWrJl27Nhh1TncqdWrV2vKlCmmY46OjipTpoxKlSolNzc3xcfHKyoqSmFhYXkKTOvatasOHTok6dbjXKFCBZUqVUqJiYk6efKkKaTp9OnTev755/XNN9+oTp06xnPg4uKiSpUqqVixYoqOjtaJEydM33MWLFigPn36qGvXrnl5GIC7zvrvnAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAsduLECVPdvHnzu75n0aJFNXDgQO3fv19JSUkKCwvTvn37tGnTJm3evFnHjh1TXFycNm7cqJIlSxrzwsPDNWbMGKv2WrNmjSSpTp06Cg0NVVhYmHbt2qXt27crODhY0dHROc5ftmyZpFthNCdOnFBYWJi2b9+u3bt368qVK9q4caMKFy5sjE9LS9P//d//GSEwWenVq1emQJqXX35ZSUlJOnr0qDZt2qSjR48qKSlJo0ePNo0LDQ3Vc889Z/H5//3330pNTZWbm5t+/vlnxcTEaP/+/dq4caOOHz+umJgYNW7c2DRn0qRJWa7l7OysTp06GXV8fLx+/PFHi/qYMGGCKayjX79+WY6bO3euNm/ebDrWo0cPJSQk6MSJE9q8ebMOHz6sxMREIzgnOTlZ+/bts6iPjA4fPqyUlBQ5OTlp+vTpunHjho4cOaJNmzbp8OHDiouL08KFC01zSpQooSZNmuiHH35QUlKSrl69qmPHjmn79u3asGGD9uzZo/DwcF27dk1PPvmkae7//d//ZdlHYGCg0tLSMgWatG3b1jie8SswMDDTOhlDWlxcXDR16lTdvHlTQUFB2r59u7Zs2aKgoCDFxcWpT58+pvnPPvus4uLisuzx+++/N247ODho+vTpSkxM1KlTp7R161bj3ENCQpSYmKhdu3bpqaeeUqlSpbJczxZun2vt2rV14cIFhYaGatu2bdq5c6ciIiL01VdfmcZv2rRJzz//vPHY9e3bVwkJCQoMDNTmzZu1b98+xcfHq1evXqZ577zzTo59hIWFqW/fvqbnr3Pnzrp48aIuX76sPXv2aOPGjTp48KDi4uK0aNEiFSxY0NTXzJkzTWu2bNnSeK7btm1rui+710RaWppatmxpGuvg4CBvb2+NHj1ap0+fVkJCgkJCQrR3715t3LhRW7du1cmTJ3Xjxg0tX77cdD07ePCg5s2bl+O5p5eSkqI9e/ZIkmrUqKHw8HCdO3dOW7du1b59+xQdHa2lS5fKxcXFmJOcnKzHHnssx3XbtWtnCqRxdHTU1KlTFR8fr0OHDmnz5s06efKkEhIS9PTTT5vm7ty5U++//77F52ALGQOMevfurRs3bigkJEQ7d+7Uhg0btHPnTp06dUrXr19XaGioXnzxRXl7e1u8x9q1ayVJHTp0UExMjIKCgrRlyxbt3r1bMTExev31103jFy5cqKeeekpRUVGSpHHjxikxMVHHjh3Tpk2bdOjQIcXFxalp06ameRm/BwH5CaE0AAAAAAAAAAAAAAAAAAAAAAAAAAAAwF2UMYCiSpUqd33PiIgILViwQI888kiO49q0aaPz58+rRIkSxrH//ve/Vu/XsGFDHTp0SGXLls10n4eHR67zS5QooXPnzqlatWpZ9hgYGKhChQoZx6Kjo/XGG29kudb58+f1yy+/mI59/fXX+uyzz7Ic/+WXX2r69OmmY4sXL1Z4eHiufd9WqFAhnT17Vj169Mh0n5ubm3bt2iUfHx/jWGxsrAICArJcK2OfH3zwgUU9pA92cXBw0JQpU7Icdzto5rZBgwbp559/lrOzc6ax06dPzxTkkRcODg7auHFjpr1vy/ga+fPPP7Vz507169cvy75u8/T01MqVK/Xqq68axyIiIrR69eo77jkr0dHRevfdd426UKFCOnXqVKZwittcXV21ZMkS05ykpCS99NJLmcYGBAQoOTnZqEeMGJHt43Vb48aN9euvv+rw4cPWnopV6tevr8OHD6t06dKZ7hs1alSmAKTvvvtOkvTGG29o0aJFWT6Hy5YtU7ly5Yw6NjZWW7ZsybaH7t276+bNm0b9ySefaO3atfL19c1yfN++fXXy5EkVKFDAOJb+ebAlf39/RUVF6csvv1SlSpVyHPt///d/On/+vOl6lpdAl5o1a+rYsWOma/dtzzzzjHbv3i0nJyfj2JkzZ7INuPrnn3+0f/9+o3ZwcNDq1auzfF07Ozvrl19+0bBhw0zHP/zwwxyDwmztzJkzxu2qVavqp59+MgXxZFS2bFl9/vnnRmCMpXr06KH169dn+X1s6tSpppC5lJQU49ozf/58TZs2LdMcFxcX7dixw7TeuXPnFBERYVVfwL1CKA0AAAAAAAAAAAAAAAAAAAAAAAAAAABwFyUkJJjq7EIU7MXZ2dn0x/NxcXE6deqUVfP/+eefO+phzZo1OQYK+Pr66uuvvzYdmzdvXpZjX375ZVPdtGlTjRgxIsf9x44dq3r16pmOZRUckp3vvvsuy3CInPpatmxZluOqVKliCrY4efKkzp8/n+Paa9asUXR0tFE3bNhQnp6emcatWLFCsbGxRu3t7a358+fnuPaYMWNUp06dHMfk5rnnnlPLli3vaI2cfPrppypYsKBRz549+67s88ILL5iCN1asWCF/f/9c502aNEl+fn5G/dNPP2UaExgYaKqfeOKJO+jUdpycnLRp06Ycx2QVXOTn56ePP/44x3mvvPKKqV66dGmW44KCgkyhKa1atdJrr72W49qSVK5cOb399ttGHRMTo5UrV+Y6727z9PTUqFGjjDokJMSqQBdHR0dt2LAhxzF169Y1hTVJ2QdcjRs3zlQ/++yz6ty5c47rf/PNN6bvZTdv3tSkSZNynGNLKSkpxu2GDRvelT3c3d31888/5zgmq+CZJk2aaNCgQTnO69Onj6nObR/AXgilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAO6i5ORkU+3j42OnTrL39NNPm+pFixZZPLdNmzZZBqBYqmLFimrQoEGu4wYOHKjChQsbdVRUlEJCQjKNyxjWkDHMJjsZg0wsDdopXLhwpoCBrAwbNsxUHzp0KNuxEydONNVjx47Nce0JEyaY6ilTpmQ57ssvvzTVI0eOzHHd22bMmGHRuOxkFVpiayVLljRuHzhw4K7s8fvvvxu3ixcvrq5du1o8t2/fvsbt+Pj4TEFDRYsWNdVr1qzJY5e21aJFC3l4eOQ4plixYnJ3dzcdyxg4k5X+/fub6uyet/fee89UZ3wd5+Stt96Sg4ODUf/www8Wz72bOnbsaKpXrVpl8dxWrVrlGoIlSf/5z3/k5ORk1BmDj247fPiwcdvBwcHia2bG61RWYUt3S/rnNH1gkS317t071zEtW7aUo6M5tiOroJqMevbsaaq3bdtmXXPAPUIoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAHAXOTs7m+qoqKh7tnd4eLhGjhypqlWryt3dXU5OTnJwcMj0VaRIEdO8U6dOWbxHjx497qjHjIE4OWnVqpWpXrJkialOSkpSdHS0UXt4eOiRRx6xaO3mzZubgjWioqIyBQplpXbt2hatX7p0aVOQwrVr17IdO2jQILm5uRl1+jCUjKKjo01hHj4+Pnr00UezHHvkyBFTPX78+NzaliQ9+uijcnFxsWhsRt7e3ipWrFie5krSnDlz1LhxYxUtWlQFChSQo6Njlq/h9AFF6V8DthIREaHY2FijbtiwoVXzmzVrZqp/++03U50x4ObLL7+0KNzibuvWrZtF47y8vEz1888/n+ucYsWKWfSe2Lx5s3HbxcVFdevWtagn6db1t1ChQkadUxiULQQFBalfv36qUKGCChUqlO01N+PjevToUYv3GDJkiEXjHB0dVb16daNOS0vT+vXrTWOOHDlius75+flZHDI2evRo0/N37tw5i+bZQvoQpxMnTqh3796Kj4+36R7PPfecReNcXV2N246OjmrTpk2uczIGsUVGRlrXHHCPEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAA3EXp/2BduhUUc7clJCSoe/fuKlWqlObMmaNTp04pPj5eqampFs235g/kswtAsVSXLl0sHtu6dWtTvXPnTlO9d+9eU12uXDmrevH3989xvayULVvW4vUdHf/35903btzIcWzfvn2N2wkJCZo9e3aW48aPH6+0tDSjzimw4urVq8btggULZgoSyUmpUqUsHptemTJl8jRvzpw5KlSokEaOHKk9e/YYIUHpzzU7SUlJedozJxmDgdauXZtl2Eh2Xz179jTNP3/+vKl2c3NT8+bNjTotLU3jx49XwYIF1ahRI02YMEHHjx+3+XnlpmbNmhaNK1iwoHHbwcHB4teWJe+JsLAw43ZSUpJVj7uDg4MprCR9sJAtRUREqFmzZqpcubIWLVqkc+fOKSEhweJrrjXfF5588kmLx2YM5frnn39M9YYNG0x1rVq1LF5buhWCdVtiYqLF53unXnnlFVO9fPlyeXh4qGrVqho5cqQCAgLueI/69etbNK5AgQLG7fQBSDkpUaKEqY6Li7O8MeAeIpQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAuIs8PDxMdVBQ0F3dLyEhQVWqVNEff/xxR2tYytrgl4xq1Khh8dhKlSqZ6qioKFMdGhpqqjP+4X9uMo7PuF5WPD09rdrjttzCVT755BM5ODgY9bRp07Ict3jxYuO2o6Oj3n///WzXTE5ONm5bGp5wW/rwCWvk5fF5/fXXNXLkSKteh+ndjWCMkJAQm64XERGR6di6desyhf8kJSVp7969mjx5smrWrKmCBQuqdu3amjZtmun5vFssfd7Tv1bTB81YI7v3xM2bN/O0XlZyC4PKi8uXL6tSpUqZQrKskT44JyfWBP5ImYO2Ll++bKovXrxoqq0J2ZKUqZf0AUJ304QJE9S1a1fTsbS0NJ06dUpz5sxR+/bt5eTkpHLlyumFF17IdN6WKFasmNVznJ2dLRqX8T1iSdgWYA+E0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAB3UbVq1Uz1tm3b7up+PXv21Pnz503HKlWqpBdffFFLly7ViRMnFBkZqZSUFKWlpRlf6VnzB/Jubm531K81wTFFixY11XFxcaY6Y9CHu7u7Vb1kDBC6cuWKVfNtycvLS40aNTLqs2fP6tSpU6YxK1asMD0GLVu2lKurq0Xru7i4WNVPXp/nAgUKWDV+69at+uSTT0zHPDw89PTTT+urr77Srl27dPHiRSUmJppev+kDi+5GwENeQi1yklVwjpubm86fP6+XXnop29CgpKQkHT16VOPHj5ebm5v+/e9/27Sv/MiWz+fdeG106NBBMTExpmO1a9fW66+/rl9//VVnzpzR1atXTa/XzZs356kvawN/MobGXLt2zVRfvXrVVBcpUsSq9TNeF8LDw62afydWr16tuXPnZvs9JDU1VSEhIfrqq6/k6+urzp075znoCnhYWRazBAAAAAAAAAAAAAAAAAAAAAAAAAAAACBPnnzySa1bt86oT548edf2iouL05o1a4za0dFRv//+u7p165bjvHsZJJDR5cuXVbZsWYvGRkZGmuqMITLFihUz1devX7eql4whN8WLF7dqvq198sknatu2rVGPHTtWv//+u1FPmjTJNH7atGkWr52UlGRVL/Hx8VaNz6sRI0aY6h49eujnn3/Odd7dDpvw9PQ01X379tWiRYtsvo+jo6NmzZqlWbNm6e+//9b8+fO1fft2hYaGKjk52TT25s2bmjp1qnbu3KmAgACb95Ifubu7Z3qf2tOBAwd09OhRoy5YsKC2bdumBg0a5Dgvr4FXWYUZ5SQ6OtpUZwyd8fb2NtUZQ2tyk/G64Ovra9X8OzVkyBANGTJEx48f1+zZsxUQEKDTp0/rxo0bpnFpaWn666+/VLZsWYWEhNxxmBrwsLAuBgsAAAAAAAAAAAAAAAAAAAAAAAAAAACAVQYMGGCqr127pt27d9+VvWbPnm2qhwwZkmsgjSQdO3bsrvRjCWtCeoKCgky1j4+Pqfbz8zPVly9ftqqXjOMzrnevtWnTRiVKlDDqP//80wiluHz5so4cOWLcV7JkSTVt2jTH9ZydnY3bGUMbchMVFWXV+Lw6ceKEcdvLy8uiQBpJio2NvVstSbrz11ZedOrUST/++KPOnDmjmzdvateuXXruuefk7u5uGrdx40Z99dVXd70fe0n/uk1MTLRjJ5nNnDnTVE+bNi3XQBpJOn36dJ72S0tLU0xMjMXjQ0JCTHX664kklSpVylSfP3/eqn4yht6ULl3aqvm2UqNGDc2aNUuHDh1SfHy8QkNDNW7cuEz9REZG6sknn7RLj8D9iFAaAAAAAAAAAAAAAAAAAAAAAAAAAAAA4C7y8vJS9erVTcfGjRt3V/bau3evqR4zZoxF81auXHk32rHI6tWrLR67ZcsWU50xhKVhw4amOjg42KpeMgY4ZFzPHl544QXj9s2bNzV9+nRJmV9Do0aNynUtb29v43ZiYmKmQImchIeHWzw2ryIiIpSSkmLUzZs3t2heUlKSVUEdefHoo4+aansEOTVu3Fg//vij4uLi1K5dO9N9t18XD6KiRYsat5OTk3X27Fk7dmOW8XWQ/v2ak3/++SfPe/76668Wjz148KCp7tChg6lu3769qT569KhVvaQPqypYsKAcHfNHhEXZsmU1bdo0XbhwQf/5z39M9wUEBNinKeA+lD/e0QAAAAAAAAAAAAAAAAAAAAAAAAAAAMADLGNgxKZNm+4olEBSliEc6QMCJKlChQoWrbV8+fI76uVOWBOIkzGU5tlnnzXVLi4upuCVuLg4HThwwKK1d+7cqevXrxu1j4+PnJ2dLe7tbnn77bdNfXz22WeSpBUrVhjHnJ2dNWHChFzXql27tqmeNm2aRT2sX79eiYmJFo29ExlDgYoVK2bRvA8++CDPe6YPwclJtWrVVKhQIaO+ePGizp8/n+d979T69evl4OBg1GFhYXbr5W5r0aKFqZ48ebLN9yhQoICpTkpKsmhebGyscdvR0dHiUJZNmzZZ3lwG//3vfy0al5qaquPHjxu1g4NDpnCl2rVrm64voaGhFgc8zZ49W2lpaUZdvnx5i+bda+PHj1eZMmWMOiUlxerAMuBhRSgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAcJd169ZNlSpVMh176qmnFB4enqf1Dhw4oNKlS2c67unpaaq3bt2a61orVqywa6BFUFCQRcExP/zwgykswcfHR/7+/pnGtW/f3lSPHDnSoj5Gjx5tqjt16mTRvLvN0dFRnTt3NuoLFy7onXfeUXx8vHGsQ4cOFgXoZDzHr7/+2qIexo4da2G3d8bX19dUBwUF5TonOTlZM2fOtGqf9GEu165ds3hextdWv379rNrXlhwdHeXm5mbUqampduvlbnvzzTdN9cKFCxUdHW3TPTJeO8+ePWvRPHd3d+N2amqqIiIicp3z/vvvm96/1tqyZYtF+7zxxhum0KWqVatmOa5u3brG7bS0NIuvmRnDoPr27WvRPHtIH0oj3QosA5A7QmkAAAAAAAAAAAAAAAAAAAAAAAAAAACAe2DLli0qUKCAUcfFxalKlSo6evSoVetMmDBBDRs21PXr1zPd17x580xjc3L27Fn179/fqv3vhq5duyo5OTnb+y9fvqzhw4ebjg0ZMiTLsZ999pmp3rlzp+bMmZPj/p999pn27dtnOjZr1qwc59xLGc8pYxjEp59+atE6vXr1koeHh1FHRUVp6NChOc756quvdOjQIQs7vTOlS5eWk5OTUe/cuVNRUVE5zmnXrp3VAROurq7G7TNnzlg877vvvjMF2mzatEn//ve/rdo7JCRECxcuzHR806ZNOnnypMXrBAUFma4BRYoUsaqP+0njxo1Vq1Yto05KSlLdunWtCnZJTU3VtGnTsr2/evXqpnrp0qUWrVunTh1T/eKLL+Y4fuvWrXrvvfcsWjs7KSkp6tChQ45jjh49mum68Pbbb2c5NuPjsmTJEv399985rj969GhdvHjRqAsUKKCJEyfmOMdWwsLCtGrVKovHp6am6tixY6ZjNWrUsHVbwAOJUBoAAAAAAAAAAAAAAAAAAAAAAAAAAADgHvD19dWiRYtMoRZxcXGqXbu2unbtquPHj2c7Nzo6Wu+88468vLw0efJkpaamZjlu2LBhpvV3796tZ599NsvxCxcuVI0aNZSQkHAHZ2Ub4eHhKl++vE6dOpXpvq1bt6pKlSq6ceOGcaxIkSKaMmVKlmuVLVtWPXr0MB0bNWqUxo4dm+X4l19+Wa+88orpWN++fVWiRAkrz+LuqVSpkqpUqZLlfX5+fqbAjtzMmDHDVM+bN0+9e/fO8jUyfvx4vfDCC9Y1e4dq165t3E5JSVH16tUVFBSUaVxISIhq1aqlrVu3Wr1HpUqVjNvXr19X+/btdeDAgVznlShRQm+88Ybp2NSpU9WoUaMcw6VSU1M1Z84cPfLIIypXrlym50C69X6sXr26qlWrpqlTp+b4vty6davq169vOta1a9dc+7+f/f7776ZQr9DQUJUsWVKzZ8/Ocd7u3bvVs2dPubm5afz48dmO69Onj6mePHmyvvrqq1wDj1599VVTvXTp0mz3mTJlitq2bZvt9dsahw8fVp06dRQREZHpvuXLl6thw4ZKSUkxjlWsWDHbALIOHTqYXk9paWnq2rVrlmFXycnJ6tWrV6bH/e2335aj472Jrzhz5oyeeOIJlShRQi+//LLCwsKyHRsWFqY6deqYnsfy5cvfs16B+52zvRsAAAAAAAAAAAAAAAAAAAAAAAAAAAAAHha9evXSn3/+qW7duik5Odk4vnbtWq1du1bu7u4qVaqUvL295eLiooiICEVERCgyMtKi9T09PdWjRw/9/PPPxrGlS5fq559/VrVq1eTj46Po6GgFBQXp+vXrxphBgwZpwYIFtjtRK/Tu3VvLli3ThQsXVLVqVZUuXVr+/v5KSUnRuXPndOXKFdN4BwcH/fzzzzmGCixfvlxlypRReHi4pFshCzNmzNCXX36pKlWqyMfHR1FRUTp16pSSkpJMc/38/LRo0SLbn+gdeueddzRgwIBMxzMG6uRm6NChWrBggbZs2WIcW758uVxcXFSlShUVL15c0dHRCgwMVGJioiTJ2dlZdevW1b59++7oHCzx7bffqkmTJkZ95coVVa5cWX5+fipXrpySkpIUGhqqixcvGmOKFCmiokWL6syZMxbt8cYbb5gCOgICAjKFvEhSjRo1dOzYMdOxyZMn6/Dhw1q1apVxbO/evapdu7Y8PT1Vrlw5eXt76+bNm4qJiVFYWJiio6OVlpZmUW+BgYH697//rX//+9/y9PRUmTJl5OXlJRcXF0VHR+vcuXO6du2aaY6bm5vmzJlj0fr3qwoVKujnn3/WU089ZYS6xMXFafTo0RozZoz8/PxUvHhxFSxYUDExMbpy5YouXbpkus7mpG7duipVqpTxukpMTNQLL7yQZSjTtm3b1Lx5c2NegwYNTO+NadOm6YsvvlC1atXk6elpXGtuv58kqV+/fvrxxx+tfhycnJxUv3597dmzR0eOHFGJEiXk7++vsmXLKiEhQUFBQYqOjjbNcXZ21l9//ZXjugEBASpdurTxfSElJUWvvfaa3n77bVWpUkVFihTRlStXdOrUKVPYjSQ1adJE77zzjtXncqeuXLmizz//XJ9//rkKFSqk0qVLy8fHR+7u7rp+/bpCQ0ON7wHp/fDDD/e8V+B+RSgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAcA916tRJx44dU48ePXT06FHTfdevX9fp06dzXcPHx0dfffVVlvetWLFClSpVMgV03Lx5U0eOHMly/GOPPab58+fbLZTmp59+Ur169XTo0CFJUlhYmMLCwrIc6+DgoCVLlqhDhw45runo6KizZ8+qdu3aCgoKMo4nJSVleszTq169ug4ePJiHs7j7+vfvr5EjR5rChFxcXDR27Fir19q8ebMaNGig/fv3G8dSUlJ04sQJnThxwjTW0dFRP/74Y7avN1tr3LixJk2apEmTJpmOh4aGKjQ0NNN4Dw8P7d+/X506dbJ4j379+umnn37Sb7/9luO4jIFFt/3+++966623NGXKFFPYTExMjA4fPpzr/i4uLhb1GRMTo5iYmBzHuLu7a/fu3XJ1dbVozftZ9+7dtXv3brVr106xsbHG8Zs3b+rMmTO5hhI5ODjkeP/q1avVtGnTbJ/32zKGsmzcuFH+/v66evWqcezGjRs6cOBAlvNHjBih/v375ymU5vZ+VapUUVhYmNLS0hQcHKzg4OAsxxYoUECbN29WpUqVclzT09NTp0+fVu3atU0haDdu3DCuzVnp0KGD1q9fn6fzsKUbN24oKCjIdL3PyNHRUf/973/VsmXLe9gZcH/LPv4PAAAAAAAAAAA8kAICAuTg4GB8ZfzBPR4e4eHhevfdd9WqVSuVKFFCBQoUML02AgIC7N0icF8YPHiw6b1z7tw5e7cEAAAAAAAAAAAAALgPVKlSRUeOHNH69etVr149FShQINc5jo6OqlOnjr7//ntFRkaqT58+2Y4NCgpS79695eiY/Z8Te3t7a/r06frzzz/zdA62dPDgQY0ePTrHsI7KlSvr+PHjeuaZZyxa09XVVadPn9bHH3+swoUL5zjW09NTM2bM0PHjxy0ODLGHrl275lhbY9++fZo4cWKO51uuXDkdOXLE4sfcVt59910tWrRIRYoUyXaMs7OzunTpoosXL6pChQpW77Fy5UotW7ZM9evXl7u7e66BJRlNnjxZ586d06OPPipnZ+dcxzs5OalatWr65JNPtH379kz3v/nmm+rbt69KlChhUS+urq4aMGCAoqKiVKNGDat6v581aNBA0dHReuutt3J8faRXrFgxDRgwINff63nkkUd06dIlDRo0SL6+vhY9r9KtYKSwsDC1b98+x+fO19dXS5Ys0ddff23Rutlxc3PThQsX1KNHDzk5OWU7rmHDhjp//ryaNm1q0bq+vr66fPmyXn755VxDjooXL65FixbZJZCmQYMGGjt2rCpWrJjj+d/m6OioFi1a6OzZsxo0aNA96BB4cDikpY9es0LDhg0VHh4uX19f7d2719Z95bu9Ld3Tno/Lbeff2GyXfW2h7JTWeZh1weZ9APeXMnmfmnLcdm0A9yOndP+hnbTNfn0A+YFLizxPHVG1oA0bAe4/cwIT01VZJ/EDD4/S/7uZkv2nRwAPBadad32LS5cu6eDBgwoODlZ0dLQSExPl4eEhLy8vFS9eXPXq1VPZsmXveh+4PwUEBKh9+/ZG/e677xJM8xBaunSp/vWvf+nGjRvZjtmwYYPatWt375oC7lODBw82fVLc2bNnVb58efs1BAAAAAAAAAAAAAD53aGfpJ+H2buL7PX8Vqp7bwM4btu0aZPWrVun8+fPKzw8XGlpafLx8ZG/v7+efPJJNW/e3Oo14+PjNX/+fG3atElRUVHy8fFRuXLl1KdPHzVo0OAunMWd++GHH7RlyxYFBwerUKFCqlKlil566aU7/r24o0ePav78+QoODtbVq1fl7e2t8uXL61//+td9E+rh5+en8+fPG/WZM2fyFMiS0dKlS/Xnn38qLCxMXl5eqlSpkoYPH65y5crd8dp36p9//tGSJUt0/vx5OTo6yt/fX02bNs134RJbtmzRL7/8orCwMEVGRsrZ2Vne3t6qWbOmOnToYNX7NzU1VWvXrtXWrVsVGBioa9euSboVnlStWjV17949T9eDB1F4eLi+++47BQYG6vLly7p586YKFy6scuXKqUWLFurWrZvc3NzuWT9RUVGaO3eudu3apZiYGBUvXlwVK1bU4MGDValSJZvvl5ycrG+++Ub79u3T+fPn5enpqVq1aumll16Sj4/PHa29fft2470XGxurYsWKqXLlyho+fHi++j3l3bt3a926dTp69KiioqKUlJSkwoULq2zZsurYsaMef/xxiwOGAJjxzgEAAAAAAACA+8SpU6f07bff6pdfftHp06dzHV+8eHG1adNGffr0Uffu3VWoUCGL9sn4h/W3Pfvss1q8eLHVfUu3/ji/UqVKyionPbc/3M+un/QcHByMUJ4KFSqocePG6tKlix599FGrP7kEeBhs2bJFzz33nFJTU+3dCgAAAAAAAAAAAAAAANJp06aN2rRpY9M13dzcNHr0aI0ePdqm695N/fv3V//+/W2+bq1atTRt2jSbr3uvHDlyxBRIU6lSJZsE0khSnz591KdPH5usZWsdOnRQhw4d7N1Grlq1aqVWrVrZZC1HR0d169ZN3bp1s8l6DzJfX1+9+eab9m7D4OPjo/Hjx9+z/Zydne/a9b158+b3RfhR48aN1bhxY3u3ATyQCKUBAAAAAAAAgHzuwoULGj9+vJYsWWJVgMSVK1e0YsUKrVixQoULF9aYMWP02muvycvLK099rFy5UjExMfL09LR67sKFC7MMpLGVtLQ0xcbGKjY2VqGhodq0aZOmT5+uqlWr6rPPPlOXLl3u2t7A/Wjs2LGm60nr1q01cOBAlS1b1vRpIPXq1bNHewAAAAAAAAAAAAAAAACy8MILL5jqd955x06dAACAhwGhNAAAAAAAAACQj/3xxx8aOHCgoqKisrzf3d1dxYoVU7FixZSYmKhLly4pMjIyU3hNbGysPvzwQ33xxRcKDg7OU7DMjRs3tGzZMg0ZMsSqeWlpaVq4cKHV+9lCYGCgunbtqkmTJundd9+1Sw9AfnPixAnt3r3bqNu1a6f169fL0dHRjl0BAAAAAAAAAAAAAICHild5e3eQs/zeHx5Khw4d0qZNm4y6cOHCGjhwoB07AgAADzpCaQAAAAAAAAAgn/r+++/1r3/9SykpKabjtWrV0tChQ/Xoo4+qTp06meYlJSVp06ZNWrNmjVasWKHg4GDjvujoaCUlJVnVh6OjoxFys3DhQqtDabZs2aIzZ85kuV5eDBgwINMP0tPS0nTt2jUdP35cv/76q/bt22e6f9KkSSpbtqzVvQMPou3bt5vqYcOGEUgD3KH58+dr/vz59m4DAAAAAAAAAAAAAO4f/k2k5/+Wos/Zu5PMvMrf6g/IJ+Lj4/X999/r1VdfNR2fOHGinToCAAAPC0JpAAAAAAAAACAf2rVrl4YMGWIKpPHy8tKsWbPUr1+/HAMkXFxc1LFjR3Xs2FFTpkzR/Pnz9dFHH5nCaazRvn17rV+/XpK0efNmnTt3TuXLl7d4/oIFC4zbvr6+KlasmI4cOZKnXiSpYsWK6tixY7b3T5w4UQsXLtTw4cOVmJhoHP/3v/+t3r17y9PTM897Aw+CwMBAU12rVi07dQIAAAAAAAAAAAAAAB5q/k0IfwFy4ODgkO19ZcqU0euvv34PuwEAAA8jPvYSAAAAAAAAAPKZqKgoPfPMM7p586ZxrGzZstqyZYsGDBiQYyBNRgUKFNCwYcN08uRJjRo1Kk/99O3bVwUKFJAkpaWlaeHChRbPvXHjhpYtW2bUzz33nJycnPLUhzUGDhyoL774wnQsMjLSqt6BB1V0dLSpJqgJAAAAAAAAAAAAAAAAuH+4urpq8+bN9m4DAAA8BAilAQAAAAAAAIB8ZtKkSQoODjbqAgUK6LffflOtWrXyvGbBggX11Vdfafny5XJxcbFqbtGiRfX4448b9ffff2/x3F9++UUxMTFGPWjQIKv2vhNDhgxRtWrVTMf++uuve7Y/kF8lJCSY6pw+UQkAAAAAAAAAAAAAAABA/lCoUCF17txZFy5cUIUKFezdDgAAeAg427sBAAAAAAAAAMD/REZGat68eaZjb731lurXr2+T9f/v//4vT/MGDRqkX3/9VZJ0+vRpbdu2TS1atMh13oIFC4zbjzzyiOrWrZun/fPCwcFB3bp108mTJ41jhw8fvmv7BQcHa//+/Tp//rxiYmLk4OAgd3d3lSpVShUrVlStWrXk6upq9brR0dE6cuSITp48qatXryopKUleXl4qUaKEGjdurHLlyt2Fs7G/s2fP6ujRowoJCdG1a9fk7OwsHx8flStXTs2aNZOHh4dN9rlw4YK2bt2qsLAw3bhxQ8WLF9cjjzyihg0b2jysJS0tTYcOHdLx48d1+fJlXb9+XcWKFVPZsmXVunVrm52TJX3cDXFxccZjGR4eLldXV7Vt21YNGjTIcV5ISIh27dqlS5cu6dq1a/Lx8ZGvr69atmyp4sWL27zPI0eO6OjRozp//rxSUlJUvnx5tWvXTiVKlMhxXnJysnbs2KHDhw/r6tWr8vT0VOXKldWuXbs8vbfvVEJCgjZt2qSzZ88qIiJCHh4eqlq16j19LdlSQkKCjh07puPHj+vKlSu6fv26ChcurKJFi6pOnTqqXbu2HB1t/3kbJ0+e1M6dOxUWFiYnJyeVKFFCTZo0UY0aNWy+lzUOHjyoPXv26PLlyypYsKB8fX3VokULlS9f3ibrR0dHa9u2bbp48aKuXLkiV1dXFS9eXPXr11fNmjVtsgcAAAAAAAAAAAAAALgzd+v3fAAAACxFKA0AAAAAAAAA5COzZ89WfHy8Ubu5uenVV1+1Y0e3PP744ypatKgiIyMlSQsXLsw1lObChQtat26dUQ8cOPCu9piVihUrmuqIiAibrp+amqq5c+fqyy+/1KFDh3Ic6+LiooYNG+r//u//NHr0aBUqVCjbsfv379eSJUv0119/6eDBgzn+ckHFihU1ZswYDRs2LMc1rbVnzx41btzYqFu1aqXNmzdbvc6CBQs0ePBgo37hhRf0xRdfZBqXkJCgP/74Qz///LP++ecfhYeHZ7umk5OTHn30Ub355ptq166d1T1J0u7duzVu3Dht3rw5y8e3fPnyevvttzVkyBBJ0uDBg00hS2fPnrU4HOLKlSv6+OOPtWTJEl28eDHLMS4uLurSpYs++OADm4c3BQQEqH379tnen92nJm3YsMH0+LZr104bN2406tuP27Fjx/Thhx9q5cqVpuuXJI0ZMybLUJrU1FTNnz9fM2bM0JEjR7Lc39HRUU2aNNFbb72lJ554Itv+0zt37pzpfAYNGqT58+dLkn744QdNnz5dBw4cyDSvQIECGjhwoKZNmyZvb2/TfTdv3tT06dP16aef6sqVK5nmFi5cWBMmTNBrr70mZ+e7/6O3a9eu6e2339aCBQsUGxub6X5XV1cNGjRIH330kYoWLZrp+X/33Xc1adKkLNe2ZmxWypcvr+DgYElSuXLldO7cuRzHnz9/XkuWLNEff/yh7du3KzExMdux3t7e+te//qXXXntNpUuXtqifnF4Pf/31lyZMmKA9e/ZkObdGjRr6z3/+Y/Frz5prRE6P8+LFi/Xee++ZAtXSa9q0qT755BO1atXKor4y+v333/XJJ59o27ZtSk5OznKMv7+/XnnlFY0ePVoFCxbM0z4AAAAAAAAAAAAAAAAAAOD+Z/uPEwQAAAAAAAAA5NnKlStNde/evVWkSBE7dfM/BQoUUN++fY166dKlOYYHSLcCIFJTUyVJzs7O6tev313tMSsFChQw1dn9AX5eXL16VW3atNGIESNyDaSRpKSkJG3fvl3jxo3ThQsXsh33xRdfqEGDBpo6daoOHDiQ66fdnDlzRmPGjFGjRo10+vRpq88jO40aNVLNmjWNeuvWrTp79qzV66QPaZBuhUJkpVWrVurVq5cWLVqUYyCNJKWkpOivv/5S+/bt9eKLL1r9vE6ZMkXNmjXTpk2bsn18z507p6FDh+qZZ55RUlKSVeunN2/ePFWqVEkzZszINpBGuvX6+O2331S/fn29//77ed7vXvvxxx9Vv359LV68OFMgTXYuXLigxo0ba8iQIdkG0ki3gmt27NihJ598Ut26dcsygMUSKSkpGjhwoAYMGJBlII10K3hm3rx5atmypSl4JiIiQq1atdKbb76ZZSCNJMXGxuqNN95Q3759lZKSkqceLXXw4EHVqFFDX3zxRbaPR0JCgubMmaN69erp6NGjd7WfO3Ho0CH5+/vr9ddfV0BAQK7fU65evapPP/1UNWvW1Jo1a+5o7/Hjx6tz587ZBtJI0vHjx/Xkk0/qgw8+uKO9LJWUlKT+/fvrueeeyzaQRpJ27typdu3aGeE6lrp8+bLat2+vJ598Ups2bcrxuhkSEqKxY8eqbt26CgoKsmofAAAAAAAAAAAAAAAAAADw4CCUBgAAAAAAAADyibi4OO3fv9907PHHH7dTN5kNHDjQuB0dHa3ffvstx/Hpw0g6d+6sEiVK3LXeshMWFmaqixYtapN109LS9NRTT2nr1q2m4w4ODipdurQaNGigpk2bqmbNmvLy8rJq7YSEhEzHChcurGrVqqlJkyZq0KCB/Pz8Mo05duyYWrdurcuXL1u1X07SB8ikpaVp4cKFVs0PCQlRQECAUdeoUUONGzfOcmxW5126dGnVrl1bzZo1U506dbIMaPryyy81fPhwi3uaNm2a3nzzTSMw6TYvLy/Vq1dPdevWNe2zbNkyvfjiixavn97EiRM1dOjQTOEhnp6eqlWrlpo0aaLy5cub7ktNTdW7776rMWPG5GnPe2n16tUaOHCgEdrj6OioSpUqqXHjxipXrpycnJwyzTl79qxatGihffv2mY47OjqqYsWKatSokcqVK5dp3po1a9S+fXtdvXrV6j5ffvllff/990ZdvHhxNWjQQHXq1FHBggVNY48fP24EaN24cUOPPfaYdu3aZdzv7++vxo0bq1q1anJwcDDNXb58uaZMmWJ1f5Y6fvy4OnbsmCncyNnZWVWqVFGjRo1M14YLFy6oS5cuNr0m2FJSUlKmUCgXFxdVqlRJ9evXV5MmTVSlShU5Ozubxly7dk3du3fXhg0b8rTvG2+8oWnTphl14cKFVatWLTVq1EjFixfPNP6dd97Rzz//nKe9rDFo0CD9+OOPRu3t7a26deuqQYMGmb6PpKSkaOjQodq9e7dFa586dUrNmjUzXY+lW9+zypcvr0aNGqlOnTry8PAw3R8YGKjmzZsrMDAwT+cEAAAAAAAAAAAAAAAAAADub4TSAAAAAAAAAEA+sX37dqWkpJiONWrUyE7dZNa4cWPVqFHDqHMKKNm9e7eOHz9u1OnDTe6ljH+AX6FCBZusu3z5cm3evNmo3d3dNW3aNF28eFEXLlzQ3r17tWPHDh09elRXr15VSEiIFixYoJ49e2YKWMhKwYIF9cwzz2jhwoUKDg5WTEyMTpw4oZ07d2rv3r0KCQlRRESEZs+erdKlSxvzwsPDNWzYMJucoyT1799fjo7/+1FC+mAPS3z//fem0IncXgf+/v567bXXtH79el27dk0XLlzQ4cOHtX37dh06dEhXr17VwYMHNWrUKFPgyXfffadffvkl1352796tN99803Ssdu3aWrt2rSIiInTgwAEdPHhQkZGRWrNmjWrVqiVJ+vbbb7Vp0yZrTl3fffedPvzwQ6N2cHDQwIEDtXv3bl29elVHjhzRzp07dfbsWV24cEFvvvmmChQoYIyfNWuWfvrpJ6v2zE69evX0999/G1+PPfaY6f4ffvjBdP/tr3r16uW47vPPP6/U1FQVKVJE06dP16VLl3T69Gnt2rVL586d04ULF/Tcc88Z45OTk9W3b1+FhIQYx5ydnfXGG28oNDRUQUFB2r17t86dO6fTp09r6NChpv327t2rUaNGWXXuGzdu1FdffSVJRsDM5cuXtXfvXh06dEgRERF67733TAEzf//9t/744w+9+uqr2r9/vxwdHfXCCy/o7NmzCg4O1q5du3TixAmFhISoZ8+epv0++uijuxICk5ycrP79+ysiIsI45u7urqlTp+rSpUsKDAzU7t27FRISolOnTun555+XJJ0/f15vvPGGzfuxpbZt22rGjBk6cuSIrl+/rtOnT2vfvn3auXOnAgMDFRsbq19//VVNmjQx5qSmpqp///6Ki4uzaq9NmzbpP//5jySpadOm+uuvvxQVFaUjR45o9+7dunTpkjZs2KDq1aub5r388stKTk6+85PNxvfff68lS5ZIkrp06aLt27crMjJSBw8e1N69exUREaFffvnFdL1PSUmxKDArPj5eTz31lM6ePWscq1ChgubMmaPIyEidPXtWu3fvNq6xa9asUYMGDYyxV65cUZ8+fZSYmGjDMwYAAAAAAAAAAAAAAAAAAPcDQmkAAAAAAAAAIJ8IDAw01Z6enjYLUbGV9KEia9euzTZ8YcGCBcZtb29vPfnkk3e9t4x27NiRKUikQ4cONll72bJlpvr333/XuHHjVLJkySzH+/n5aeDAgVqxYoXOnDkjX1/fbNfu2rWrQkJCtHTpUg0YMED+/v5ZjitatKhGjhypI0eOqGHDhsbx3377TceOHcvDWWVWunRpderUyaiDgoK0ZcsWi+enDy5ydHRU//79sx07e/ZsnTlzRp988ok6dOggT0/PTGMcHBxUt25dffXVV1qzZo0KFixo3DdlypRc+xk5cqQp+KlDhw7avXu3OnfubAq5cXJyUpcuXbR79261bdtWkkyBDrk5c+aMKSyiUKFC+uOPP7RgwQI1atTIFPQj3XqcJ0+erHXr1qlQoULG8ZdeekkJCQkW75sdb29vdezY0fgqVaqU6f6WLVua7r/95e3tneO6ly5dkq+vr3bs2KGxY8eqWLFipvtLlixpChL57LPPtHPnTqN2cXHR77//ro8//tgUtiFJlSpV0rfffqtvvvnGdHzp0qX69ddfLT73c+fOSbr1WK5du1aNGzc23e/h4aF33nlHkyZNMh1/7bXX9M0338jJyUlLly7VF198ofLly5vGlC1bVj/99JPpunLjxg0tWrTI4v4sNXv2bO3bt8+oPT09FRAQoNdff10+Pj6msZUrV9a8efOMMB5rXrv3kr+/v44cOaKAgAC98sorqlWrVpahXa6urnrqqae0fft2U1BRWFiY1UFZtx+LwYMHa+vWrerUqZNpTwcHB7Vr106bNm0yvSYvXLigP/74w9pTtNiZM2ckSRMnTtSaNWvUrFkzU1CSk5OTnn76aW3YsEFubm7G8V27dungwYM5rv3aa6+ZQuKeeOIJHTp0SMOHD8/0Hnd2dlaXLl20bds29ejRwzh+4MAB4/UEAAAAAAAAAAAAAAAAAAAeHoTSAAAAAAAAAEA+ERUVZaozBjzkB/379zcCNZKTk7MMX0hKStLixYuN+plnnjGFh9wLgYGB6tOnj+mYk5OTnnvuOZutf1v16tXVvn17i+f6+fnJw8Mj2/tr1aqlEiVKWLyet7e3Fi9ebAo6mT9/vsXzc5M+iEgyB83kZMeOHabHqWPHjipTpky241u3bm0KhslNp06d9Prrrxv1rl27cgzj2bp1qynUo2jRolq2bJlcXV2znVOoUCEtX74813CWjKZOnar4+Hij/u9//6uuXbvmOq9Nmzb65JNPjPry5cv64YcfrNr7Xps/f76qV6+e67iUlBR99tlnpmOTJ09Wly5dcpw3bNgwjRw50nRs+vTpVvXYvHlzzZw50xTykdH48ePl5eVl1CdPnlRaWpr+/e9/q1evXtnOc3Jy0ocffmg6tmbNGqv6s8Tnn39uqmfNmqVGjRrlOGfUqFE5BkHZW4kSJVSrVi2Lxzs6OurLL79UpUqVjGPfffed1fs2atTICBzKTvHixTVx4kTTsbvxvKb31FNP6f33389xTNWqVfXSSy+ZjuXUV2hoqObOnWvUdevW1bJly3L8HiRJBQsW1A8//GAKYvrss89MoV4AAAAAAAAAAAAAAAAAAODBRygNAAAAAAAAAOQTGUNpihQpYtX8TZs2ad26dbl+bd26Nc89lilTRo8++qhRZxVQsmrVKtO5ZAw1uRvS0tJ07do17dy5U//+97/VsGFDhYSEmMY8//zzqlatmk32u3HjhnG7QIECNlnzTlSpUkVNmjQx6m3bttls7aefflqenp5GvWzZMiUkJOQ6b8GCBab6brwOMgZu5HTeGV+rr7/+unx8fHLdo1ixYho3bpzFPUVFRZn2at68uZ599lmL5w8bNswUSrRixQqL595rrVq1UufOnS0a++effyo0NNSoy5UrpzFjxlg096OPPpKbm5tRb9myRcePH7e4z/fee88U2pQVV1dXPfbYY6Zj7u7uGj9+fK7rN2/e3PSc7d+/3+LeLLF9+3adOnXKqGvUqKGBAwdaNHfy5Mm5nvv9xMXFRb179zbq/fv3m67Hlnj//fctum4/88wzpjp9qNXdMHnyZIvGZQxcy6mvL7/8UsnJyUY9bdo0i0Pi3Nzc9Oqrrxp1cHCw9uzZY9FcAAAAAAAAAAAAAAAAAADwYHhwfgsVAAAAAAAAAO5zsbGxptrd3d2q+T179lSnTp1y/erXr98d9Zk+XGT//v06evSo6f70YSRVq1ZV8+bN72i/jN577z05ODiYvhwdHeXl5aVmzZpp6tSpiouLM81p0aKFZs2aZbMeSpcubdw+duyYzUMo8qJChQrGbVv2U6hQIVM4Q3R0tFauXJnjnMTERC1dutSoPT091aNHD5v1dFv6c5ZyPu8tW7aY6ueee87ifTKG3+QkICDAFJIxYMAAi+dKt0KO2rdvb9Tbtm1TamqqVWvcK3379rV47MaNG031wIED5ezsbNFcHx8fPf3006ZjmzZtsmiul5eXOnbsaNHY2rVrm+pOnTpZHA6Wfu6VK1csCm6yVFavXQcHB4vm+vn5qW3btjbrJT9I/75PTk7WkSNHLJ5bpEgRi4OUfHx85O/vb9TpQ5VsrU6dOqpZs6ZFY2vXrm167+TU1+rVq43bvr6+Fr8XbssY1LR582ar5gMAAAAAAAAAAAAAAAAAgPsboTQAAAAAAAAAkE8ULlzYVF+/ft1OneSsR48epl7Th9BcuXJFa9asMWprAzlszcHBQaNHj9bff/8tV1dXm63bqVMn43ZKSoq6dOmiuXPnKj4+3mZ7SNKlS5f0+eefq1+/fqpTp45KliypQoUKZQrlcXBw0OLFi4158fHxpmCUOzV48GBTvXDhwhzH//7777p69apR9+7dW4UKFbJ4v127dumtt95St27dVLFiRfn4+KhAgQKZzrlgwYKmeREREVmuFxcXpxMnThi1n5+f/Pz8LO7H399fZcqUsWhsxtCGRo0aWbxP+v1ui4mJ0YULF6xe415o0qSJxWN37txpqjt06GDVXo8++qip3rFjh0XzGjRoYHGAS9GiRTPNtVTGuTExMRbPzc2ePXtMdYsWLayab+14e4iPj9eSJUs0YsQINWvWTKVLl1bhwoXl6OiY6X0/YsQI09zs3vdZadCggRwdLf/xaIkSJYzb165ds3ietay5ThQoUEBeXl5GnV1fV69eNQX2WHvukvlaJEnHjx+3aj4AAAAAAAAAAAAAAAAAALi/WfYRlLDa5cuX1bBhw7u2/t69e+/a2gAAAAAAAADsw8fHx1TfzT+AvxNubm7q1auXvvvuO0nSjz/+qI8//lhOTk5atGiRbt68KelWIIw9QmkcHR1Vo0YNde7cWcOHD1e1atVsvsfIkSM1a9YsXbp0SdKt/yc8bNgwjRkzRh07dlT79u3VsmVLPfLIIypQoIDV60dERGj8+PFauHChUlJS8tRjdHS0VUEwOWnZsqUqV66s06dPS5L++usvXbp0SSVLlsxyfMbQmkGDBlm0z+bNm/Xiiy/q0KFDeeozOjo6y+Ph4eFKTU016ho1ali9do0aNSwKh8kY2mBNcEt2oqKirArRuVcqVKhg8djg4GBTXbduXav2qlevnqkOCQmxaF7x4sUt3sPNzc1mc20ZCpXxdWft6zcvr/d75ebNm/r000/10UcfKTY2Nk9rZPe+z0r6kBlLuLu7G7dt+ZxmlJe+bofxZNfXyZMnlZaWZtSrV6+2OKApO1FRUXc0HwAAAAAAAAAAAAAAAAAA3F8IpbExDw8PSVJqaqrCw8Pt3A0AAAAAAACA+0nGUJrIyEir5t/+A/WMAgIC1L59+zz3lZWBAwcaoTRhYWFat26dOnfurAULFhhj2rZtq3Llytl0X0kaMGCABg4caDrm4OAgd3d3FSlSRP7+/qYggbvBx8dHq1at0pNPPqmLFy8ax+Pj4/Xbb7/pt99+k3Tr/xm3bt1aPXv2VK9eveTl5ZXr2kFBQWrXrp3Onz9/Rz0mJibe0fyMBg4cqHfeeUeSlJycrB9//FFjx47NNO7KlStas2aNUVesWFGtWrXKdf05c+Zo1KhRphAFa2V3zhlDK4oUKWL12pY8d5L171tL5NeAKk9PT4vHXr161bjt6OiY6XqXm2LFimW7Xk5cXV2t2sdWc+/kdZzRnb5+LX3t3ms3btxQ9+7d9c8//9zROtZc6+7kOb2b7sZr7WG6FgEAAAAAAAAAAAAAAAAAgLuDUBobe/311zVt2jTFxcXZuxUAAAAAAAAA95mqVaua6mvXruncuXMqX768fRrKwe3AmeDgYEnSwoULVbp0ae3fv98YM2jQoLuyd8WKFdWxY8e7srY1GjVqpCNHjmjKlCmaN2+eoqKiMo2Ji4vTmjVrtGbNGo0dO1Zjx47VhAkTVKBAgSzXTEpKUrdu3TIF0lSpUkVt27ZVtWrVVKZMGbm7u6tQoUJycHAwxkybNk1//fWXbU8ynQEDBujdd981AhAWLlyYZSjNokWLlJycbNQDBw409ZmVDRs2ZAqkcXZ2VqtWrdS0aVOVK1dOJUqUkKurqwoWLGia26lTp1x7zxha4eLikuucjDLum52MASK2kJqaavM1bSG713FW0v/cxM3Nzeq9MgZNxcbGWr3G/epOX7+WvnbvtdGjR2cKpClevLjatWunevXqyc/PT56enipUqJCcnJyMMX/99ZemTZt2r9u97zxM1yIAAAAAAAAAAAAAAAAAAHB3EEpjY927d1f37t3t3QYAAAAAAACA+1Dz5s3l5OSklJQU49iePXvyZSiNg4ODBg4cqA8++ECS9Ouvv5pCI9zd3dWrVy97tXfP+Pj4aOrUqfrwww8VEBCgDRs2aOPGjdqzZ49u3rxpGhsbG6v33ntP69at019//ZVlMMfXX3+twMBAoy5ZsqTmz5+vLl265NrLvHnz7vyEclC+fHm1bdtWAQEBkqSDBw/q0KFDqlu3rmncggULjNu3Xye5ee2110yBNI8//ri+/vprlS1bNsd5GcM6slOkSBFTnZdg+ZiYGIvGZXxev/vuu1zPIzf16tW7o/n5gYeHh65duyZJio+Pt3r+9evXTXXhwoVt0tf9IKvXr5eXl8XzLX3t3ksHDhwwXSsKFCigqVOnavTo0bmG7gQFBd3t9h4IGa9F7du311tvvXVHa3p7e9/RfAAAAAAAAAAAAAAAAAAAcH8hlAYAAAAAAAAA8gkPDw/Vr19fe/bsMY6tXr0634a7pA+liY+P17fffmvc16NHD3l4eNirtXvOxcVFjz32mB577DFJ0o0bN7Rjxw6tXr1aixYtUlhYmDF269atGjdunL766qtM6yxZssRU//LLL2revLlFPURFRd3BGVhm0KBBRiiNdCuAZvr06UZ95MgR7d+/36hbt26tChUq5LhmYGCgaU7t2rX1888/5xpMIVl+zj4+PqY6/fNhKUvnFCtWzFTXrFlTTZo0sXq/B423t7cRSpOamqqrV69aFXARERGRab2HRVavX2tCaax5vTs4OFg8NiuWBg799NNPpiCq9957T6+88opFc+/Fte5BkPFa5Orqqo4dO9qpGwAAAAAAAAAAAAAAAAAAcD8ilAYAAAAAAAAA8pGnnnrKFErz008/aebMmfL09LRjV1mrXLmyWrRooW3btmW6b9CgQXboKP8oVKiQ2rdvr/bt22vy5Ml677339NFHHxn3z507V1OmTDE9r6mpqdq9e7dRP/LIIxYH0kjS0aNHbdN8Dnr16qUXX3xR169flyQtWrRIU6dOlZOTk6RbITXpWfI62LFjh6keOnSoRYE0kuXnXKpUKXl7e+vq1auSboXnJCcny9nZsh+TJCcnW7xXxhCe06dPE0ojqVy5cjp37pxRHzx4UO3atbN4/sGDBzOt97CoWbOm/v77b6Pev3+/atasafH8AwcOWDzW1dXVVN+4ccPiucnJycZ7LDfp3/eOjo4aOXKkxfvci2vdgyCraxEAAAAAAAAAAAAA5DcHLx9UaFyovdvIxM/DT/VK1LN3GwAAAIDdEUoDAAAAAAAAAPnIqFGj9PHHHys+Pl6SdP36dX322WeaOHGinTvL2sCBAzOF0pQtW1YdOnSwU0f5T4ECBfThhx9q48aN2rJliyTp5s2b2rlzpzp16mSMi4yMVHJyslFXq1bN4j0CAwN14cIF2zWdDQ8PD/Xs2VPff/+9JCk8PFx//fWXunbtqpSUFP3444/GWDc3N/Xu3TvXNS9dumSqrTnvf/75x6JxDg4OatSokRHscePGDa1fv16dO3e2aP5ff/2lhIQEi8a2b99e8+bNM/X43HPPWTT3QdasWTNt3LjRqP/55x+rQmkyPtfNmjWzVWv5XsZQo1WrVqlfv34WzU1JSdEff/xh8V4ZA9Ayvj9zcuDAAdM1LCfp1y1evLi8vb0tmpeammp6HSF7ZcuWVeXKlY0wmlOnTik0NFR+fn527gwAAAAAAAAAAAAAbjl4+aD6r+lv7zay9UPXHwimAQAAwEPP0d4NAAAAAAAAAAD+p2jRonr++edNxz788EMdOnTITh3lrE+fPipYsKDpWP/+/eXoyP9+zqhly5amOiIiwlSnpaWZ6qSkJIvX/uqrr/LemJUGDx5sqhcsWCBJ+vvvv3Xx4kXjeI8ePVS4cOFc18vreScmJuq///2vRWMl6cknnzTVX375pcVzv/jiC4vHduzYUc7O//tMgCVLligyMtLi+Q+qtm3bmuoffvjB4gCTq1ev6pdffjEda9Omjc16y+8ee+wx03V25cqVFodQrVixQuHh4Rbv5efnJwcHB6Pev3+/xXOXLl1q8dj073trrnW//fabzp8/b/H4h12XLl1MtTXXMgAAAAAAAAAAAAC420LjQu3dQo7ye38AAADAvcBfBQAAAAAAAABAPjNp0iT5+fkZdVJSkp588kmdOHHCjl1lzcvLS1FRUYqNjTW+PvjgA3u3lS9lDKHx9vY21UWLFjWFmezYscOi0I4DBw7c01Ca9u3by9/f36hXrlypmJgYLVy40DRu0KBBFq3n6+trqrds2WLRvIkTJ+rSpUsWjZVuhSUVKlTIqH///XetWLEi13lLlizRmjVrLN6nZMmSGjBggFFfv35dL7zwgsXzH1SPPfaY6XVz9uxZiwMyJk6cqPj4eKNu3bq1qlevbvMe86tixYqpR48eRn3jxg2NHj1aqampOc67cuWKxo0bZ9VeHh4eqlq1qlEfOnRIJ0+ezHVecHCw5syZY/E+6d/3V69e1bFjx3KdExcXp9dee83iPSC9+uqrpu8rn3/+ufbt22fHjgAAAAAAAAAAAAAAAAAAwP2EUBoAAAAAAAAAyGeKFi2qpUuXqkCBAsax4OBgtWzZUkuWLFFaWppV6wUGBtq6RRM3Nzd5eHgYX+n/AP5BFBMTo379+mn//v0Wzzl37pyWL19u1I6OjnrkkUdMY5ycnNS0aVOjvnjxoqZPn57juqdPn9ZTTz2lmzdvWtzLnXJwcDCFriQkJGju3Ln69ddfjWNlypTRo48+atF6LVq0MNVff/21Tp8+neOcOXPm6JNPPrG8ad0KUMoYaNGvXz8tWbIk2zmLFi3S4MGDJd06b0u9/fbbcnNzM+qlS5dqxIgRSkpKsniNqKgoffjhh/r9998tnpOfOTk5acyYMaZjb7zxhtavX5/jvP/+97+ZQpcexmCSiRMnysXFxah/++03DRo0SLGxsVmOP336tB577DGFhoZa9dqVpG7dupnqV155RSkpKdmOv3Llinr27JltL1nJ+L4fP358jiE78fHx6tmzp86cOWPxHpAqVqyoIUOGGPWNGzfUvXt3bd++3ap1/vnnHw0fPtzW7QEAAAAAAAAAAAAAAAAAgHyOUBoAAAAAAAAAyIeaN2+ub775Ro6O//vfuFFRUerbt6/q16+vzz//XMePH89yblpams6ePauvv/5arVq10ogRI+5V2w+F1NRULVq0SA0aNFDjxo31n//8R/v27csycCQmJkbz5s1T8+bNde3aNeP4E088IV9f30zjBw4caKrffPNNjRs3TpcvXzYdj4iI0PTp09WoUSOFhITIwcFB1apVs9EZ5i5jnxMmTNCNGzeMesCAAabXbk4qV66s5s2bG3VsbKzatGmjZcuWKTk52TT24MGD6tOnj0aOHKm0tDTVqFHDqr7ffvtt1axZ06gTExPVt29ftWzZUp9++qlWrlypX3/9VdOnT1eLFi3Ur18/JSYmytfXV08++aTF+1SsWFHz5s0zHfvmm29Up04dffvtt7p06VKmOWlpaQoKCtL333+vnj17qmzZspo4caIiIyOtOsf8bMyYMabgpcTERHXt2lUTJkzQxYsXTWPPnDmjESNGaOjQoaYgrj59+uipp566Zz3nFzVr1tTEiRNNx3744QdVq1ZN48aN05IlS7R69WrNnz9fAwcOVJ06dXTgwAFJsvp7wJAhQ0zv37Vr1+rpp5/OFBYVGxur7777Tg0aNNC+ffvk4+MjLy8vi/bo37+/aY8//vhDTzzxhI4dO2Yal5CQoOXLl6tevXr6+++/Jcnq9/3DbsaMGapfv75RX7x4UW3atNG//vUv7dixI9N1VpLi4uK0ZcsWTZgwQdWrV9ejjz6qv/766162DQAAAAAAAAAAAAAAcF+aOXOmHBwcjK+ZM2fauyWreXl5Gf3n9vtA6c+1Xbt296Q/AMC99WB/XC0AAAAAAAAA3McGDx4sb29vDR48WNHR0cbxgwcP6uWXX5YkeXh4qHjx4ipWrJjS0tIUGxur8+fP6/r161muWaxYMb3zzjv3ov2Hwp49e7Rnzx698cYbcnFxUdmyZeXt7S0nJydFRkbq3LlzSklJMc0pXry4Pv/88yzX+9e//qXZs2cbYRJpaWmaPn26ZsyYoSpVqsjLy0uRkZE6e/asad0333xTFy5c0MmTJ+/auaZXtWpVNW/eXNu3b5d0KzgivUGDBlm13ieffKJ27drp5s2bkm6FJjzzzDPy8PBQlSpV5OjoqPPnz5vCXNzd3fXjjz+qQYMGFu9TsGBBrV27Vm3bttXZs2eN49u2bdO2bduynFOoUCEtX75c3377rem4s3POP2J59tlnFRYWptdff12pqamSpMDAQA0fPlzDhw+Xn5+fihUrJmdnZ0VHRys8PFyxsbEWn8v9yMnJSYsWLVL79u0VEhIiSbp586YmT56sKVOmqEKFCvLx8dGVK1d07ty5TPMbNGig2bNn3+Ou848JEyYoODhYc+fONY5dvHhR06dPz3bOSy+9pJ49e+rrr782juX22q1Vq5ZGjx6tL774wji2atUqrVq1ShUqVFDx4sV17do1nTlzxnjPOjk56fvvv9fo0aNN36+yU716dY0cOVJfffWVcWz16tVavXq1/Pz8VKpUKcXFxencuXOKj483xrRp00YDBgzQsGHDct0DtxQqVEi//fabunXrpsOHD0uSkpOTNX/+fM2fP1/u7u7y8/NTkSJFFB8fr6tXr+rChQumMCgAAAAAAAAAAAAAgO1s2bJFrVu3vuN1XF1dTR8iBQAAANwNln1MKQAAAAAAAADALp566ikdOnRIffr0kYODQ6b74+LidPbsWe3evVt79uzRyZMnswyk8fb21rhx43Tq1Ck9//zz96L1h05SUpLOnDmjvXv3ateuXQoKCsoUSFO9enVt2bJFfn5+Wa5RoEABrVy5UlWqVDEdT01N1cmTJ7Vz506dPn3atO7YsWP14Ycf2v6EcpFd8EyTJk1UvXp1q9Zq0aKFvv32WxUoUMB0PC4uTvv379fevXtNgTTe3t5atWqV6tevb3Xffn5+2rx5s7p3757r2PLly+uff/5Ry5YtFRcXZ7qvSJEiuc4fO3asVq9erVKlSmW6LzQ0VPv379fu3bt16tSpLANpChYsqBIlSuS6z/2kYsWK2rp1a6YwodTUVAUFBWn37t1ZBtJ07dpVAQEB8vb2vked5j8ODg765ptv9J///EeFChXKcayzs7M++OADzZo1K0+v3WnTpunxxx/PdPzs2bPatWuXTp48aQTSuLq6avHixerWrZsVZyPNmDEjy/dhaGiodu3apWPHjpkCadq3b6+VK1fmGqqDzMqWLavt27erf//+mf4tcf36dZ04cUI7d+7U4cOHdf78+SwDafz9/e9VuwAAAAAAAAAAAAAAADkKDg5Wu3btjK+lS5fauyUAAB5YhNIAAAAAAAAAQD7n5+enJUuW6Pjx43rttddUsWJFi+aVLFlSTz/9tJYsWaKLFy9q2rRp8vLyurvNPgS8vLy0f/9+TZo0SS1btlTBggVznVO3bl3NmjVLhw4dUtWqVXMc6+/vr927d+vFF1+Uq6trtuOaNWumP//8U9OnT88ysOhu69OnT5b9DRw4ME/rDRo0SJs2bVKbNm2yHePq6qrnn39eR48eVbt27fK0jySVKVNGv//+u/755x8NHTpU1atXV+HChVWgQAGVLl1a3bp107fffqvjx4+rWbNmkqSoqChjvqOjozw8PCzaq3Pnzjpz5oxmzZqlunXr5vpceXh46PHHH9fs2bN18eJFq4M+7gdly5bV7t27NXfuXNWqVSvbcQ4ODmratKlWrlyp1atXq3Dhwvewy/zJwcFB48eP18mTJ/Xhhx+qWbNmKlmypJydnVWkSBE1atRIb7zxhgIDA/X2229LMr92JctCaVxdXfXrr79q+vTpKlasWLa9dO/eXfv371fv3r2tPhcXFxetXLlSM2bMkK+vb7bjypcvry+++ELr1q3je9gdcHd31/fff68DBw6ob9++Fj2W1atX15gxY7Rt2zZt2rTp7jcJAAAAAAAAAAAAALCYoyN/HoyHV2hoqDZu3Gh8/f333/ZuCQCAB5ZDWlYfdWeBhg0bKjw8XL6+vtq7d6+t+8p3e9vzfK11/o3N9m4hz8pOaZ2HWRds3gdwfymT96kpx23XBnA/cqrxv9tJ2+zXB5AfuLTI89QRVXP/A1zgQTYnMDFdFWa3PoD8ofT/bqYctV8bQH7glH3Igq1cvHhRhw4dUnBwsK5evaqkpCQVLlxY3t7eKlq0qOrUqaNy5crd9T4gJSYm6tixYzp9+rQuXryouLg4OTg4yNPTU+XLl9cjjzyiMmXy9v9w4uLitHnzZp0+fVrXrl1ToUKF5Ofnp2bNmsnf39/GZ5J/nDt3Tlu3btXFixeVmJgoLy8vVatWTS1atJCbm5tdeipevLgiIiIk3QrJOHv2bJ7WuXLlinbu3Knw8HBFRkYqNTVVnp6e8vX1VY0aNVSlShUVKFDAlq3neyEhIdq5c6cuXbqkmJgYeXt7q1SpUmrRooVKlChh7/bue+PGjdP06dONOiAgQG3btrV4fkpKinbu3KmjR48qIiJCzs7OKleunFq3bq1SpUrZpMfk5GTt3r1bhw4dUmRkpJycnOTr66tHHnlE9erVs8keMEtNTdW+ffsUGBioiIgIxcTEyM3NTV5eXqpUqZJq1qyp4sWL27tNAAAAAAAAAAAAAA+wVWdW6c3Nb9q7jWx93Ppjda/Y3ebrbtmyRa1b/+/vKIsUKaLo6Gib7wM8yDK+j4YMGaK5c+fasaP/mTlzpl599VWjnjFjhl555RX7NZQHXl5eunbtmiSuUQAAydneDQAAAAAAAAAArFeqVCmbhQHgzhQsWFD169dX/fr1bb62h4eHunbtavN187vy5curfPny9m7DcPDgQSOQRpIaNWqU57WKFy+u7t1t/wtL9zN/f/8HOmTJ3tavX2/cdnR0VIMGDaya7+TkpBYtWqhFi7wH2ubG2dlZzZs3V/Pmze/aHjBzdHRUo0aN7uh6BgAAAAAAAAAAAAAAAAAAHmyO9m4AAAAAAAAAAAAgP5s1a5apbtmypZ06AayzefNmHThwwKjr1KmjwoUL268hAAAAAAAAAAAAAAAAAAAA3DcIpQEAAAAAAAAAAA+NtLQ0q8avXbtW3333nVG7uLioX79+tm4LyJW1r92YmBgNGzbMdOz555+3ZUsAAAAAAAD/j737joribPs4/qMICIiIiqgo9t67sWGLJcYWTewlaozpzTRTjOZJ0Viex8TERGNvUWNMNGossXcRe+xYUERUQEREyvuHx3kZdoFdRFDz/Zyz5+w1c933fc0yu+ZkZ68BAAAAAAAAAADAY8w5pwsAAAAAAAAAAADILmPGjFFoaKhef/11lS5dOs28xMRETZ06Va+//rqpGUifPn1UsGDB7CgVMDl79qz69++vDz74QK1bt5aTk1OaucHBwerbt6+OHTtmbMufP7/69euXHaUCAAAAAAAAAAAAAICHVFxcnCZPnqwDBw7o0qVL8vHxUc2aNfXiiy/Ky8vrvuY+e/aspk+frtOnT+vy5cvy9PRU0aJF9dxzz6lRo0ZZdATSzp07tWTJEoWEhCgyMlKFCxfW999/L3d3d6v5ERER+u6773T06FFFRkaqWLFiqlWrloYOHSpHR8csq+thdPHiRf3www86efKkIiIi5Ofnp2eeeUadOnVKd9zWrVs1b948nTlzRg4ODipRooSGDRumKlWq3Fc9ERER+umnn3Ts2DGFhYXJzc1NhQsX1tNPP6327dvf19yZFRMTo2XLlmnbtm0KDQ1VTEyMvLy8VKhQIbVt21ZPP/10lp4nW7Zs0dKlS3UQR7j2AAEAAElEQVT69GnduXNHJUuWVJ8+fVS/fv37mvfea/vPP//o8uXLcnd3V9GiRdW5c2e1bNkyi6rPGklJSVq2bJnWrl1rvOYuLi7y9vZW+fLl1axZMwUGBt7XGseOHdOMGTN08uRJRUdHq1ixYurTp0+G865cuVK//vqrzp8/L1dXV5UpU0avvfaaAgIC7qseAHiU0ZQGAAAAAAAAAAD8a9y6dUuTJk3SpEmTVKdOHTVt2lRVq1ZVgQIFJElXr15VUFCQ/vjjD505c8Y01t/fX+PGjcuJsgFJ0qZNm7Rp0ybjopfatWuraNGicnd3V1RUlE6dOqV169Zp/fr1FmN/+OEHeXt7Z3/RAAAAAAAAAAAAAAAgWzk7OysxMVGSVLp0aZ08eVLx8fF66qmntH79eiUlJZnyFyxYoPfee08tW7bU77//nmZzl7SMHTtWX3zxhSIjI63unzRpktzc3PTiiy9q3LhxGTb4GDx4sKZNm2bEmzdvVuPGjTV58mR9+OGHioqKshgzatQoi6YRFy5cUPv27XXw4EGr67z66qvq3LmzFi9eLMn665ZSu3bttGrVKiOePHmyhg0blu6xpJSUlCRXV1clJCRIklxdXRUXF2fz+LQ4ODgYz5s1a6YNGzbo0KFD6tq1q06cOGGRP3v2bHl5eWn+/PkWjWCWLFmiF154QdeuXbMYN3nyZJUuXVqrV69O92Zg1syZM0dvv/22wsPDre7/4Ycf5OzsrGeffVbTpk2Tm5ub1Txvb2+rf/9p06aZzpmU7r0mKR06dEijRo3SunXrrB5ryrocHR0VGBiomTNnyt/fP83cjCxcuFDDhg3T9evXLfZ9++238vb21tSpU/XMM8/YNe/q1as1ZMgQnT9/3ur+b7/9Vrly5VLv3r31008/ydk5a1sLWDv/0hIdHa3u3btr3bp1xnstvXlLly6tKVOmqEWLFhb7t2zZoiZNmhjxoEGDNHXqVK1bt079+/dXaGioxZhp06bJz89PK1asUK1atUz7Jk2apA8++EA3b960GDd+/HjVrl1bf/31l3x8fNKtGwAeRzSlAQAAAAAAAAAA/0p79uzRnj17bMotWrSoli9fTlMPPBQuX76smTNnaubMmRnmOjg46Ouvv1a3bt2yoTIAAAAAAAAAAAAAAPCwOXPmjGrUqKHo6Oh089atW6cSJUooJCTEpsY0ERERqlGjhtXmD6nFxcVp4sSJWrhwoQ4cOGDcQMpW7du318qVK23OX7dundq0aZNu44vExEQtWbJERYsWtdq8JbWJEyeqQoUKRjx27Fi7mtJ88cUXRkMaSerYsaPNY+2xdOlSdevWzaL5UErR0dHq0KGD5s+fr+eee06S9NZbb2nChAnpzn3q1ClVqVJFR44cUcmSJTOsJT4+XnXq1EmzMVBKCQkJmjdvnv744w/t3r1b5cuXz3BMZrVu3VphYWE25SYlJWn9+vUqVaqUfv31V3Xo0MHu9V599VV9++236eZERkaqW7duGjp0qH744Qeb5u3Zs6cWLFiQYd6dO3c0Y8YMLV68WHv27Hmgr21aDh06pLp169rciCk5OVknT57Ub7/9ZrUpjTUTJ07Um2++mW5OWFiY6tWrp+3bt6tu3bqSpC5duui3335Ld9zevXtVtmxZnTlzRl5eXjbVAwCPC5rSAAAAAAAAAACAfw0/Pz85OTlleKeVexwdHdWtWzeNHz9eRYsWfcDVAWlzc3NT/vz5dfXqVZvHVKxYUWPGjMnUxTAAAAAAAAAAAAAAAODRl5iYqNq1axsNaVxcXFSqVCkVLFhQMTExOnbsmGJjY438K1euqH379tqwYUO684aFhalMmTK6efOmabuXl5cCAgKUL18+xcbGKiQkRBEREcb+S5cuqVy5cgoLC5OLi4tNx/DFF1+YGtIULFhQxYsXl6urq8LCwnTmzBlT/qFDh6w2pPH29lapUqWUO3duXbx4USEhIUpOTtbFixfVrFmzDOsoX768SpUqpdOnT0u62+znxIkTKlu2rE3H8d1335ni8ePH2zTOHteuXTM1pMmTJ4/KlCkjT09PhYaG6syZM0pOTpZ0t+lH37599dRTT2nq1KmmhjSFChVS8eLFlStXLp05c0aXLl0y9sXFxal169Y6efJkurXEx8erWLFiCg8PN213d3dXQECAChQooNu3b+v8+fOm+W/cuKHq1avr3Llz8vX1NY11dHS0+zXJaIyjo6Py5cunIkWKKE+ePMqVK5eioqJ0/vx503U6d+7cUefOnXXixAmbGvLc8/fff+v333834rx586p06dJyc3PThQsXdO7cOVP+lClTlDdvXn399dfpztu5c2ctW7bMtM3BwUElS5ZU4cKFdfPmTZ06dUo3btww9sfExKhatWo6fvy4AgICbD6GrNCkSROLhjT58uVTsWLF5O3trcTEREVHRys0NFTXr183zlNbHT16VNOmTTNiHx8flSxZUm5ubjp37pzOnz9v7EtMTFTr1q0VGRmpF1980dSQpmjRoipWrJjRFCflOXDt2jV17Ngxw89HAHjc0JQGAAAAAAAAAAD8a7z44ot65plntGrVKm3dulWHDh1SSEiIrl+/rri4OOXJk0c+Pj4qW7asAgMD1aVLF9MdjoCc4ufnp7CwMG3cuFEbNmzQ3r17derUKV2+fFk3b96Us7OzfHx8VLhwYTVq1EitW7dW+/btM3UxDgAAAAAAAAAAAAAAeDyEhIRIutusYsSIERo9erRFzptvvqmJEyca8caNG3XhwgX5+/unOW/9+vVNDWlKly6tBQsWqE6dOha5Bw4cUPv27RUaGipJun79utq2bav169fbdAz3GtIULVpUf/75p6pVq2baHxcXJ2fn//+5dNu2bU0NaXLnzq0FCxaoY8eOpnHR0dFq27attm/frj179thUy0cffaTnn3/eiN966y398ccfGY7bs2ePwsLCjLh8+fLpvr6ZdfDgQUmSq6urZs2apWeffda0/+zZs6pVq5auXbsm6W6jlT59+hjHULRoUa1atUpVqlQxjfvzzz/VsWNH43U9deqU1q1bp5YtW6ZZS7NmzUwNaQoWLKjZs2erTZs2Frlnz55Vhw4ddOjQIUnS7du31bhxYx0/ftyUd6/uLVu2qEmTJsb2QYMGaerUqem8MmZ+fn6qXLmyXnvtNYvzIqWLFy/qmWee0Y4dOyTdbWbSoUMHHT582Oa17jWkcXV11Zw5c9StWzfT/rCwMDVv3lz//POPsW3s2LEaMGCAKlasaHXOWbNmWTSkadKkif788095enpa5A4ePFh37tyRdLdZUJMmTSya4TxIkyZNUmRkpBEXLFhQf//9typXrmw1PyEhQTNmzNC4ceOUJ08em9bYtm2bpLtNf3777TcFBgaa9gcFBalRo0ZGY5yoqCgNHDhQM2bMkCRVrlxZq1atsnhffv/993rppZeM2JbPRwB43NCUBgAAAAAAAAAA/KsULFhQffv2Vd++fXO6FMAuzs7OatmyZboX9AAAAAAAAAAAAAAAgEdHVFSUHBwc7B63cOFCi4Yj6Vm+fLnat29vdd+ECRN0+PBhrVmzxtj2ySef6Oeff7aa/+GHH5oaWrRs2VJr165Nc+1q1arpwoULKl68uM6fPy9J+vvvv3X27FkFBATYVL+/v7/Onj1r9eY8bm5uxvNp06YZzW8kKVeuXDp48KBKly5tMc7Ly0vbtm1Ts2bNtGnTJpvqGDhwoF555RXFxsZKklavXq2kpKQMbxo0fPhwU/zRRx/ZtF5mODs76+jRoypZsqTFvoCAAG3bts10k657zU2KFCmikJAQU4Ofe9q3b6/Ro0frww8/NLaNHj06zWtYFi5caDRyke424Tly5Eiar1NAQIAOHjyo+vXra9euXZKkEydOaM2aNWrdurUNR22fffv22ZRXpEgRbd++XU899ZT+/PNPSdKRI0d05swZq69vWpycnLR3716rTVj8/Px09OhRVahQQceOHZMkJScnq0ePHtq/f7/V+V577TVT/NRTT2n58uVWc/v166cqVaqoXr16RlOh8+fP69tvv9Urr7xi8zHcj0WLFpniAwcOyM/PL818Z2dnDR48WIMHD7ZrHQ8PD507d05eXl4W+2rVqqVFixbp6aefNrbda0hTs2ZNBQUFWZ1z2LBh2rp1q+bOnWtsGzlypF1NkADgUcetEQEAAAAAAAAAAAAAAAAAAAAAAAAAAIBHRHJyss25Xbp0SbMhzT0//fSTKd6+fXuauZMmTTKee3l56a+//rKpjtWrV5vi1I1a0rN+/foMG79I0ldffWWKP/roI6sNaVLX5eLiYnMtPXv2NJ7fuXNH48aNSzc/Pj5emzdvNmJ3d3f16dPH5vXs9dFHH6XbMKV8+fIqU6aMxfb169dbbUhzzwcffGDaf+jQoTRz3333XeO5s7Ozdu3aZdPfb82aNXJycjLilE1wctKiRYtMzaMmT55s1/h3333XakOalFKf4wcOHFBERIRF3i+//KKoqCgj9vT01O+//57u3LVq1dLbb79t2pb6vfIgXblyxXju4eGRbkOa+zF37lyrDWnu6dChg7y9vU3bnJycMmxKNXHiRFO8devWzJYIAI8kmtIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAjyFbGmgEBATI3d3diC9evGg1b82aNYqJiTHiQYMG2dRsRJIqVqxoakaxbds2m8aVLVtWZcuWtSn31KlTxnNnZ2d98sknGY5xc3NTu3btbJpfkr755htTg5L//ve/6eaPHDlSiYmJRty9e3eb17KXk5OTPv744wzz6tata4pLlSql8uXLZziucOHCxvPIyEirORERETp37pwRP/nkk+k2CknJy8tLVapUMeIjR47YNO5Bc3d3l4eHhxFv3LjR5rHOzs76/PPPM8wrUqSIGjZsaNpm7dz64YcfTPHgwYNteg9+/fXXypUrlxGHhoYqNjY2w3FZIeVrFxsba7XZzv3y8vJSp06dMsxLfZ43atRInp6e6Y4pUKCA6RjS+nwEgMcVTWkAAAAAAAAAAAAAAAAAAAAAAAAAAACAbJY3b14lJyfb/Xjuuedsmt/T09PUCCY9+fLlM57fvn3bas6cOXNM8fPPP2/T3PcULVrUeH758mWbxrRo0cKmvE2bNik5OdmIy5QpY3NdAwcOtDnX29tbtWvXNuLQ0FAdOHAgzfypU6ea4m+++cbmtezl7+9vU4OS1K9No0aNbJrf19fXeJ6y0U5KP//8synu2rWrTXPfk7IBUWxsrJKSkuwab4/4+Hh9+eWXql69ury9veXs7CwHBwerj5TNmMLDw21eo2LFijY3bho0aJAp/vvvvy1yDh48aIpHjBhhcy01atQwxYsWLbJ57P1IeX4lJyerUqVKCgoKytI1UjYzSk+xYsVMsa0Nqby9vY3naX0+AsDjiqY0AAAAAAAAAAAAAAAAAAAAAAAAAAAAwGMmb968Nue6uroaz9NqOLJ3715TXLVq1TSbeFh7pByf1hqpNWzY0Ka8nTt3mmJbm1RItjemuGfs2LGm+O2337aat2XLFl25csWIq1WrpgIFCti1lj1SNo1JT8oGG5JUunRpm8Z5eHhkmLNhwwZTPHjwYLvOkcWLF5vGX7hwwaba7PXJJ5/Iw8NDH374oQ4cOKCoqCibz8mbN2/avE7qRjDp6dSpkyk+ffq0RU5kZKTx3MXFxa7zqUGDBqZ4+/btNo+9H6NHj5azs7MRX7lyRbVr11aBAgX09NNPa+rUqaamP5mRutlMWvLkyWOKK1WqZNM4Wz4fAeBxRVMaAAAAAAAAAAAAAAAAAAAAAAAAAAAA4DHj5uZmc66Dg4PxPDk52WpOVFTUfdeU0RqpFSlSxKa8sLAwU2xrgxbpbnOPlMefkcDAQBUsWNCI//77byUkJFjkvfvuu6Z49OjRNq+RGe7u7jblOTqaf15ua/MiW16jiIgIm+ayVWhoaJbOJ0ldu3bV6NGjrf7NbGHPOFubpUiSj4+P6TW+deuWRU7Khii2/r3vKVWqlClO2TDpQfLy8tLcuXMtzrurV69q+fLlGjJkiPLkyaN8+fLpySeftGhsZAtPT0+b8lLX4OPjY9M4Wz4fAeBxRVMaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAOmKjY3N9jVz585tU17qBh72NOSRbGu4ktLLL79sPE9MTNTnn39u2h8bG6udO3casZeXlzp27GjXGo+iGzduZOl8KZuwZIVp06Zp6dKlpm358+dXjx499PPPP2vfvn26fPmy7ty5o+TkZOORsnGPPU1JvL297aov5XkYHx9vsT/l2i4uLnbNnboBS3R0tF3j78ezzz6r48ePq169emm+1yIjI7VmzRo1b95cRYsW1datW7OtPgBA2mhKAwAAAAAAAAAAAAAAAAAAAAAAAAAAACBdqZtgXL9+3dS4w95HVipQoIBFbfZISkqyK3/EiBFydnY24u+//960/6OPPjLN2adPH7vmf1Slbga0YsWK+zpHGjdunKX1ffDBB6Z4+PDhioiI0Pz58zVw4EDVqFFDvr6+pr+tJN25cydT60VGRtqVn1HTmYya1qTn2rVrptjLy8uu8ferdOnS2rlzp2JiYjRhwgQFBgYqf/78VnMvXryoJk2aaOHChdlaIwDAknPGKQ+v8PBw1a5dO9vWAgAAAAAAAAAAAAAAAAAAAAAAAAAAAP6NvLy8FBYWZsQnTpxQ3bp1c7Ci/1e8eHFTfPr0aZvHnjlzxu71nJ2d1aZNG61YsULS3d8h79y5U/Xr15ckzZgxw8h1cHDQ119/bfcaj6LUTUZCQkJyphArIiIidOXKFSMuW7asxowZY9PY27dvZ2rNCxcu2JwbGRlpakqTO3duixwnJyclJCRIkmJjY+2qJfV7omDBgnaNzyru7u5644039MYbb0iSEhISNHv2bE2ZMkW7du0yXoPk5GT169dPXbp0sdqgBwCQPRxzuoDM8PT0lHS362BYWFi2POztcAgAAAAAAAAAAAAAAAAAAAAAAAAAAAA8LipUqGCKly5dmkOVWOrcubMpPnLkiM1j582bl6k1//vf/5rid955R5K0evVqXb9+3dhep04d47fRj7tatWqZ4rVr1+ZQJZa2bdtmip966imbxh04cECJiYmZWnPfvn025y5btswUlypVyiLH29vbeB4fH6+IiAib59+xY4cpbtiwoc1jHyRnZ2cNHDhQO3bsUEhIiNzc3Ix98fHx+vbbb3OwOgDAI9mUZvjw4SpTpoz8/Pyy7eHo+Ei+VAAAAAAAAAAAAAAAAAAAAAAAAAAAAMB96927tyletGhRDlViqUCBAnJ3dzfiK1eu6MSJEzaNnT59eqbWLF26tMqUKWPE27ZtU1xcnEaMGGHK++qrrzI1/6No2LBhpvjvv//O8jVSNi2RpISEBJvGXbp0yRQXKlTIpnGjRo2yrTArjh49qqSkJJtyp06daoqbN29ukVOtWjVT/J///MfmWoKDg03xc889Z/PY7FK8eHG99957pm2bN2/OoWoAAJLknNMFZEaHDh3UoUOHbF2zdu3aCgsLy9Y1AQAAAAAAAAAAAAAAAAAAAAAAAAAAgIdBt27d5Orqqtu3b0uSTp48qZUrV6pdu3Y5XNldbdu21a+//mrEzz77rPbt25fumN9//12nTp3K9Joff/yx+vfvL0lKSkrS22+/raCgIGN//vz51aJFi0zP/6gpWbKkChYsqCtXrkiSIiMjNX78eL311ltZtoafn58pjoiIsGlcgQIFTPGRI0cyHBMeHq5ly5bZXlwqCQkJ+vjjjzNsHhMWFqZt27aZtr3++usWecOGDdP69euNeOrUqRo3bpwcHR3TnX/EiBG6c+eOEfv7+1s093lY1KlTxxTf+7wBAOSM9P+FAQAAAAAAAAAAAAAAAAAAAAAAAAAAAPCv5+joqGHDhpm2de7cWYcOHbJrnsWLF+vo0aNZWZokacKECXJwcDDi4OBgvfjii2nmHzp0SN27d7+vNfv16ycPDw8jnjx5spKTk4140KBB9zX/o+jrr782xcOHD9eaNWvsmmPDhg3asGGD1X3+/v6mv/OBAwdsmrN58+am+Ndff1VCQkKa+UlJSWrYsGG6ObYYM2ZMhud7ixYtlJSUZMTVqlWzaKIj3W0MlTdvXiOOiYlRx44d0507ODjY4m/y4Ycf2lJ6lpgzZ45dr+H8+fNNcfny5bO6JACAHWhKAwAAAAAAAAAAAAAAAAAAAAAAAAAAACBD48aNU9GiRY04Pj5e1atX16uvvqr4+Pg0x128eFEvv/yyChQooO7du9vcSMQexYsXt2hCM2XKFJUsWVKTJ09WWFiY4uLitGnTJnXp0kXVqlVTfHy8nJ2dTU1O7NWrVy+r2x0dHfXZZ59let5H1cCBA1W7dm0jTkpK0pNPPqmePXsqMjIyzXGRkZH68MMPVaxYMTVv3lyrV69OMzdlw5bz58+rZ8+eOnHiRLp1+fj4yM/Pz4hv3rypihUr6tq1axa5wcHBKlasmE6fPp3unLZISEhQrVq1tGTJEot94eHhqly5sqlpjYODgxYsWJDmfP/73/9M8YoVKxQYGKjY2FiL3Llz56p+/fpKTEw0thUrVsyiudSD9Morryh37txq27Zths2J3nvvPc2bN8+07e23336Q5QEAMuCc0wUAAAAAAAAAAAAAAAAAAAAAAAAAAAAA/zZRUVGZboayZcsWNWrUKIsrypijo6OCgoJUunRpxcTESLrbdOTbb7/Vd999p8KFC6tw4cLy9PRUTEyMrl+/rkuXLunWrVvZUt/kyZO1e/du7dmzx9gWEhKil19+WS+//LLVMdOnT9eAAQOMxh1OTk52rfnNN99o6tSpSk5ONm1v3Lix3Nzc7DyCx8O2bdtUokQJXbp0ydi2YMECLViwQL6+vipatKi8vLwUGxuryMhIXbx4UTdv3rR5/sGDB+vLL7+0mDu11q1b66+//jLisWPHqm/fvkZ88uRJFShQQCVLllTRokUVGxurc+fO6cqVK0ZOyZIlde3aNUVFRdlc3z0dO3bU77//rri4OHXr1k3e3t4qXbq03NzcdOHCBZ07d87ivBk+fLgqVqyY5pz9+vXTr7/+qmXLlhnbNm7cKE9PT5UsWVJFihRRbGysTp48qejoaNNYFxcXbd682e7juF8JCQlavXq1Vq9eLScnJ/n6+qpQoULKmzevEhMTFRYWprNnz+rOnTumcZ07d5a/v3+21wsA+H80pQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAeIakbWWQnX19fhYaGqkGDBjp69KipposXL+rixYsZzpE7d+4HVt/u3bv19NNPa/ny5enmOTk5adq0aerTp4/69etnbPf09LRrPS8vL9WrV087d+40bf/mm2/smudx4uLionPnzqlVq1bauHGjaV94eLjCw8MznCO9c+SLL77QypUrFRwcnO4c8fHxprhPnz7666+/NHv2bGNbcnKyTp8+rdOnT1uM9/PzU3BwsIoXL55hvdY0b95c/v7+mjx5siQpMjJSe/fuTTN/yJAh+vrrrzOc97ffflPPnj1NjXjSOw7p7nm9Z88eBQQE2HkUWSsxMVGXLl0yNSyyJjAwUEuXLs2mqgAAaXHM6QIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAPDq8vLx05MgRLViwwOaGHR4eHmrZsqW2bNmijh07PtD6/vjjD23ZskVNmzaVu7u7HBwcJN1tROPj46P+/fvrwoUL6t+/v+Lj401Nfry8vOxe77333jPFfn5+qlu37v0dxCPO2dlZGzZs0Lp161ShQgXjb5AeNzc3NWjQQMuWLdMnn3ySbu6+ffs0efJkVahQQblz57ZpfkmaNWuWxowZk27TG1dXV/Xu3Vvnz5/P1PmQ0nfffafZs2crb968aeZ4e3tr0aJF+vHHH22ed/78+Vq1apX8/f3TzcuVK5f69eunq1evqnz58jbPn1WmTJmiJ554Qu7u7jblFy5cWHPmzNHff//9gCsDANjCITmTrRBr166tsLAw+fn5pduR7XHxKB3vhfc353QJmeb/VZNMjArN8jqAR0vRzA9NPJpxDvA4c6r4/8/jt+VcHcDDwOWJTA8dWs41CwsBHj1Tjt9OEWXc0R94vBX5/6eJh3OuDOBh4FQ5pysAAAAAAAAAAAAAAAAAgIfW8tPL9cHmD3K6jDR92eRLdSjVIafLeKTExcVpzpw52rVrl8LDwxUTEyNPT0/5+vqqXr16at++vYoUKZLxRDngl19+0XPPPWfEr7zyiiZNmmTXHD169NDChQuNePTo0froo4+yrMbHQUJCghYtWqTNmzfr8uXLioqKkoeHh/Lnz69atWqpXbt2Kl26dLbWtHTpUv3++++6dOmS3NzcVLx4cTVv3lxdunR5IOtt2rRJS5cu1ZkzZ5SQkKCAgAD16dNHDRs2vK95IyIiNGXKFB09elRXrlyRu7u7ihQpoo4dO6pNmzZZVP39Cw8P1++//66goCCFhobq5s2bcnV1Vf78+VWvXj1169ZNfn5+OV0mACAFmtLY6FE6XprSAP82NKUBMo2mNMD/oykNkGk0pQFSoikNYKApDQAAAAAAAAAAAAAAAACkiaY0eJg0a9ZMmzZtMuK1a9eqZcuWds2RO3duxcXFSZKcnZ11+/ZtOTo6ZmmdAAAA2Y3/mgEAAAAAAAAAAAAAAAAAAAAAAAAAAADwrxMUFKTNmzcbsYuLi90NacaPH280pJGkNm3a0JAGAAA8FvgvGgAAAAAAAAAAAAAAAAAAAAAAAAAAAACPhZEjRyo8PDzDvKCgIDVp0kTJycnGtqeeesqutRISEvTpp5+atk2aNMmuOQAAAB5WNKUBAAAAAAAAAAAAAAAAAAAAAAAAAABAtinmWSynS0jXw14f0jd+/Hj5+fmpQoUKeu+997Rp0yZFRkYqKSlJZ8+e1eTJk9WgQQPVrl1bsbGxxjg3NzfNmDHDpjWSkpK0bt06lS5dWjExMcb2xo0bq2TJkll9SAAAADnCOacLAAAAAAAAAAAAAAAAAAAAAAAAAAAAwL9Hdd/qmtNujs7HnM/pUiwU8yym6r7Vc7oM3Kfk5GQdO3ZMY8aM0ZgxYzLMd3R01JIlS+Tl5ZVuXsGCBRUREWF1n7OzsxYtWpSpegEAAB5GNKUBAAAAAAAAAAAAAAAAAAAAAAAAAABAtqruW53mL3ggcuXKZVe+r6+vVq5cqVq1amWYe+fOHavbHRwcNGPGDPn5+dm1NgAAwMPMMacLAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICscOnSJY0ZM0ZPPPGE8ufPL2dnZ9N+JycneXl5qVGjRlqwYIEuX75sU0Maa3LlyqWqVatq586d6t27d1aUDwAA8NBwzjgFAAAAAAAAAAAAAAAAAAAAAAAAAAAAAB5+Li4uGj58uIYPH57lc0dGRmb5nAAAAA8rx5wuAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADw8HDO6QKQ9fy/apLTJWSzojldAPDocqqY0xUADw+XJ3K6AuCRNeX47ZwuAXiIFMnpAoCHh1PlnK4AAAAAAAAAAAAAAAAAAAAAAAAAQCY55nQBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICHB01pAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAG55wuAFnvwvubc7qETPP/qkkmRoVmeR3Ao6Vo5ocmHs26MoBHkVPF/38evy3n6gAeBi5PZHro0HKuWVgI8OiZcvx2iuhijtUBPByK/P/TxMM5VwbwMHCqnNMVAAAAAAAAAAAAAAAAAAAAAAAAZJpjThcAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAHh40JQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGCgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEBTGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgaY0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADTWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAaa0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADDSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYaEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADA453QBAAAAAAAAAAAAAAAAAAAAAAAAAAAA+HeJ3bdP8Rcu5HQZFlz8/eVes2ZOlwEAAADkOJrSAAAAAAAAAAAAAAAAAAAAAAAAAAAAINvE7tunsz175XQZaQqYP4/GNAAAAPjXc8zpAgAAAAAAAAAAAAAAAAAAAAAAAAAAAPDvEX/hQk6XkK6HvT4AAAAgO9CUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgoCkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAUxoAAAAAAAAAAAAAAAAAAAAAAAAAAADgIXD48GF9+eWXeumll9SlSxc9+eST6t69u1588UV9//33CgsLs3vOiRMnysHBwXhMnDgx6wsH/sW8vb2N95e3t3dOl2M3ez4jAgMDTbkAgMebc04XAAAAAAAAAAAAAAAAAAAAAAAAAAAAAPxb7dmzR6+88or279+vuLi4dHNfeuklubq6qnbt2nrllVfUs2fPbKoSAAAA/zaOOV0AAAAAAAAAAAAAAAAAAAAAAAAAAAAA8G9z7tw51apVS3Xr1tXOnTszbEhzz+3bt7Vt2zb16tVLXl5eGjNmzAOuFMh5H3zwgQIDAxUYGKguXbrkdDkAAPwrOOd0AQAAAAAAAAAAAAAAAAAAAAAAAAAAAMC/yfr169WuXTvFx8db7HNxcVHhwoWVN29eeXt76/r167p27ZoiIiJ0+/ZtU+6NGzf03nvvqU6dOmrRokV2lQ9ku0WLFunUqVOSJCcnpxyuBgCAfwfHnC4AAAAAAAAAAAAAAAAAAAAAAAAAAAAA+LdYvHixWrVqZdGQpnHjxtq1a5du376tkJAQ7d+/Xxs3btSBAwd04cIFxcXFafPmzercubNFU47k5OTsPAQA/1IbNmxQcnKy8QAAPN5oSgMAAAAAAAAAAAAAAAAAAAAAAAAAAABkg4sXL6pXr16mZg4eHh46ePCgNm/erLp166Y7vnHjxlq6dKmuXbumZ5555kGXCwAAgH8xmtIAAAAAAAAAAAAAAAAAAAAAAAAAAAAA2aBx48a6c+eOEXt5eSkkJERVqlSxax4vLy8tXrxYa9euVe7cubO6TAAAAEDOOV0AAAAAAAAAAAAAAAAAAAAAAAAAAAAA8Lhbvny5zpw5Y9r2559/qkCBApmes2XLlrp48aKSkpLutzwAAADAhKY0AAAAAAAAAAAAAAAAAAAAAAAAAAAAwAP21ltvmeJWrVqpUaNG9z2vt7f3fc+Rk4KDgzV//nydP39eERERcnFxUeHChdW4cWN1795d7u7uNs8VHh6uP/74Qzt37tTly5cVGxurvHnzqkiRIurcubNatGiRpbVv3bpVS5Ys0alTp5SYmCh/f3/16tVLTZs2zdJ17PHLL79ow4YNCgkJkaOjo2rWrKlXX31Vvr6+aY6JjY3Vjz/+qK1btyoqKkr58+dXkyZN9MILL8jZ+f5+jr5y5UqtXLlSFy9e1I0bN1SwYEFVrFhRQ4YMSbemB2nPnj1as2aNDh48qGvXrkmSChQooAoVKqhfv34qXrx4lq0VFxenyZMn6+DBg7p06ZIKFCigmjVr6uWXX5abm9t9zb169WotX75coaGhio2NVaFChVSxYkW98MIL8vHxyaIjyBpnzpzRokWLtG/fPl2/fl2JiYny8PBQ0aJFVbNmTXXu3Pm+GnQlJSXp559/1q5du3Tu3Dm5ubmpfv36evXVV+Xp6ZnmuIiICE2ZMkV79+5VTEyMChYsqHbt2qlPnz6ZrgUAHic0pQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAeoMjISJ04ccK0bdy4cTlUjaWCBQsqIiLCiK9cuWJXg4hNmzapWbNmRly1alUdOHAgzfzY2FgNGzZMCxcu1O3bt63mTJ06VQMGDFDRokX15ptv6u2337aat3XrVv3nP//Rli1bdOPGjTTXnDRpkpydndWxY0dNnz5dXl5eGR7X4MGDNW3aNCPevHmzGjdurCVLluiFF14wGpqkNGXKFHl5eemHH35Qz549M1zDXoGBgdq4caMRJycnS5Jee+01/fTTT4qLizPlr1ixQp9//rnatm2rFStWyNHR0dgXFxenrl27atWqVcY89yxYsECvvfaa3n33XX3xxRd21Xjt2jX16dNHa9asUUJCgtWcjz76SMWKFdOUKVPUrl07qzkTJ07Um2++abE9MTFRDg4Oaa6f+lgSEhL03Xff6aefftKxY8fSrEmSPv74Y+XPn1+ffvqpXn311TTzMhIXF6f27dtrw4YNFvXMnTtXw4cPV+vWrbVs2TK7mtPEx8dr4MCB+uWXX9I8jg8++EABAQGaMWOGAgMDM30M1qR1/qVl7ty5euuttxQeHp5u3pAhQ+Th4aGOHTtq3rx5VnPKlCmjU6dOSZKcnJyUkJCghIQE9enTR0uWLLF4PZYtW6YRI0ZowIAB+vnnn037IiIi1KFDB+3cudNinXnz5mnw4MEaP368XnrppXTrBoDHnWPGKQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAya+bMmaY4X758qlatWg5VY2nYsGGm+J133rFr/LvvvmuKR48enWbuypUrlS9fPs2aNSvNhjQphYaG6qOPPkpzf6tWrbRy5cp0G9Lck5CQoF9//VVFihRRUFBQhvnWvPjii+rWrZvVhjT3REdHq1evXvrqq68ytYa9qlevrkmTJlk0pElp1apVKl++vBFfvHhRfn5+WrlyZZqNRRITE/Xll19qwIABNtcyZ84c+fr6auXKlek2f5Gk8+fPq3379hoyZIjN82fGkiVL9MYbb+jw4cMZ1iRJV69e1WuvvaYmTZpkar0LFy6oUKFC+vvvv9N8bZOTk/XXX3+pQIECOnfunE3zHjp0SPny5dO8efMyPI6zZ8+qefPmGjx4sN31Z5UePXqoT58+GTakuefmzZtasmSJzfPHxsYqICBACxcuTPP1SE5O1vTp09W8eXNjW3BwsPz9/a02pLnn9u3bevnllzVq1Cib6wGAxxFNaQAAAAAAAAAAAAAAAAAAAAAAAAAAAIAHaPny5aY4ZXOQh8Enn3wiZ2dnI160aJHNY2NiYrRr1y4j9vLyUqdOnazmTp8+XU899ZTi4+NN293c3FShQgU1btxYDRo0UJkyZeTm5mbnUUhOTk7y9fVVtWrV1KhRIzVp0kRVq1ZV3rx5TXk3b95UkyZNFBMTY9f8P/30k6ZMmWLEvr6+qlOnjho3bqyAgACL/A8//FBHjx61+zjs0a5dOx04cECS5ODgoFKlSqlRo0aqU6eO8uTJY8o9efKknn/+eSUkJKhq1aqKioqSJLm4uKhixYrG65UrVy7TuJkzZ2rlypUZ1jJu3Dj17dtXiYmJxjYHBwcVLlxYderUUdOmTVW5cmWLv+3UqVP1/PPPW8zn4OBg24tgp9y5c6tEiRKqXbu2mjVrpgYNGqhUqVIWx71lyxZ17tzZ7vkbNmyo6OhoSZKjo6NKly6txo0bq1q1anJ3dzfl3rx5U5UqVcrwXDxx4oRq1qyp2NhY0/Y8efKoRo0aatSokUqVKmXxmk2bNk09e/a0+xju1/jx47Vw4ULTNmdnZ5UsWVINGjRQYGCg6tSpoxIlSmTqvS5JdevW1cWLFyXdfe+XK1dOjRs3Vs2aNS3m3LBhg8aPH6+IiAg1bNjQaIiVO3duVa1aVU2bNlWFChXk5ORkGjdy5EgdO3YsU/UBwOOApjQAAAAAAAAAAAAAAAAAAAAAAAAAAADAA5S6qUHDhg1zqBLrnJ2d1bp1ayOOjY3V3LlzbRo7YsQIJScnG3Hv3r2t5h09elRDhgwx5Xp5eWnevHm6deuWjh49qs2bN2v79u06ceKEbt26pR07dqhNmzby9PRMc/1ChQqpc+fO2rhxoxISEnT58mXt379fW7Zs0aZNm3TgwAFFRkbq0KFDKlu2rOkYu3fvbtMx3jNr1ixJUsmSJXXkyBFdvnxZu3fv1ubNmxUSEqLjx48rX758Rn5ycrKGDBli1xr2WrVqlSSpRYsWio6O1qlTp7Rlyxbt3r1b0dHRGj58uMUxdOrUSdeuXZMkvfPOO7p9+7aOHDlivF4xMTGqX7++adxLL72Ubh179uyxWKtv376Kjo7WxYsXtXv3bm3cuFGHDh3SrVu39M0335gagEyfPt2i8c3rr7+u5ORkJScnq3Tp0sZ2JycnY7u1R2ouLi4qXLiwPvjgA126dEmxsbE6c+aM9uzZow0bNmj79u06deqU4uPjNWnSJLm4uBhjly1bpt27d6d77ClFRUXpwoULkqTAwEDduHFDJ0+e1ObNm7V//37dvHlT48aNk6Pj///M/+bNm3rqqafSnbdZs2ZKSEgwYldXV82ZM0fR0dHat2+ftmzZolOnTikyMlINGjQwjV2wYIF++eUXm48hK4wePdoUv/nmm7pz545Onz6t7du36++//9bu3bt15swZ3bp1S0eOHFHfvn3l6+tr0/yJiYk6cuSIJKlXr16Ki4vTsWPHtHnzZgUFBenWrVvq1q2baczIkSPVsmVLxcXFycnJSRMmTFBsbKwOHDigjRs36ujRo4qIiFDJkiWNMcnJyRo0aNB9vhoA8OiiKQ0AAAAAAAAAAA+5kJAQOTg4GI8BAwbkdEnIIVFRURo7dqxatmypwoULy9XV1XRuzJgxI6dLhO7eVSfl32XkyJE5XRKQI0qUKGG8D0qUKPHA1pkxYwafhQAAAAAAAAAAAAAeejExMaa4VKlSOVRJ2v773/+a4tRNJdJyr1GLJDk4OOirr76ymte5c2clJiYacZEiRXTp0iX17Nkzzbnr16+vVatW6fLly2nmhISEaOnSpWratGm6dVauXFnHjx9XlSpVjG1r1qxRfHx8uuNSq1Chgk6fPq2KFSta7Ctbtqz27dsnBwcHY9uOHTvsmj8zunTponXr1llt3jNmzBhTE6TExET9+eefku5+5z527FiLMS4uLtqxY4dpvpCQEEVERKRZQ8eOHY2GMA4ODlq8eLFmzZqVZkOht99+Wxs3bjS9Vq+88koGR5o5Xbp00cWLF/XFF1/Iz88v3dxXXnlFR44cMTXMee211+xes23btvr777/l7u5use+tt97SsmXLTNvuNQSyZvz48bp06ZIROzs7a+/evVYbQHl5eWn79u1q3ry5afuLL75o9zFkVmxsrCIjI424ZcuWGj9+fLpjKlasqFmzZun8+fN2rfX+++9r7ty5cnZ2tti3aNEiBQQEGPGNGzd04MABOTg4aP369XrjjTcsxnh7eys4ONg0X3a8hwHgYUVTGgAAAAAAAAAAgEfAhg0bVKpUKb377rtav369wsLC7L4gBgAAAAAAAAAAAAAAADkjLi7OFGfUGCMnlC1bVqVLlzbiY8eO6cKFC+mOWblypan5RO3ateXl5WWRt3v3bh0/ftyInZyctHv3bqsNO6xxdMy6n0TPnz/feJ6YmKhffvnF5rGOjo7avn17ujkBAQGqU6eOaY2goCD7C7WRh4eHfv3113RzrDWeqVevnvr375/uuOeee84Up7XOn3/+aWqa0q9fPz3zzDPpzi1JjRo1Mq1x+vRpnT17NsNxD1rp0qXVtm1bI963b59d43Pnzq0//vgj3ZwOHTqoQ4cOpm1vv/221dxvvvnGFH/66aeqXLlyuvOvWrVKuXPnNuLr169r6dKl6Y7JKinf65IUGBj4QNbx9/fXl19+mW7Om2++abHtueeeS7eJlZeXlxo1amTEiYmJCg4OznSdAPAooykNAAAAAAAAADxkSpQoIQcHB+OxYcOGdPNHjhxpyr/3SHlnE3vFxMTI09PT6ryZrSf1w9PTU/7+/mrQoIFeffVV/fbbb0pISMh0zcDj7OTJk+rQoYOuXbuW06UAAAAAAAAAAAAAAAAgE1JfG+Xj42PTuGXLltl0PVauXLmypM6PP/7YFL/11lvp5o8YMcIUf/XVV1bzUjeO6Nq1q4oUKZKJCu9flSpVTE1uli9fbvPYhg0bytvbO8O8lA1NJGndunU2r2Gv7t27Z5jTqFEji8Y+1hrVpNa1a1dTvG3bNqt5//nPf4znDg4O+vbbbzOc+55Ro0aZ4mnTptk89kFKeQ3m7du3FRYWZvPYHj16yNnZOcO8KVOmmOKtW7da5MTExJga/ri4uOijjz7KcG4XFxeLpkOTJ0/OcFxWKFCggCnevHnzA1nnjTfeyDCnd+/eFtsmTZqU4bhWrVqZ4jVr1thcFwA8TmhKAwAAAAAAAACPqR07dujEiROZGrtkyRLdvHkziysyu3nzpkJDQ7Vz5059++236tKli0qUKKHZs2c/0HWBR9GHH35oek9Wq1ZN3377rVasWKE1a9YYjzZt2uRglQAeJyEhIaaLFwcMGJDTJQEAAAAAAAAAAADAIy11gwpbb06UlJRkU15ycrLdNVnTv39/ubu7G/Eff/yRZm5kZKSCg4ON2MfHRy1btrSau2PHDlOcsolJVomJidF7772nSpUqKU+ePHJyckqziU/K1/XcuXM2r/HUU0/ZlFezZk1THB4ebvMa9urVq5dNeW5ubsZzR0dHNW3aNMMxtWrVMsVXr161mnfw4EHjef78+eXp6WlTTZJUtmxZOTg4GHFajW+ySlBQkDp37qzixYvLzc1Njo6OVs+R1I1f9u/fb/Ma7733nk15RYoUUcGCBY341q1bFp8NCxcuNMWp/ybpSd1kyp5juB/+/v5ycXEx4r/++ktvvvlmlt+4cODAgRnmFChQwHR+5cmTx6JpjjWVK1c2xQ/yPQwADzOa0gAAAAAAAADAY2zWrFmZGjdz5swsrsQ2oaGh6tevnwYNGpRlF0kAj7ro6GgtW7bMiMuXL6+dO3fq5ZdfVvv27dWqVSvjUbhw4RysFAAAAAAAAAAAAAAAAGlJ2RBEksLCwnKokoz17NnTeB4XF6fvv//eat67775rus5r0KBBac4ZERFhPM+VK5fKli2bBZX+v+eff15eXl4aM2aMjh49qpiYGJsb+kRFRdm8TqVKlWzKy58/vymOjIy0eQ17pW6Ak5ZcuXIZz3Pnzm3TGF9fX1McExNjNe/GjRvG84iIiDSbAaX1SHke2dqwyV6nTp1ShQoVVLt2bS1btkznz5/X7du3bb5W8cKFCzblOTo6qnz58jbXVaZMGVO8fv16U7xz505T3KBBA5vnLlKkiKk5zPXr120ee7+6d+9uiidOnCg3NzdVq1ZNb731lvbs2XNf8zs4OMjHx8emXEfH/2+pkDdvXpvGZOd7GAAeZjSlAQAAAAAAAIDHTMovz+bMmWN3c5ezZ89qw4YNVufLjCeffFJr1qyxeCxZskRjxoyxereVn3/+WZ988sl9rQs8Lvbu3av4+Hgj7tevn8VFSgDwbzZgwAAlJycbjwEDBuR0SQAAAAAAAAAAAABgwcPDwxSfPn3apnFdunQxfSea8uHk5PQgStU333wjBwcHIx47dqzVvPnz5xvPHR0dNWrUqDTnTEhIMJ67u7tnQZX/r3bt2po+fXqmb4SW8tqcjKRuVJGW1NfdPcibtBUoUMDuMc7Ozjbl2XIcKRsOZYW0Gt/cj0OHDqlSpUo6duxYpuewtS5XV1e75vXz8zPFoaGhpjj161uqVCm75k/ZgCgxMdGusfdjzpw5qlq1qmlbYmKiDh48qAkTJqhu3bpGg6oPP/zQ7r97Zq9ttfXvk53vYQB4mNGUBgAAAAAAAAAeM82bNzeeh4SEaNOmTXaNnz17tvHlmYuLixo1anRf9RQuXFitWrWyeHTt2lXDhw/Xxo0btXLlSuXLl8807uuvv9aZM2fua23gcXD8+HFTXLly5RyqBAAAAAAAAAAAAAAAAJlVrlw5U7x9+/YcqiRj3t7eqlOnjhGfOXNGJ06cMOUsWbLE1ESiUaNG6d5oKWVDB3ubdqTnzTffVFBQkGlb0aJFNXDgQM2ZM0eHDh3SlStXlJiYmGZDH5pN3J9z585l6XxJSUlZOp8ktWjRwtR8yMHBQXXr1tXHH3+sFStW6OzZs4qKijKdIxMmTDDNYet5kitXLrtqy5Mnjym+fv26KY6OjjbFqa+1zEjK91t2n+sHDhzQ559/rrx581rdn5CQoJMnT+rLL79U3rx51a9fv2ytDwCQMZrSAAAAAAAAAMBjpm3btvL19TXimTNn2jV+1qxZxvMOHTrIx8cny2pLS9u2bfXLL7+Ytt25c0fffffdA18beNhFRkaaYi8vr5wpBAAAAAAAAAAAAAAAAJn29NNPm+Jjx47lUCW2+eabb0zxW2+9ZYpHjhxpiseOHZvufA4ODsbz27dv319xKUyePNkivnDhgn7++Wf17t1blStXVoECBeToaP5J9YNofPJvlT9/flNcrFgxU3MXex8nT57M0vqmT5+uK1euGHHevHl17tw57dq1S6NGjVL79u1VvHhxi+uyrl27lqn17ty5Y1f+jRs3THHqpjOp60rdtCYjKd9vKd+H2WXEiBGKjIzUjh07NGjQIJUrV85qY6qkpCTNnj3booEXACBn0ZQGAAAAAAAAAB4zzs7O6tWrlxEvXrxYt27dsmnstm3bTHe06d+/f5bXl5ZWrVqpdevWpm1//fVXtq0PPKzi4uJMcU5cGAAAAAAAAAAAAAAAAID7069fP1N8/fp1BQcH50wxNmjatKnp5mirV682GrmEh4fr0KFDxr5ChQqpfv366c7n7OxsPI+Njc2SGtevX6/4+Hgjbt68uYYNG5bhuJiYGCUnJ2dJDZACAgJMcUxMTA5VYt3PP/9sipcvXy5/f/8Mx507dy5T69nbdCksLMwUFy1a1BQXKFDAFJ8+fdqu+VNeP+rk5GTX2KxUv359TZ06VceOHVNcXJz++ecfDR061OLGiSdOnLDpfQwAyB7OGacAAAAAAAAAAB41/fv318SJEyXdvYvG0qVLTY1q0jJz5kzjecGCBdWuXTuLL2QfpA4dOmjNmjVGfPjwYSUlJVncpSYrhIWFKSgoSCEhIYqOjlZSUpLc3d3l6+urUqVKqUqVKvL09LR73piYGB0+fFj//POPrl69qlu3bilv3rwqUKCAatWq9djexePChQs6fPiwzpw5o6ioKEmSj4+PihYtqoYNG1rcvSWzrl69qo0bNyo0NFQ3btyQj4+PqlatqgYNGjyQL8yPHTum/fv368qVK4qKipKPj4+KFCmixo0bW3wZ/qA8qAtgbt++ra1bt+rChQu6dOmSnJycVLduXTVr1izdceHh4dq2bZvCwsJ07do15c2b17iwqFixYlle56lTp7Rv3z6Fhobq1q1b8vf3V9OmTVW8ePF0xyUnJ2vPnj0KDg7WlStX5OHhoRIlSqhFixbKkydPltdpr/3792vPnj0KDw+Xq6ur/Pz89MQTT6hEiRI5XZpdEhIStGPHDh06dEjXrl2Tl5eXihUrpsDAQOXNm/e+509KStKuXbt04sQJhYeHKzExUb6+vipZsqSeeOIJ5cqVKwuO4sHJrs/GlG7duqVNmzbpn3/+UUxMjPLly6cSJUqoWbNm8vDwyPL17kd4eLg2b96sM2fO6M6dOypQoIAqVar0wD7TbXXu3Dlt27ZN586dU3JysgoWLKgaNWqoZs2aWdIYLD4+Xjt27FBISIiuXLmipKQkFSxYUGXLls3xYwcAAAAAAAAAAADw4Pj4+KhMmTI6efKkse2dd97R2rVrc7Cq9L388sv69NNPJUl37tzRuHHjNHz4cL3zzjumPFsaSBQsWFAXL1405jpx4oTKli17X/Vt3rzZFPft29emcQsXLryvdWHJzc3NuPlWZGTkA7vuLzNCQkKM5y4uLmrcuLFN43bv3p2p9ZKSknTs2DGVL1/epvyUnwmS1KJFC1Ncv359/fTTT0a8Y8cOm2sJCwszNW56ENeqZFb58uX1ww8/6IcfftDgwYM1bdo0Y9/8+fP1/fff52B1AIB7aEoDAAAAAAAAAI+hGjVqqGrVqjp48KAkadasWRk2pYmLi9Mvv/xixD179sz2H/uXKlXKFCclJen69evKnz9/lq2xaNEiTZgwQdu3b083z8nJSdWqVVPnzp31yiuvpNuA5Pjx45o/f75WrVqlPXv2KCEhIc1cPz8/DRs2TK+++mqWfsEbFhYmf39/JSYmSpKKFy+ukJAQu388v3HjRgUGBhrxU089peXLl1vkJSQkaO3atVq8eLHWrl2rs2fPpjmng4ODGjRooHfffVedOnXK1A/6jx8/rrffflurVq2y+vr6+vrqnXfe0ZtvvilnZ2eNHDlSn332mbH/77//Nh1XemJiYjRu3DjNnDlTZ86csZrj5OSkJk2aaNSoUWrSpIndx5OekJAQlSxZMs39zZs3t7p9+vTpGjBggBEPGDDA1GjqzJkzKlGihC5cuKDRo0frl19+UWRkpGmOTp06pdmU5rffftNXX32lXbt2pdkop2rVqnr77bfVt29fmy8qSXk+NGvWTBs2bJAkrVy5Ul988YW2bNlidUznzp31v//9z+KuRcnJyfrpp5/0xRdfWD0vXV1d9eqrr+qzzz6Tu7u7TTVmpfnz5+uzzz7TsWPHrO6vX7++vvnmm3QvftmxY4caNmxoxK1bt9Zff/1ldy3/+9//9Prrrxvxu+++q6+//tqUs2HDBtM59+mnn2rkyJG6c+eOxo8fr3HjxunKlSsWc7u6uqpLly765ptvLO7eZIvw8HCNGjVKCxYs0NWrV63m5MmTR126dNGoUaMs7riV0p49e9SoUSPjAhsnJyf9/fffNr93U79ORYoU0f79+y3uQiVlz2djWu/tqKgoffbZZ/rxxx918+ZNi3EuLi4aNGiQRo0aZbX2e0qUKGG17pkzZ5rWTS31Z1B6jh8/rvfff1/Lli0z7qKXUv78+fXhhx/q1Vdftem/RWbMmKGBAwfaXEtanzu7d+/W+++/r/Xr11sdV6xYMY0aNcrm40zt0KFDGjVqlFauXJnmXeG8vb3Vp08fffzxx6Y7DgIAAAAAAAAAAAB4PHzzzTfq3LmzEa9bt05bt25Vo0aNcq6odHz00UcaPXq0cb3Of//7Xw0fPlxLliwxcpydnTVixIgM52rYsKFp3IcffqhFixbdV33h4eGmOPV1JGmh2UXWq1ixovbt2yfp7vU7kyZNMl1vkRWcnTP3s/jY2Fjjuaurq81j/vnnn0ytJ0ljx47V1KlTM8y7ePGi6dqb3LlzW1yr2LNnTw0ePNiIg4KCbK5j1KhRprh69eo2j81OU6dO1fz5842/1b2bPwEAct7D0WIOAAAAAAAAAJDl+vfvbzxfu3atLl26lG7+smXLTA0qUo7PLtZ+eJ5egxd73L59W127dtWzzz6bYUMaSUpMTNS+ffv06aefpvsl7vLly1W+fHmNHDlSO3bsyLDesLAwffrpp6patWqm76RijZ+fn9q0aWPE586dM35kb4/UDQfSOg969Oihdu3aadq0aek2XZDuXmSwfft2denSRd26dbPaLCE9s2bNUrVq1bR8+fI0X9/w8HC9++67atmy5X19Ib18+XKVLl1aI0eOTLMhjXT3/NiwYYOaNm2qoUOHZtl5+qCtXbtWVapU0Y8//mjRkCYtUVFRatOmjbp06aKdO3em2ZBGkg4ePKgBAwaoQYMGGX7mpOfDDz9U+/btrTakke6eU0uXLlW9evV0/PhxY/utW7f09NNPa+jQoWmel7dv39Y333yjNm3a2H0u3o/4+Hj16dNHvXr1SrMhjSTt3LlTgYGBmjFjRpo5DRo0ULVq1Yx47dq1pjs62SrlHZQcHBw0ZMgQm8ZFRkaqefPmev/99602pJHuvs4LFixQpUqVtHLlSrvq+uWXX1SmTBl99913aTakkaQbN25o1qxZKl++vL777rs08+rUqaOvvvrKiBMTE9WrV690574nKChIw4cPN2JHR0fNnTs3zaYu2fnZmNKRI0dUvXp1TZgwIc154uPj9f3336tBgwaZOl+yyuLFi1WjRg0tXbrUakMaSbp69arefvttdenSxbiL2oP2v//9Tw0bNkyzIY0knT9/XgMHDtQLL7yQZu3WJCQk6NVXX1X16tW1aNGiNBvSSHffX99++63KlCmjFStW2HUMAAAAAAAAAAAAAB5+nTp1srjxSvv27RUREZFDFaXP0dHRdE1SaGioPvnkE1OTjxYtWtjULOSjjz4yxUuXLtXFixfvq77UzTv27t2b4Zjg4GCb8mCf1A1oRo4cmeXXVHl4eBjP7928zRZubm7Gc1uvzxg0aJBd1wakNn/+fJvGDxs2zBRba1Dl7u6uwoULG3F8fLw+//zzDOdOSEiwuB7wlVdeyXBcTsnKmxcCALIOTWkAAAAAAAAA4DHVu3dvOTk5Sbr7BeycOXPSzU/55WPlypVVq1atB1qfNdYuMkh94UBmDR48WEuXLrXY7uvrq5o1a6pBgwaqXLmyChYsaNe81n4snzt3bpUrV061a9dWnTp1VLJkSTk6mv+XfGhoqAIDA3X06FH7DiQdqRvIzJo1y67xsbGxWrx4sRHny5dPHTt2tJpr7bgLFiyoSpUqqX79+qpevbrVpg2//vqrOnXqZPMX9gsWLNDAgQN1+/Zt03YPDw9VrlxZNWvWNH0ZvWnTJj333HPpNk5Jy48//qjOnTtb3EHJ3d1dFStWVL169VSmTBmLv+WPP/6obt26ZWrN7LRv3z516tTJ1LQnICBAderUUenSpa02hbp27ZoCAwP1119/WewrXry46tSpo1KlSlm8Jrt371ajRo0ybMphzdixY/Xll18asbe3t6pXr67q1aubLiyRpEuXLqlLly66c+eOkpKS1K1bN1MTh8KFC6t27dqqXLmyxcVPW7Zs0RtvvGF3fZnVv39/zZ0714jz5cunatWqqVatWvL29jblJiYmavDgwek2rnrxxReN58nJyZo2bZpd9ezYsUOHDh0y4sDAQJUpUybDcUlJSerevbu2bt1qbMufP79q1qypihUrmi7ikaTo6Gh17dpVf//9t011/fTTT+rZs6du3Lhh2u7p6anKlSurRo0aFq/X7du39corr1jc2SmlN998Ux06dDDiCxcuaMCAAenWEhMTox49eig+Pt7Y9vHHHyswMDDNMdn12ZhSSEiIWrZsaXq/BQQEqG7duqpYsaLx3wL3nDp1Sl27ds2RZlorVqxQjx49dOvWLUl3m9GVK1dO9erVU4kSJazmv/vuuw+8rh9++EGvv/66ccFa7ty5VbFiRdWtW1dFihSxyP/pp580ceJEm+aOjY1Vhw4d9O2331r8ff38/FSjRg3VqlVLvr6+pn03btxQp06d7vuugAAAAAAAAAAAAAAePlu2bDFdxxAdHa0SJUqYvsd/mPz3v/81xaNHjzbF48ePt2meGjVqqEKFCkacmJiounXrmhrcpMfad+otW7Y0xend1Ea6+1q3aNHCpvVgn/79+5uuvYuMjFSDBg3suhYiLi4u3e/jU19bsHz5cpvmLV26tPE8KSlJn332Wbr5Cxcu1IIFC2yaOy2xsbFpXnt3z8qVK/X777+bto0bN85qburrJz777LMMrz1s166d6f3l4+OjTp06pTsmqxw4cEA7d+60OT8mJsZ0/aiLi8uDKAsAkAk0pQEAAAAAAACAx5Sfn5+efPJJI549e3aauWFhYaamE6mbm2SXDRs2mGJ/f3+rjTLstXv3blNTHmdnZ33wwQcKCQnR5cuXFRQUpO3bt+vQoUMKDw9XWFiYFi1apH79+il37twZzu/k5KSnnnpKU6ZM0YkTJxQTE6Njx45pz5492r17t06fPq3IyEjNmTNH5cqVM8bFxsaqV69eWdbMpGPHjqZmDUuWLLH5og3p7t2HUjaCeO655+Tq6ppmfoECBfTSSy9pxYoVunLlisLDw3X48GHt2LFDwcHBunLlik6cOKEPPvjA1Khi3bp1FherWHPu3DkNGTLEdGFCQECAfvnlF129elWHDh1SUFCQrly5oi1bthh3iVm9enW657s169at07Bhw0x30Hn66ae1YcMGRUVF6ciRI9q5c6dOnDihK1eu6Ouvv1aePHmM3GXLlmnMmDF2rZkWPz8/rVmzxnj07dvXtP+bb74x7b/3SHlXKmuGDRum2NhYubm56aOPPtKFCxcUEhKi3bt36+TJkwoPD9dbb71lGvPCCy8oODjYtG3IkCE6efKkzp49q927d+vUqVM6f/683n//fdMFU2fOnFGvXr3suivRyZMnNWLECElS3bp1tW7dOkVERCg4OFjBwcG6evWqJk+ebDovjxw5oh9//FFjxozRn3/+KUnq2bOnDh06pIsXL2rPnj06dOiQLl++bHFnoWnTpunAgQM215dZs2fPNi5Uadu2rbZv366rV69q//792rt3ryIiIrR06VJT84vExMR070zUu3dveXp6GvH06dPteq1/+uknUzxkyBCbxs2aNUtr166VJNWqVUt///23rly5oqCgIB05ckRXrlzRlClTTJ9FcXFx6tWrlyIjI9OdOygoSC+//LLpPV+8eHH98ssvioiI0KFDh7Rv3z5FRERo1apVqly5smn8p59+qpUrV6Y5/4wZM1S0aFEjXr58uSZMmJBm/osvvqgTJ04YcdOmTfXxxx+newzSg/9sTG3QoEEKCwtT7ty59cknnyg0NFQhISHatWuX8Td577335ODgYIzZt2+fxTlwz9y5c7VmzRqLZnZPPvmk1c8eWz+DoqKi1LdvXyUmJsrf318///yzrl69qmPHjmnnzp06c+aMjh8/rqeeeso07rvvvtPhw4ftfl1sdfLkSeNubRUrVtSSJUt07do1HTlyRLt27VJoaKj27t2rhg0bmsZ9/PHHunr1aobzDxs2TKtXrzZiT09Pffzxxzp9+rQuXbqkffv2ae/evbp8+bKCg4PVrVs3IzcxMVGDBg3SyZMns+hoAQAAAAAAAAAAADwM/P39NWfOHNP3uDdv3lTVqlXVtGnTdG9iI0nx8fEaP368ChcubNe1AplVunRplS1b1uq+YsWKWXx/n57ff//ddHOVixcvqnDhwlq4cGGaY4KCgtSuXTv5+flZ7AsMDDR9937hwgU1btzY6k1l1qxZo+LFi+v69es21wv7LF682HRe7927V35+flqyZEm649asWaMnn3xSefLk0QcffJBmXuomL71799bcuXNNNxyyZujQoab4s88+07fffms197XXXlPPnj3Tnc9WK1asUKtWrayej5MmTdLTTz9t2ta0aVNVq1bN6lxvvPGGChcubMQJCQmqWbOm1fdOTEyMGjdubFzjc8/333+fmcPIlPXr16tBgwYqXry4RowYke51Q0ePHlW5cuVMn2d169bNhioBALagKQ0AAAAAAAAAPMZSNpc5ePCg9u3bZzVvzpw5xhd6Tk5O6tOnT7bUl9LZs2ctviDNqrvSLFq0yBT/+OOP+uKLLxQQEGA1v1ChQurWrZtmzpyp8+fPq3r16mnOXbt2bZ06dUrLly/XCy+8oDJlysjR0fJ/v+fJk0e9e/fW/v371b59e2N7cHCwVq1alckjM3Nzc9Nzzz1nxDdu3NCvv/5q8/iZM2ea4vSaE40YMULnz5/Xd999p/bt26tAgQJW88qUKaMvvvhC27dvl4+Pj7F93LhxSkhISLeeN998UzExMUZctWpVBQUFqXv37qamJA4ODmrUqJE2bdpkXBBw5syZdOdOKTIyUn369DEaYTg6OmratGn6/fff1axZM1OjFenuHWPeffdd7dixw3R3n08++URhYWE2r5sWNzc3tWrVyniUKlXKtL927dqm/fceKS86sOby5cvy9PTUmjVrNHr0aFNzDkny9vZW06ZNjXjJkiUWF4RMnTpVP/74o+nuQZJUpEgRffnll1q2bJmpkdS2bds0adIkm489NDRUd+7cUdeuXbV161a1aNHCdCGSq6urhg0bph9//NE0buzYsRo1apQkacKECZo3b57FBU8+Pj6aPHmy6bxOTk7Wzz//bHN9mXX69GlJd5tYrFy5Ug0aNDBdgOPk5KTOnTvr77//lru7u7F9165d2r9/v9U5vby8TBfAhIaGGk15MnLjxg3T523+/PnVtWtXm8aePXtWktS+fXvt2LFDgYGBpmPx9PTUCy+8oF27dsnX19fYHhYWZjQcsiY5OVkDBgzQnTt3jG3Vq1fX/v37Ld7zTk5OatOmjfbu3Wvx78SgQYN069Ytq2vkz59f8+bNM51T77//vvbs2WORO336dM2dOzfdsdZkx2djaqdPn5aPj482btyozz77zNTcSJLy5cunr776yuJOdanfR/c0atRIrVq1Mhp93VO4cGGrnz22fgZFRkbq+vXrqlWrloKCgjRw4EBTcy9JKlu2rJYtW6a2bdsa25KSkjR16tQMX4fMCg0NVXx8vNq0aaM9e/aoa9eupgsWpbsNmNauXWv674HY2FiLxj2pLVy4ULNmzTLi0qVLKzg4WKNGjVLJkiUt8qtXr65FixaZmpzduHFDb7/9dmYPDwAAAAAAAAAAAMBD6rnnntPq1astbti1efNm1atXT66uripRooRq1KihwMBANWzYUFWqVJGPj4/c3Nz09ttvW1ynUqJEiQdW7yeffGJ1+xtvvGHXPGXLlrW4iUp0dLR69Oih3Llzq1KlSmrSpImeeOIJlStXTu7u7qpdu7ZWrVplutlWSm+++aYp3rp1qzw9PVWhQgU1a9ZMtWrVkre3t5588klFRUVJkho3bpzhNQCwX9OmTS1uEHTlyhV169ZNbm5uKl++vJ544gk1bdpU1atXV+HCheXs7GzcKCejayb69+9vuslcdHS0+vTpI1dXVzk4OJgeKfXu3dt0XUNycrJeffVV5c2bV3Xq1FHTpk1VsWJF5cqVS5MmTTJuMmfr9TSp5c2b17g2a926dfLw8FC5cuXUpEkT1ahRQ56ennrttddMTVg8PDy0YsWKdOfduHGj6Vq227dvq0ePHsqbN69q1aqlJk2aqHTp0vLy8tLWrVtNY3v06KFnn302U8dzP86fP68vvvhC+fLlk6enp8qXL68GDRqoefPmql27tgoUKKBKlSrp0qVLxhgnJyfNmzcv22sFAFhHUxoAAAAAAAAAeIx16tRJefPmNeLUTUesbbflh+VZLSwsTJ07d9bt27dN2/v165cl8x8/ftx47unpade8+fPnNzUeSa1kyZJpNrexxs3NTbNnzzb9XaZPn27z+IykbiST8sfw6QkNDdW6deuMuFy5cmrQoEGa+Q0bNrT40X56atSoYfqRfWhoqP76668088+fP6/ffvvNiF1cXLRkyRJT84bUHB0dNX36dJUpU8bmuiTphx9+MF2k85///EfPP/98huMqVaqkGTNmGHF8fHyad9B5WHzzzTdq3LixTbnjxo0zxa+88ooGDRqU7pj27dtbNL6YOHGiXXflKlWqlGbNmmVx0VVK/fr1U8WKFY347NmzunXrlnr16pXhxU7/+c9/TI2jVq5caXNt96NTp05G45y0lCtXTq+++qppW3r1pb6LU+qLttIyf/583bx504j79u1ravqSkcKFC2vBggXp/o3Kli1r8fkzffp04+Ku1NasWaODBw8asbu7u37//Xd5e3unuYarq6t+/fVX053QLl26lO5FKU2bNtWnn35qxPHx8erRo4eio6ONbf/884/F32HGjBkWjZysedCfjWmZNm1ahneIeu+99+Tv72/EwcHBunz5st1r3Q8vLy/9+uuv6f676uTkZHGB2oN+nxYvXlwLFy40NYVKzd3dXV999ZXNdSUnJ2vkyJGm8atXr7Zo6mXN8OHD1b17dyP+448/TP8tAwAAAAAAAAAAAODx0Lp1ax07dkzVqlWz2BcfH6+zZ89q//792rhxo3bs2KHDhw/r+vXrRsOMezw9PfW///1PJ0+efGC19unTRx4eHqZtLi4ueuutt+yea+DAgfrjjz8srjuIi4vT0aNHtWXLFm3fvl0nTpxI88Y0KX3xxRdq0qSJaVtiYqKOHTumTZs2ad++fabrFSpXrqyNGzfaXTds8/rrr+uPP/6wuBbl9u3bOn78uLZv367NmzfrwIEDCgsLs7iuKKNmQfPnz7d607iMbN++3eKajujoaO3du1ebN2/WP//8Y2qK8+WXX1qcV/bYsWOHvLy8JN29Ic+JEye0ZcsW7d+/33TdjnS3Ic3hw4fl6emZ7pxly5bV3r17TY157h3Hvn37tGXLFp0+fdriM2LAgAGaP39+po8lq9y8eVPHjx/Xzp07tWHDBgUFBenq1aumnFy5cumvv/5S8eLFc6hKAEBqNKUBAAAAAAAAgMeYm5ub6e4W8+fPt7ibSFBQkA4dOmTEqZuaPCgxMTEKDg7W559/rmrVqik4ONi0v3Xr1mrZsmWWrJXy4gRHR8dMfSmdlXx8fNSuXTsj3rZtW5bN3bBhQ5UrV86I161bp9DQ0AzHzZkzR0lJSUb8IM6DHj16mC4aSO+4U9fz/PPPq2zZshmu4erqqs8++8zmmhITEzVp0iQjLl68uN5++22bx7dv3141a9Y04iVLltg8NrsVL15cQ4YMsSn3yJEj2r59uxF7eHhYNJtJy1tvvaVixYoZ8dmzZ+1qsvH+++9bXMRkzdNPP22KHRwcTA0g0lK0aFHVrl3biE+ePKmYmBib68usL774wqa85557zhQHBQWlmVu7dm3VqVPHiP/880+b3u+pm9fYel7c89FHHylPnjwZ5rVp00YtWrQw4lu3bqV5gcvUqVNN8ZtvvmnTxSV58+a1+Lv/+OOP6Y4ZMWKEqa5Tp07phRdekHT3ArfnnnvOdPHPG2+8oQ4dOmRYS2bZ89loTb169dS5c+cM85ydnS3u4LV371671rpfL774ok2N3CpUqGC66PLEiRMP9H36/vvvm5rFpaV169bKly+fEaf3/ly9erX++ecfI3799ddtakhzz0cffWQ8T05O1tKlS20eCwAAAAAAAAAAAODRUbJkSe3fv1+7du1SvXr1bL4Riqurq+rWravffvtNN27csLj5yoOQ8nona7E9OnTooGvXrqlbt27p3hTnnoCAAIsbLKW0adMmvfHGG3J2dk4zx8PDQ2+99ZYOHTqU49ePPe46dOigyMhIDR06NN0bxNzj4OCgIkWK6KWXXtK5c+fSze3UqZNOnz6tLl26qECBAhk2sbknICBAZ8+eVY0aNTLM+/vvv/X+++/bNG9a/P39dfnyZTVp0kQODg5WcxwcHNS6dWuFh4fbfGO8atWq6dq1a+rZs2e657t093qxdevWZelN82zVq1cvDRkyRP7+/mkef0q5cuVSu3btFB4ebrquBwCQ89L/1wYAAAAAAAAA8Mjr37+/0YAgPDxcq1atMv24fubMmcZzLy8vm37Ybo+ZM2ea1rBFuXLlsvTOHEWKFDGeR0dH648//lDHjh2zbP7MKFmypPE8NDRUV65cUcGCBbNk7n79+hk/Zk9KStKcOXP03nvvpTtm1qxZxnNHR0f17ds3S2pJycPDQ76+vrp06ZIkad++fWnmbtmyxRT37t3b5nW6dOkiDw8PizvKWLN//35dvHjRiHv06GHThS4pPfnkk8ax/PPPP4qIiFCBAgXsmiM7PPfcczZfUJP6blBdu3aVt7e3TWNz5cqlvn37mpqwbNq0yaYLkRwcHPTMM8/YtE6VKlVMcfXq1W1qXCRJVatW1e7duyXdfY9cuHBBFSpUsGlsZlStWlWVKlWyKbdKlSpydnY2GoidP38+3fyhQ4dqz549ku42WZo+fbqpmUVq+/fvN/Il6YknnrC5Nunu37dHjx425/fv31/r16834g0bNujFF1+0yNu0aZMpHjhwoM1r9OzZU2+++abRgGzv3r2KjY1N86ImR0dHzZkzR9WrV9eVK1ckSQsXLlTLli0VFBSkAwcOGLm1a9fW119/bXMtmWHPZ6M1qRsZpSf1hVUZnV9Zzd5a7/0tkpKSFBoaqvLly2d5TQ4ODqYGfulxcnJS1apVjfP1ypUrun37tsXd3aS7TaJSsvff1WrVqsnPz09hYWGSpM2bN2f4bzkAAAAAAAAAAACAR1fdunW1c+dOSdKhQ4f022+/6cKFCwoLC1NsbKzy5s2rggULqmLFiurevbv8/Pzsmv+NN97QG2+8cV817tixwxRPmDDhvubz9PTUokWLJN29bmDJkiUKDQ1VZGSkcufOLX9/fzVu3FjPPPOMTc16JkyYoLFjx2revHn666+/FB4erjx58qhEiRJ66qmnLBpdpL65W1qmTp1qcbMdWzRu3FjJycl2j7PFhg0bMjUuMjIyU+Mycxxubm764Ycf9MMPP+jUqVOaPXu2Tp8+rfDwcCUnJ8vLy0ulSpVSkyZN1LZt2wwbrKQUEBCgX3/91e6afH19tW/fPl24cEFTp07VgQMHFBMTo8KFC6t06dJ64YUXTO8te9431l5bNzc3bdq0SbGxsfruu+90+PBhXbp0Sfnz51fNmjX16quv2tyIKvW88+bN07x587Ry5UotX75cFy9eVGxsrAoVKqSKFStq6NCh8vHxsXlOe47VlvPP19fXdGOpDRs2aOPGjfrnn390/fp1JSYmysvLSyVKlFDbtm3VunVrm9Y+efKkTXmp2fp+T+lBvocB4FFCUxoAAAAAAAAAeMw1atRIpUuX1qlTpyTdbT5yrynNnTt3NG/ePCO3e/fuyp07d47UeU+3bt30ww8/KH/+/Fk2Z+vWrTVjxgwj7t27t0aOHKlBgwbZ3GjDFpGRkVqyZIm2bNmi/fv369KlS4qOjlZsbGyGYyMiIrK0Kc0nn3yipKQkSdLs2bPT/SH7nj17dOTIESNu3ry5ihUrZvN6hw8f1uLFixUUFKQjR47o+vXrio6O1p07d9IdFxERkW5N9zg7O6tu3bo215M7d27VqFFDW7duzTB38+bNprhOnTo2r3NP8eLFTfHRo0fVpEkTu+d50OrVq2dz7r2LnO6x9+4zLVu2NDWlSX1RUlpKlixp88UQqT8jatWqZXN9qcdGR0fbPDYz7DmvcuXKJW9vb+P9ERUVlW5+z5499fbbbxvHMG3aNI0YMSLNOwzda1J2z5AhQ2yuTbrbKMOeC1YCAwNN8a5duyxyQkJCdPnyZSMOCAhQ6dKlbV7Dy8tLderUMd7PiYmJ2r17t5o1a5bmmMKFC2vWrFlq3769cfHIK6+8ovj4eCMnT548WrhwoVxcXGyuJaUH8dlojT3nl6+vrynO6PzKSrly5VL16tVtzs+uWkuUKGHXf3NYqyv1Nsn874uHh0emGl8VK1bMaEpz9OhRu8cDAAAAAAAAAAAAeDRVqVLF4mY9Oe3QoUO6cOGCEZcuXdp0U6771bRpUzVt2vS+53F2dla/fv3Ur1+/LKgKWaV06dIaOXJkTpdh8Pf3z9Z63N3dNXz48Acyd7t27Wy6WVhOCwwMtLiOCADwaLDtdpgAAAAAAAAAgEdayi/Zf//9d+OuHH/++afpx+/9+/fP7tLk4OCgMmXKaOjQodqzZ48WLVqUpQ1ppLvNdipVqmTEMTExeuedd1SoUCE9+eST+uqrr7RlyxbFxcVlav6bN2/q3XfflZ+fnwYPHqwZM2Zo3759xp2KbJHZu9BYU6xYMTVv3tyIDx8+rL1796aZP2vWLFNs63lw8OBBNWvWTFWqVNHIkSP1+++/6+TJk7p69WqGTRektI85ISFB4eHhRlyqVCm5urraVNM9FStWtCkv9Y/8n332WTk4ONj1ePnll01zXLt2za5as4s9FwKdPXvWFFerVs2utVI3njh37pxN4+xpzOTu7p5lY2/dumXz2Myw1rAiPR4eHsbzjGrz8PBQ3759jTgkJERr1qyxmnvr1i3NnTvXiL28vPTss8/aVZu9F70VL15cXl5eRnz27FmLOwjd7/kmZe6ca9u2remCn5QNaSRpypQpdjXHuedBfTamxZ7zK+W5JT34cz8lHx8fOTk52ZyfXbXez/tTSruulP++3Lx5U46Ojnb/+7J7925jjof13xYAAAAAAAAAAAAA/w6pr4/55JNPcqgSAAAAZCea0jyCpkyZktMlAAAAAAAAAHjE9O3bVw4ODpKk27dva+HChZKkmTNnGjklS5ZU48aNs3ztJ598UmvWrDE91q5dq23btunw4cO6du2aTpw4oR9++EG1a9fO8vUlKVeuXPrjjz9Uvnx50/b4+HitWbNGH3zwgZo0aSJvb281b95cEydO1OXLl22aOyIiQg0bNtTYsWN1+/btTNd4P2OtSd1YJuXfOqU7d+5o/vz5Ruzp6amuXbtmOP/y5ctVp04dbdq0KdM1pnXMqRsy5M2b1+65vb29bcq7evWq3XNnJCoqKsvnzAopG4Nk5Pr166a4QIECdq3l4+MjR8f//xoq9XxpcXNzs2udrBqbuklKVnvQtQ0dOtQUT5061Wre4sWLTe+v3r17WzToyUhmmob5+PgYz5OSkhQdHW3af7/nm7Uxtp5z//nPf1S/fn2L7c8//7x69uxpdx0P8rMxLQ/zuZ/S/dQpPbhaH0RdN2/ezPJ/1x/Wf1sAAAAAAAAAAAAAPP4OHDhg+h48T548ppukAQAA4PHlnNMFwH4//vijxcXFAAAAAAAAAJCekiVLqkmTJsbFAbNmzVK3bt20YsUKI6dfv35G45qsVLhwYbVq1SrL57VXqVKlFBQUpIkTJ2ry5MkKDQ21yLl9+7Y2bNigDRs26L333tPQoUP1n//8R3ny5Elz3u7du+vgwYOmbcWKFVPz5s1VqVIl+fv7y9PTU7lz5zY16Zg1a5Zmz56ddQeYSteuXfXSSy8pJiZGkrRgwQKNGzdOuXLlMuX9+eefioiIMOJu3brJw8Mj3bmPHz+ubt26KT4+3tjm4OCgevXq6YknnlCpUqXk5+cnNzc3ix/79+nTJ8OGP6l/yO/i4pJuvjWurq425aVugJMVkpKSsnzOrJD6b5+ee+fNPRmdE6k5ODgod+7cunnzpiTpxo0bdo2HfapWraonnnhC27ZtkyQtW7ZMV65cUcGCBU15P/30kykeMmSI3WvZ28RGsjx/YmJiTM2m7vd8szbG1nMuMTFRcXFxFtsDAwPtruFBfzbi0fEg/m3JzgZCAAAAAAAAAAAAQHZw8ffP6RLS9bDXlx1iY2M1e/Zsvfnmm6btH3/8cQ5VBAAAgOxGUxoAAAAAAAAA+Jfo37+/0ZRm27ZtGj16tPHDeQcHh3/F3Wvc3d314Ycf6v3339fWrVu1fv16bdiwQTt37tStW7dMufHx8Zo0aZL++usvbdq0Sb6+vhbz/f7779qwYYMR58mTR99//7169uxpakBjzbp167LkmNLi4eGhbt26acaMGZKkK1euaOXKlerYsaMpb+bMmaa4f//+Gc79/vvvmxrH1KtXTzNnzlSFChUyHGtL46OUzSoky4YVtoiOjrYpL3WDja+++kq1a9e2e72UKleufF/jHwaenp6m+ObNm8qfP7/N45OTk03vqfQaOyFrDB061GhKEx8fr5kzZ+qdd94x9h87dkybN2824tq1a6tmzZp2rxMbG2v3mHvNie5JfX5ZO9/udw1bz7m33npL+/fvt9j+6quvqnHjxipZsqTNNTzoz0Y8OlL/2+Lj46OFCxfmUDUAAAAAAAAAAADAw8m9Zk0FzJ+n+AsXcroUCy7+/nLPxHfqj4v0vsMuWrSohg8fno3VAAAAICfRlAYAAAAAAAAA/iW6d++uV155xWgU8d///tfY16hRI5UqVSqnSst2jo6OatKkiZo0aaJPP/1Ud+7c0Z49e7Rq1SrNmzdPJ0+eNHKPHTumAQMG6M8//7SYZ8GCBaZ4ypQp6tmzp001XLt27f4Owgb9+/c3mtJIdxvQpGxKc+3aNa1YscKIAwIC1KxZs3TnjImJMY0pVKiQVq1apXz58tlU0/Xr1zPM8fT0VK5cuXTnzh1J0sWLF22aOyVbxxQoUMAUlyxZUq1atbJ7vcdN6r/n1atXVbx4cZvHX7t2TUlJSWnOh6z37LPP6o033jDeY1OnTjU1pZk6daopf8iQIZlaJyIiwu4xKT/vHB0d5eXlZdpv7Xy737psOeeWLl2qyZMnW90XFRWlnj17asuWLXJ2zvgr1ez4bMSjw9vbW87OzkpISJAk3bp1i39bAAAAAAAAAAAAACvca9b8Vzd/edS4ubmZbogDAACAx1/6t2kFAAAAAAAAADw28uTJoy5duljd179//2yu5uGSK1cuNWzYUJ999pmOHz+u7777To6O//+/0FeuXKmjR49ajNuxY4fxPH/+/Hr22WdtXvPw4cP3V7QNmjVrpoCAACNevny5qfHB/PnzFR8fb8T9+vVL905HkhQUFGQa07NnT5ubLpw8eVK3b9+2KbdSpUrG8ytXrig0NNSmcfcEBwfblFeyZElTnLIh0b9ZyvNGkvbv32/X+NT5qedD1nNzczN9lh87dkybNm2SJMXHx2vmzJnGPg8PD/Xq1StT6xw6dMiu/LNnzyo6OtqIAwICLD5n7vd8szYmo3Pu3LlzGjRokGnbtGnTVLFiRSPeuXOnRowYYdP62fXZiEeDg4OD6Ry8detWphqsAQAAAAAAAAAAAMDDIHfu3GrTpo1CQ0MtrrUBAADA442mNAAAAAAAAADwL9KvXz+LbW5uburevXsOVPNwcnBw0EsvvWTRsGHLli0WuZcvXzaelylTRk5OTjatER0drb17995foTZwcHAw/c3j4+O1YMECI541a5Yp39r5kVrKY5ak8uXL21zP+vXrbc6tV6+eKV6+fLnNY48cOaJTp07ZlNu8eXNTbE+Nj7MGDRqYYntfl9T5qefDgzF06FBT/NNPP0mSli1bpitXrhjbe/TooTx58mRqjYMHD+ratWs252/cuNEUp35vS1KJEiVUqFAhIz579qxOnz5t8xo3btzQnj17jNjZ2Vl16tRJMz8hIUG9evUyNekaNGiQnn/+eS1cuFBubm7G9rFjx2rNmjUZ1pBdn43ZKWVzNklKTk7OoUoeTfz7AgAAAAAAAAAAAOBRlZycbHrExsZq1apV8vHxyenSAAAAkM1oSgMAAAAAAAAA/yKtWrVSkSJFTNs6d+6svHnz5lBFD69GjRqZ4oiICIuclD/Qj4+Pt3nun3/+WXFxcZkvzg79+/c3xTNnzpQk/fPPP9q1a5exvVGjRipTpkyG86VuSmDrcScnJ+v777+3KVeSOnbsaIp/+OEHJSUl2TT222+/tXmdevXqKV++fEa8fv16HTlyxObxj6tmzZqZ4qVLlyoqKsqmsXfu3NHs2bPTnQ8PRoUKFUyv9eLFi3X9+nVNnTrVlDdkyJBMr3Hnzh1Tc6uM3PvMuSetcyH19hkzZti8xvz583Xr1i0jrlOnjtzd3dPM//TTT7V161YjrlSpkv73v/9JkqpWrarx48cb+5KTk9W3b1+LpjOpZddnY3by8PAwxbGxsTlUyaOpbdu2ptief5sAAAAAAAAAAAAAAAAAAHgY0JQGAAAAAAAAAP5FnJycdOrUKd24ccN4zJo1K6fLeiilbkKTsnHJPX5+fsbzw4cPKzIyMsN5Q0ND9dlnn913fbYqXbq0GjdubMQ7d+7U8ePHLf7uqZvXpCXlMUvSli1bbBr3/fffKzg42KZcSWrXrp38/f2NODg4WBMmTMhw3NatW/Xjjz/avE6uXLn0xhtvGHFycrKGDh2qO3fu2DzH46hixYp64oknjDgmJkaffvqpTWP/+9//6ty5c0ZcokQJtW7dOstrhHUvvvii8TwuLk6ff/651qxZY2yrWrWq6tevf19rfP7557px40aGeatXr9b69euNOHfu3OrZs6fV3MGDB5vi8ePH68KFCxmuER0drZEjR5q2pdd0Z926dfrqq6+M2M3NTQsWLDA1sRk2bJieeeYZI758+bL69u1r0Xgmpez6bMxOXl5ecnJyMuIzZ87kYDWPns6dO5uave3cufOhbUAEAAAAAAAAAAAAAAAAAIA1NKUBAAAAAAAAgH8ZNzc3eXp6Go9cuXLldEkPXO/evbVx40ab869fv66pU6eattWuXdsiL2XTjvj4eH3wwQfpznvlyhV16NDBpuY1WSl1w5kZM2Zozpw5Ruzm5qZnn33Wprlq164tFxcXI/7111+1bdu2dMcsX75cb731lh0V322glLrRxPDhwzVx4sQ0G0OsXbtWHTp0UGJiohwcHGxe6/XXX1ehQoWMeMuWLerWrZuioqJsnuPmzZv63//+p2nTptk85mH39ttvm+L//e9/GTaxWr16tUaMGGHa9sYbb8jRka+kskvXrl1VsGBBIx4/frzpPZNewxZbXbp0ST169Ei3edPJkyfVr18/07b+/fvL29vban6rVq1UrVo1I75586Y6deqU7vswPj5e3bt316VLl4xthQsXVq9evazmh4eHq0+fPkpKSjK2TZgwQVWrVrXInTp1qgICAox4zZo1GjNmTJq1ZNdnY3bKlSuXypUrZ8TBwcE6depUDlb0aHFyctLo0aNN215//XX99NNPds1z/PhxvfDCCwoNDc3K8gAAAAAAAAAAAAAAAAAAyBBXAAMAAAAAAAAAHnsrVqxQYGCgKlWqpE8//VTbt29XbGysRd6tW7e0aNEi1a9fX2fPnjW2V69eXXXr1rXIT91w4YcfflD//v1NYyXpxo0bmjZtmqpVq6bg4GBJUsWKFbPgyGzz7LPPKnfu3EY8YcIEnT9/3og7d+6svHnz2jSXh4eHnnnmGSNOTExUu3bt9OOPPyouLs6Ue+LECb300kvq1KmTbt++LV9fX+XPn9/mugcNGqRWrVoZcXJyst58801Vr15dX3zxhX799Vf98ccf+vbbb9WmTRu1bt1akZGRcnd3V58+fWxeJ2/evFq0aJGpQdPvv/+uypUra9y4cTp37pzVcefPn9fixYvVp08fFSlSRK+//rrpdX3Ude3a1fS3Tk5O1oABA/Tiiy/q9OnTptxLly7pww8/VIcOHRQfH29sf+KJJ/TKK69kW82QXFxcNHDgQKv73Nzc7HpvWHOvUcuff/6phg0bauPGjaamNzdv3tRPP/2kevXqKTw83NheqFAhffHFF2nO6+DgoOnTp5veh0FBQapRo4aWLFliOq+SkpK0Zs0a1alTR3/99Zdpnp9//llubm4W8ycnJ6tfv34KCwsztj3zzDN68cUXrdbj7e2tefPmydnZ2dj20UcfaefOnVbzs/OzMTs9+eSTxvPExEQ1bdpUn332mZYuXao1a9Zo7dq1xiNlcyDc1aNHDw0dOtSI79y5oxdeeEEtW7bU8uXLdfPmTYsxd+7c0f79+zVx4kQ1adJEFSpU0E8//ZRuEygAAAAAAAAAAAAAAAAAAB4E54xTAAAAAAAAAAB4PBw9elSjRo3SqFGj5OTkJH9/f/n4+MjFxUWRkZE6ffq0xY++3d3d9fPPP1ud78knn1T79u31559/GttmzZqlWbNmqVSpUipYsKAiIyN15swZU0OFXr16qWzZsvrss88ezIGm4uXlpc6dO2v+/PmSZNEgoX///nbNN3r0aK1YsULR0dGSpOjoaA0dOlSvv/66ypUrJ1dXV126dEkXLlwwxjg5OWnGjBkaNmyYrl69avNaixYtUqtWrbR3715j28GDB3Xw4EGr+ffWOXz4sGl7ysYS1jRp0kSzZs3SwIEDjdcnNDRU77zzjt555x0VLlxYvr6+cnV1VVRUlMLDw3X9+nWbj+NR9eOPP+rkyZPav3+/pLuNPaZMmaIpU6aoRIkSKliwoK5du6YzZ84oKSnJNLZkyZKaN2+enJyccqL0f7UXXnhBY8eONTWLkaRu3bopX7589zV3v379tGPHDq1Zs0Z79+5VYGCg8ufPr4CAAMXFxenMmTO6deuWaYyrq6vmzJmT4dq1atXSd999pxdffNE4n0JCQtStWzflyZNHJUqUkJOTk86ePWv1/ffZZ5+pbdu2VuceO3asVq9ebcQBAQGaOnVquvU88cQT+uyzzzRixAhJUkJCgnr06KHg4GCrjbyy87Mxu7z00kuaMmWK8bl48eJFjRw50mru9OnTNWDAgOwr7hExadIkXb9+Xb/88ouxbf369Vq/fr2cnZ0VEBAgHx8fJSQkKDIyUqGhoab/ZgAAAAAAAAAAAAAAAAAAIKc45nQBAAAAAAAAAADkhMTERJ09e1b79u3Tzp07dezYMYuGNEWLFtXatWtVq1atNOeZO3eu6tWrZ7H99OnTxrwpf1zeo0cPTZ8+PesOxEZpNZ4pXLiwWrdubddcpUuX1qJFi+Tp6WnaHhcXpwMHDmj37t2mpgtubm6aO3eu2rVrZ3fd3t7eWrt2rU2NcwoUKKDffvtN3bt3V0xMjGmftQYSqfXo0UNbtmxRuXLlLPZdunRJ+/fv165du3Ts2DGrDTGcnJxUpEiRDNd5lPj4+Gjjxo1Wz5GQkBDt3r1bp06dsmhIU7duXW3dulUBAQHZVSpSKF26tFq1amWxfciQIfc9t6OjoxYtWqTGjRsb265evaqgoCAdOXLEoiFNnjx5tHjxYqv1WDNkyBDNnz9fefLkMW2/ceOGDh48qODgYIv3n6urq7799lt98sknVufcuXOnPvroIyN2dnbW/Pnz5e3tnWE977//vlq2bGnEISEheuGFF6zmZudnY3YpV66cZs+ebXFMsF2uXLm0cOFCff3118qdO7dpX0JCgk6dOqXdu3dr3759Fk3s7ilQoIDFWAAAAAAAAAAAAAAAAAAAHjSa0gAAAAAAAAAAHnu7du3SmDFj1LJlS3l4eGSYX6ZMGY0ePVrHjh1Tw4YN08319vbWpk2b9PHHH6fb+KRy5cqaN2+e5s+fLxcXF7uP4X61atXKasOU3r17y8nJye75nnzySe3evVtPP/10mjnOzs7q1q2b9u/fr+eee87uNe7x9vbWjBkztHv3br3++uuqUqWK8uXLJ2dnZ/n6+qpFixaaMGGCTp06pQ4dOkiSrl27ZprDlqY0klS7dm0dOXJEs2bNUoMGDTJ8bVxdXdWiRQt98803On/+fJrNKh5lefPm1V9//aVff/1V9erVk4ODQ5q5VapU0fTp07Vjxw4VLlw4G6tEas8//7wpLl++vJo2bZolc+fNm1fr16/Xl19+qQIFCljNcXFx0bPPPqsjR44Y70tbPfvsszp58qRefvll+fj4pJmXJ08e9evXT//8849efvllqzlRUVHq2bOnqenYqFGjMvxsv8fR0VFz5syRr6+vse2XX37Rjz/+aDU/Oz8bs0u3bt10/PhxffXVV2rTpo2KFSsmT0/PdD8LYOndd9/VmTNn9M4776h48eIZ5vv5+alPnz769ddfdfHiRRUqVCgbqgQAAAAAAAAAAAAAAAAA4P85JCcnJ2dmYO3atRUWFiY/Pz/t3bs3q+t66DxMx1u7du10a7jw/uZsrCZr+X/VJBOjQrO8DuDRUjTzQxOPZl0ZwKPIqeL/P4/flnN1AA8DlycyPXRoOdcsLAR49Ew5fjtFdDHH6gAeDikaHSQezrkygIeBU+WcrgDpSExM1NGjR3XixAmFhobqxo0bku42NyhatKhq1KihkiVLZmruuLg4bd++XUePHtX169fl4uKiIkWKqG7duipXrlxWHsZD5dKlS9q8ebMuXLig2NhYeXl5qUyZMnriiSfk7e2dIzXVqVPH+H/pLi4uunHjRqaaAUVFRWnHjh26ePGiIiIidOfOHeXJk0e+vr6qUKGCypcvLzc3t6wu/6F2+fJlbdu2TWFhYbp+/bq8vLxUqFAh1a9f36ZGC8gen3zyiUaPHm3EY8eO1TvvvGP3PBs2bFDz5s2N+NNPP9XIkSONOCEhQdu3b9fBgweN88Hf31/NmzfPkvd/YmKidu3apRMnTig8PFxJSUkqWLCgSpUqpSeeeEK5cuW67zUelIfxsxEPj5MnTyo4OFhXrlzR9evX5ezsrLx586p48eKqWLGiSpQokdMlAgAAAAAAAAAAAAAAAAD+5ZxzugAAAAAAAAAAALKTk5OTqlSpoipVqmT53G5ubmrevLmpgcO/QeHChfXss8/mdBmGiIgIHThwwIirVauWqYY0kpQ3b161adMmq0p7LBQqVEhdunTJ6TKQjsTERE2fPt2IXVxc1L9//weylrOzs5o0aaImTTJz44GMOTk5qWHDhmrYsOEDmf9Betg+G/FwKVOmjMqUKZPTZQAAAAAAAAAAAAAAAAAAkCbHnC4AAAAAAAAAAAAgK33//fe6c+eOETdq1CgHqwGy359//qkLFy4YcZcuXVSwYMEcrAgAAAAAAAAAAAAAAAAAAACPGprSAAAAAAAAAACAh1ZycrJd+fv379cXX3xh2vb8889nZUnAQ2/MmDGm+OWXX86hSgAAAAAAAAAAAAAAAAAAAPCooikNAAAAAAAAAAB4aM2dO1f9+vXT/v37M8xdsmSJAgMDFRcXZ2xr0aKFqlWr9iBLBB4qM2bM0JYtW4y4Tp06atKkSQ5WBAAAAAAAAAAAAAAAAAAAgEeRc04XAAAAAAAAAAAAkJaEhATNnj1bs2fPVqVKldSiRQtVr15dvr6+cnZ21rVr13Tw4EGtWLFChw8fNo3NkyePpk6dmkOVAw/epUuXjPM+PDxca9eu1axZs0w5o0aNyonSAAAAAAAAAAAAAAAAAAAA8IijKQ0AAAAAAAAAAHgkHDlyREeOHLEpN2/evFqyZIlKliz5gKsCcs7q1as1cODANPd369ZN7dq1y8aKAAAAAAAAAAAAAAAAAAAA8LigKQ0AAAAAAAAAAHhoFShQQK6urrp9+7bNY5588klNnDhRFStWfICVAQ+3+vXra9q0aTldBgAAAAAAAAAAAAAAabp0Oko3Im7ldBkW8hTIrcKl8uZ0GQAAAECOoykNAAAAAAAAAAB4aHXo0EHh4eFavXq1tmzZogMHDujMmTO6evWqbt26JXd3d/n4+KhEiRJq1qyZnn76adWpUyenywaynYODg7y8vFSlShX16NFDQ4cOVa5cuXK6LAAAAAAAAAAAAAAArLp0Okq/jtmb02Wkqeu7tWlMAwAAgH89mtIAAAAAAAAAAICHmpeXl7p3767u3bvndCnAQ2XAgAEaMGDAA10jMDBQycnJD3QNAAAAAAAAAAAAAMC/z42IWzldQrpuRNyiKQ0AAAD+9RxzugAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwMODpjQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANNaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABprSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACATClTpowcHBzk4OAgZ2fnnC7Hblu2bDHqd3Bw0ODBg9PMHTx4sCl3y5Yt2VgpAPwfe/cdHVW1v3/8SSEJSWghQIBA6BCaQqSDFEGlKIpUpVgQFK9eFMGGKBZUEPXa8FKugKBUUUGKtASClADSe0JJCAFCSEJIT+b3Bz/ONyeTMhMCCfB+rZW1Zp/Ze5/POVO8d83m2bcWoTQAAAAAAAAAAAAAAAAAAAAAAAAAAADATZY99KBs2bIFmid7IEKnTp0KtU4AAABAkm6/mDEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAO4iX331lX777TejHRgYWGS1AADuDoTSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABQjP32228KCgoq6jIAAHcRx6IuAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAoLibOXOmLBaL8de+ffuiLgkAbhpCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABkJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGQmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAbnoi4AAAAAAAAAAAAAAAAAAAAAAAAAAAAAwK2zZs0arVixQmfPnlViYqIqVaokf39/jRgxQl5eXoV2nu3bt2vp0qU6deqUYmNjVblyZU2bNk3u7u75jo2MjNQPP/ygEydOKDo6Wj4+PnriiSfUu3fvPMdt2bJFP//8s06ePCkHBwfVqFFDL774oho3bnxD17Jo0SJt3LhRkZGRSk1NVeXKldWsWTM999xzNl2PrVauXKnVq1crNDRUJUqUUPXq1fXss8/q3nvvvaF5U1NTNXfuXO3cuVNnz55VRkaGfHx81Lp1az399NNycXEp0LwHDhzQ0qVLdfDgQcXFxcliscjT01N+fn4KCAjQY489Jk9Pzxuq3V5r1qzRypUrFRYWpoyMDPn7+2vUqFGqXbt2rmPS09M1e/ZsbdiwQdHR0SpTpowCAgL0r3/964br37p1q5YsWaLw8HDFxsaqfPnyqlWrloYPH66aNWve0NwFdfjwYa1atUp79uxRdHS0MjIyVL58edWsWVODBw+Wv79/oZ0rMzNTM2bM0M6dOxUeHq6yZcuqYcOGeuWVV1S2bNkbmnv79u1asGCBIiIiFB8fL29vb9WtW1fDhw+Xr69v4VxAIYmKitLixYu1fft2xcTEKDU1VR4eHqpUqZKaNm2qRx99VNWrV7+hcyxatEiBgYE6deqUHB0d1axZM7388suqWLFirmMSExM1ffp0bdmyRXFxcSpfvrw6dOigESNGyNmZOArgbudgsVgsBRkYEBCgqKgo+fj4aNeuXYVdV7FTnK43ICAgzxoi3tx8C6spXL6fdijAqLOFXgdwe6la8KEZhwuvDOB25JTl/xin/l10dQDFgUvbAg8dWc+1EAsBbj//PZaSpRVZZHUAxUOV/3uYcbDoygCKA6dGRV0BAAAAAAAAAAAAAAAAABRbx3ZEae3/DhV1Gbnq9mxD1WvpU+jzBgcHq0OH//t3lGXKlFFsbKzd8wwfPlyzZs0y2h07dlRgYGC+41JTU/XMM89o0aJFSk9Pz7Wfn5+fZs+erU6dOtldy+bNm9W+fXt9//33evvttxUXF2c15tSpU/Lz85MkOTg4WF3HgQMH1KdPHx0/fjzHc5YuXVq//PKLevToYTq+dOlSjRgxQjExMTmOq127ttasWZNnMEl2MTExevLJJ7V27VplZmbm2q9Ro0ZasGCBTcE3nTp1UlBQkNG+/k+9//Of/2j8+PFKSEjIcZyPj4/mz5+vLl262Fy/JO3evVtDhw7VoUOHlNs/K3dwcFDLli31yy+/2BySMmXKFH388cc5vsbZlS1bVs8884y++OILu2rPTW7vu8mTJ+ujjz7SlStXchwXEBCgwMBAq5CZYcOGaf78+crIyLAa4+DgoCFDhmjOnDl21ZicnKwRI0Zo0aJFSklJybVf+fLlNXXqVA0bNizH57N/b9jq+j3Jau7cufrmm2+0f//+PGuSJE9PT73yyiv6+OOPbTpfnTp1FBoaKklycnIyvmP69++vX3/9Ncd7K117TVauXJlnaEpOXn31Vf3www9KTk7OtU/FihX19ddfa8CAAfnOl/0+P/fcc5o5c2aOfXN7/+Vm/fr1evbZZ3XmzJl863Bzc1O7du20bt26HJ/P7fvjlVde0YwZM3K9Hw8//LD+/PNPOTo6GseSk5PVp08frV69OsfvBicnJ40bN06TJk3Kt24Ady7H/LsAAAAAAAAAAAAAAAAAAAAAAAAAAAAAuF0dOHBA5cqV088//5xnII0knT59Wp07d9bw4cMLdK4ePXropZdesimsJLtly5bpnnvuyTWQRpLi4+PVq1cvLVy40Dj22muvqW/fvrkG0khSaGioGjdurJMnT9pUy5o1a1SpUiWtWbMmz0AaSTp48KCaNGmijz76yKa5s+vevbtGjx6dayCNJEVFRalr165atGiRzfM+//zzCggI0MGDB3MNpJGuBVts375dderU0bx58/Kdt3Xr1ho3bpzNr3FsbKzmzp1rc90F0atXL73xxhu5BtJI0q5du+Tn56fExERJUmJioqpXr665c+fmGppisVg0d+5c3X///TbXEhgYqLJly+qnn37KN/zl0qVLevrpp/Xggw/aPH9BnD59WsOGDdPOnTvzrUmSEhISNGnSJNWrVy/P92VukpOT5evrq8WLF+d6b6Vrr4mvr6+2b99u07xRUVGqUKGCvvrqqzwDaSTpwoULGjhwoLp27WpX7YXprbfeUteuXW0KpJGu3bf169fbdY577rlH33zzTZ73Y/Xq1apfv77RjoyMlI+Pj1atWpXrd0NGRoY++eQTPf3003bVA+DO4lzUBQAAAAAAAAAAAAAAAAAAAAAAAAAAAAC4OY4fP65mzZpZhdGUKlVKtWvXloeHh86dO6eTJ0+awglmzZqlq1ev6pdffrH5XJMmTdKqVauMdoUKFVS9enW5uroqKioqz0CYmJgY9e3b1wiAKVWqlOrUqSNPT0+dPXvWVJ/FYtGQIUPUs2dPzZw5U19++aUxT6VKlVS9enWVKFFCJ0+e1Llz54znkpOT1a1bN504cSLP61i7dq26d+9uFdbg5eWlmjVrysXFRREREQoPDzc9/+677yolJUUffvhhPnfq/zz77LNavXq1JMnBwUFVqlRR1apV5ezsbFX/9et+8MEHVbZs2Tznffjhh7VmzRrTMWdnZ1WvXl0VK1aUk5OTzp07p9OnTxuhIZmZmRoyZIg8PDz0+OOP51pv9gARFxcXY14XFxdduXJFFy5c0Pnz55WammrzvSiojz76yHStvr6+qlatmjIyMhQaGqpLly4Zz8XExKh79+4KCgpSs2bNjNfQyclJtWrVUqVKlXT16lUdOXJESUlJxrjNmzfrq6++0ujRo/OsZenSperXr5/Ve+f6Z8HDw0OxsbEKDQ3V1atXjefXrl2rjh07KigoyDTOwcHB7vthyzhXV1dVrFhRFSpUkKenp9LT0xUdHa0zZ86Ywk2OHz+u9u3ba8+ePXadv0OHDjp79qxRS/Xq1VW1alUlJSUpLCzMFGiUlpam9u3b68iRI6pdu3auc8bHx6tOnTqm+yZJJUuWVJ06dVSmTBlduHBBoaGhpiCc9evXq3Xr1tq2bZtd13CjVq5cqU8//dR0zNHRUVWrVlXlypXl7u6uxMRExcTEKDIy0ghLskf37t21b98+Sdfuc82aNVW5cmWlpKTo6NGjppCmEydO6Nlnn9X06dPVpEkT4zVwcXFR7dq15e3trdjYWB05ckRpaWnGuDlz5mjAgAHq3r17QW4DgNucY1EXAAAAAAAAAAAAAAAAAAAAAAAAAAAAAODm6NixoymQxtXVVfPmzVN8fLz++ecfBQcHKzQ0VLGxsWrdurVp7IIFC7Ro0SKbz3U9kKZq1arau3evLly4oJ07d2rLli0KDQ1VYmKiqlatmuPY/fv3KzMzU66urlq4cKHi4+O1e/dubdq0SaGhoTp58qS8vLyM/mlpaRo8eLDGjBljnHP//v2KiorSjh07tGXLFkVGRurPP/+Uk5OTMS40NFTr16/P9RqSk5P1+OOPm0JFSpUqpfXr1+vSpUvauXOn/v77b505c0bh4eGqU6eOafzHH3+snTt32nzPfvzxR0lSQECAIiMjFRERoe3btxv1//3333JzczP6p6am6t///neec2YPaXFxcdHkyZOVlpam0NBQbd261XjdExISNGDAANP4gQMHKiEhIce5f/rpJ+Oxg4ODpk6dqpSUFB0/flxbtmzRxo0btXPnTp05c0YpKSnasWOHevfurcqVK9t8T+x1/VobN26ss2fPKjw8XH///be2b9+u6Ohoff/996b+mzZt0rPPPqtjx45JkgYNGqTk5GQdO3ZMmzdv1u7du5WYmKi+ffuaxk2YMCHPOiIjIzVo0CDTe+ehhx7SuXPnjM9CUFCQ9u7dq4SEBP38889ydXU11fXVV1+Z5mzXrp0sFossFos6duxoeu768Zz+2rVrZ+rr4OCgcuXKadSoUTpx4oSSk5N15swZ7dq1S0FBQdqyZYuOHj2qpKQkLVmyRKVKlTLG7t27V7Nmzcrz2rPKyMgwPgP+/v6KiorSqVOntGXLFu3evVuxsbFauHChXFxcjDHp6el68MEH85y3U6dOpkAaR0dHTZ48WYmJidq3b582b96so0ePKjk5WY899php7Pbt2/XBBx/YfA2FIXuAUb9+/ZSUlKQzZ85o+/bt2rhxo7Zv367jx4/r6tWrCg8P17/+9S+VK1fO5nNcD7Tq0qWL4uPjFRoaquDgYIWEhCg+Pl5jx4419Z87d6569+6tmJgYSdLrr7+ulJQUHTp0SJs2bdK+ffuUkJCgVq1amcaNGjWqAHcAwJ2AUBoAAAAAAAAAAAAAAAAAAAAAAAAAAADgFouLi5ODg4Pdf/aEQ3zxxRc6d+6c0XZ2dtauXbv01FNPWfUtXbq0tm7dqs6dO5uOv/DCC3Zdl6+vr86cOaOmTZtaPefm5iZnZ+dcxzo7O+vw4cPq37+/1XN+fn76+++/Tcd+//13ZWZmqkqVKjp16pQaN25sNa5Hjx768MMPTceyt7MaNWqUKfjCw8NDYWFh6tKli1VfX19fHT9+XHXr1jWOWSwWPfnkk7nOn5Nu3bpp586d8vHxsXquTZs2RtjPdb///nuuc8XGxuq9994z2iVLltTx48etwimuc3Nz04IFC0xjUlNT9fLLL1v1DQwMNAUcjRw5Uq+99lruFyapRYsW+u2337R///48+92oZs2aaf/+/apSpYrVcy+++KLVe/56GNCbb76pn3/+Ocf35eLFi+Xn52e0r1y5ouDg4Fxr6NWrl9LS0oz2559/rtWrV+f4ukrXwnCOHj2qEiVKGMeyvg6FqXr16oqJidF3332n2rVr59n3iSeeUEREhEqWLGkcK0igS8OGDXXo0CFVrFjR6rn+/fsrJCTEFBgVFham+fPn5zjXhg0b9M8//xhtBwcHrVy5Msf3tbOzs5YtW6bnn3/edPyjjz5SZmam3ddRUGFhYcbjevXqadGiRaYgnux8fX31zTffGIExtnr88ce1fv16eXp6Wj03efJktWnTxmhnZGRo5cqVkqTZs2drypQpVmNcXFy0bds203ynTp1SdHS0XXUBuDMQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAADcgT7//HNT+7333lOjRo3yHLN69WpTGMXly5e1bNkym8+5YcMGOToW7J8wjx8/XjVr1sz1+fr166tOnTo5njOvsJu33nrL9PyBAwdy7btgwQJT+6effpK3t3deZWvTpk2maz5+/LiOHj2a55jrSpYsqdWrV+fZp1OnTvL19TXacXFxSk1NzbHvSy+9ZAreWLp0qapXr55vHe+//76qVatmtBctWmTV59ixY6b2I488ku+8t4KTk5M2bdqUZ5+vvvrK6li1atX0ySef5Dlu9OjRpvbChQtz7BcaGmoKTWnfvr3GjBmT59zStbCl8ePHG+34+Pg8Q4duldKlS+vFF1802mfOnLEr0MXR0VEbN27Ms0/Tpk316quvmo7lFhj1+uuvm9oDBw7UQw89lOf806dPNwUCpaWl6f33389zTGHKyMgwHgcEBNyUc3h4eOjXX3/Ns09OwTMtW7bUsGHD8hw3YMAAUzu/8wC4MxFKAwAAAAAAAAAAAAAAAAAAAAAAAAAAANxhEhISdO7cOaPt4uJiCr/IjYuLi1VYwffff2/TOevWrau6devaV+j/5+TkpHfffTfffi1atDC1a9Wqpfr16+c7rnLlysbj2NjYHPts3bpVSUlJRrtChQp6/PHH853bx8dHnTt3Nh37z3/+k+84SerXr59NIT6tW7c2tYODg3Pst3z5cuNxhQoV1L17d5vqkKRBgwYZjxMTExUREWF6vnz58qb2qlWrbJ77Zmrbtq08PT3z7OPt7S0PDw/TseyBMzkZPHiwqb1nz54c+02cONHU/u677/Kd+7q3335bDg4ORnvevHk2j72ZunbtamqvWLHC5rHt27dXxYoV8+332WefycnJyWhnDz66bv/+/cZjBwcH/fDDDzbVkf07JaewpZsl62uaNbCoMPXr1y/fPu3atbP6jskpqCa7Pn36mNp///23fcUBuCMQSgMAAAAAAAAAAAAAAAAAAAAAAAAAAADcYmXKlJHFYrH777nnnrNp/oULF5razZs3t7m27EEOe/futWlcly5dbD5Hdr6+vjaFs9SpU8fUbteunU3zZw3IyMjIyLHPggULTO1u3brZNLckvfLKK6b25s2bbRr35JNP2tTP39/f1I6MjLTqEx0drStXrhjtgIAAm+a+LnvwzR9//GFqZw+4+e6772wKt7jZevToYVO/smXLmtrPPvtsvmO8vb1N4SJxcXE59sv6eru4uKhp06Y21SRJzs7OKlmypNHet2+fzWMLIjQ0VE899ZRq1qypkiVLysnJSQ4ODlZ/2e/rwYMHbT6Hrd9Tjo6OatCggdG2WCxav369qc+BAweUnp5utKtVq6bSpUvbNP+oUaNMr9+pU6dsGlcYsoY4HTlyRP369VNiYmKhnsPW7w83NzfjsaOjo+6///58x2T/b8alS5fsKw7AHYFQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAOAOs337dlM7e+BIXqpUqSIXFxejffnyZZvGtWnTxuZzZJc1NCYv2YNFateubdM4Dw+PfPvs37/f1O7atatNc0vWwShnz561aVyzZs1s6pf9unMKiFi+fLmpvXr16hzDRnL769Onj2l8RESEqe3u7m56jS0Wi8aNGydXV1fdd999euedd3T48GGbrqcwNWzY0KZ+rq6uxmMHBwere5qbrGFJSUlJOfbJGhKUmppq1313cHAwhZVkDRYqTNHR0WrdurXq1Kmjn3/+WadOnVJycrIyMzNtGh8VFWXzuR599FGb+957772m9oYNG0ztjRs3mtqNGjWyeW5J8vLyMh6npKTYfL03avTo0ab2kiVL5OnpqXr16umFF15QYGDgDZ/D1u+PEiVKGI+zBiDlJft3ckJCgu2FAbhjEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAA3GGio6NN7Vq1atk1PmtwQUZGhk1jqlSpYtc5snJ3d7epX9aAEEkqU6aMTeMcHBzy7RMbG2tqN2jQwKa5JcnZ2VlOTk5GOzk52aZxtobxZL9ui8Vi1efMmTM2zWWr7O8hSVq3bp0qV65sOpaamqpdu3Zp0qRJatiwoVxdXdW4cWNNmTJF6enphVpTTrKGjuQl63sg+/20VU73XZLS0tIKNF9Ocgu+uREXLlxQ7dq1rcKq7JE1OCcv9gT+SFL16tVN7QsXLpja586dM7V9fX1tnluyDnTKGiB0M73zzjvq3r276ZjFYtHx48f13//+V507d5aTk5P8/Pz00ksvWV23Lby9ve0e4+zsbFM/W75zANz5CKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAA7jDx8fGmdrly5ewa7+rqajy2NYwga5DN7ejq1aumtr2BD1nDHm5FGEt2BQm1yEtmZqbVMXd3d0VEROjll1/O9fVOTU3VwYMHNW7cOLm7u+uNN94o1LqKo8IM7LgZ4R9dunSx+k5o3Lixxo4dq99++01hYWG6fPmyLBaL8bd58+YC1WVv4E/20Ji4uDhT+/Lly6a2rUFU12UPvIqKirJr/I1YuXKlZs6cmWv4VGZmps6cOaPvv/9ePj4+euihh2wOtAKAW4FQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAOAOU7p0aVM7e7BDflJSUozHDg4OhVJTcefh4WFqR0dH2zU+axBN1oCaWyX7az5o0CBTyIi9fzNnzszxPI6Ojvr666+VmJiov/76S08++aRq1qyZ4zWnpaVp8uTJ6tSp08245GLJw8Pjhu57bGxsodazZ88eHTx40Gi7urpq165d2r9/vyZPnqzevXurZs2aVuEwFy9eLND5cgozykv2680eOpM9UCt7aE1+EhMTTW0fHx+7xt+o5557TufPn9ehQ4f08ssvq0mTJjkGOlksFv3111/y9fW1qhkAigqhNAAAAAAAAAAAAAAAAAAAAAAAAAAAAMAdxtvb29QOCwuza3xSUpLx2MnJqVBqKu6yh3IcOXLE5rHp6enKyMgw2m5uboVVls2qVatmal+4cOGmn7Nbt26aP3++wsLClJaWph07dujJJ5+0CvgJCgrS999/f9PrKSpZA3myBjoVB1999ZWpPWXKFDVv3jzfcSdOnCjQ+SwWi+Lj423uf+bMGVO7YsWKpnblypVN7YiICLvqyR56U6VKFbvGFxZ/f399/fXX2rdvnxITExUeHq7XX3/dqp5Lly7p0UcfLZIaASA7QmkAAAAAAAAAAAAAAAAAAAAAAAAAAACAO0yrVq1M7W3bttk8NioqSqmpqUa7XLlyhVZXcdakSRNTe926dTaPXblypaldtWrVQqnJHg888ICpfejQoVteQ4sWLTR//nwlJCSoU6dOpuemTp16y+u5VcqXL288Tk9P18mTJ4uwGrPs74OXXnrJpnEbNmwo8Dl/++03m/vu3bvX1O7SpYup3blzZ1P74MGDdtUSExNjPHZ1dZWjY/GIWPD19dWUKVN09uxZffbZZ6bnAgMDi6YoAMjGOf8uyOrChQsKCAgo8hry4vtph1tUSXFx6/9HOXDHcPIv6gqA4sOlbVFXANy2/nuseKU3A0WraNKygWLJqVFRVwAAAAAAAAAAAAAAAAAAAHBXGzRokIYPH260d+/ebfPYDz74wNS+5557Cq2u4uzJJ5/U119/bbTXrl1r89hvvvnG1O7Q4db/e9/69eurZMmSSkpKkiSdO3dOERER8vX1veW1SNL69evl7Owsi8UiSYqMjCySOm6Ftm3batmyZUZ70qRJmjFjRqGeo0SJEqZ2amqqXFxc8h135coV47Gjo6PNoSybNm2yr8As/ve//2no0KH59svMzNThw4eNtoODg1W4UuPGjeXs7Kz09HRJUnh4uOLj41W6dOl85582bZrx/pOkGjVq2HgFt9a4ceP09ddf6+zZs5KkjIwMnT59Wn5+fkVcGYC7XfGI8boNeHp6Srr2H7aoqKgi/cvMzCziuwEAAAAAAAAAAAAAAAAAAAAAAAAAAIDizN3dXZUrVzbaqamp+uijj/Idl56erjlz5piO/etf/yr0+oqjVq1aqWTJkkb74sWLpqCR3Fy4cEEbN240HXv11VcLvT5bdO7c2dR+6qmniqQO6VoAiru7u9G+k/+N9FtvvWVqz507V7GxsYV6juwhLCdPnrRpnIeHh/E4MzNT0dHR+Y754IMPlJiYaF+BWQQHB9t0njfffFMZGRlGu169ejn2a9q0qfHYYrHohRdesKmODz/80NQeNGiQTeOKQtWqVU3thISEIqoEAP4PoTQ2Gjt2rOrUqSMfH58i/7M1fQ4AAAAAAAAAAAAAAAAAAAAAAAAAAAB3r3HjxpnaEydO1OHDh/Mc0717d1MYhZeXl3r37n1T6iuOnnzySVN7yJAh+YZrdOzY0RSsUb9+fdWtW/em1JefH3/8UQ4ODkZ706ZNeuONN+ya48yZM5o7d67V8U2bNuno0aM2zxMaGqqrV68a7TJlythVx+2kRYsWatSokdFOTU1V06ZN7Qp2yczM1JQpU3J9vkGDBqb2woULbZq3SZMmpnZ+IVNbtmzRxIkTbZo7NxkZGerSpUuefQ4ePKgvvvjCdGz8+PE59s1+XxYsWKC1a9fmOf+oUaN07tw5o12iRAm9++67eY4pLJGRkVqxYoXN/TMzM3Xo0CHTMX9//8IuCwDs5lzUBdwuevXqpV69ehV1GZKkgICAPJ+PeHPzLaqk8Pl+2sHuMZYjb+XfCbiDOTT4pOCDYxcXXiHA7ahsP+OhZVn7IiwEKHoOjwcXeOzIeq6FWAlw+/nvsZQsrcgiqwMoHqr838PMsKIrAygOHGsVdQUAAAAAAAAAAAAAAAAAAGj06NGaPHmyEcyQnp6uZs2aac6cORowYICpb0JCgh5++GFt2bLFdHzatGm3rN7i4Ntvv9WCBQuMMJWrV6+qdu3aWr58ue6//35T38jISHXu3FnHjh0zjjk4OGjevHm3tOasKlasqDfffFOffPJ///Zw8uTJWr9+vebMmWMKTskqMzNTM2bM0LRp07R3717de++9Gjp0qKnP3LlzNWvWLNWrV0/PPfecXnnlFbm5ueU435YtW9S9e3fTseztO83y5ctVv359paWlSZLCw8NVqVIlTZ48WS+++GKu40JCQvTJJ59o5cqVSklJ0dixY3PsN2DAAE2aNMloT5o0Sd7e3ho6dKg8PT1znf/VV1/V7NmzjfbChQtVvXp1TZ482arvp59+qvHjxyszMzO/y83X/v371aRJE23cuFHe3t6m55YsWaLBgwebwpxq1aqlwYMH5zhXly5d1KxZM/3zzz+SJIvFou7du2vy5Ml67bXXTH3T09M1cOBALV261HR8/PjxcnR0vOHrskVYWJgeeeQRVahQQQMHDtSbb76pKlWq5Ng3MjJS3bp1U0JCgnGsRo0at6xWAMgLoTQAAAAAAAAAAAAAAAAAAAAAAAAAAADAHSooKEgNGzZUenq6JCklJUUDBw7UiBEjVLt2bXl4eCgyMlInT56UxWIxjR04cKD69+9fFGUXGTc3Ny1btkwPPfSQcT/i4+PVsWNHlS9fXjVr1pSLi4vCw8MVHh5uNf6dd97Rfffdd6vLNpk0aZL279+vFStWGMd27dqlxo0bq3Tp0vLz81O5cuWUlpam+Ph4RUZGKjY21ur1z82xY8f0xhtv6I033lDp0qVVtWpVlS1bVi4uLoqNjdWpU6cUFxdnGuPu7q7//ve/hXqdxU3NmjX166+/qnfv3kaoS0JCgkaNGqV///vfqlatmipUqCBXV1fFx8fr4sWLOn/+vPHZzE/Tpk1VuXJlI2QqJSVFL730kl566SWrvn///bfatGljjGvevLl2795tPD9lyhR9++23ql+/vkqXLq2YmBgdP35cKSn/t3HvU089pfnz59t9H5ycnNSsWTPt3LlTBw4cUMWKFVW9enX5+voqOTlZoaGhio2NNY1xdnbWX3/9lee8gYGBqlKlihEYlZGRoTFjxmj8+PGqW7euypQpo4sXL+r48eOmsBtJatmypSZMmGD3tdyoixcv6ptvvtE333yjkiVLqkqVKvLy8pKHh4euXr2q8PBwRUVFWY0rymArAMiKUBoAAAAAAAAAAAAAAAAAAAAAAAAAAADgDlW3bl3t2rVLrVu3VlJSknE8Pj5e//zzT67jnn76af3444+3osRip1u3bvrzzz/16KOPmgJDLl26pEuXLuU6buLEiUUSfJGT5cuX6+2339ann35qCpuJj4/X/v378x3v4uJi03ni4+MVHx+fZx8PDw+FhITIzc3NpjlvZ7169VJISIg6deqkK1euGMfT0tIUFhamsLCwPMc7ODjk+fzKlSvVqlUrpaam5tkveyhLUFCQqlevrsuXLxvHkpKStGfPnhzHjxw5UoMHDy5QKM3189WtW1eRkZGyWCw6ffq0Tp8+nWPfEiVKaPPmzapdu3aec5YuXVonTpxQ48aNTZ/DpKQk7du3L9dxXbp00fr16wt0HYUpKSlJoaGhCg0NzbWPo6Oj/ve//6ldu3a3sDIAyJ1jURcAAAAAAAAAAAAAAAAAAAAAAAAAAAAA4OZp2rSpYmJiNGjQIDk7O+fZt3r16lq/fv1dG0hzXffu3XX+/Hl169ZNjo55/5Pshg0bau/evcUmkOa6SZMm6dSpU3rggQfyfd0lycnJSfXr19fnn3+urVu3Wj3/1ltvadCgQapYsWK+4SmS5ObmpiFDhigmJkb+/v4FuobbUfPmzRUbG6u3335bZcqUsWmMt7e3hgwZolOnTuXZ795779X58+c1bNgw+fj42PS6SpKnp6ciIyPVuXPnPF87Hx8fLViwQD/88INN8+bG3d1dZ8+e1eOPPy4nJ6dc+wUEBCgiIkKtWrWyaV4fHx9duHBBr7zySr4hRxUqVNDPP/9cJIE0zZs312uvvaZatWrlef3XOTo6qm3btjp58qSGDRt2CyoEANs4WLJG29khICBAUVFR8vHx0a5duwq7LuQhICAgz3se8ebmW1hN4fL9tIPdYyxH3roJlQC3D4cGnxR8cOziwisEuB2V7Wc8tCxrX4SFAEXP4fHgAo8dWc+1ECsBbj//PZaSpRVZZHUAxUOV/3uYmXeCP3DHc6xV1BUAAAAAAAAAAAAAAAAAQLF1bEeU1v7vUFGXkatuzzZUvZY+RV3GTbVq1SqtWLFCkZGRSkxMVKVKleTv76+RI0fKy8urqMsrlhYuXKj169fr3LlzSktLk4+Pj5o1a6bnn39e7u7uRV2eTYKDg7Vs2TJFRkbq0qVLcnZ2Vrly5dSwYUN16dJFbdq0sXmuzMxMrV69Wlu2bNGxY8cUFxcnSSpdurTq16+vXr162TXfnSwqKko//vijjh07pgsXLigtLU2lSpWSn5+f2rZtqx49etzS91BMTIxmzpypHTt2KD4+XhUqVFCtWrX09NNPq3bt2oV+vvT0dE2fPl27d+9WRESESpcurUaNGunll1++4e+brVu3asGCBYqIiNCVK1fk7e2tOnXqaMSIEfL19S2kK7hxISEhWrdunQ4ePKiYmBilpqaqVKlS8vX1VdeuXdWzZ0+bA4YA4FYilOY2RCiNGaE0uNsRSgPcAEJpAAOhNEDBEUoDZEUoDWAglAYAAAAAAAAAAAAAAAAAckUoDQAAAFD8ORZ1AQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACA4oNQGgAAAAAAAAAAAAAAAAAAAAAAAAAAANwypbxLFnUJeSru9QEAAAC3gnNRFwAAAAAAAAAAAAAAAAAAAAAAAAAAAIC7R+VaZdRnXICuRCcVdSlWSnmXVOVaZYq6DAAAAKDIEUoDAAAAAAAAAAAAAAAAAAAAAAAAAACAW6pyrTKEvwAAAADFmGNRFwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAKD4IpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgIpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgIpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgIpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgIpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABici7oAAAAAAAAAAAAAAAAAAAAAAAAAAAAA3F0ijx1W7Pmooi7DStlKPqpSz7+oywAAAACKHKE0AAAAAAAAAAAAAAAAAAAAAAAAAAAAuGUijx3WL++OLeoycjXowykE0wAAAOCu51jUBQAAAAAAAAAAgJvPwcHB+OvUqVNRl4MikpycrGnTpqlnz57y9fVVyZIlTe+N999/v6hLxG3i/fffN713AgMDi7qkW+bUqVOma3/66aeLuiQAAAAAAAAAAAAAuO3Eno8q6hLyVNzrAwAAAG4F56IuAAAAAAAAAAAAADff/v379cgjj+j06dNFXQoAAAAAAAAAAAAAAAAAAACAYs6xqAsAAAAAAAAAANhu9erVcnBwMP117tz5huacPXu21ZwODg7y9fVVZmZmgea0WCyqVatWjvPOnj27QPVk/3N3d1eVKlUUEBCgESNGaP78+UpOTi5QvcCd7tKlS3rooYcIpAEAAAAAAAAAAAAAAAAAAABgE+eiLgAAAAAAAAAAYLucAl2CgoJ0+vRp+fn5Feq5zp49q/Xr16tbt252j920aZNOnjxZqPVkl5SUpKSkJJ07d067d+/WjBkz9Morr+jtt9/Wq6++KkdHctmB6z777DOdO3fOaNesWVOjRo2Sv7+/XF1djeO1atUqivIAAAAAAAAAAAAAAAAAAAAAFDOsyAcAAAAAAACA20RcXJx+//13q+MWi0Vz5sy5KeecO3dugcbdrHryExMTo9dff109evRQampqkdQAFEc//fST8bh8+fLasWOHXn/9dfXs2VNdu3Y1/gilAQAAAAAAAAAAAAAAuP3UqVNHDg4OcnBwkLOzc5HW8tVXXxm1ODg46Kuvvir0c3Tq1Ml0DsAexenzUhDBwcGm9//w4cNz7Tt8+HBT3+Dg4FtYKQDgTnD7/ZcSAAAAAAAAAO5SCxYsUHJyco7PzZ07VxMmTCiU8zg6OiozM1OStGzZMiUkJMjT09Pm8YmJiVqyZEmO8xVE06ZNNXXqVKvjCQkJOnPmjP766y+tXLlSFovFeG7NmjV67rnnTEEcwN3q5MmTioqKMtp9+vSRt7d3EVYE3N5q1Khh+m8OAAAAAAAAAAAAAAC2Cg4OVocOHYx2mTJlFBsbW3QFAQAAAHlwLOoCAAAAAAAAAAC2mT17tvHYwcFB7dq1M9qhoaHavHlzoZync+fOxuOrV69q6dKldo3/9ddfdeXKFaPdpUuXG6qnXLly6tq1q9XfY489pldeeUUrVqzQ9u3bVa1aNdO4efPmacuWLTd0buBOcOzYMVO7UaNGRVQJAAAAAAAAAAAAAAAAgLvBV199pU6dOhl/AADg9kQoDQAAAAAAAADcBo4dO6Zt27YZ7Xbt2unNN9809ZkzZ06hnKtx48Zq1qxZgefN2r958+a3JACjRYsWWrVqlVxdXU3Hv/zyy5t+bqC4y76bVunSpYumEAAAAAAAAAAAAAAAAAB3hd9++01BQUHGHwAAuD0RSgMAAAAAAAAAt4HZs2eb2oMHD9bDDz8sb29v49jixYuVmJhYKOcbNmyY8TgwMFDh4eE2jYuIiNCGDRtynOdma9SokYYOHWo6tn79emVmZt6yGoDiKDk52dR2cHAookoAAAAAAAAAAAAAAABwM5w4cUIWi0UWi0Xp6elFWsvo0aONWiwWi0aPHl2k9QB3s5kzZ5o+j+3bty/qkgAAtxnnoi4AAAAAAAAAAJC3zMxM/fTTT0bbxcVF/fv3l7OzswYMGKDvvvtOkhQfH69ly5bpqaeeuuFzPvnkkxo7dqzS0tJksVj0008/6e2338533E8//WSEwJQoUUJPPvmkPvrooxuux1a9evXSjBkzjHZsbKzCw8Pl5+dX6OeKiYnR7t27deLECcXFxSk9PV3u7u7y9vZWzZo11ahRI5UrV87ueZOTk3Xo0CEdPnxYFy9e1NWrV1WqVCmVL19eTZo0UePGjeXoeOdlzl+4cEEHDhxQaGioYmNjlZ6eLi8vL/n4+KhVq1by8fEplPNcuXJFQUFBOnPmjC5fvqwyZcqoYcOGateunVxdXQvlHFmdOXNGO3fu1Pnz543z+fj4qF27doV2TfmxWCw3Zd6MjAxt27ZNp06d0rlz55SRkaFGjRqpV69eeY6LjY3Vli1bFBkZqejoaHl6eqpixYpq1qyZ6tWrV+h1nj17ViEhIYqIiNCVK1dUqVIltWvXTvXr18937IEDBxQSEqLz58/LxcVF1apVU5cuXVS+fPlCrzM/EREROnjwoE6ePKm4uDhJkpeXl6pWrao2bdoU6PsmJ7GxsQoMDFRERITi4+Pl5eWlxo0bq3Xr1nJ2LvyfFo8ePaq9e/fq4sWLiouLk5eXl6pUqaL27dvLy8urUM+VlJSkTZs26ciRI0pISFC5cuVUo0YNdezYUR4eHoV6LnvExcVp06ZNOnHihJKSklS+fHnVqVNH7du3L5TvJYvFon379unw4cO6cOGCrl69Km9vb/n6+qpDhw7y9PQshKsAAAAAAAAAAAAAAAAAAAB3GkJpAAAAAAAAAKCYW79+vSIiIox2jx49jPCBwYMHG6E0kjR79uxCCaWpUKGCHn74YS1fvlySNHfuXJtCaebMmWM87t69u7y9vW+4FnvUqlXL6lh0dHShhtKsX79en332mdavX28E8OTEwcFB9evXV+/evfXSSy+pWrVqufaNiIjQggUL9Oeff2rr1q1KSUnJtW+5cuX0zDPPaMyYMapSpcoNXUtWqampqly5smJiYiRJ7u7uioqKUqlSpeya59SpU6pVq5YRhNKoUSMdOHDAqp/FYlFwcLAWLVqktWvX6ujRo3nO26RJE40ZM0ZPPfVUgYIxzp07p7Fjx2rp0qVKTk62er506dIaNWqU3n33Xbm7u2v27Nl65plnjOd//PFHPf300zadKzU1VdOmTdP06dN16NChHPs4ODgoICBA7777rh599FG7ryc/Dg4OuT73zDPPmK7tuvfee0/vv/++0X7//fc1ceJEo71x40Z16tRJMTEx+uSTTzRv3jxFRUWZ5rjnnntyDaXZtGmTJk6cqE2bNuW6I1edOnU0atQovfTSS3JxccnrEg01atTQ6dOnJUl+fn46deqUJGnbtm364IMP9NdffykjI8NqXOfOnfXdd9/J39/f6rmlS5dqwoQJOb5+Tk5OGjJkiD7//PObGk6Tnp6udevWacmSJVq3bp1xjTlxcHBQ69atNW7cOPXu3TvP1z83ERERev3117Vs2TKlpqZaPV+hQgWNGTNGY8aMueFwmoSEBE2dOlVz5szRyZMnc+zj5OSkDh066IMPPlCHDh1smvfpp582/Xfo5MmTqlGjhuLi4jRx4kRNnz5dV69etRrn4uKi5557Th988IFN/906deqUatasabSHDRum2bNn59q/U6dOCgoKMtrXvx/PnTund955R/Pnz8/xnnt4eGj06NF66623ChSac/HiRX3yySdasGCBzp07l2MfFxcXPfzww/rwww/VtGlTu88BAAAAAAAAAAAAAAAAAADuXHfeVqoAAAAAAAAAcIfJ/g/dBw8ebDxu3bq1ateubbQ3bNhgCrC5EcOGDTMeHz16VNu3b8+z//bt203BIlnH3yolSpSwOpZb+IW9LBaLXn75ZXXt2lVr167NM5Dmev8jR47os88+05o1a3Ltt2/fPlWvXl1jx45VYGBgnoE0knT58mV98cUXatiwoVatWlWga8mJi4uLBg4caLQTExO1ZMkSu+eZO3euEbgg5f4+GDt2rO6//359++23+QbSSNL+/fv19NNPq3Pnzrpw4YJdNa1du1b+/v6aP39+joE0khQfH69PP/1ULVu21NmzZ+2aP6vt27erQYMGGj16dK6BNNK198fOnTvVu3dvPfroozkGZRRHe/bsUZMmTfT5559bBdLkJjU1VUOHDlXHjh21YcOGPD+TJ06c0GuvvabGjRvryJEjBa5z2rRpat++vVatWpVjII10LWSndevW+vvvv41jmZmZGjFihPr27Zvr65eRkaHZs2erXbt2On/+fIFrzM/AgQPVvXt3zZo1K89AGuna+2nr1q16/PHH1bdvX7vfTytXrlSjRo20cOHCHMNRpGsBJ2+++aY6d+6suLg4u+bPasWKFapdu7bef//9XANppGv3OTAwUPfff79GjhxZ4O/yQ4cO6Z577tGXX36Z6325HiTVunVrI9ToZtu8ebMaN26sH3/8Mdd7fvXqVX388cfq3LmzERhmq1mzZql27dr68ssvcw2kka5d+x9//KFmzZrpgw8+sOscAAAAAAAAAAAAAAAAAADgznZj2xgCAAAAAAAAAG6q+Ph4LVu2zGiXKVNGvXr1MvV56qmnjH9InpmZqblz5+rtt9++4XM/8sgjKleunC5fvizpWthIq1atcu0/Z84c47GXl5dVnbdCZGSk1bHy5csXytwTJkzQt99+a3Xcy8tL1apVU8mSJXX16lVFR0fnGQCQXWpqqinERboWEFOtWjWVLl1aJUqU0OXLl3Xy5ElTKENcXJx69eqldevWqXPnzgW/sCyGDRum77//3mjPnTtXzzzzjF1zzJ0713js5ORkClHKKqdwmHLlysnHx0elS5dWSkqKzp8/b3Uvg4OD1aVLF4WEhKhkyZL51hMUFKTevXsrKSnJdNzNzU01atSQh4eHzp49awSsHDx4UD169NALL7yQ79zZLV++XAMGDLA6l4uLi2rWrKkyZcooPj5eJ06cML2Wy5cvV5cuXRQUFCQ3Nze7z3urhIeHa8CAAaZQoKpVq8rHx0dXrlzRmTNnrMakpKQY79PsKleurKpVqyo+Pl4nT55UWlqa8dzx48fVvn17rV27Vs2aNbOrzkWLFumll14yPleenp6qUaOGXFxcFBYWptjYWKNvfHy8+vTpo0OHDsnLy0ujRo3SjBkzjOe9vb1VrVo1paen6/jx46b37dGjRzV48GCtXbvWrvpsldNnpEKFCqpQoYJKlSql5ORknT17VtHR0aY+v/76q+Li4vTXX3/J0TH//Sk2bNigJ554wup87u7uqlmzpkqUKKHTp08b/y0IDg5Wv3791KZNG7uvafr06Ro1apRVUJC7u7v8/PxUqlQpxcTEKCwszBT8NX36dJ0/f17Lli2Tg4ODzec7deqUBg0aZApQ8vPzU8WKFZWQkKBjx46ZagkNDVWfPn20Y8cOOTvfvJ9Rd+3ape7duxshOY6OjqpZs6bKly+vy5cv68SJE6b/LoSEhOjpp5/WH3/8YdP87777rj766COr46VLl1a1atXk4eGhCxcumAJ4MjMz9d577+nSpUv6z3/+c2MXCAAAAAAAAAAAAAC4bZ05c0YzZszQiRMndPnyZfn6+qpbt24aMGDALath0aJFWrt2rcLDw1WuXDnVqVNHzz//vKpXr37Dcx84cEBLly7VwYMHFRcXJ4vFIk9PT/n5+SkgIECPPfaYPD09C+EqbLdmzRqtXLlSYWFhysjIkL+/v0aNGmXarC679PR0zZ49Wxs2bFB0dLTKlCmjgIAA/etf/7rh+rdu3aolS5YoPDxcsbGxKl++vGrVqqXhw4erZs2aNzR3QR0+fFirVq3Snj17FB0drYyMDJUvX141a9bU4MGD5e/vX2jnyszM1IwZM7Rz506Fh4erbNmyatiwoV555RWVLVv2hubevn27FixYoIiICMXHx8vb21t169bV8OHD5evrWzgXUEiioqK0ePFibd++XTExMUpNTZWHh4cqVaqkpk2b6tFHH73hz+SiRYsUGBioU6dOydHRUc2aNdPLL7+sihUr5jomMTFR06dP15YtWxQXF6fy5curQ4cOGjFixE1d7wMAdzO+XQEAAAAAAACgGFu0aJEp4KJv375ydXU19Rk8eLARSiNdC4cpjFAaFxcXDRw4UNOmTZMkLVy4UF9++aVcXFys+qakpGjBggVGe+DAgTn2u9kCAwNNbRcXF1WtWvWG5z179qw+++wz07GRI0fq3//+d44/aF++fFl///23VqxYoV9++cWmc3Ts2FGPPfaYunXrpvr161v9QJqcnKw1a9Zo0qRJ2rFjh6RrP4APHjxYR48eLZTFEC1btlSDBg105MgRSdcCXU6fPi0/Pz+bxm/ZskWhoaFGu1u3bqpcuXKu/UuVKqW+ffuqZ8+eatu2bY59z549q3nz5unTTz81wkQOHjyoN998M9/ghPj4eA0ePNj0GSpfvrw++eQTDRo0yHTP9u7dqwkTJuiPP/7Qvn37NGXKFJuu+bqDBw9aBdJ06NBBb7zxhh544AFT2MyVK1e0YMECvfvuuzp//rwkaceOHXr11VeNz9uNyhqU8tdff5muZ+zYsXrwwQetxtSqVSvPOceOHasLFy7IyclJI0aM0GuvvaY6deoYzyclJWnLli2mMW+//bZVIM1jjz2miRMnqmnTpsaxmJgYzZo1S++9955xDy9duqR+/fppz549Nr+/4+LiNGLECFksFtWrV0+fffaZevToYXwfZWRkaOnSpXrhhReMkJXz58/rk08+UUBAgP773/9Kkh588EF98MEHatmypRGCcvXqVX3++eeaOHGiERiybt06LV++XI888ohN9dnL29tb/fv3V8+ePdWyZUt5e3tb9Tlx4oT+97//6csvvzSCZdavX6///Oc/evXVV/OcPzY2Vk899ZQpkKZixYqaMmWK+vXrZwQ/ZWZmasOGDXrttde0f/9+rV27VsePH7frWtavX68XX3zRFDbzyCOPaMyYMWrXrp3pOy8mJkYzZ87URx99pCtXrkiSfv/9d02ePFlvvPGGzed87rnnFBUVpZIlS2rs2LEaOXKkqlSpYjx/+fJlffbZZ5o8ebLxmv7zzz+aMWOGXnzxRbuuzx79+/fX1atX5eXlpfHjx2vo0KGmALXIyEi98847mj17tnFs+fLlWrVqlbp3757n3D/++KMpkMbBwUFDhgzRyy+/rObNm5uCiiIjI/Xtt9/q888/N0Khvv76a7Vr1079+/cvpKsFAAAAAAAAAAAAANwKderUMdbtODk5mTZMyi7rhjAdO3ZUYGCgjh49qh49eigsLMyq/6xZszRkyBC9+uqrVmuocvLVV1+Z1ix8+eWXGj16dL7j3n//fX3yySdKTU21eu6jjz5SrVq1tHLlStWvXz/fubKbMmWKPv74Y8XFxeXbt2zZsnrmmWf0xRdf2H2enAwfPlyzZs0y2ps3b1b79u01efJk09qI61atWqUvvvhCAQEBCgwMtFo3M2zYMM2fP99qU6AlS5bo7bff1pAhQ0wbzNkiOTlZI0aM0KJFi5SSkpJjn0mTJql8+fKaOnWqhg0blmOf4OBgdejQIcfn8tqI6Po9yWru3Ln65ptvtH///lxrul6Xp6enXnnlFX388ce59rNF//799euvv1rdW0l67733FBAQoJUrV+YZmpKTV199VT/88EOOm1RJ0sSJE1WxYkV9/fXXhR4Aldv7Lzfr16/Xs88+m+MGZVm9/PLLcnNzU7t27XLcsEySOnXqpKCgIKN9fX3OK6+8ohkzZljdjz///FMfffSRHn74Yf3555+mdS7Jycnq06ePVq9ebbUJ4IIFC/TKK69o3LhxmjRpUp51AwDsl//2iAAAAAAAAACAIpP9x+HBgwdb9albt65atmxptI8dO6atW7cWyvmz/nh86dIl/fnnnzn2W758uRHwkH3crRIXF2cESlzXunVrI1ThRvz+++/GP9iXpAkTJuiHH37IdYeVcuXKqWfPnpo2bZoiIiL00EMP5Tp39erVdeDAAQUGBmr06NFq1KhRjjt2uLm5qXfv3tq6dauGDx9uHI+MjNRPP/10A1dnlvW1s1gsds2d/f2a1/vgmWeeUUREhP73v//piSeeyDW8pmrVqnrjjTe0Z88e0047M2bMUExMTJ71TJw4UREREaa5QkJC9Pzzz1st1rjnnnv0+++/G4EXJ0+ezHPurNLT0zVw4EBTIM3EiRMVFBSknj17mgJppGthPM8//7x27dqlunXrGsd/+OEH/fPPPzafNy9du3Y1/ho2bGh6rmHDhqbnr//lF0pz/vx5OTs7a8GCBfr+++9NgTSSVLJkSXXt2tVoh4SE6MsvvzT1mTBhgpYtW2YKpJEkLy8vjR07Vps3b1bp0qWN46GhoRo/frzN1x0bG6u4uDi1a9dOISEheuyxx0wBWU5OTurfv7+WLVtmGjdz5kxjMdTo0aO1evVqtWrVyrQYxsPDQ++9954mTJhgGpt10UZheueddxQeHq7vvvtOPXr0yDGQRrq2qGzSpEnaunWrvLy8jONTp07Nc4GZdC00KCoqymj7+flp9+7dGjp0qOm709HRUV27dlVISIi6dOkiSTp16pTN1xIbG6vBgwcbgTSOjo6aNWuW/vjjD3Xs2NHqO8/Ly0vjxo3Ttm3bVKFCBeP4hAkTTPXmJywsTF5eXgoKCtLEiRNNgTTSte/qTz/9VB9++KHp+PTp020+R0GEhYWpRo0aRhhV1kAaSapSpYp+/PFHPf/883bVFRYWpn/9619Gu2TJkvrzzz81Z84c3XfffaaFOtfPM2nSJK1bt870er/88su5LoICAAAAAAAAAAAAANx5li5dqkaNGuUYSHNdWlqaJk+erEaNGpk2pCks9913nyZOnJhjIM11YWFhatSokZYsWWLX3K1bt9a4ceNsCqSRrq1zmDt3rl3nsFevXr30xhtvWAXSZLVr1y75+fkpMTFRkpSYmKjq1atr7ty5OYamSNfWe82dO1f333+/zbUEBgaqbNmy+umnn/IMf5GurSF8+umnc9wQqzCdPn1aw4YN086dO/OtSZISEhI0adIk1atXTwkJCXafLzk5Wb6+vlq8eHGu91a69pr4+vpq+/btNs0bFRWlChUq6Kuvvsp3LcaFCxc0cOBA0/qrW+2tt95S165d8w2kuS45OVnr16+36xz33HOPvvnmmzzvx+rVq03hU5GRkfLx8dGqVausAmmuy8jI0CeffKKnn37arnoAAPkjlAYAAAAAAAAAiqnQ0FAFBwcb7WrVqqljx4459s0eVmPvTie5adWqlenHvdzmzXq8QYMGppCcW+HKlSvq37+/zp8/bzo+dOjQQpn/2LFjpvaoUaNsHuvp6alq1arl+nzFihXVqFEjm+dzdHTUd999p9q1axvHfvzxR5vH52fw4MGm4AJbQ2mSk5O1ePFio12mTBk99thjufYPCAgwhY/kx8/PTzNmzDDaSUlJWrBgQa79ExMTrcJC5s+fbwq2ycmnn36a6249uVmyZIkOHDhgtEeOHKkJEybkubuPdC0kZ+nSpab7PXXqVLvOfau9/vrr6tu3r019v/zyS9MigF69emnixIl5jgkICLAK3pg5c6bNi4Kka0EjixYtyvP91bFjRz388MNGOzY2VlFRUWrfvr2mTp2a52v35ptvqkyZMkb7r7/+ynMxSkG1adPGKtAoL/fee68mT55stM+ePau//vor1/6xsbGaPXu20XZwcNDixYtVtWrVXMe4urpq6dKlpqAYW/zwww+mMJmPP/5Yzz77bL7jGjZsaKoxNTVV3377rV3nnjVrllq0aJFnnzfeeEO+vr5Ge8+ePVb/PSlMTk5OWrx4sel7PCefffaZ6T3w119/5bmwb/LkycZCNEn63//+p+7du+dbz/3336/PP//caF+4cEHz5s3LdxwAAAAAAAAAAAAA4PZ35coVDRw40Fj74OrqqsaNG6t9+/Zq0KCBnJycTP0PHTqU7+/w9rr//vu1a9cu0zEnJyfVr19f7du3V+PGjY1NiTIyMjRo0KA8w1yyevbZZ60CRFxcXFSnTh21bdtWnTp1UkBAgKpVq2ba+Ohm+uijj0ybw/n6+qpNmzZq2bKl1cY2MTExxm//zZo1U3h4uKRr96du3bpq3769mjVrZrVx2+bNm/XVV1/lW8vSpUvVpUsXq+CXChUqKCAgQPfff7+aNm0qDw8P0/Nr167NcS1jfuulcpPfOFdXV1WrVk3NmzfX/fffr7Zt26pevXpWa2uOHz+u9u3b233+Dh066OzZs0Ytfn5+atu2rZo1a2ZaJyRdC2hq3769QkND85wzPj5ederUUXR0tOl4yZIl1aRJE7Vv31716tWz+oytX79erVu3tvsabtTKlSv16aefmo45OjqqWrVqatmypTp16qSWLVuqTp06cnd3L9A5unfvrn379km6dp9r1aqldu3a6b777lOpUqVMfU+cOKFnn31W6enpatKkibF+zMXFRf7+/urQoYOaNGmiEiVKmMbNmTNHq1atKlB9AICcEUoDAAAAAAAAAMVU1n+IL0lPPvlkrj++Dhw4UM7OzkZ74cKF+e6sYauswS4rV67UpUuXTM9fuHBBq1evzrH/zZSYmKjDhw/rP//5j5o0aWIVvuDv769hw4YVyrmSkpJM7ew/ZN5qLi4u6tevn9H+559/rGosKF9fXz3wwANG+9ixY9q2bVu+437//XfFxsYa7f79+9sVqGGLBx54QJUrVzbaf//9d571ZA0yefjhh3MNdcou+4/r+cm6gMPd3V2ffPKJzWObNGmi3r17G+3ff//9pgScFAZ3d3e99dZbNvWNjY3V0qVLjbaDg4PNgTsDBgwwLay4evWqfv75Z5vrfPHFF1WlSpV8+z366KNWx95//31TSFBO3NzcTLs9JSUl6ejRozbXdzMNHDjQtFAlr8/IL7/8Yvre6Nu3r02LxsqWLat33nnH5poyMjL0zTffGO3q1atrzJgxNo/v0aOHmjVrZrSzvq/y07JlyzzDsa5zdnZWnz59TMeyL3YrTE888YTuu+++fPuVK1fO9F5LTEzUkSNHcuwbExNj2qWtTZs2GjhwoM01Pf/886pYsaLRtuc+AwAAAAAAAAAAAABuX7t371Z6erqka5sVJScna//+/dq8ebMOHz6s1NRUDRgwwGrMF198USjnnzlzpjZv3mw69vjjjys5OVlHjhzR5s2btX//fqWkpOi1116TJKWnp2v37t02zZ91Q67r61dSUlJ0/PhxbdmyRRs3btTOnTt15swZpaSkaMeOHerdu7dpjVJhW7NmjSSpcePGOnv2rMLDw/X3339r+/btio6O1vfff2/qv2nTJj377LPGxmqDBg1ScnKyjh07ps2bN2v37t1KTEy02mhqwoQJedYRGRmpQYMGmTadeuihh3Tu3DlduHBBO3fuVFBQkPbu3auEhAT9/PPPcnV1NdWVPfimXbt2slgsslgsVmu1rh/P6a9du3amvg4ODipXrpxGjRqlEydOKDk5WWfOnNGuXbsUFBSkLVu26OjRo0pKStKSJUtMgSZ79+612swsLxkZGdq5c6eka2sOo6KidOrUKW3ZskW7d+9WbGysFi5caAotSk9PN63pyEmnTp109epVo+3o6GhsOLRv3z5t3rxZR48eVXJystX6lu3bt+uDDz6w+RoKw+jRo03tfv36KSkpSWfOnNH27du1ceNGbd++XcePH9fVq1cVHh6uf/3rXypXrpzN57i+zrRLly6Kj483Nm4MCQlRfHy8xo4da+o/d+5c9e7dWzExMZKufUelpKTo0KFD2rRpk/bt26eEhAS1atXKNM6eTQcBAPkjlAYAAAAAAAAAiiGLxWL6QVySBg8enGv/ChUqmH7kjI2N1e+//14otQwZMsQIaUhLS9Mvv/xien7+/PnGwgRHR0cNGTKkUM57XVBQkBwcHKz+PDw81LBhQ40ePVqnT582jalQoYKWL19uCuq5EdkDLubNm1co896ImjVrGo/T09N14MCBQps7e5jPnDlz8h2TNQwhpzkKS40aNYzH//zzT679goODTe2nnnrK5nO0bdvWdH/zcunSJe3YscNo9+rVy64f2iWZPrsJCQl5XldR6tmzp0qXLm1T361btyo1NdVoX9/Zx1bPPvusqb1p0yabx2YNbMpL48aNTe1y5cqpS5cuNo1t0qSJqX3mzBnbirvJPDw8TMEieb2XAgMDTW17AsWeeuopm79f9+7dq8jISKM9cOBAu4O9sn5Gjhw5YrWDVG6yL4rLy7333mtqX99Z7Ga4GXUFBgaaQobs/W9xiRIl1LlzZ6P9999/KzMz0645AAAAAAAAAAAAAAC3r++++05TpkyxOu7o6KgFCxboxRdfNB23Z0ObvFwPmrlu2LBh+vXXX3NclzB16lSrEJS8BAYGGuvaJGnkyJFW58uuRYsW+u2337R//36bz1MQzZo10/79+3PceOnFF1+0Wuv0448/SpLefPNN/fzzzznen8WLF8vPz89oX7lyxWoNVVa9evVSWlqa0f7888+1evVq+fj45Nh/0KBBOnr0qGndx3vvvZfr/DeievXqiomJ0XfffafatWvn2feJJ55QRESESpYsaRwrSKBLw4YNdejQIdPam+v69++vkJAQ02ZRYWFhmj9/fo5zbdiwwbRux8HBQStXrrQKXZGubaa0bNkyPf/886bjH3300S1duxEWFmY8rlevnhYtWmQK4snO19dX33zzjREYY6vHH39c69evl6enp9VzkydPVps2bYx2RkaGVq5cKenaJo85fUe5uLho27ZtpvlOnTpl8/oiAED+CKUBAAAAAAAAgGJo48aNpqCVpk2bWgUoZJf9h+jZs2cXSi3VqlVTp06djHb28JGsgSWdO3eWr69voZy3oDp16qSQkJB8f4y2R7du3UztMWPGaPz48YqKiiq0c0hSYmKiFixYoJEjR6p169aqUqWKSpUqJUdHR6tQnpEjR5rGFuaPqH369DGFjyxcuNAUMJLd+fPnjR18JKlOnTpWu9fk5dSpU5o8ebL69esnf39/VahQQa6urjmGEW3dutUYl9c1X9+95rq2bdvaXI89/YODg027Bd133312nUe6togiq8OHD9s9x63QsmVLm/tu377d1LY17OW6Bx54wNTetm2bTeNKlChhFRiTm/Lly5vazZo1k4ODQ4HGxsfH2zSuoA4ePKiJEyeqd+/eqlu3rry9veXi4pLjZ+TcuXPGuLw+I1nDlBwcHKx2p8qLt7d3vv9Nui77bma38jNiz7myLyiKi4uzeay9bkZdhX2f4+PjdfbsWbvnAAAAAAAAAAAAAADcfpo1a6ZRo0bl2ef77783/YadnJysadOm3dB5ly5dqitXrhjtcuXK5bvm7d///rfNa0OOHTtmaj/yyCN213gzODk55btBU07hO9WqVdMnn3yS57jRo0eb2gsXLsyxX2hoqCk0pX379hozZkyec0uSn5+fxo8fb7Tj4+MLbfO8G1G6dGlTcNKZM2fsCnRxdHTUxo0b8+zTtGlTvfrqq6ZjH374YY59X3/9dVN74MCBeuihh/Kcf/r06aZAoLS0NL3//vt5jilMGRkZxuOAgICbcg4PDw/9+uuvefbJKXimZcuW+W6Sl32TqPzOAwCwHaE0AAAAAAAAAFAMZf9xffDgwfmOeeyxx0y7Paxdu9YUTnAjsv6gFxISYgQC7N27V3v37s2x361UvXp1DR48WBs3btTGjRtNO74UhrZt25qCadLT0/Xxxx+ratWq6tChg95//32tX7/etEjCHmlpafrss8/k4+OjQYMGafr06dq+fbvOnTunhIQEU+hJbmJjYwt07pyULFlS/fr1M9qXL1/W8uXLc+0/f/5804/SQ4cOtek8p0+f1mOPPaZatWrpjTfe0JIlS3TkyBFFR0fnGYJzXV7XnDVMwdXVVTVr1rSppuv8/f1t6pc9HGPcuHE5BoXk9dezZ0/THPbuHnOr2HMPs4ZqSdcWZdijVq1aKlWqlNEODw+36XPg5eVl2pEoL+7u7qZ2hQoVbK4v+9ikpCSbx9pj//796tixoxo3bqz3339ff/zxh06cOKFLly6ZdqrKTW6fkczMTNNrVL16ddP9toWtC7yyf0b69+9v92fkpZdeMs1h62ckp52rcuPh4WFq36zXVLo5dWW/zy1btrT7Pmdf1FNcv4sAAAAAAAAAAAAAAIUrpwCUnGQPLZk1a9YNnfe7774ztV944QWbxn355Zc29cu+6dCqVatsK+wma9u2rWmdX068vb2t1gxkD5zJSfZ1hnv27Mmx38SJE03t7K9FXt5++23Txk/z5s2zeezN1LVrV1N7xYoVNo9t3769Tes5PvvsM9PapOzBR9ft37/feOzg4KAffvjBpjreffddU3vRokU2jSsMWV/TrIFFhSnrmsTctGvXTo6O5viDnIJqsuvTp4+p/ffff9tXHAAgV4TS3IZGjBhR1CUAAAAAAAAAuIkSEhJMuzQ4OjrqySefzHecu7u7Hn/8caOdkZGhn376qVBqeuKJJ0w/cs+dO1eSNGfOHOOYp6en1Q97haFp06Zau3at6W/dunUKDg7W/v37dfHiRZ0+fVo//fSTOnXqVOjnv+7nn39W69atTccyMzMVHBysiRMnqmvXrvLy8lKbNm308ccf69SpUzbNm5SUpIcfflhvvvlmgUNtJCklJaXAY3OSPWAo62udXdbnHBwcNGTIkHzn37Fjh+655x79/vvvNoWN5CSv4JqsYRylS5c2/Whui7Jly9rU79KlS3bNa4u4uLhCn7MwlC5d2ua+ly9fNrW9vb3tPl/WhUEZGRk2fT7c3NzsPk9hjC3oezgvK1as0H333Zfvzlh5ye17IT4+3lRz9kVYtrB1TFF+Rorba3rdzajrbvouAgAAAAAAAAAAAAAUHldXV91///029X3ttddM7dwCOWx14MABU3vcuHE2jXvggQfk4uKSb7/u3bub2t99951N4RY3W48ePWzql3390rPPPpvvGG9vb9M6qdx++9+8ebPx2MXFxa4Np5ydnVWyZEmjvW/fPpvHFkRoaKieeuop1axZUyVLlpSTk1OOm/Fkv68HDx60+RzPPfecTf0cHR3VoEEDo22xWLR+/XpTnwMHDig9Pd1oV6tWzeZ1V6NGjTK9frauQSwMWdcCHTlyRP369VNiYmKhnsOWdbCSeW2No6OjTd9RzZs3N7VvxloaALhbORd1AbDfyJEji7oEAAAAAAAAADfR4sWLdfXqVaNdr149HT58WIcPH853bK1atUztOXPm2PxjfV48PDz0xBNPGGE08+fP18SJEzV//nyjT/bgmsJSrlw5q11MioK3t7c2bdqkmTNn6osvvtCJEyes+qSnp2vbtm3atm2bJkyYoCeffFKff/65KlWqlOu8o0aN0oYNG0zHKlSooE6dOumee+4xfpS+/oP6dX/99ddNXSTRvn171apVS2FhYZKk1atX6+LFi6pQoYKp3969e00LCzp27KgaNWrkOfelS5fUo0cPq0UPTZs2VYcOHVSnTh1VqVJFJUuWlJubm+mH9jFjxti0kCFrGIcti1Cyc3V1talf1vCbwpKZmVnocxaGEiVK2Nw3ISHB1C7Id0P2MVeuXLErGOd2duzYMfXt29cUvOTg4KCWLVuqbdu2qlWrlnx8fOTm5mYVcDJ48GCdP38+z/mzvz7u7u5212jra3o3fUaKEvcZAAAAAAAAAAAAAFAQPj4+Nvd1dnaWp6ense4g+/oDe2Xd9MjV1dXmTaQkqXLlyjp9+nSefdzd3dWmTRtt3bpV0rUAkXHjxmn8+PFq0qSJHnroIQ0ePFj+/v4Fqr+gGjZsaFO/rOuXHBwcbL4/jo6OysjIkHRtw7ScREZGGo9TU1Pt3vArqxvZiC0v0dHR6tWrl7Zv316g8VFRUTb3ffTRR23ue++995oCbzZs2KAHHnjAaG/cuNHUv1GjRjbPLUleXl5GoEpKSooyMzPl6Oho1xwFMXr0aI0fP95oL1myREuXLlWdOnXUpUsXDRw48IY3DWzWrJlN/bKuU8sagJSXihUrmto3+v0EAPg/hNIAAAAAAAAAQDEzZ84cU/vIkSPq1q1bgeY6dOiQQkJC1KJFixuua9iwYUYoTXh4uMaNG6cLFy6Ynr/TlShRQi+++KJefPFF7dy5U+vXr1dgYKD+/vtvxcfHm/pmZmZq3rx5WrdunQIDA1W/fn2r+fbs2WN6vUuUKKHJkydr1KhR+QaphIaGFs5F5cLBwUFDhw7V+++/L0lKS0vTL7/8oldeecXUL/v71Zb3wccff2zaiaRu3bqaN2+eWrZsme9YW8MzypQpY5yjID8wZ389ba1n9OjR6tmzp93nyyp7uNTtyNPT09TOGrRlq+xjSpUqdUM13U7efPNNU7BSy5YtNWfOHNNOS7mxZZFQ9kCZguxqZOtrmv0z8umnnyogIMDu82Vl72Kdu0H2+/zjjz/K19f3hua85557bmg8AAAAAAAAAAAAAKD4K1++vF39S5UqZazFsVgsNxSYkZ6ebjy2NXjiOi8vr3xDaSRp3bp1qlOnjs6dO2ccS01N1a5du7Rr1y5NmjRJLi4uqlu3roYNG6ZXX31Vzs43959ee3l52dQv6xqQgt5ji8WS4/G0tLQCzZeT3IJvbsSFCxdUt25dm9dw5cTW9TD2BP5IUvXq1U3trGsoJZnea5LsXr9RtmxZ09q6yMjIG14DYot33nlHW7Zs0apVq4xjFotFx48f1/Hjx/Xf//5Xjo6O8vX1Va9evfTee+9ZBcHkx9vb2+66bP08Zv+M5PbeBwDYj1AaAAAAAAAAAChGTp48qU2bNhXqnHPmzCmUUJrOnTurWrVqCg8PlyT95z//MZ6rXr36De+Ccbu57777dN999+mNN95QZmam9u7dq9WrV2vhwoXau3ev0S8qKkp9+/bV3r17rX74XLRokenHz4kTJ2r06NE2nT8mJqZQriMvQ4cO1cSJE40a58yZYwqlSU9P188//2y0PTw81Ldv33znXbhwofHYzc1Nq1evtjmIxdbrzrpjTHx8vBISEqyCUvKSdTegvGT/obxy5crq2rWrzee5U5UrV87UzrpQwlZZxzg5Od01oTQJCQn6888/jXalSpW0evVqq3uam6y7iOWmTJkycnBwMD7b0dHRdtdp62ua/TNSs2ZNPiM3Qfb73LBhQ5uCvgAAAAAAAAAAAAAAdzdbN4i6ztXV1dSOjo62O5giJ/lt4JWdrXW7u7srIiJCo0eP1syZM3MMUElNTdXBgwc1btw4vfPOO3r11Vf12Wef2VXP7aYwAztuRvhHly5drAJpGjdurO7du6tdu3Zq2rSpypUrZwqTCQ4OVocOHeyuy97An+wBNnFxcaZ29rU7ZcqUsWv+7O/tqKioWxJKI0krV67UrFmz9Pbbb1uF7UjXNuo7c+aMvv/+e02bNk3dunXT77//Ljc3t1tSHwCgaBQsGg8AAAAAAAAAcFPMmTOn0H+k/eWXX5SamnrD8zg4OGjIkCE5PjdkyBDTzix3G0dHRzVr1kxvvfWW9uzZo6VLl5p27zlw4IDWrFljNW7btm2mOV544QWbz3nw4MEbK9oGNWvWNP1Qv3v3btN516xZo/PnzxvtPn365Bv8cubMGVPgy8MPP2xzIE1SUpJOnjxpU9+GDRsajy0WiykoyBZ79uyxqV/NmjVN7RMnTth1njuVn5+fqW3v/Q8LC9OVK1eMdvXq1e+a75jdu3ebvrMHDRpkcyDNiRMnlJKSkm8/R0dH02sUHh5u985S+/bts6kfn5Fbg/sMAAAAAAAAAAAAACiIxMREu/pnX5eQfROVgrJ3fZs9dTs6Ourrr79WYmKi/vrrLz355JOqWbOmnJ2drfqmpaVp8uTJd9UGbR4eHrJYLAX+i42NLdR69uzZY1qj5urqql27dmn//v2aPHmyevfurZo1a1qFw1y8eLFA58vMzLSrf/brzR46k32dT/bQmvxkf2/7+PjYNf5GPffcczp//rwOHTqkl19+WU2aNDGthbzOYrHor7/+kq+vr93fIwCA2wuhNAAAAAAAAABQTFgsFs2dO9d0LDAwsEA/9D7wwAPGHDExMVq+fHmh1Dh06FC7jt+t+vTpozFjxpiOBQcHW/XLGuhSoUIFm4MnMjMzFRQUdGNF2mjYsGGmdtb3aPb3a/a+Ocl6zZJUv359m2vZvHmz0tLSbOrbsmVLU3vFihU2n+fy5cs5vl456dy5s6m9YcMGm89zJ2vdurWpbe99yd4/+3x3shv5jNhzn1u0aGE8tlgs2rRpk81jo6OjbQ7G4jNya3CfAQAAAAAAAAAAAAAFERMTY1f/rJsMOTg4yNGx4P9MOWsoTFJSkl1j7a37um7dumn+/PkKCwtTWlqaduzYoSeffFIeHh6mfkFBQfr+++8LdI7bQdZ7b8sGSLfSV199ZWpPmTJFzZs3z3dcQTfwsVgsdm3mdObMGVO7YsWKpnblypVN7YiICLvqyR56U6VKFbvGFxZ/f399/fXX2rdvnxITExUeHq7XX3/dqp5Lly7p0UcfLZIaAQC3BqE0AAAAAAAAAFBMbNq0SSdPnjTaVapUUYcOHQo016BBg0zt2bNn30hphvr166tVq1amY61bt1a9evUKZf47Sbt27Uzt6Ohoqz4Wi8V4bM9uP3/88YfdP1YXVL9+/eTu7m60582bp8zMTMXGxuqPP/4wjlerVs0qFCEnWa9Zsu+67Vno8cgjj5jac+bM0dWrV20aO2PGDJsXW1StWlWNGzc22qGhoVq1apXNdd6pWrduLRcXF6MdHBxs18KP//3vf6Z2x44dC6224q6gnxGLxaJp06bZfJ7sO3plD5nKy/z585Wenm5T35YtW5oCtzZs2KBDhw7ZfC7YpmvXrqYFYwsWLNClS5eKsCIAAAAAAAAAAAAAwO3g3LlzNvdNT09XQkKC0fb09Lyhc2ddT5CSkmIVxpGXqKioGzr3dS1atND8+fOVkJBgtZZi6tSphXKO4qh8+fLG4/T0dNO6xaKWfV3JSy+9ZNO4G9nA57fffrO57969e03tLl26mNrZ19DZuvHTdVkDl1xdXW8o+Kkw+fr6asqUKTp79qw+++wz03OBgYFFUxQA4JYoHv8lAgAAAAAAAABozpw5pvaAAQMK/INinz59TIEQq1ev1oULF26ovuuCgoJ05coV448fFHOWPYQm6yKK63x8fIzHly9ftimoISEhQWPGjLnxAm1UqlQp9enTx2hHRkZq3bp1WrRokZKTk43jQ4YMsen9mvWapWthJbZYuXKlfv/9dxurlpo0aaLWrVsb7XPnzunNN9/Md9zx48f10Ucf2XweSRo7dqypPXr0aMXFxdk1x52mbNmy6tu3r9G2WCx6/fXXbRq7ZMkSbd261Wh7enpaBW3dyQr6GZk2bZr27Nlj83kGDRokNzc3o71kyRKFhITkOy42NlYff/yxzecpUaKERo8ebbQtFotGjhyptLQ0m+dA/ipVqqQhQ4YY7atXr9q8KAsAAAAAAAAAAAAAcPdKSUnRpk2bbOr75Zdfmto3upFZ1o2gJGnKlCk2jVu/fr3NG07ZY/369XJwcDDakZGRhX6O4qJt27am9qRJkwr9HCVKlDC1bd2Y6cqVK8ZjR0dHm9dQ2vo+zkn2DbRyk5mZqcOHDxttBwcHPfDAA6Y+jRs3Nm0sFB4ervj4eJvmnzZtmmlDqxo1atg07lYbN26cqlatarQzMjJ0+vTpIqwIAHAzEUoDAAAAAAAAAMVAYmKilixZYjo2cODAAs9Xrlw5PfTQQ0Y7PT1d8+bNK/B8Wbm6usrT09P4c3V1LZR5i7OXXnpJy5cvN/3gm5eUlBR9/fXXpmMBAQFW/bL/uD9u3DhlZmbmOm9iYqL69OmjsLAwm+ooLMOGDTO1586daxWilL1PbqpXr276QTokJEQLFy7Mc8yOHTs0ePBgG6v9Px9++KGp/e2332rcuHG5LnDYvXu3unbtqitXrpgWmOTnqaeeUqNGjYz2sWPH1L17d7sWpqSlpWnOnDlWu8jczl599VXTopDff/8938CfPXv2aPjw4aZjw4cPV+nSpW9KjcVRQECAKVTs119/1d9//53nmBUrVui1116z6zzlypUzfW4tFov69++f5/s2JSVFffv21cWLF+0617///W9VqlTJaAcHB6tv3752hTddvXpVX3/9tWbNmmXXue8m48ePl7u7u9FeuHChRo4cafOiLunablsfffSRli9ffjNKBAAAAAAAAAAAAAAUQ6+++qpN/aZOnWpqP/fcczd03lGjRpnaP/zwg03j7F0jYStHR0fT7+55reO63b311lum9ty5cxUbG1uo58i+3ufkyZM2jfPw8DAeZ2ZmWm0Ol5MPPvhAiYmJ9hWYRXBwsE3nefPNN5WRkWG0cwtmatq0qfHYYrHohRdesKmO7OvdivNGXlnXAErXNtsDANyZCKUBAAAAAAAAgGJgyZIlph0+ateurZYtW97QnNlDbbKHiMB2W7Zs0aOPPqqaNWvq9ddfV2BgYI67l6SlpWn16tVq166dQkJCjOM+Pj7q1auXVf/BgwebQjv+/PNPPfLIIzp06JCpX3JyspYsWaJ77rlHa9eulST5+/sX1uXlq0uXLvL19TXaS5YsMYVktG7d2q6dj4YOHWrV/uSTT6zuaUREhMaPH6/7779fly9flpubm127v3Tt2tVq8cuUKVPk7++vCRMmaPHixfrzzz81ffp0PfHEE2rZsqXOnDkjR0dHPf/88zafx8nJSUuXLlWZMmWMY1u3blXjxo313nvv6dixYzmOO3/+vFasWKGRI0eqatWqevrpp0076dzu7rvvPqtFS++++6769u2rAwcOmI5fvnxZn3/+udq1a2cKKqldu3a+QTZ3Gg8PDz3xxBNGOyMjQ927d9f06dOVnJxs6nv8+HGNGjVKvXv3VkpKiipWrKjy5cvbfK5JkyapYsWKRvvUqVNq3ry5fvrpJ9O5MjMztX79erVs2VLr16+XZN9OTGXKlNHixYtNO2D98ccfatSokaZOnaozZ87kOC48PFxLlizR4MGDVaVKFf373/9WeHi4zee929SqVcsqtGf69Olq0qSJZsyYofPnz1uNsVgsCg0N1U8//aQ+ffrI19dX7777ri5dunSrygYAAAAAAAAAAAAAFLHdu3dr+vTpefZ55ZVXTL87u7m56cUXX7yh8/bt21eenp5GOyYmxmozo+y+//577du3z6b5N23apKNHj9pcT2hoqK5evWq0s64FutO0aNHCtAlXamqqmjZtalewS2ZmpqZMmZLr8w0aNDC189u47LomTZqY2v/617/y7L9lyxZNnDjRprlzk5GRoS5duuTZ5+DBg/riiy9Mx8aPH59j3+z3ZcGCBca6v9yMGjVK586dM9olSpTQu+++m+eYwhIZGakVK1bY3D8zM9NqjeOtXM8IALi1CKUBAAAAAAAAgGIge2DMgAEDbnjO3r17m3Zu2bdvn/75558bnvdudvr0aU2dOlWdO3dW2bJlVa1aNd17771q06aNGjVqpNKlS6t79+7atWuXMcbJyUkzZ85UyZIlreZr0KCB1S4oK1euVKNGjVS9enW1atVKjRo1Uvny5dWvXz+dOHFCknT//ffftB1/cuLo6KghQ4YY7ZSUFNPzw4YNs2u+119/3RRyk5qaqrffflvly5dX48aN1apVK9WqVUvVq1fXxx9/bJzv66+/lp+fn13n+v7779W9e3fTsbCwMH344Yfq37+/evXqpZEjR+rXX381drGZMmWK2rRpYxrj7Oyc53nq16+vZcuWqVy5csaxy5cv64MPPlD9+vVVoUIFNWnSRK1bt1bDhg1VsWJF+fj46JFHHtH06dN18eJFu67rdvHxxx+ra9eupmNLly5VkyZNVLVqVbVo0UINGjRQpUqVNHbsWNPClvLly2vRokWm3Y/uFh9++KFpt6j4+HiNHDlS5cqV0z333KOWLVuqWrVqqlevnqZNm6bMzEw5OTlp9uzZpsVa+fHy8tL8+fPl6upqHDt//ryGDh2q8uXLq0mTJmrevLkqVKigrl27Gou6HnzwQatwqfx06NBBc+fOlZubm3Hs7Nmzev311+Xn56cqVaro3nvvVatWrdSgQQN5eXmpevXq6tevn+bPn59jEBisDRw4UFOnTjUFnh07dkwjRoyQj4+PqlevrubNm6tly5aqV6+eypQpozp16mjo0KFatmyZkpKSirB6AAAAAAAAAAAAAEBReeGFF/TGG29YHc/MzNRTTz2lb775xnS8sDYZ+vLLL03tWbNmqV+/fsrMzLTqO27cOL300ks2zz137lw1aNBA9evX1+TJk602A8pqy5YtatasmelY9jVHd5rly5ebNhgKDw9XpUqVNG3atDzHhYSEqE+fPnJ3d9e4ceNy7Zd9DeSkSZP0/fffKyEhIc/5s2+CtXDhwlzP8+mnn6pjx445vl/stX//fjVp0kTR0dFWzy1ZskQBAQHG+jLp2uZBgwcPznGuLl26mN5PFotF3bt3twq1kaT09HT17dvX6r6PHz/etP7jZgoLC9MjjzyiihUr6pVXXlFkZGSufSMjI9WkSRPT61ijRo1bVisA4NbjGx4AAAAAAAAAitiZM2e0ceNG07FBgwbd8LweHh7q1auX6Vj28BsUnMViUUREhPbu3att27bp0KFDVgsXypUrp6VLl6pnz565zvPll19avU7StR/5d+zYoUOHDpmCOjp37qzff/8935CUwpZb8Iyrq6vdIUpeXl76448/VKlSJdPx9PR0HTx4UDt27NDJkydlsVgkXQvF+eKLL/T888/bXbeLi4uWLVum119/Pd975uHhoZkzZ+q1116zWvxgy85HnTt3VkhIiFq0aGH1XHR0tA4cOKDt27fr8OHDOYbQODg4qFq1avme53bi6uqqP//8M8cFGJGRkdq5c6eOHj2qtLQ003N169ZVcHCwmjdvfqtKLVZq166txYsXWwXMJCcna9++fQoJCVFERIRx3M3NTfPnzy/QYqiuXbtqyZIlKlWqlOl4YmKiDhw4oH/++UcxMTHG8fbt22vRokVycHCw+1wDBw5UcHCw6tWrZ/XcuXPntHfvXu3YsUNHjx7V5cuXrfo4OTmpSpUqdp/3bvPaa69p5cqVqly5stVz4eHh+ueffxQSEqLjx4/rypUrVn1cXV1VsWLFW1EqAAAAAAAAAAAAANz14uLi5ODgYPdfly5dCuX8zZs3l7OzsywWiyZPniw3Nzc1bdpU999/v/z9/eXq6qqff/7ZasyYMWMK5fzDhw9X+/btTceWLFkiFxcX+fv76/7771fTpk3l5uamKVOmSLq2uZQ9a0qOHTumN954QyVLllSZMmXUsGFDtW3bVp06ddK9996rsmXLqn379qbf0N3d3fXf//63UK6xuKpZs6Z+/fVXU5hIQkKCRo0aJRcXF9WuXVutW7dWx44d1axZM/n6+qpEiRJq2bKlli1bZrWxWXZNmzY1rV1ISUnRSy+9pFKlSlm9n7du3Woal/31nTJlitzd3dWsWTN17NhRTZo0kZubm9566y0jKOapp54q0H1wcnLSfffdJ0k6cOCAKlasqBo1aqh9+/a67777VK5cOfXr1890vc7Ozvrrr7/ynDcwMNC0GVdGRobGjBkjd3d33XPPPcZnzM3NTUuXLjWNbdmypSZMmFCg67kRFy9e1DfffKOqVavK3d1dderUUcuWLdW5c2e1bNlSlStXVtWqVXXo0CHTuHnz5t3yWgEAtw6hNAAAAAAAAABQxObOnWuEb0hSo0aN1Lhx40KZO3u4zc8//2wV/oD8LV++XN9++6169uypsmXL5tu/SpUqGjt2rI4dO6bevXvn2dfFxUW///67vvzyS/n4+OTar0aNGvr222+1bt06m2oobPXr11erVq2sjj/yyCMqV66c3fM1a9ZMu3bt0uDBg+Xk5JRjHwcHB3Xr1k3btm2z2gHHHq6urpoyZYr279+vt956S82aNZO3t7ecnJzk5eWldu3a6cMPP1RoaKiee+45STKFcEi2hdJI18JEduzYoT/++ENdunSRi4tLnv2dnJzUpk0bffDBBzpx4oQ+/PDDgl1kMebi4qKffvpJgYGB6tKlS57hQLVr19bUqVN14MABNWjQ4BZWWfw8+OCDCgkJ0SOPPJJrH2dnZ/Xt21d79+61Oxwqq169eungwYPq169fru9Zb29vffLJJ9q4caPNn4ecBAQE6NChQ5o7d65at26d6+f/OldXV3Xp0kWff/65wsPDNWLEiAKf+27y0EMPKSwsTF9//bWaNm2ab4iQp6enevbsqWnTpuncuXPq0aPHLaoUAAAAAAAAAAAAAFAQmZmZhTJPqVKl9MsvvxjBJCkpKdq/f782b96sI0eOKD093dTf399fISEhhXLu6zZv3qxmzZqZjmVkZOjIkSPavHmz9u/fbwSCODo6av78+Vab79gqPj5ehw8f1tatWxUUFKS9e/cqLi7O1MfDw0M7d+6Um5tbwS7oNtKrVy+FhIRY3c+0tDSFhYVp+/bt2rRpk/bs2aOzZ89avR/yW4+wcuXKfNdPSTKCZa4LCgqyWpOWlJSkPXv2aNOmTTpw4IApJGbkyJF64YUX8j1PboKCgoyNkiwWi06fPq0tW7Zo165dio2NNfUtUaKEgoODVbt27TznLF26tE6cOKHy5ctbXce+ffuMz1j2a+/SpYu2b99e4GspLElJSQoNDVVISIgCAwMVEhKiqKgoUx9HR0fNnj1b7dq1K6IqAQC3goMl6790sENAQICioqLk4+OjXbt2FXZduAERb24u6hIKzPfTDnaPsRx56yZUAtw+HBp8UvDBsYsLrxDgdlS2n/HQsqx9Hh2BO5/D48EFHjuynmshVgLcfv57LGvCe2SR1QEUD1X+72FmWNGVARQHjrWKugLcRBaLRceOHdPx48d15swZxcfHKyMjQ6VKlZKPj4+aNm2qevXqmXaQsVV6erpCQkK0b98+Xbp0SU5OTvLx8dG9996re+655yZcTfEQExOjTZs26fTp07py5Yo8PDxUs2ZNtW3bVhUrViySmvr27WvagebUqVPy8/Oze57ExERt27ZN4eHhunTpkpKSkuTp6Slvb2/Vr19f/v7+ph1x7gaxsbEKDg5WZGSkLl26JA8PD1WqVEn33nuv6tevX9TlFUvnzp3T5s2bFRERocTERJUuXVp16tRR27ZtCz2k6vLlywoMDFR4eLiuXLkiLy8vNW7cWG3atMkzUKig4uLitG3bNkVGRio6OlppaWkqVaqUKlasqAYNGqh+/fp3xSKvm+3ixYvavn27oqKidOnSJWVmZqp06dLy8fGRv7+/6tatqxIlShR1mQAAAAAAAAAAAADuIoc2b9Sqb6cWdRm56v6vMWrYoXOhzxscHKwOHez/d5TZdezYUYGBgUa7Tp06Cg0NlXRtk6Ts4SFZZQ0SuT7P4cOH1b17d50+fTrHMSVKlNDo0aM1efLkfGv76quvTBtQffnllxo9enS+4yZMmKDPPvtMqampOT7v5+enVatWyd/fX506dVJQUJDxXE7/VDo0NFTvvvuu1q9fr4sXL+bYJys3Nzf169dPM2fOtClIxVbDhw/XrFmzjPbmzZvVvn3+/47Jntc0K2dnZyPopHbt2jpx4kS+YzIzM/Xuu+/qu+++swrpyYm3t7e6d++ujz76SNWrV8+zb2xsrEaPHq01a9YoOjo6x+vI6Z4kJyerR48eCgwMzPW18/Hx0VdffaUBAwZYfbaee+45zZw5M8dxud3bPn366I8//rAKirkuICBAK1eutGtNW2Zmpl599VVNnz5dycnJufarUKGC/vOf/1htRJgTe67VlvdfYmKi3n33Xf322286ffp0rtd/naOjo1q3bq1ffvklz9ffls9pTsqWLWu8D8uUKWMVDJSbnL7bAAA3jlCaOxChNMDdhVAa4AYQSgMYCKUBCo5QGiArQmkAA6E0wG0tLS1Nvr6+unDhgqRrP/hffwwAAAAAAAAAAAAAAIAbd7eG0hQHeQU3nD59WtOnT1doaKhiY2NVuXJlPfjggzYFZRSWhQsXas2aNYqMjFTZsmVVu3ZtjRgxokAbSl2XmZmp1atXa8uWLTp27JgReFG6dGnVr19fvXr1Ups2bQrrEm5rUVFR+vHHH3Xs2DFduHDB2GDIz89Pbdu2VY8ePeTu7n7L6omJidHMmTO1Y8cOxcfHq0KFCqpVq5aefvpp1a5du9DPl56erunTp2v37t2KiIhQ6dKl1ahRI7388svy8vK6obm3bt2qBQsWKCIiQleuXJG3t7fq1KmjESNGyNfXt5Cu4MaFhIRo3bp1OnjwoGJiYpSamqpSpUrJ19dXXbt2Vc+ePW/K5lYAgOKJb3wAAAAAAAAAAIAsFixYYAqhadu2bRFWAwAAAAAAAAAAAAAAANwafn5++vjjj4u0hgEDBmjAgAGFOqejo6N69OihHj16FOq8dyIfHx+99dZbRV2GwcvLS+PGjbtl53N2dtaoUaNuytxt2rS5LcKPWrRooRYtWhR1GQCAYsKxqAsAAAAAAAAAAAC4WSwWi139IyIi9Nprr5mOPfvss4VZEgAAAAAAAAAAAAAAAAAAAAAUe4TSAAAAAAAAAACAO9amTZvUu3dvbdmyJd++gYGBatOmjaKjo41j9erVU8+ePW9miQAAAAAAAAAAAAAAAHedspV8irqEPBX3+gAAAIBbwbmoCwAAAAAAAAAAALhZLBaL/vjjD/3xxx+qUaOGunXrpubNm8vHx0dubm66fPmyDh8+rDVr1mjHjh2msU5OTpo7d66cnJyKqHoAAAAAAAAAAAAAAIA7U5V6/hr04RTFno8q6lKslK3koyr1/Iu6DAAAAKDIEUoDAAAAAAAAAADuCqdOndKMGTNs6uvi4qIff/xRrVq1uslVAQAAAAAAAAAAAAAA3J2q1PMn/AUAAAAoxhyLugAAAAAAAAAAAICbpUyZMvL09LRrTKtWrRQYGKgnn3zyJlUFAAAAAAAAAAAAAAAAAAAAAMWbc1EXAAAAAAAAAAAAcLM0a9ZMFy9e1Lp167Rp0yb9888/CgsL08WLF5WUlCQXFxeVL19evr6+6tChg7p3765OnToVddkAAAAAAAAAAAAAAAAAAAAAUKQIpQEAAAAAAAAAAHc0Nzc39erVS7169SrqUgAAAAAAAAAAAAAAAIAiZbFYiroEAAAA3CYci7oAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEDxQSgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADA4FzUBQAAAAAAAAAAAAAAAAAAAAAAAAAAAODuknI6TukxKUVdhhVnL1e5+pUp6jIAAACAIkcoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAG6ZlNNxujhtX1GXkasKLzYlmAYAAAB3PceiLgAAAAAAAAAAAAAAAAAAAAAAAAAAAAB3j/SYlKIuIU/FvT4AAADgViCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgIJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAglAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAECxMXz4cDk4OBh/wcHBRV1SsVS2bFnjHpUtW/amnCPr69CpU6ebcg7cHe6Ez7Wzs7NRf506dXLtFxwcbLrW4cOH38IqAaDwEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAA3GTZQwqu/02dOtWuebIHO7z11ls3qWIAAADczQilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAIrI+++/X9QloAg9/vjj6tSpkzp16kTAEG5YcHCw8X7q1KmTgoODi7okAMBtjFAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAoIgkJCRowoQJRV0Gisjy5csVFBSkoKAgLV68uKjLwW1u586dxvspKChIO3fuLOqSAAC3MUJpAAAAAAAAAAAAAAAAAAAAAAAAAAAAgCL0+eefKzMzs6jLwG0mNjZWFotFFotFsbGxRV0OgP+vffv2xmfTYrFo5syZRV0SABQIoTQAAAAAAAAAAAAAAAAAAAAAAAAAAABAEUpKStKYMWOKugwAAADAQCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAcIv5+fmZ2t9//71SU1OLqBoAAADAzLmoC0Dh8/20Q1GXcEs5NPikqEsAbl9l+xV1BUCx4fB4cFGXANy2/nsspahLAIqRKkVdAFB8ONYq6goAAAAAAAAAAAAAAAAAAACKtXLlyqlq1ar6+++/JUmpqal64YUX9L///a+IKwMAAAAIpQEAAAAAAAAAAAAAAAAAAAAAAAAAAACKxM8//6yaNWvKYrFIkubOnauvv/5anp6eRVzZjVuxYoXWrFmjc+fOKS4uTh4eHqpVq5a6du2qHj163PTzr1q1SqtWrVJkZKSuXLmiChUqyN/fX88//7wqVqx4Q3OfOXNGM2fOVGhoqKKjo1W6dGlVq1ZNgwYNUosWLQrpCm6uyMhIffvttzp27JgSExNVo0YNtWvXTk899VShnufgwYOaN2+eTp8+rejoaJUpU0Y1atTQsGHD1Lhx40I9lz2OHj2q2bNn68SJE4qPj1e1atU0ePBgderUKc9xq1at0q+//qrw8HC5urqqTp06euWVV+Tn53dD9Zw+fVo//vijwsLCdP78eXl6eqpq1aoaMGCA2rVrd0NzF9SFCxe0fPlybd++XefPn1diYqLKlCmjKlWq6LHHHlOXLl0K9XwrVqzQX3/9pZMnT8rZ2Vm1atXSiBEjVL9+/Rua93b6vCYnJ2vp0qUKCgrS+fPnlZCQIDc3N5UvX17+/v7q1q2b7rvvvhs6x/bt27Vo0SKdPHlSiYmJxn2+99578xy3cOFCrVy5UlFRUfLw8FCjRo3073//W97e3jdUD4DizcFy/X+l2ikgIEBRUVHy8fHRrl27CrsuAAAAAAAAAAAAAAAAAP+PvfsOi+J63wZ+0wWkiIhYQcWCBXvHgr13Y+89lmgsXzUxlkST2I0lsWLvJcYuFlSsCIpiBwQEQToovez7hy/zY9jCLCwCen+ui+vaZ/acM8/szswOsOcZIiIiIiIiIiIiIqKvUPyjMEQfeVXQaShVYlB1GNfPW9ESRdzc3NCqVSshrlevHh49eoSOHTviypUrwvKBAwfi6NGjSscZP348du7cKcTz58/H77//rrCtnZ0dfH19AQA6OjpIS0uTlGv2ddy6dQuOjo459vP19cWoUaNw9+5dZGRkKG2nra2NWrVqYdWqVejcubPG1h8VFYXhw4fDxcVF5bZWqFABW7duRdeuXXMcM6u9e/di9uzZiIiIUNrG0NAQ06dPx59//qm0Tdu2bXHjxg211m1mZoaYmBjRMnNzc8TGxip9XpG3b9+iW7duePnypcLnDQwMMGXKFKxbtw4AoKWlJTzXpk0buLq65riOjIwMzJ07F//88w8SEhKUtjMxMcHChQsxf/78HMdUV/bjbdy4cdixYweuXr2KUaNGITg4WGE/a2trnDt3Dg0aNBAt37hxq49WvQABAABJREFUIxYsWID4+HiF/Ro2bIjLly/DwsJCrTxXrVqFFStWqHzvihUrhsmTJ2PNmjXQ1tZW2EZXVxfp6elqrTvzNcnq9u3bWL58Odzc3PDx40eV/XV1ddGrVy84OzvD1NQ0x/UpO65XrVqFpUuXKn1ty5Qpg2PHjqldnEdTx2tWWV/nKlWqwMfHR2E7ZfufMkFBQejfvz/c3d2RU/kHHR0d2Nvb4+jRo7C3t5d7fv369Zg1a5YQr1u3DjNnzsT+/fsxc+ZMREZGKhzXzs4OV69eRcWKFUXLFyxYgLVr1yIlJUVhv06dOuHcuXPQ1dVVmTcRFU08sr9CdfbUKegUcu3pqKdq95EFrsmHTIiKDq2Ks3PfOf6C5hIhKoqM/++PRinr7AowEaKCpz9L8R9ApJhUzUCDmRAVPVtfJ2eJ3hdYHkSFQ1nhkexKlwLMg6jgaXW4WNApEBERERERERERERERERERERERURGxb98+lC1bVihEcOLECURFRald3KKg/fTTT1ixYoWkthkZGXj69Cnmz5+vsChNbuzfvx+jR4+WVJjj3bt36NatG8aPH4/t27dLGt/R0RG3b9/OsV1iYiJWrlyJvXv34tmzZ4XqfTx//jx69uypsmBQcnIy1q9fj+vXr+Px48dqr+PVq1do2rSpUCxHlY8fP2LBggU4cOAAPDw8oK+vr/b61JG9WIcioaGhaNKkCe7evYvGjRsDAPr27Yt///1XZT8PDw9UrVoVb9++lVSgJSIiAvXq1VNaHCerpKQkrF+/HkeOHMGTJ09gaWmZY5/c6tChA5KSkiS1TUtLw8mTJ3Hp0iXcvHlTrpCPFD179sTZs2dVtgkJCYGjoyP+/PNPzJs3T9K4Rel4vXTpErp37y65qFB6ejq8vb1x+/ZthUVpFJkxYwY2btyoso2Pjw+qV6+ON2/eoHz58gCARo0awcPDQ2W/y5cvo3r16kIBNCL6urAoDRERERERERERERERERERERERERERERERERERERFRAbG2tkavXr1w+vRpAJ8LtgwfPhznz58v4MykGzp0KA4dOiS33MzMDBUrVkSJEiUQHx+P0NBQhIaGSi6+INWaNWswZ84c0TItLS1YW1ujXLlyMDIyQmRkJHx9fUUFN3bs2IH09HTs2rVL5fj16tWDl5eXaJmOjg6qVKkCKysrxMbGwsfHB4mJicLzoaGhqFSpEkJCQmBkZCTqq62trfY25qZPVu7u7goL0pQsWRK2trbQ09NDQEAAQkJCAABeXl747rvv1FrHw4cP0bx5c6SlpYmWlyhRAjY2NjAzM8PHjx/h5+eHmJgY4Xlvb2/UrFkTPj65v+lyTl68eIGdO3cKsYWFBSpVqoRixYohMDAQ7969E55LT09Hx44dERMTg8mTJ4sK0pQrVw4VKlSATCaDj48PIiMjheeioqLQq1cvuLq6qswlNDQUdnZ2iI+PFy03NTWFjY0NSpQogYSEBPj7+yMiIkJ4PiQkBNWqVUNoaKhcAR8tLS11Xg4AOe9TOjo6KFmyJKytrWFiYgJtbW3ExMQgMDBQVHQoPj4erVq1wocPH1C8eHHJ69+wYYOoII2lpSVsbW2ho6ODwMBAYV/M9L///Q8WFhYYP368ynE1fbzmp7i4OPTs2VPunFiqVCmUL18epqamSElJQWxsLIKDgyUVe8ru7NmzuHr1qhBbW1ujYsWK0NXVhZ+fH0JDQ4XnkpKS0KZNG/j6+qJDhw5CQRotLS3Y2tqibNmySE5OxqtXr/Dx40ehn5+fH8aPH48dO3aonR8RFW55u/ogIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIqI82b17t6hAxMWLFxEUFFSAGUm3adMmuYI01apVg5eXF2JiYvDkyRPcuHEDDx8+RFBQENLS0nDw4EHUqVMHhoaGeV7/w4cPMXfuXNGyESNGIC4uDu/fv4e7uztu3LgBb29vJCYmYvXq1dDR0RHaOjs748KFC0rHX7hwoVyBi4EDByIlJQWvXr3CrVu38OTJEyQkJGDFihWi9zEuLg7t2rWTG/PatWuQyWSQyWSiXKpUqSIsz/4TFRWl9muTVffu3UUFaYyNjXHlyhVERETg4cOHuHv3Lt6/f4/nz5/D2toaAHDs2DHJ4yclJaF9+/aigjQNGzbE69evERUVhUePHsHV1RUeHh6Ijo7G1atXYWZmJrT19fXFxIkT87SNqty5cwfA50JJ169fR2RkJB4+fAg3NzcEBgbCw8MDxYoVE9rHxsZizJgx2Lp1KwCgVq1aePfuHYKCgnD37l3cu3cPERER2LJli2g9N27cyPHYbdq0qaggTZUqVeDu7o7Y2FjheHF3d0d4eDi8vLxQrlw5oW10dDS6dOkiN2ZqaipkMhnWrVsnWr5u3Tql+9S2bdvkxildujT69OmDGzduIC0tDR8+fICXlxfc3Nxw8+ZNPHnyBDExMfD29kbVqlWFfgkJCRg4cKDK7c7u+PHjAD4X47l16xbCw8Ph7u6Oe/fuye2LmaZOnSoqaJRdfhyv+WnOnDlITU0V4qpVq+Ldu3cICwuDp6cnXF1dcefOHTx79gwxMTGIj4/H6tWrUaFCBdH+qkpmQZpy5crB29sbISEhuH//Pm7fvo2QkBCcOXNGdB7y8/PD1KlThX5OTk6IiYmBn58f3Nzc4O7ujri4OLnz7u7du+UKUhFR0ceiNEREREREREREREREREREREREREREREREREREREREBcjc3ByDBw8WYplMhuHDhxdgRtKkpaXhxx9/FC3r0aMHXr16BQcHB6X9hgwZgidPnsDNzS3POfTq1QsymQwAoKWlhePHj2Pv3r0oXry4wvazZ8/GjRs3oKWlJSybNm2awrZpaWlYtWqVaNmPP/6Io0ePiopZZFqwYIFQaCPT/fv3NbKdefHHH38gPDxciA0MDPDy5Uu0b99erq29vT3evn0LS0tLtdYxdOhQxMXFCfGkSZPw8OFDUeGSrNq1a4egoCCYmpoKy5ydnZGSkqLWetVhbGyMwMBAtG3bVu65Bg0ayBXh2b17NwCgfv368Pb2Rvny5eX6TZkyBcOGDRMtW7JkidIcFi5ciMDAQCFu3749fHx80KhRI4XtHRwcEBQUhAoVKgjLrl+/joCAAKXryAt/f3+cOnUKrVu3VtmuVq1aeP36NWrXri0sc3FxUfv9MzIygq+vLxwdHeWeU7QvpqSkYNSoUQrHKorHa2bhFwDQ09PD8+fPFe5nmYyMjDB79mwEBgaq9RlRtmxZ+Pv7o1atWnLP9ejRA7/++qtoWWaxpb59++LatWui4zTTypUr0bx5cyFOT0/Hpk2bJOdEREUDi9IQERERERERERERERERERERERERERERERERERERERWw7du3Q1dXV4hv3LiBN2/eFGBGOZs7dy5SU1OFuGzZsjhz5ozk/ooKRajj/PnzCAkJEeKRI0eif//+OfZr2bIlBg0aJMR+fn4Ki3z88ssvSEtLE+IKFSpgzZo1Ksfu27cvevfuLVo2c+bMHHPKTxs3bhTFq1evVln4olixYjh79qzk8ZOSkvDff/8JcaVKlfDPP//k2K948eLYt2+fEKelpWHFihWS16uuAwcOKCyukalHjx4wNzcXLdPR0cHNmzdVjrt+/XpRfPv2baVts74XpqamuHz5ssqxM126dEkUz507V1K//Hbo0CHhcXp6Oo4ePapW/x07dqgsgKRoX7xw4QIyMjLk2hbF4zU2NlZ4bG1tLfoM0KRr166pHHvBggVyzxsbG+PkyZMqx125cqUovnDhQu6TJKJCiUVpiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIgKmJGREcaNGydaNmzYsALKRpojR46I4i1btnzR9S9fvlx4rKWlhU2bNknuu2zZMlG8c+dOuTbHjx8XxUuXLpU09o4dO0TxkydPJOelaZ8+fcL79++F2NDQENOmTcuxX9OmTWFraytpHevXr0d6eroQL168WHJ+vXr1QrFixYRYnWI46jA1NZUrPqJI9erVRXHLli1RvHhxlX0sLS1hbGwsxFlf76xcXFzw6dMnIR43bpzkwkz29vawtrYW4jt37kjql99q164t2gZ13j8zMzMMGTIkx3bZ98XU1FScOnVKrl1RPF4NDAyExx8+fBAV1dGUypUry+3XipQpU0YUf/fddzn2cXR0FL3/hb2QGhGpj0VpiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIgKgU2bNomKFLi7u+Px48cFl5AKGRkZCA0NFeJixYpJKvqhSU+fPhUelyxZMsfiIVlVrVoVWlpaQqyoyEdAQIDwWFtbG2PGjJE0tqWlJcqXLy/EqampePHiheTcNOnYsWOiuGHDhpL79unTR1K77IVI1C2mVLJkSeGxn5+fWn2lql27tqR2FSpUEMVdu3aV1M/c3Fx4nJycrLDN/v37RfHYsWMljZ2pXLlywuMPHz6o1Vddnz59wv/+9z/UrFkTJiYm0NHRgZaWlsKfjIwMoV9gYKDkdTg6Okpum31f/O+//+TaFMXj1cHBQXickpKCmjVr4u3btxpdR8uWLSW1s7KyEsVSCgYBEBWVio+Pl54YERUJLEpDRERERERERERERERERERERERERERERERERERERFQI6OrqYvr06aJlI0aMKKBsVPP09IRMJhNiW1vbL57Dx48fhccRERFKi2Yo+8maf1RUlGjsjIwMpKSkCHHWwilS1KpVSxRfvXpVrf6akr3YTpMmTST37dKli6R2r1+/FsV6enpqvQ/BwcFC36SkJMn5qSN7sRllTExMRHHNmjUl9ctaTCo9PV1hGw8PD1Fcp04dtV6nrP2VrUMTxo4dC1NTU6xcuRIvXrzAp0+fRIVnVImNjZW8HnWK0mTfF729vUVxUT1e165dK4rfvHmDypUro0yZMvjuu+9w6NAh0XblRpUqVSS1MzY2FsX169eX1E9PT094nNdciajwYVEaIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIqJC4s8//4ShoaEQe3t7w83NrQAzUszf318Uly5d+ouuPyIiQqPjffr0SRQHBQWJYnNzc7XGK1++vCgOCQnJVV55lf11Uqd4kL29vaR2CQkJ6qSkUmpqqsbGyqp48eKS2mlri6ffW1hYSOqnpaUlPM5a7CgrdQq25ETZOvKqYcOGcHZ2zvX46hQlsbOzk9w2+74YFxcniovq8Wpvb4+VK1fKLQ8NDcWxY8cwdOhQGBgYoFSpUujXrx8eP36s9jrMzMwktcu6DwOApaWl2uvKr/2SiAoOi9IQERERERERERERERERERERERERERERERERERERERUS2tramD9/vmjZmDFjCigb5cLDw0Wx1KIfmhIYGKjR8TIyMkTxhw8fRHGxYsXUGi97IYjo6OjcJZZHHz9+FMUlSpSQ3NfKykpSO3UKkeTkay5qocniPflh1qxZ8PT0FC0rV64cxowZg/3798Pb2xvh4eFIT0+HTCYTfnR0dIT26rx/Ugv+APL7YmJioiguysfr3Llz8eDBA5VFoCIiInDq1CnUr18f1atXx5s3b75YfkT0bWNRGiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiKiQuSXX36BiYmJEPv4+ODChQsFmJG87EUiPn369EXXX7JkSVFcoUIFUaEMdX98fHxE45UuXVoUJyUlqZVfbGysKFanGIwmZd2PAPWKbYSFhUlql7Uoiba2dp7eh7S0NMn5FTX6+vqiODo6Ok+vlaZt2bJFLg4KCsKuXbswbNgw1KpVC5aWltDWFpcoyF7QSaqoqCjJbbPvi4aGhqK4qB+vjRs3xvPnzxEZGYlff/0VzZs3h7m5ucK2r1+/hr29Pe7fv/9FcySibxOL0hARERERERERERERERERERERERERERERERERERERFTK//vqrKJ44caLaY2hpaeVq3dkLNChSqVIlURwaGpqrdeWWjY2NKNZ0UZzy5cuL4piYGLX6BwUFieIyZcrkNaVcsbS0FMX+/v6S+7548UJSu6wFQjIyMr7qwjJ5YWpqKorfvHlTQJnIu3btGlJSUoTYyckJU6ZMybHfp0+fcl0gx8/PT3Lb7Pti9tfyazleLSws8PPPP+POnTuIjo5GfHw81q1bhzp16ojapaeno2vXrgWSIxF9W1iUhoiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiokPnhhx9gYWEhxEFBQThy5IhaY2QvFiJVcHBwjm3q1asnKnoTEBCgVm6aUKxYMeFxTEyMWtuYE21tbejr6wtxZGSkWv2fPXsmitu3b6+RvNTVokULUfzgwQPJfS9duiSpXYUKFUTxyZMnJa/jW1KjRg1RfOrUqQLKRN6tW7dE8YgRIyT1U/eclNXNmzclt82+L9auXVsUfy3Ha3ZGRkaYOXMmnjx5ggcPHkBHR0d4Ljo6Gm5ubgWYHRF9C1iUhoiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIioEFq9erUonjFjhlr9zczMhMcymQyhoaGS+nl7e+fYRltbG2XKlBHipKSkL15kw97eXngsk8mwceNGjY5vY2MjPM7IyICzs7OkflFRUQgKChJiPT09Ua5ZaWv/33RvTRbVyTRw4EBR7OHhIbnvv//+K6ld9+7dRfHff/8teR3fkmHDhoniY8eOaXwdBgYGojg1NVVSv7CwMFFcvnx5Sf3y8l6rU1Al+77Yq1cvuTZf4ngtSI0bN0a/fv1Ey86dO1dA2RDRt4JFaYiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiICqExY8bA2tpaiMPCwnD79m3J/evVqyeKt23blmOfo0eP4uPHj5LGz15kY9q0aZJz04QffvhBFC9ZsgRpaWkaGz97QZfFixdL6jdhwgRRnP19yEpXV1d4HB8fLz05iYoXLy4qHpSYmCipkIi7uzvevn0raR0//vgjtLS0hPjGjRt49eqV+sl+5QYMGCAqGuPj44MLFy5odB2lSpUSxVILUVlYWIhiKcWLHj9+rFaRo+xiY2Nx9OjRHNtl3xf19PTQt29fuXZf4ngtaLVr1xbFSUlJBZQJEX0rWJSGiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiKiQ2rRpkyh++fKl5L6DBg0SxX/99ZfK9hERERg9erTk8VesWAF9fX0hfv/+PXr27Cm5f0ZGhuS2iowaNUpUhCMmJgbNmjVTa9ykpCSsX79e4XNLly4VFY159+4dZs+erXK8s2fP4uTJk6Jl69atU9re3NxceBwREaHRojqZpk+fLop//PFHvH//Xmn7lJQUdO/eXfL4lpaW6NKlixDLZDI0bdpUckGUTFu2bEFcXJxafYoSbW1tTJkyRbSsT58+8Pb2Vmuc48eP48WLFwqfa9KkiSh2c3OTNGb79u1F8ebNm1W2j4uLQ7t27SSNrcrYsWMRFRWl9HlF+2LXrl2hrS1fJuFLHK+atmfPHrXaZy9i5ODgoMl0iIjksCgNEREREREREREREREREREREREREREREREREREREVEh1b9/f9jY2OSqr6OjI8zMzIQ4MjISTZs2RVJSklzb06dPw9bWFomJiZLH19XVlSvocvbsWdjb2+PJkydK+x0/fhwODg5wdHSUvC5VY2lpaQmxh4cHrK2tceLECZX9XFxc0KlTJ5iYmGDBggUK2+jq6mLu3LmiZWvXrsWQIUMUFr5ZuXIlevfuLVrWtGlTtGzZUmke9evXFx5nZGSgbt26uHnzpsrc1bVgwQJR8Z6kpCRUr14drq6ucm1fvXoFW1tbhIeHq7WOo0ePonjx4kIcGxsLGxsbLF26VGW/N2/eYOTIkTAxMcHUqVMRHR2t1nqLmjVr1qBcuXJCnJKSgrp162L69OlISUlR2u/9+/eYOnUqLC0tMXDgQKXHV8WKFWFgYCDEDx48wIwZMxAUFKQyr7Zt26JYsWJCHBQUBEdHR4XnChcXF1SsWFEj71V8fDwqV66M27dvyz2naF/U09NTWsjlSxyvmjZu3DgYGhpiwIABcHd3V9ouLS0Nw4cPx71794RlOjo6GDFixJdIk4i+Ybo5NyEiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiooKyfft2dOrUKVd9f/vtN0yfPl2IHzx4ABMTE1SrVg2WlpaIi4uDr68vPn78CADQ1tZGu3btcOXKFUnjT5kyBbdu3cKhQ4eEZS9fvkTdunVhbm6OihUrokSJEkhISEBoaChCQkKQlpYGAKhXr16utimr1q1bY926dZg5c6awLDw8HAMGDICBgQFsbGxQsmRJ6OrqIjY2FmFhYQgPD0d6errQXldX+ZTrFStW4Pz58/Dy8hKWHT58GMeOHYOdnR2srKwQGxuLN2/eyBX0MTU1xbVr11Tmv3z5cpw/f16Inz9/jjZt2si1s7S0VLtQTFbnzp1Ds2bNhOIcnz59gpOTEywtLWFrawtdXV28e/cOwcHBQp+BAwfi2LFjksYvXrw4bt++jUaNGiE1NRXA54IrS5YswbJly1ChQgVYWVnByMgIHz9+RGRkJEJDQ5GcnJzrbSqKtLW14enpiSpVquDTp08APhcj2rRpEzZv3owyZcqgTJkyKF68OD59+oTo6GiEhISoVSyqW7duOHXqlBBv3LgRGzdulGs3ZcoUbNmyRYhnzZqF33//XYhv376N4sWLw87ODqVLl8bHjx/h5+eH2NhYoY2joyPu3r0rOp6kGjBgAI4fP47Y2Fg4OjqiVKlSsLW1hY6ODgIDA/H+/Xu5Plu2bIG5ubnSMfP7eM0PSUlJOHHiBE6cOAE9PT1YW1ujVKlSMDU1RXJyMkJCQhAYGChXWGf27Nkqz11ERJrAswwRERERERERERERERERERERERERERERERERERERUSHWsWNHVKtWDa9fv1a777Rp03DhwgVR4ZO0tDQ8f/5crq22tjb27t2L69evq7WOgwcPonz58li1apVoeUxMDGJiYtTOWV0//PADqlSpggEDBoiKnCQnJ0t6zXR0dFQ+//jxY7Rs2RJ37twRlqWnp+PVq1d49eqVwj6lS5eGt7c3jIyMVI5dr149zJgxA3/99ZfKdpmFXnKrcePGOH36NHr37i0qbhEREYGIiAi59nXq1MHRo0ehpaUleR0ODg7w8fFBs2bNEBISIizPyMhAQEAAAgICchxDT09P8vqKKisrKwQHB6NZs2Z48eKFsFwmk+H9+/cKi7FkZ2hoqPS5w4cPo3LlyqICQ4qkpKSI4hUrVsDNzQ23bt0Slqnaz2vVqoUbN25AX18/x3wV+eGHH5CYmIhz584B+FxMSlXhpd9//x3jx4/Pcdz8PF7zW2pqKt69e4d3796pbDdq1Cj8+eefXygrIvqWaRd0AkREREREREREREREREREREREREREREREREREREREpNru3btz3ffcuXOYN28edHV1lbapXLkynj9/jmHDhuVqHStXrsTz58/RsGHDHAuZ6OjooEGDBli3bl2u1qVIjx49EBMTg0mTJkkqLKGlpYWyZcvi+++/R2BgYI7tb9++jd27d6NkyZIq2xkaGmLOnDkIDQ2FpaWlpNw3bNiA69evo0WLFjA1NVWrEIw6evToAR8fH9SoUUNpG319fXz//fd48uRJrtZRsWJFvH//Hn/99ResrKwk9TE3N0fv3r3h7e2NsmXL5mq9RY2pqSmeP3+Ow4cPo2LFipL6GBsbo3379nBzc0OvXr2UttPX10dQUBCWLl2KypUrw8DAQHJeN2/exMyZM1WeK4yNjfHjjz/C29sb2tp5K1dw9uxZ/P777yqL7FhbW+PWrVuYP3++5HHz83jVpPXr16NBgwaS36PKlSvjypUrefo8ICJSh5ZMJpPlpmPDhg0RGhoKa2treHh4aDovyoM6e+oUdAq59nTUU7X7yALX5EMmREWHVsXZue8cf0FziRAVRcZdhYcp6+wKMBGigqc/yyfXfSdVk/6HKaKv0dbXyVminKuRE33d/u8fQLIrXQowD6KCp9XhYkGnQEREREREREREREREREREREREVGjFPwpD9JFXBZ2GUiUGVYdxfWnFNIqajIwMHDp0CJcvX0ZoaChMTU1RtWpVTJo0CTY2Nhpfz61btxASEoKEhASYmJjAzs4O7du3R+fOnTW2LmV8fX2xb98++Pn5ISwsDDKZDKampqhcuTJatWqFLl26qCy8oUpAQAB27NgBX19fREREwNTUFBUqVMDgwYPRtGlTDW9J/nj//j02btyI169fIyEhATY2NmjRogVGjhyp0fXExMRgz549ePr0KT58+ICkpCSYmJigXLlyaNq0Kbp16wYLCwuNrrMoSkpKwv79+/HgwQOEhYXh06dPKF68OKysrNCkSRN069btixbsSUtLw8GDB3H58mWEhYXBxMQEtra26N69O9q1a5cv6/zvv//g4uKCt2/fQkdHB5UrV8bEiRNhb2+fp3GLyvEaEBCAM2fOwMvLC6GhoUhISIChoSGsrKzQsmVL9O/fH+bm5gWdJhF9Y1iU5ivEojRE3xYWpSHKAxalIRKwKA1R7rEoDVFWLEpDlIlFaYiIiIiIiIiIiIiIiIiIiIiIiJRjURoiIiIiosJPu6ATICIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIqLCg0VpiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiOiL0bUwKOgUVCrs+RERERERfQm6BZ0AEREREREREREREREREREREREREREREREREREREX07DGzMUGqKA9Kikgs6FTm6FgYwsDEr6DSIiIiIiAoci9IQERERERERERERERERERERERERERERERERERER0RdlYGMGA5uCzoKIiIiIiJTRLugEiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiKjwYFEaIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIhKwKA0REREREREREREREVEB8Pf3h5aWlvAzevTogk6pyPHy8sLUqVPh4OAACwsL6OjoiF5T+rbwmKL8lJqaipo1awr71+bNm5W2dXV1Fe2LS5Ys+XKJUoHK+r63bdu2oNOhQqht27aSr1V2794tart79+58zW3JkiWi9bm6uubr+r4FXbp0EV7PH3/8saDTKfK+9DHxLfgWPrf4OwIRERERERERERERERERUd6wKA0REREREREREREREREVKRkZGZgzZw7q1auHLVu24OnTp4iOjkZGRkZBp0ZEX6mNGzfixYsXAIAqVapg4sSJBZwREREVditWrBCKD23atAmvXr0q4IyIiIiIiIiIiIiIiIiIiIiI1MOiNERERERERERERERFyMWLF0V3eNbS0oKTk1OO/WxtbeX6afon+121XV1dJfUrVqwYrK2t4eDggNGjR2P79u2Ii4vLp1eQiL4Gf/zxB9asWVPQaRDRNyIiIgLLli0T4sWLF0NPT68AMyIioqKgQYMG6NevHwAgNTUVs2fPLuCMiIiIiIiIiIiIiIiIiIiIiNTDojRERERERERERERERcju3bvllt24cQMBAQFfPhkNSU5OxocPH/D06VPs2bMHEydORNmyZbFgwQKkpKQUdHpESi1ZskRUYMnV1bWgU/omREVF4ddffxViLS0tjBw5EgcPHsTly5fh4uIi/BARacLKlSsRGxsLAKhYsSKGDBlSwBkREcnLWojU1ta2oNOh/+9///uf8PjcuXO4d+9eAWZDREREREREREREREREREREpB4WpSEiIiIiIiIiIiIqImJjY3H69Gm55TKZDHv27CmAjPJPfHw8/vjjDzRr1gzR0dEFnQ4RFSJHjx5FUlKSEC9atAh79uzBkCFD0LFjR3To0EH4ISLKqw8fPmDz5s1CPGvWLOjq6hZgRkREVJQ0btwYbdu2FeJffvml4JIhIiIiIiIiIiIiIiIiIiIiUhO/LUdERERERERERERURBw+fFhUiCGrvXv3qpzcduDAASQmJua4jg8fPmD48OGiZS4uLpLyK1GihMrnS5cujf3798stT0xMRHBwMK5fv45Tp04hNTVVeO7Ro0fo06cPrl+/Dm1t1lmnr4utrS1kMllBp1Hk3L17VxRPmjSpgDIhom/Bxo0bkZCQAAAwNDTE6NGjCzYhKtT4uU6aNHr06C96zlmyZAmWLFnyxdb3LZk8eTJcXV0BfP79+vHjx6hXr16B5kT0reDv3UREREREREREREREREREecOiNERERERERERERERFxO7du4XHWlpaaNGiBW7fvg0A8PX1xa1bt9CqVSuFfVu2bClpHf7+/nLLOnTooHauihQrVkzlWJMnT4aPjw/69euHp0+fCstv3ryJQ4cOYdiwYRrJg4iKttevXwuPzc3NUbZs2QLMhoi+ZklJSdi6dasQDxgwAObm5gWXEBERFUl9+/ZFyZIlERkZCQDYsGEDnJ2dCzgrIiIiIiIiIiIiIiIiIiIiopzxtrJERERERERERERERcDr169x7949IW7ZsiXmz58varNnz54vnZbG2dnZ4dKlSyhVqpRo+bp16wooIyIqbGJiYoTHpqamBZcIEX31jh07hoiICCEeMWJEAWZDRERFlb6+PgYNGiTEhw4dEl3TEhERERERERERERERERERERVWLEpDREREREREREREVATs3r1bFA8fPhxdunSBpaWlsOzYsWNISEj4wplpXpkyZTBz5kzRMk9PT+Gu8kT0bUtKShIea2lpFWAmRPS1279/v/C4RIkScHJyKsBsiIioKOvbt6/wODk5GcePHy/AbIiIiIiIiIiIiIiIiIiIiIik0S3oBIiIiIiIiIiIiIhItYyMDOzbt0+I9fX18d1330FXVxeDBg3C5s2bAQBxcXE4deoUhg0bVlCpakyPHj3w008/CbFMJoO3tzfatGmTL+tLS0vD7du34ePjgw8fPqBYsWKoUqUKWrVqBQsLC42vLyYmBnfu3EFISAjCw8NRrFgxlCpVCvXr10fNmjU1vj4vLy88fPgQYWFhMDAwgLW1NVq0aAFbW1uNjP+lt8fb2xsvXrxASEgIPn36hNKlS2PkyJHQ09NT2icoKAjPnj3D27dvERsbCwCwsLBAuXLl0Lx5c5QoUULjeX4pjx8/xvPnzxEWFoakpCRYWVmhQoUKcHR0hKGhoUbXlZGRgQcPHsDLywuRkZEwNjZGmTJl0Lp1a1hbW2t0XcrIZLJ8GTc4OBgeHh54//49IiMjYW5ujr59+6Js2bIq+33J1z89PR13796Fn58f3r9/DwMDA9SoUQNt27bNcV1xcXFwc3PD69evkZCQAEtLSzg4OKBp06YFUtwnMjISN27cQHBwMD5+/AgLCwvUqVMHzZo1g46OzhfPp6C8evUKT548QUREBKKiooTzp729PerWrQt9ff1cjRsYGIgHDx7gw4cPiI2NhYWFBaytrdGyZUuUKlUqTzknJibCy8sLz58/R3R0NBITE2FoaAhTU1PY2tqiRo0aqFChQq7Hl8lkePLkCV68eIGwsDDEx8fD0tIS5cuXR6tWrVC8ePE85S9VWFgYrl69KsTdunWDrq7m/r2e35/NX0paWhru3bsHb29vREVFwdTUFBUqVEDbtm1hZmaW5/EzP3fevHmDsLAwpKenw8rKCpUqVUKLFi1UfvYXVenp6Xj48CF8fX0RERGBjx8/onjx4ihXrhxq1qwJe3v7L3rezjwmX758ifDwcMTGxsLIyAjW1tawt7dHnTp1cn3efvXqFR49eiQ61suWLQtHR0eN7D/Z5edx9/btW9y/fx/BwcFITU2FtbU1GjVqhNq1a+c98SIkISEBbm5uePfunfB7gZWVFWrVqoW6detqfH2xsbG4efMmfHx8kJiYiJIlS8LOzg6Ojo4wMDDI9bhRUVHw9PSEj48PYmNjkZaWBiMjI1haWqJSpUqoVauW2r8/ZJ4XM38XOXjwIMaPH5/rHAu7N2/ewN3dHe/fv0daWhrKlCmDRo0aoVatWgWd2hdRFK/xCpvU1FQ8ffoU3t7eiIyMRHx8PAwMDGBiYgIbGxtUq1YNVapUyfc8Xr9+DS8vL4SEhODjx4/Q1dWFsbExypUrhypVqqBmzZoavUYkIiIiIiIiIiIiIiIiIips+B9RIiIiIiIiIiIiokLu6tWrCAoKEuJu3boJE+CGDx8uFKUBgN27d38VRWkqV64stywiIkLj60lOTsaKFSuwZcsWhePr6OhgwIABWLlyJSpWrAh/f39UqlRJeH7UqFHYvXu35PWdOXMGq1evxp07d5CWlqawTcWKFTFz5kx8//33kiZSurq6wsnJSYgXL16MJUuWAAAOHTqEpUuX4tWrVwr7Nm3aFKtXr4ajo6PkbcjqS25PWloa/v77b2zduhXPnj2T69e/f3+Ym5sLcVpaGq5cuYLjx4/jypUrCAgIULpOLS0tNGvWDPPmzUPv3r1VTvTOnl9WypZnyl5MJS/708ePH/Hnn3/C2dkZ79+/V9imWLFi6NKlC3799VfJE6J3796NMWPGCLGzszNGjx6NjIwM/P333/jjjz9E56NMWlpa6NSpE1avXq3xyddLlizB0qVLFT4XEBCg9P3K/npnbdemTRu4uroCAM6dO4e1a9fC1dUVGRkZoj7lypVDnz595Mb+0q9/UlIS/vzzT2zfvh3BwcFy/czNzbFgwQLMmTMH2traouc+fPiARYsWYd++fUhKSpLra2tri/Xr16N3796Scsyr169fY/bs2bh48aLC84aVlRXmzJmDWbNmQVdXV+79v379Otq2bauRXJTtE1KMHj0ae/bsEeK3b99KLmoQERGBVatW4dChQ3j37p3SdsbGxujYsSMmTJiArl275liEIiMjA7t378a6devg7e2tsI22tjaaNGmChQsXomfPnpLyzeTj44Nly5bh5MmTiI+PV9m2bNmy6Ny5MyZOnIhmzZpJGj88PBy///47Dh8+jJCQEIVt9PX1hePKwcFBrfzVdeHCBaSnpwtxTud4qTTx2Xzv3j00b95ciDt27IjLly+rnctff/2FH374QYjnzZuHP//8U9RG2edyamoq1q5dizVr1iA8PFxubAMDA/Tt2xerV69GuXLl1M4tLCwMy5Ytw+HDhxEZGamwjYmJCfr27Ytly5bBxsZG6VgPHz5Ey5YtkZKSAuDz9d3169fRqlUrSblkf53Kli0LLy8vWFpayrXNy3nl2bNnWLFiBc6dOycUrVDE2toavXv3xpQpU/KlyEemwMBArFixAqdOnUJYWJjSdiVKlEC3bt0wadIkSa9pcnIyNm7ciH/++Qe+vr4K2+jq6qJNmzZYsmSJ5OvUgromvnv3LubMmYM7d+4ofL5WrVpYtmwZ+vXrp/bYyq4Lssr+eZRJ1TUSoPi6My+fuV5eXli8eDEuXbqk8HoD+HxdNXbsWMybN09ygbG2bdvixo0bQpx5fRcSEoKffvoJBw4cEI7trIyNjTFz5kwsWLAAxsbGktYFfP7d+88//8TVq1flrguz0tLSQvXq1dG7d29MnTpVUjE2XV1dODo64ty5cwCAW7duIS4uDqamppLzKwouXbqEBQsW4NGjRwqfr127Nn777Tfh2lPZe5xbhw8fxpAhQ4R4woQJ2LZtm9rj/Pjjj1i3bp0Qb9myBVOmTFHZp6he46mSmJiIsmXLIiYmBgBgZmaGkJAQtYtvPnnyRPSZ1aRJE9y/f19h29DQUPz22284ePAgoqOjVY5raWmJdu3aYezYsejcubPCNrn5vTs5ORnr16/H9u3blX5WZTI0NETz5s0xcOBATJ48WWVbIiIiIiIiIiIiIiIiIqKiSDvnJkRERERERERERERUkLJPlhk+fLjwuFmzZqI7Q1+7dk1hwYiiRk9PT26ZsqInuRUYGIh69eph2bJlSgvepKen48iRI3BwcBBNlFNXWFgYnJyc0KtXL9y8eVPltgQGBuLHH3+Eg4NDjpOflElJScHw4cMxdOhQpZNvAeD+/fto27atWoV1gC+/PdHR0XBycsKMGTMUFqRRZPDgwejatSt27typsiAN8Hni4927d9G3b18MGDAgx2ILBe3GjRuws7PD8uXLlRZEAYCkpCT8+++/qFevHn766adcry8uLg6dOnXCtGnTlJ5fZDIZLl26hKZNm+LSpUu5XteXJJPJMH36dPTo0QPXrl1TOfE4qy/9+n/48AEtWrTAkiVLFBakAYCYmBj873//w5AhQ0Tb4e7ujrp162L79u1KJ4j7+/ujT58+2LBhQ65zlGrv3r1wcHDA2bNnlZ43wsLCMG/ePLRv315lUYaiauvWrahUqRJWrlypcrIyAMTHx+Pff/9F9+7dczyPBQcHo3Hjxhg3bpzSycrA50nN9+7dQ69evdCtWzd8/PhRUt779u1D7dq1sW/fPknnyPfv38PZ2RmbNm2SNP7OnTtRpUoVrFu3TmlBGuDz59t///2H+vXrY9myZZLGzq3s57I2bdrkaTxNfjY3a9ZMVJTnypUr8Pf3Vzun7du3C4+1tLQwYcIESf1iYmLg5OSE+fPnKyxIA3yezH348GHUrFkTFy5cUCuvo0ePws7ODps3b1ZakAb4XCBs7969qF69uqhIY3aNGjXCH3/8IcTp6ekYOnSoyrEzeXp6Yu7cuUKsra2NAwcOKCxIk1upqan4/vvv4eDggIMHD+Z47gsNDcXWrVvzrZiYTCbD0qVLUbVqVWzdulVlQRrg83XagQMH0Lp16xzHfvbsGWrWrIm5c+eqvC5MS0vD1atX0apVK4wdOxapqalqbweQ/9fEy5Ytg6Ojo9KCNMDnbe7fvz9mzJiR52IbhZFMJsP//vc/NGjQAKdPn1Z6vQF8/qz69ddfUbVqVbi5ueV6nbdu3ULt2rXh7OyssCAN8PkzdPny5XByckJUVJSk7Zg+fTo6dOgAFxeXHK8LZTIZXr58iT///FOta9+snyWZRTS/FjKZDNOmTUOXLl2UFqQBAG9vb/Tp0wezZs3Klzz69euHUqVKCfHhw4fV/v0uOTkZe/fuFWIjI6Mci/8W1Wu8nBgaGmLkyJFCHBsbi2PHjqk9TtZrDgCYOHGiwnYuLi6oUaMGNm/enGNBGuBzIaCjR4/i119/VTsnZTL/VjR//nxJf8NITEzEtWvXMGXKFI3/3YqIiIiIiIiIiIiIiIiIqDBgURoiIiIiIiIiIiKiQiwuLg6nTp0SYjMzM/To0UPUJuvkqIyMDNHkqaJKUaGHkiVLamz80NBQODk54eXLl6LlWlpaqFy5Mho3bozKlSsLdyyPjY1Fz5495dpL8ebNGzRr1gyurq5y67K1tUWjRo1Qp04dFC9eXPT869ev0bx5c7x+/VrtdY4aNQoHDhwQ4hIlSsDBwQENGjSAubm5qG16ejrGjx8Pd3f3Qrk9aWlp6NWrl2jiaub2ODg4wMzMTGE/RRNiS5UqhZo1a6Jp06aoW7euwgnlJ0+eRO/evSUXKPnSzp07hy5dushNEC9WrBhq1KiBBg0aiCZhAp/f4xUrVmDcuHFqry81NRU9evTA1atXhWVWVlZo0KABHBwcYGxsLGqfkJCAAQMG5Di5sjBYuHChqGCGoaEhatSogYYNG8La2lphny/9+iclJaF79+6iib02NjZo3LgxqlatKpyjMh09elQovPDy5Ut07NgRHz58AADo6OigWrVqaNy4McqXLy+3rlmzZuVpgnhODh8+jDFjxiA5OVm03NjYGLVq1UL9+vVF5/mbN29i0KBBX9UE/pkzZ2Ly5Mn49OmTaLmOjg4qVaqExo0bo06dOnL7UE7evn2LFi1awNPTU7RcW1sblStXRqNGjWBjYyPX78KFC3Bycspxwq+LiwtGjRol994ZGRmhZs2aaNasGerXrw9bW1toa6v/7+dFixZh/PjxcpOnTU1NUatWLTRp0gS2trai5zIyMrB48WL88MMPaq9Pqlu3bgmPzc3NYWdnl6fxNP3ZPHnyZOGxTCbDzp071crn3r17osntbdu2lbSNGRkZGDhwIG7fvi0sK1myJOrXrw97e3sUK1ZM1D4uLg79+vXD9evXJeW1fft2DBkyRG5/KF68OGrVqoV69erJvV7JycmYNm2aykJFs2bNEl1DBwUFYfTo0Spz+fTpEwYPHiwqerFo0SK0bdtW0rZIERsbiw4dOuDvv/+Wu/YwNDREtWrV0LRpU9jb28tdW+WH1NRUfPfdd1iyZIlcsQ99fX1UqVIFTZo0Qa1ateTeh5w8fPgQrVq1gp+fn2i5np4eqlatikaNGqFs2bJy/ZydndGrVy+lxUdUyc9r4hUrVmDx4sVy75uFhQXq16+PmjVrwtDQUFi+ceNGLF++XO1tKMxkMhlGjx6NlStXyr0OlpaWaNCggcLzQmhoKDp16pSrQoYeHh7o2rWrUGhGW1tb2C8VXRu5u7vneKwDwC+//KKwkJqFhQXq1q2LZs2aoU6dOihTpozaOWfVsGFDUXzz5s08jVeYTJs2TWGBsMx9oVatWqLfHdavX48VK1ZoPA99fX2MGTNGiD9+/IjDhw+rNcapU6dEhcsGDRoEU1NTpe2L6jWeVJMmTRLF2QvM5CQxMRH79+8XYhMTEwwePFiu3bNnz9CzZ0+54mwGBgaoXr06mjRpgoYNG8LOzk5hIWNNSExMRIcOHeT+9qOtrQ0bGxs0bNgQTZo0QY0aNb7I5zIRERERERERERERERERUWHAojREREREREREREREhdjRo0eRmJgoxAMGDICBgYGozfDhw0Xxnj17vkhu+Sl7wRMAqFSpksbGnzRpkmhCrK6uLubPn4+goCD4+vriwYMH8PX1xbt37zBv3jzo6uri48eP+P7779VaT0JCAnr37o23b9+KtmPr1q2IjIzE27dv4e7ujidPniA6OhoXLlxAgwYNhLbh4eEYNGiQXCEAVfbt2ydMuuvSpQvu3r2LyMhIeHl5wcPDAxERETh16pRo0m96ejqmTZtWKLdnx44dQqGMDh064Pbt24iIiICXlxe8vLwQHR0NFxcX0aTfTJaWlvj+++9x7tw5hIeHIywsDM+ePcO9e/fw+PFjhIeH482bN1iwYIFosuzVq1exYcMGhfnUrVsXLi4ucHFxwYgRI0TPrV69WnhO0U9evXv3DsOHDxcV3ClZsiS2b9+O8PBwvHjxAh4eHggLC8OdO3fg6Ogo6r9r1y78888/aq3zjz/+EAozDBs2DE+ePMGHDx/g4eEBLy8vREZGYteuXaJJmp8+fcK8efPysKViI0eOFL2OpUuXFp4rXbp0rl7v58+fY+XKlQCAqlWr4tixY4iKisKLFy/w8OFDhISE4OnTp3BwcBD6FMTr/+eff8LDwwPa2tqYNm0a/P394e/vjwcPHuD169cIDAxE//79RX2WL1+O4OBgDBw4ELGxsTA3N8e6desQHh6OV69e4cGDB3j37h3u37+P2rVrC/1kMlm+FfgIDAzEhAkTRBPWbWxscPToUURGRsLb2xuenp4IDw+Hm5sbWrZsCQC4dOkS9u3bly85fWkbNmyQO69UrlwZu3fvRnh4OPz8/PDgwQM8efIEYWFhCAwMxPbt29GuXTuV46alpWHIkCEIDAwUlmV+pr179w6+vr5wd3eHv78/fHx8MH78eFF/Dw8PTJkyReU6Zs6cKSoO5OTkBFdXV8TFxeHZs2e4e/cuPD098fbtW3z69Al37tzBzz//jMqVK+f4ujg7O+O3334TYi0tLYwcORLu7u6Ijo6Gt7c37t+/j7dv3yI4OBgLFiwQTUL+66+/cPTo0RzXo66wsDAEBQUJca1atfI0Xn58Ng8bNkw0GdrZ2Rnp6emSc8o+oXzChAmS+u3duxdXrlwBADRo0ADXr19HeHg4PD098fz5c4SHh2Pr1q2igh9JSUkYOnQoYmJiVI7t6emJqVOnis4VFStWxNGjRxEREQFvb288evQIERERuHjxotz7snjxYly4cEHp+Lt370a5cuWE+OzZs1i3bp3S9pMnT8abN2+EuHXr1li0aJHKbVDXmDFj5IpStGnTBhcvXkR0dDRevXqFe/fu4fnz54iNjYW3tzeWL18Oe3t7jeaRad68eTh+/LhoWb169XDixAlERUXBx8cH9+/fh7e3N6Kjo/HmzRusX78ejRs3Vjnux48fMXDgQFGBBCMjI6xcuRKhoaF4/fo13N3dERwcDC8vL/Tp00fU/+LFi/jll1/U2pb8vCbOPM9llXk8REREwNPTE8+ePUNERAR27twpFF1bunQpfHx81NqOnMybN0/tayQXFxeNXKtt2rRJriBqq1atcPv2bYSFhcHDw0M4L2zfvh0WFhZCu8TERAwdOlRhMVJVvvvuO8THx8PCwgJr165FWFiYsF++fv1aYcGpM2fOqDw3BAcH488//xQtmzRpEp4/f47IyEg8fvwYd+/exZMnT/D+/XtERUXh7NmzmDx5stICmcrUqVNHFD98+FCt/oXVqVOnsGXLFtGyVq1a4c6dO8K+4O3tjcjISBw5cgQVKlQAACxZsgS+vr4az2fixImiAkXqFlFR5zOyKF/jSVWzZk20atVKiN3c3NQq2Hv8+HHRNcCQIUPkipsCn89nWf9WUK9ePZw9exZxcXF4+fIl7t+/j4cPH+LNmzeIj4+Hp6cnli9fLndc5cWmTZtEn/2lSpXCtm3bEBERAX9/fzx8+BD379/HixcvEBcXBx8fH/zzzz/o1KmTXFEsIiIiIiIiIiIiIiIiIqKvhZYsl7cXbNiwIUJDQ2FtbQ0PDw9N50V5UGeP5v7Z/qU9HfVU7T6ywDX5kAlR0aFVcXbuO8cr//Id0TfBuKvwMGVd3u5yS1TU6c/K/RfhJ1UzyLkR0Vds6+usE6rVm0RB9PX5v4lcsitdCjAPooKn1eGixsZq1aqVUJADAK5fv462bdvKtWvatCkePHggxHfu3EHz5s3VXp+/v79c8Zdc/hkZrq6ucHJyEmIbGxv4+/vn2C81NRUNGzbE06f/9/fCihUrIiAgIFd5ZHfmzBn06tVLiPX09HDq1Cl0795dZZ9+/fohLS1NtHzUqFHYvXu30n5TpkwRFaHo2bMnDh48qPKO2snJyRgyZAhOnTolLFu7di1mzZqlsH321znTokWLsGzZMqXref36NerXr4+EhARh2ePHj1G3bt1CuT0zZ85UOXE8u7t376J+/fqiYjOqPH78GO3bt0dUVBQAoFy5cvD394eurq7SPkuWLMHSpUuFWNnxqUz24y2n/alnz544e/asEFeoUAG3bt2CjY2NwvYZGRkYPXq0qKiHkZER3rx5I5qAndXu3bsxZswY0TItLS1s27ZNbqJjVrdu3ULbtm2FQgJ6enoIDg5GqVKllPbJLVtbW+F8IPW8AkDhJMFWrVrh/Pnzku5yX1Cvv7a2Ng4fPoyBAwcqXU/nzp2FIhEAYG9vjxcvXsDa2hrXrl1TWrwgPDwctWrVQnh4uLDs0aNHqFevnsL2udW/f3+cPHlSiOvUqQNXV1fR5PSsMjIyMHz4cBw6dEjuOVXHmbrHVNZ9ok2bNgoLoikzevRoURG4t2/fwtbWVmFbb29vNGjQAKmpqcKyAQMGYP/+/XKF5hTx8PCAnZ2dwonva9aswZw5c4RYX18fp0+fRpcuyq/Ht2/fjokTJ4qWnTp1Sq4IBPC5gFPWwh9OTk64cuUKtLVzvvdJRkYGfHx8UK1aNYXP+/n5oU6dOsLnkKGhIU6cOIGuXbsqbJ/p5s2b6NKli1C0z8rKCgEBAZLP91JcuXIFHTt2FOKxY8di586dkvt/qc/miRMniibO//fff+jZs2eO+X38+BFlypRBfHw8gM8FtoKDgxXuj8q2pVu3bvj3339FRYKyevPmDRwdHREWFiYs+/7777F582aF7WUyGerWrSu6/qtbty5cXV1FBW6ySk5ORrdu3XDt2jVhWZkyZeDr66uwWB3wef9p166dUMBHX18ft2/fRqNGjUTtnJ2dMXbsWCEuWbIkvLy8REVtFFHnvLJ161ZMnjxZ1HflypWiY1oZmUym9L3JrUuXLsmdO2bMmIF169ZJOuavXr2K9u3bK3xu+vTp2LRpkxCbmZnh+vXrqF+/vtLxfv75ZyxfvlyItbW14e7uLip4mNWXOu4yMjLg4OCAZ8+eCctyOh4CAgLQsmVLBAcHyz2n6net7NcFzs7OcgVXssrtNVImda5tg4KCUK1aNVEB1VGjRsHZ2VlpYQZ/f384OjqKXofevXvj33//VZpT27ZtcePGDdEyW1tbXLlyBVWqVFHaL/v5sU+fPqLfR7LasmULpk6dKsS//PKL6HVQ5dOnT4iOjhaKrEhRsmRJ4XeO4sWL4+PHj5L7FkbJycmoVKkSQkJChGXDhw/Hnj17lJ47IiMj0bp1azx//lzuOU0dE506dRIVqnzy5Imk4iV+fn6ws7MT8qhVqxa8vb0Vti3K13iZpH5uHTx4EMOGDRPiH3/8EWvWSPu+Vps2bUQF2Nzd3eU+d2NjY2FpaSn8zaVGjRrw9PRU+nme3cuXL1GjRg2Fz6nzO0KTJk3g7u4OADAwMMDjx4+Vjqsoh+rVq7M4DRERERERERFRLgQGBuZ4c4OCYG5ujooVKxZ0GkREREREBS7nbw4RERERERERERERUYHw9fUVFaSpUKEC2rRpo7Dt8OHDRXHWifJFSWpqKsaPHy+akAwAI0eO1Ng6/vrrL1H8008/qSxIA3wuRjF//ny11vPu3Tvs2LFDiB0cHHDs2LEci18YGBhg//79ouIGGzZsECZPS9G7d2+Vk28BoFq1apg+fbpo2YULyosYF+T2tGjRAmvXrpXcHgCaN2+uVoGCevXqYeXKlUIcHByMy5cvq7XO/PTq1SucO3dOiLW1tXH8+HGlBVEy2+zatUs0+TIhIQF///23WuueMWOGyoI0wOfiLlmLpqSmpuLq1atqredLK1GiBI4ePSqpIE1Bvv5z585VWpAmcz2//fabaNmLFy8AfP4sUFaQBgBKlSolVyBK1XkgN969eyeabK6vr48TJ04oLUgDfN4mZ2dn2Nl9HYWkV6xYIZqs7OjoiCNHjkiarAx8vlmIosnK6enp2LBhg9y6VE1WBoAJEyaICmEAUDqp+PXr16J40qRJkopTAJ/fR2UFaQBg5cqVoiIQu3btyrEgDQC0bt0aq1evFuKwsDDs379fUk5SvX37VhSXL18+z2Pmx2fzpEmTRHHWAgyqHDp0SChIAwAjRoyQvD8Cnwu/HD58WGkBDgCoWrUq9u7dK1rm7OyM2NhYhe1dXFxE139GRkb477//lBakAT5fY5w8eRLW1tbCspCQEBw8eFBpn9atW2Px4sVCnJKSgsGDByMuLk5Y9vLlS7n3Yffu3TkWpFFHWloaVqxYIVq2YMECSQVpgM9FBDRZkAaAXBGOoUOHYsOGDZKPeWUFaWJiYrBr1y7Rsp07d6osSAMAv/32m+ickJGRoVaBQiB/jjsXFxdRQRopx4ONjQ0OHz4sMeuiYcuWLaKCNHXr1sWOHTtUFmOwtbXFsWPHRG3+++8/vHnzRvJ6dXR0cOzYMZUFaQDgzz//FP0ucPnyZaF4YnbZP+u+//57yfkUL15crYI0AETFCT99+oTIyEi1+hc2x48fFxWkqV69Onbu3Kny3FGyZEmcOHFC5XGTV7n9jNyxY4eoMM6ECROUti3K13jq6t+/PywtLYV47969SElJybHfq1evRAVp6tWrJ1eQBvh87ZW1CPDo0aMlF6QBILlwTE6yng+cnJzUGrdGjRosSENERERERERElAuBgYHYtWsXTp48Weh+du3ahcDAwIJ+iYiIiIiIChyL0hAREREREREREREVUtnv3Dx06FClE1wGDx4MXV1dIT5y5AiSkpLyMz2NSUpKgq+vL3bs2IH69evLTWAuVaoUZs+erZF1BQcHiwplmJub43//+5+kvgsWLFA4YUyZzZs3iyZVrVq1SvLkNCMjI1GhiICAADx8+FDyurNPclZm0KBBotjT01Np24LcnmXLln2RyV2DBw+Gjo6OEN+5cyff1ynVzp07RZMjhwwZgiZNmuTYT1dXF6tWrRIt2759u2gsVQwNDbFo0SJJbdXZnwqDqVOnigoZqFJQr7+xsbGkglhNmzZF6dKlRcscHR3RqVOnHPv27t1bFD969EhSblLt379fNAl87NixqFq1ao79DAwM5AokFEUhISE4evSoEGcWK5Ja5EGVS5cu4d27d0JsY2ODH374QVLf5cuXw8jISIjd3NyEYkZZZS04AEBjk8ejoqJEn/fNmzfH4MGDJfefMGECrKyshPjEiRMayStT1tcV+Fx0Iq/y47O5YcOGoond58+fR3BwcI7ryD4xX9WEe0V+/vlnmJiY5Niuc+fOaNeunRAnJibi0KFDCttmLXwHALNmzZJ050kzMzMsWbJEtGzbtm0q+/z000+ivHx9fTFx4kQAn69LBw0aJCraM3PmTPTo0SPHXNRx8uRJ0ZeYK1euLLcdX9KDBw9w9+5dITYxMcHmzZs1MvbBgwdFBahatmyJ/v37S+qbvSjh0aNHlRY2UiQ/jrvsBXakHg+Ojo7o06ePpHwKO5lMhp07d4qWrV69WvT7qDLNmzcXvd4ymUzu+Felf//+CgtaZFeiRAnRdVBCQgJevnypsG1+fdYpk/0zJftnTlGT/ff3pUuXQl9fP8d+NWrUwOjRo/Mpq8/XuFlf6/379+f4N5K0tDTR32EMDAwwYsQIhW2L+jWeugwMDETvV0REBE6dOpVjv+zHt7Jrji99HCqTNY+CyoGIiIiIiIiI6FsTExNT0CmoVNjzIyIiIiL6EliUhoiIiIiIiIiIiKgQkslk2Ldvn2jZ8OHDlbYvVaqUaNJdTEwMTp8+nW/55UZAQAC0tLTkfgwNDWFnZ4cJEybg2bNnoj5GRkY4c+YMzM3NNZLDnTt3RMUg+vfvj2LFiknqa2RkhH79+kle1/nz54XH1tbW6NChg/REAbliErdu3ZLUr06dOqhZs6aktrVr1xZNHlU1IbKgtqd06dKiieP5ydjYWFTkQNPFOfLixo0bonjs2LGS+3bs2BHly5cX4g8fPojuAK9Khw4dULJkSUlt69WrJ4oL+wTbIUOGSG5bkK+/1PNfrVq1RPGAAQMk9atevbpowqWm7/Tm5uYmiocNGya5b9++fWFsbKzRfL60q1evIj09XYi7du0qqSiPFNn3y5EjR0oqCAAAFhYWcsURbt68KdeubNmyovjAgQPqJamEq6uraNKvsgnfyujp6cHJyUmI79y5Iyp+lFfZi14UL148T+Pl12czAEyaNEl4nJ6eDmdnZ5Xtvby8RIXhWrRoITk34PNrr04BoVGjRoliV1dXhe2y739jxoyRvI4hQ4bA0NBQiD08PERFULLT1tbG/v37UapUKWHZkSNHsH37dsyaNQtPnjwRljds2BB//vmn5Fykunz5siieMmVKgU5+z57PyJEjNXb9nZfP0Bo1aqBFixZCnJKSgnv37knqm1/HXdZ9WN3jIT8LcHxJL1++RFhYmBBXrFgR7du3l9w/+z6g6PNHmewFhFSRem2a/bNu//79kteRG9k/U9QptFTYZGRkiApaGRsbyxU8VEXV3zjySldXV7SvRUdH4/jx4yr7nDt3DiEhIULcv39/WFhYKGxb1K/xcmPixImiYrHZi9xll5qaKipaZGRkpPR3gezH4dGjR5GampqHbHMnax43b97kXbCJiIiIiIiIiIiIiIiIiMCiNERERERERERERESF0vXr1xEQECDEDg4OqF27tso+2Sf3ZL3Dd1Hk4OCAu3fvomnTphobM+skbACiSa5SSG0fHR0Nb29vIW7QoIHad0yvWLGiKJZ6d/NGjRpJXoeenp5owrGyCZEFvT1ZJ77lxrNnz7B06VL07t0bVatWhaWlJfT19RUWSco6CTEiIiJP69WU5ORkPH78WIj19PTg6Ogoub+2traoeAMAyRO61dmfshb0AQr3BFsTExPY29tLaluQr3/Dhg0lryd78aAGDRpI6qejoyM6D8TFxUlepxRZz7u6urpo3Lix5L6GhoZyE8qLmuwFuLp166axse/fvy+K1S3glb2AgKL9smnTpjA1NRXikydP4rvvvsPTp0/VWld22V8Xdc41mbJ+rsTFxSE4ODhPOWWVvaBJ1oInuZEfn82ZhgwZInqPdu7cKSrAl132CeQTJkyQnBvw+fpM2QR9Rdq2bSuKHzx4INfG398fHz58EGIbGxtUqVJF8jpMTU1Fr3F6ejrc3d1V9ilTpgz27t0rusaYNm0a/vnnHyE2MTHBkSNHoK+vLzkXqfLz3JAbRf1cpUh+HHcBAQGiYizqHg9t2rSR3LYwy/6eOjk5qXW93rp1a1GBjUePHiElJUVS3/y4Nu3YsaMonj17Nn7++WeEhoZKXpc6jIyMRHF8fHy+rOdLePXqFT5+/CjEDRo0kFz0FQCaNGkiudhKbkycOFH0e2tORVTU+Yz8Gs+bOalataool2vXrsHPz09p+9OnT4vOmQMHDoSZmZnCtjY2NqKiPvfv30eXLl1w584dDWQuXdbzQWxsLJycnHDs2LECKZBDRERERERERERERERERFRY5N9/9knOmTNnsHr1anz69CnPY3l4eGggIyIiIiIiIiIiIiqssheUkXIH8T59+qB48eLC3yBdXFwQEhKCMmXK5EeK+aJ06dJo2bIlRo8ejW7dukFHR0ej42efsC61KIW67V+9eiWaEH7+/Pk8F1aJioqS1C775MucGBsbC8VXEhMTFbYpyO2pVKlSrtfx9OlTTJs2Ldd3ho+Jicn1ujUpNDRUNFG3Ro0aak+Qr1u3Lvbt2yfEUu/4rs7+ZGxsLIqV7U+FgY2NjeR9uCBf/1KlSkleR/YJzrntq8n3LS0tTTQRtXLlyjAwMFBrDHt7e9y+fVtjOX1pvr6+ojg3xVeUyVq8DvhcHEEddevWFcWK9stixYrhf//7H3766Sdh2bFjx3Ds2DHY29ujU6dOaNOmDZo3bw5ra2vJ685emKxJkyZq5a5IVFQUKlSokOdxFFFV5EWK/Phsztp2xIgR2Lx5M4DPBV5cXFzQqVMnubaJiYk4cOCAEJuamuK7775TK7eciiRmV7FiRZiamgoFrwICAiCTyUTn4Lzuy8Dn/TlrgQAp59kuXbpg7ty5WLlyJQDIFcXYunWrWsVx1JF1Er+xsbHa16Sall/nKplMhnfv3gmxqakpbG1t1RpDyrlKkfw47t6+fSuK1T0ezM3NUaFCBdFrUhTl9Zg1MDBAjRo1hKKXycnJ+PDhg6RzeH5cm7Zo0QIdO3aEi4sLgM/XL8uXL8fvv/+OFi1aoH379mjVqhWaNGkCExMTyetXJq+fKarcvn1b8rWco6OjWgVkFMnr79fFihWDra0tfHx88pSHMhUrVkTXrl1x7tw5AMDNmzfx+vVrVKtWTa5tUFAQLl68KMRVq1aVK6yWVVG/xsutyZMn4+rVqwA+78s7duzAihUrFLZVtxDekiVLRMWWr127hmvXrsHW1hadO3dGmzZt0KJFC9jY2ORxK5SbO3cuDhw4IBQo9PPzw3fffQdzc3N07twZbdu2RYsWLVC7dm21C/USERERERERERERERERERVVLErzBa1evTrfvkhBREREREREREREX49Pnz7h5MmTQqytrY2hQ4fm2M/IyAh9+/YVCh+kp6dj3759mDdvXr7lqo7SpUtj//79cssNDAxgZmaGUqVK5XsBnexFRpTdpVsZc3NzSe0iIyPVGleK2NhYSe3yMrFQ2QTJgtweU1PTXI1/9uxZ9O/fX26CuTqSk5Nz3VeToqOjRbGlpaXaY2Tvk31MZfJjfyoM1Nmviurrn9u+mnzf8nrOBaSfdwur7AW41C2SoErW/UhbWxsWFhZq9Ze6Xy5YsAABAQHYtm2baPmLFy/w4sULbNiwAQBQvXp1dO7cGUOHDkXTpk1VrrsgP1ekyF7kKSkpKU/j5fe5dNKkSUJRGgDYsWOHwqI0x48fFx2Xw4YNk9vWnJQsWVKt9gBgYWEhFKXJyMhAXFyc6HxQkOfZ5cuX48aNG7h//75o+dixYzFkyBC185AiLi4OaWlpQmxpaZnnYn95lfVcpaWllav3QJHY2FhkZGQIcW72n8J0DZP9cy0321OyZMkiX5Qmv45ZKUVp8ut8evDgQfTs2RP37t0TlmVkZMDNzQ1ubm4AAF1dXTRq1Ag9evTAsGHD1C6wlCl70ZjsxXPyYtiwYXIFTZR5+/ZtrrchU1G41ps0aZJQlAb4/BmZWYwsK2dnZ6Snpwvx+PHjVY77NVzj5Ubv3r1hbW2N0NBQAJ8LKi9btgy6uuKvIAYEBODKlStCbG9vj5YtW6oce+jQofDz88Mvv/wiOl79/f2xdetWbN26FcDnAqOdOnXC4MGD4eTkpNHP0KpVq+L48eMYPHiwcO0CfN7Xjxw5giNHjgAASpQoAScnJwwYMAB9+vSBoaGhxnIgIiIiIiIiIiIiUpe5ubnw/3IzM7NCcxOuwqJt27a4ceOGEBfm7zNpmq6urvD/jypVqnB+OxER5Rpv2fEFZd6dWFtbG9bW1nn6ISIiIiIiIiIioq/XsWPHEB8fL8TVqlXDixcvcOXKlRx/KleuLBprz549Xzp9pYoVK4YOHTrI/bRq1QoODg75XpAGkC8yoq+vr1Z/AwMDSe3y45/7WSf0fmkFuT16enpqj/369WsMGDBAVJBGS0sLTZs2xaxZs7Bx40YcO3YMZ86cgYuLi+indOnSaq8vv2X+fyFTbiauZu/z8ePHPOVU1KmzX/H1z728nnMB6efdwir7e128eHGNjZ1131S3sAggfb/U0tLC1q1bcfHiRTg6Oiod79WrV/jrr7/QrFkzODo64uHDh0rbFvbPyewT5Av7MVunTh20aNFCiE+fPo3w8HC5dtu3bxfFEyZMUHtdmtjXsp9XC/I8m56errDoUNu2bdXOQar8PC/kVtacjIyMoK2tma+SfG2fodm3RxPHQ1H0tb2vwOciHjdv3sSWLVtgZ2ensE1aWhru3buHn3/+GVWqVMGIESPw4cMHtdeV/fXLTSGXwqIoXOt169ZNVPBoz549SE1NFbXJyMjAzp07hVhPTw+jR49WOe7XcI2XG3p6ehg7dqwQh4SEiIr+ZNq5c6fo2kzqNcfPP/+Mu3fvolu3btDR0VHYJiAgANu3b0f79u3h4OAAFxcXNbdCta5du+LZs2eYOHGi0vc1OjoaJ0+exNChQ2FjY4ONGzd+U5MYiIiIiIiIiIgo79zc3KClpSXpR09PD6VLl0bHjh2FQupERKSYnZ2d5POrqp8FCxYoXYfUMXR1dVGiRAk0atRIKL5PRFRU6ebchDTNysoKHh4eBZ0GERERERERERERFVLZC8m8fPkSHTt2zNVYz58/h7u7Oxo3bqyJ1Iq87BP+sk8IzEnWO2Wrkn3imJOTExYuXKjWurIrUaJEnvrnRVHbnvnz54smSDZp0gR79uxBjRo1cuyrybusa0r2iXBZi1ZJlb2PiYlJnnL6lvD1z728nnMB6efdwir7e/3p0ydYWlpqZOzixYsLdztLSEhQu7+6+2Xnzp3RuXNnvH37FpcvX4arqytu3ryJ9+/fy7W9ffs2WrZsif3792PgwIFyz2f/XHF2dkb58uXV3oas6tatm6f+WWWdvA58nnBd2E2aNAl37twBAKSkpGDPnj2YM2eO8PyrV69w69YtIW7YsCHq16+v9no0sa9lP68W5Hn2xx9/hJeXl9zy6dOnw9HREZUqVVI7l5woOi8UNBMTE0RHRwP4/B5nZGRopDDN1/YZmr3QgyaOh6Loa3tfM+np6WHKlCmYMmUKHj58iKtXr8LV1RV37tyRux7JyMjA/v37ceXKFbi6uqJ69eqS15P9c7NixYoayb8gFIVrPR0dHYwfPx6LFy8GAISFheH06dMYMGCA0MbFxQUBAQFC3KtXL1hZWakc92u6xlPXhAkT8McffwhFZ7Zv347evXsLz6enp8PZ2VmIDQwMMHLkSMnjN23aFOfOnUNISIjomtPPz0+urbe3Nzp37ow1a9Zg1qxZedgqsfLly2Pr1q1Yu3atcJzfvHkTXl5ewh1lM4WHh2PGjBm4ceMGjhw5orSYDhERERERERERUW6lpaUhLCxMuGFZpUqVcPXq1Xz5PxYRFT59+/YV/pfbvHlz/P777wWcEUmVnp6OmJgYeHh4wMPDAz/++COcnZ3x3XffFXRqRERqY1EaIiIiIiIiIiIiokLk7du3uHnzpkbH3LNnD4vS/H8WFhai+P3796hXr57k/oom3iuSfTJasWLF0KFDB8nrKWyK0vZ8+vRJdKf20qVL4+LFi5KL4GT+E78wyZ57ZGSk2mNERESoHJOU4+ufe8WLF4eenh5SU1MBSD+HZpWbPl+C1AnC2T93wsLCYGtrq5EcSpQoIUxYzsjIQHR0tFr7Vm73y0qVKmHSpEmYNGkSAMDPzw9Xr17FyZMncfnyZWGCckpKCkaOHImmTZvKTbjP/rlSs2ZNNGnSRHLu+S37exQUFFQwiajhu+++w8yZM4XPsR07doiK0uzYsUPUfsKECblaT/b9RoqoqCjhsba2NkxNTUXPF9R59tSpU9iyZYvC52JjYzFkyBC4ublBV1ezX6swNTWFrq4u0tLSAHzOXSaTFWhhPAsLC2HfkclkiIiIyLEggxRmZmbQ1tYWzgtF/TPU3NxcFOfmeMjNa1DYfAvXRo0aNUKjRo3wv//9DxkZGfDy8sLFixdx5MgRUSGr0NBQDBgwAF5eXpILOWW9tjExMZG7VsgLf39/jY0lhaLfr9X1JQq/jR8/Hr/++qtw3t2+fbuoKE1uPiO/xms8qWxtbdG5c2dcuHABAHDx4kUEBQUJBQYz40x9+/ZFyZIl1V5PmTJlMGrUKIwaNQrA5/3r2rVr+Pfff/Hff/8Jv2PIZDLMnj0bzZs3R7NmzfK6eSLGxsbo3bu3UHQnLi4Obm5uOHfuHA4dOiT6+8GJEyewZs0azJs3T6M5EBERERERERERZff27VtUq1YN7u7uan3niYiKpjNnzggF04OCgliU5gvRxE1csktISMCgQYPw7t07zJ49W+PjExHlJ82fFYmIiIiIiIiIiIgo1/bs2QOZTKbRMQ8dOoSUlBSNjllU1axZUxQ/evRIrf6PHz+W1C773Yh8fHzUWk9hU5S2x9PTU7S/DxkyRPIkPB8fHyQnJ+dXarlWpkwZ6OvrC/HLly/VPqazTp4FABsbG43k9i3g6583Wc+74eHhCA4OVqu/1PNubhQrVkx4nJiYqFbf8PBwSe2qVq0qih8+fKjWelTJvh9l389yoqn9snLlypgwYQIuXLgALy8vVK5cWXguKSkJmzdvlutT2D9XHBwcRPGrV68KKBPpihUrJkzaBj7nnFnoMCUlBXv27BGeMzY2xtChQ3O1Hm9vb7XaBwQEIC4uTohtbGzkiq/kdV9W1Cen/TkwMBDjxo0TLdu5cyfs7e2F+P79+/jpp5/UzkUKOzs74XF8fDxevHiRL+uRKr/OVVpaWqhQoYIQx8XFqV00ozB9hmY9vwHqHw8xMTF49+6dJlMqEHk9ZpOTk0XnVQMDA5QuXVojueUHbW1t1K9fHwsWLMDjx49x4sQJGBoaCs97e3vj0qVLksYKCwsTFbHI/nlT1Njb24vO6er+fh0UFJSr4k7qKlu2LHr06CHELi4uwrkoLCwMp0+fFp6ztbVFp06dchzzW7jGUyWzOCHw+S6fu3btEuLt27eL2k6cOFEj6yxbtiyGDx+O48eP482bN6LiyzKZDOvWrdPIelQxNTVFt27dsHnzZrx79w5jxowRPb9u3TqN/02PiIiIiIiIiIi+DWZmZpDJZHI/7969w19//SX63xIApKWlwdHRUbgxAH3bYmJihH0mJiamoNMpdFxdXUXHFX1bMv92r+7P8uXLJY2vo6OjsH94eDj279+Pli1byvWZM2dOgX9HgIhIXSxKQ0RERERERERERFRIyGQy7N27V7Qs+z9Fpf60b99eGCMqKgpnzpz50ptTKDVp0kQUnz17Vq3+//33n6R25cuXF30h5M2bN0V6AmpR2p4PHz6I4urVq0vue+3aNbXWlf2OKPn1xQV9fX3Ur19fiFNSUuDm5ia5v0wmg6urq2iZpu8g/zXj6583eTnvPn/+HL6+vppOSWBqaio8zn7uUCUjIwOenp6S2rZq1UoUnz9/XvJ6cpJ9P1L3HJa9vSb2y9q1a2Pbtm2iZYqOFycnJ5W5FDQrKyuUL19eiNUtPFFQsk4QB/5vUvjp06dFhZQGDx4MExOTXK3j6dOniIqKktz+xo0bojj7OQH4XAAga0GKgIAA+Pn5SV7Hx48fRcUAdHV10ahRI6Xt09LSMHToUFFhiHHjxmHs2LE4cuSIqGDVqlWr4OLiIjkXqfLz3JAb39q5KrdsbGxgZWUlxHk9HjQt67Vpfn6hNvt7kPk7q1S3bt1CamqqEDdo0EBUALCw69evn9ydC6VeGz59+lQUqzpXFQUlSpQQ/Z7o4+OjViE3qb9fa8LkyZOFxzKZDDt37gTwuTBw1v1x3LhxcsXTFPnWz5s9evRAuXLlhHjXrl3IyMhASEgIzp07Jyy3s7ND27ZtNb5+GxsbHDx4ULRMnd/RNMHY2Bjbtm2Dra2tsCw0NLTQFVskIiIiIiIiIqKirXz58pg+fTrevHmDuXPnip6Lj4+X+3s1EREVDpaWlhg2bBjc3Nxw+vRpuf8/Zb3pEBFRUcCiNERERERERERERESFxM2bN/H27VshLlu2rNxEJ6mGDBkiinfv3p2X1L4ajRs3Fk16vnfvHjw8PCT1ffDgAdzd3SWvq0uXLqJ406ZNkvsWRkVle7JPiE1JSZHc7++//1ZrXcbGxqI4ISFBrf7qaNOmjShW55h2cXERFREqU6YMqlWrpqnUvgl8/XOvV69eoviff/6RfLe2/D7P2NjYCI8DAwMlFxa4cOEC4uLiJLVt3749dHV1RX3fvHmjXqJKZN8v9+/fj7S0NEl9o6OjcerUKdGy1q1baySv7Hd5ioiIkGvToUMH0ety+PBhREZGamT9mpL1GiwuLk5j71t+qlGjhmi/OH78OKKjo7Fjxw5RuwkTJuR6HampqTh8+LDk9nv27BHF2fdbZcvVOc8eOnQIiYmJQtyoUSMYGRkpbb948WLcvn1biGvWrIm//voLAFCnTh2sXbtWeE4mk2HEiBFqFa6SIvt11d9//y0qivClZc9n3759GruLYl7e21evXoneKwMDAzRt2lQjeeVW1u1R93jI79/Jsl6b5ud1afXq1eUKSV2/fl1y/127doliZeeFwkzKZ50i2X/31NRnb0HKfq23efNmSf3S09Pxzz//5EdKCnXq1AmVKlUSYmdnZ6SlpQnFaYDPd7IcM2aMpPG+xWu8rHR0dDB+/HghDggIwOXLl7F7925RruPHj5dU5Cc37OzsROciqcehJunq6sp9LhVEHkRERERERERE9G1YuXKl6IY6ANT6PwURERWMXr16YfTo0aJljx49KphkiIhyiUVpiIiIiIiIiIiIiAqJ7JN2Bw0aJLrbvTr69esnutv8xYsXERYWlqf8vgZ6enpyk8ymTJmCpKQklf0SExNFd1aXYtasWaJJahs3boSnp6daYxQmRWV7rK2tRbHUu6X//fffePz4sVrrsrCwEMVZi0pp2rhx40ST+Q4cOCCpoFJ6ejrmzZsnWpZ18iBJw9c/97p27Yry5csL8ePHj7Fu3boc+92+fRvbtm3Lz9TQoEED4bFMJsOxY8dy7JOamorFixdLXkfp0qUxePBgIc7IyMC4ceMkF+ZRpVOnTqhYsaIQv337VnIhn0WLFokKFrRq1Qo1atTIc06A/ETcEiVKyLUpXbo0RowYIcTx8fGYOnWqRtavKZ07dxbFN2/eLKBM1JP1eiUpKQm//fYbXFxchGV16tTJc1GP3377DR8/fsyx3aVLl3Dt2jUhNjQ0lCucmCn7uXHt2rUICgrKcR1xcXFYsmSJaJmqojtXr17FH3/8IcTFihXD4cOHRUVspkyZgv79+wvxhw8fMGLECLnCd3nRu3dv2NraCrGfn5/cdnxJDRo0gKOjoxDHxcVp7JgcMmSIqFjKrVu38O+//0rqm/3unt999x3MzMw0kldujR07VhRLPR7c3Nwkb3duZb02jYyMlJRXbmhpaWHcuHGiZXPnzkV6enqOfR88eCD6gryWllaRvDaS8lmnyI0bN4THurq6aN++vUbzKgjZz7l///037t27l2O/VatW4enTp/mVlhwtLS1MnDhRiIODgzF//ny8evVKWNatWzeUK1dO0njf4jVedhMmTICOjo4Qb9u2TVQIT09PT+4L1pqUkpIiKhQp9TjUtNyeD4iIiIiIiIiIiHJj+fLlojg0NLSAMiEiInVkvTkOAKSlpcHb27uAsiEiUh+L0hAREREREREREREVAgkJCTh+/LhoWdYJTuoqUaKEaDJ1Wloa9u/fn+vxviazZs0STdh0d3dH7969lRbt+fDhA3r27IlHjx6pdYfvypUriyZrJiYmokePHrh7965a+V67dk00ea6gFJXtadiwoagg08mTJ3Hnzh2Vfc6ePYsff/xR7XXVqlVLFJ84cULtMaSqVq0aevToIcQZGRno37+/ygn7MpkM48ePh5eXl7DM2NhY7QJLxNc/L3R0dOQKLcydOxfr169XWuDhypUr6NGjB9LT09U676qrW7duonjZsmUIDw9X2j4tLQ0TJ06UVJAoqwULFsDAwECIb926hcGDByM5OVlS/4cPHyI2NlZuuY6ODn744QfRsvnz5+Pq1asqx9u1axe2bNkiWpa98EOmDRs2YPPmzaLJzTlZtWqVKG7YsKHCdj///LOoEMiRI0cwadIkpKSkSF5XVFQUfvvtN5w5c0ZyH6m6dOkimmh9/fp1ja8jP/Tr1w+lSpUS4rVr14qONVUFW6QKCQnB4MGDkZqaqrSNj48PRo4cKVo2atQomJubK2zfoUMHODg4CHF8fDx69+6tcN/PlJKSgoEDByIkJERYVqZMGQwdOlRh+7CwMAwfPlxUMGDdunWoU6eOXNsdO3bAxsZGiF1cXLBy5UqluahLR0cHP//8s2jZ77//jjVr1kjqL5PJNL5PLl68WHTOPXjwIGbOnCm5GI+yc4+5ublcIZexY8fiyZMnOeZz7tw5IdbW1sasWbMk5ZKfOnXqBHt7eyGWcjwEBATk6Xc7qbJem8pksny9Np0yZQoMDQ2F2NPTE5MnT1a5vwQGBmLAgAGiNr1790aVKlXyLU8ppk6dijNnzkje15OTk/HXX3+Jlin7rMsqLS0Nt2/fFuKWLVsWeJElTahevTqGDx8uxGlpaejevbuoKFlWGRkZWLNmDRYuXAgA+Xqtl93YsWOhp6cnxNnPuep+RhblazxNKFeunOh3pFOnTsHPz0+Ie/bsidKlS0sa6+DBg/j9998RHR0tef2bN29GYmKiEEs5DlV58eIFpkyZAh8fH8l93N3d4erqKsTm5uaoXLlynvIgIiIiIiIiIiJSpWvXrnLL1C1Mc/ToUcyYMQN9+vRBly5dsGjRIpXtExISsGHDBowZMwZdu3ZF7969MWXKFI3+H+LmzZv48ccfMXjwYHTs2BG9e/fG1KlTcfToUaSlpeVqzICAACxZsgQjR45E586d0b9/f8yYMUP0t3p1ZWRk4NSpU5g6dSr69OmDDh06oFu3bhg6dCiWLl0q+nuhVHfv3sWCBQswYMAAdOrUCV26dMGAAQMwb948HD9+XK3/n+dVUFAQFi1ahMGDB6NLly4YNWoU9u/fn+eC7Pv378fYsWPRpUsXDBkyBIsWLUJAQICGsv5ykpKSsHbtWowePRqdO3fGkCFDsHLlSlEB9aIkP7cnP46/os7c3BzFihUTLXv58mUBZUNEpD4WpSEiIiIiIiIiIiIqBI4fPy66k32VKlXQpEmTPI2ZfeLjnj178jTe18LKygobNmwQLbt8+TKqV6+OKVOm4MCBA7hw4QL279+PyZMno3r16sLkr0mTJqm1rnXr1qF+/fpCHBISgtatW2PMmDG4d++ewi9OfPr0CW5ubvjpp59Qo0YNtG/fHpcvX87FlmpeUdgeY2Nj9O/fX4jT09PRtWtXbNu2DUlJSaK2b968wffff4/evXsjOTkZVlZWKFmypOR11a9fH5aWlkLs6uqKdu3a4Z9//sH58+dx5coV0U9ebdmyRTSZPyAgAPXr18euXbsQHx8vanvv3j20bdsWu3fvFi1fvXo1ypYtm+dcvkV8/XNv3Lhx6NChgxDLZDLMmjULdevWxYoVK3Dy5EmcOXMGmzZtQufOndGxY0fExMTAyMhINMlZ07p164Zy5coJ8fv379GmTRu4urqKJqanpaXBxcUFjo6OwntaqVIlyeupWbMmVq9eLVp27Ngx1KpVC3v27FE4+TYoKAg7duyAk5MTGjdurHSC7g8//ICmTZsKcXJyMrp27YqffvpJVKgDAPz8/DBp0iSMHz9etH2DBg1C7969FY7/9u1bTJs2DWXLlsWoUaNw6tQpuXEzPX78GIMHDxZN1NfW1pYrRpGpcuXK2Llzp2jZtm3bUKdOHWzfvh0fPnyQ6yOTyeDr64t9+/ahX79+KF++PBYtWoTIyEiF68iL0qVLo127dkJ8/vx5lUUnCgt9fX2MGTNG4XPFihXL8zGVWajl/PnzaN68OW7cuCHan+Lj47F9+3Y0adJEVPCvdOnSWLFihdJxtbS04OzsLCoW4OnpiXr16uHEiROiL1tmZGTAxcUFjRo1krum2LVrl9wXuYDP+87IkSNFX8rt37+/0kJh5ubmOHjwIHR1dYVlP//8M+7fv690G9Q1btw4DBgwQJTjnDlz0K5dO1y+fFmuqEFGRgaePXuGFStWwN7eXun7nFsdOnSQK9K3YcMGNGzYECdPnpT7rAM+Fx/asGEDGjVqJDrPZ7d8+XLY2toKcXR0NFq0aIE1a9bInd+8vb3Rv39/LFu2TLR87ty5ouvQgqKtrY2tW7eKCmlkHg/ZPz8SEhLg7OyMRo0aITg4GLq6uqLPHU3r1KmTKJ4yZQqmT5+OQ4cO4dKlS6Lr0ufPn+dpXeXLl5cr1LRjxw60a9cO9+7dEy2Pj4/Hzp070bBhQ7x7905YbmFhgc2bN+cpD024ffs2evXqhUqVKmHOnDlwdXVV+GXb1NRUXLx4ES1btoS7u7uw3NraWlSYQxlXV1dRARJlBbSKovXr14uKj0RFRaF9+/bo0qULNm/ejDNnzuDEiRNYvnw56tatizlz5kAmk6FOnTpo3rz5F8vTysoKffv2VfhcuXLl5AoW5qQoX+Npiqq/kahT5CcsLAwLFy5E+fLlMWDAABw6dEjphITXr19j2rRpcgV38lp4Lzk5Gf/88w+qV6+ONm3aYOPGjfD29kZ6erpc2/DwcKxduxbt27cXPT9q1ChRkVwiIiIiIiIiIqL8oK0tnhKc/WY6dnZ20NLSgpaWluh/XaNHj4aBgQEGDRqEjRs34vTp07h06ZLc3zkzeXp6wt7eHsbGxpg5cyZ2796Nixcv4r///sM///yDAQMGQFdXFz169MhVEYuIiAj06NEDenp6aNOmDdatW4cjR47gypUr+O+//7BlyxYMGjQI+vr6sLOzk3wztlWrVqFEiRKwtbXF0qVLsW/fPly+fBknT57Exo0b4ejoCENDQ8yaNUtysZW4uDh07twZ+vr66NevH7Zs2YLTp0/j6tWruHDhAg4dOoQlS5bAyckJ2traqFq1qtLi7ZlmzZoFY2NjtGjRAn/88QdOnDgBFxcXXLp0CSdOnMCqVaswcOBAGBgYwMrKSu7mMFmZm5sL77mym4QAgJubm9BOS0sL48ePBwD4+vqiWrVqqFChAn777TccOXIEly5dwt69ezFixAgYGBjk6mZf06dPh56eHkaMGAFnZ2dcunQJhw8fxm+//QZbW1tUrlwZL168AAC0bdtWlJsmqTO2rq6u0M7Ozg7A55uldOzYEcbGxpg9ezb27NmDy5cv4/Dhw/jf//4HMzMzdOjQQa2b/aiTc9a/Q/v6+oq2JeuPqvc+q/zcnvw4/r4mWb8PAUDljbOIiAob3ZybEBEREREREREREVF+y14wZtCgQXkes3fv3jAyMhL+QfjkyRM8evSoUEzmLGjDhw+Hv7+/6G4/MTEx+Oeff/DPP/8o7NOnTx/MnTtX9HzWL28oYmhoiP/++w/dunXD06dPAXwubrB7927s3r0bxsbGqFChAszMzJCQkIDo6GgEBweLJpIVJkVle3799VecO3dO+NJLXFwcJk2ahB9++AHVqlWDgYEBQkJCRF/M0dHRwe7duzFlyhTJxQ309PQwY8YM/PLLL8Ky69ev4/r16wrb5/V1KF++PPbv34/+/fsLE9UjIiIwbtw4TJs2DZUqVYKhoSHevXsnKgSQaezYsUon31PO+PrnzbFjx9ChQwd4eHgIy54+fSqcS7LLPCafPXsmWp7TeVcdurq62LBhg6goxIsXL+Dk5AQrKytUrFgRycnJ8Pf3FxWOmz9/PkJCQvD27VvJ65o2bRr8/Pywbt06YZmvry9Gjx4NHR0d2NjYoGTJkkhOTkZISIjkL57o6Ojg4MGDcHJyQmBgIIDPk+ZXrFiBP/74A5UqVYKFhQXCw8Ph7+8v179Bgwb4+++/c1xPbGws9u7di7179wIASpUqBSsrK5iYmCApKQn+/v6IiYmR6zdv3jw0bNhQ6biDBw/G+/fvMXfuXOELR69fv8bEiRMxceJEVKhQAZaWltDV1UVMTAxCQ0NF70V+GzFiBFxcXAB8LqJx/fp1uaIPhdHEiROxatUquc+dAQMGoESJEnkae+TIkbh37x5cXFzg4eGBtm3bomTJkrCxsUFSUhLevn2LxMREUR8DAwPs378/x3U3aNAAmzdvxuTJk4X9wd/fHwMGDICJiQlsbW2ho6ODgIAAhZP4ly5dii5duigce9WqVbh06ZIQ29jYYMeOHSrzadGiBZYuXYqffvoJwOdrnsGDB+Px48cwMzNT2VeqnTt3Ijw8HDdu3BCWZV5LGBkZoUKFCjA3N8fHjx8RGBiIT58+ibZB0/744w+8e/cOR48eFZY9evQI/fv3h76+PmxsbGBhYYH4+HgEBQUpPO4VMTExwbFjx9CpUyfhvYuPj8ecOXOwYMECVKpUCaampggJCUFwcLBc/y5dusgVqSlIrVq1wtKlS0XXgB4eHnBychKOh+TkZPj5+YmOhyVLlsDFxUXhNmpCjx49UL16dbx69QrA57sbbtq0CZs2bZJrO2rUKLnieeqaOnUqHjx4gH379gnLXF1d0bx5c5QqVQoVKlRQel4wNDTEwYMHC1WxvoCAAKxZswZr1qyBlpYWypUrh5IlS8LQ0BBxcXHw8/OTK3Kpo6ODHTt2wNDQMMfxT506JTw2MDDAwIEDNb4NBaVkyZK4dOkS2rVrh6ioKGH5pUuXROferCwtLXH8+HFMnDhRWKajo5PvuU6ePFl0jss0ZsyYXK2/qF/j5VXnzp1ha2srl0PFihVzdc2UkJCAEydOCHdYNjc3h7W1NczNzZGSkoJ3794pfA0HDx6MPn365GYT5GRkZODmzZu4efMmgM/nq3LlygnXMWFhYQgMDJS7zqpatSp+/fVXjeRARERERERERESkSvZCDuXLl1fZ/tOnT6hevTrev38veR0LFy7E77//nmO79PR0nDt3DqVKlYKLiwtat24tafxt27ZhypQpkopSZN64ZO7cuSpvwBEREYF69epJ+j9MUlIS1q9fjyNHjuDJkyeiG1Jl5+3tjcaNG8v9j0BVvj4+Pvj3339FN0HJuu6aNWuq9X2D8PBwODs7Y+7cuZL7SHXo0CGMGDFCYYHuTGlpaVi3bh2ePn0q/P9clZSUFNjb28PPz09lu7dv36JOnTo4ePCg2nl/KW/fvkW9evVyLLx09epV4e/lRkZGXyg79eXX9uTX8fe1yXozHuDzzS+IiIoK7ZybEBEREREREREREVF+CgwMlCtiMWTIkDyPa2xsLHfH9uzFb75lP//8M5ydnXOcIK2lpYWpU6fi6NGjcncAkTIhuXz58rh79y6GDx8ud7eV+Ph4vHz5Evfv38fTp08RFBSksHBJxYoVJWzRl1EUtqdKlSo4duwYihcvLlqelJSEJ0+ewN3dXVSQplixYjhw4AC6du2q9roWLlyo8ksvmta9e3dcunQJVlZWouWJiYl4/vw5PDw85Aqi6OjoYMGCBdi5c+cXy/Nrxdc/98zNzXHlyhWMGjUqx7aWlpb4999/MXDgQFEBBkDaeVcd/fv3V1joICwsDA8fPsTTp09FRVDmzJmDFStW5Gpda9euxebNm+W+sJOeng4/Pz+4u7vjyZMnat8JqXLlyrh9+zYaNGggWp6RkQFfX1+4u7srnKzctWtXuLq65qpISXh4OJ49e4Z79+7h8ePHcoUpdHR08Msvv0j6ouKPP/6I8+fPo0yZMnLPvXv3Do8ePYK7uzvevHmjsCBN5p3h8sPAgQNFX4DKWnihMKtSpQo6dOggt3zChAl5HltbWxvHjh2Do6OjsCwyMhKenp54/vy5XOEJExMTHD9+XGE+ikyYMAGHDh2CiYmJaPnHjx/x9OlTPH78WK4gjYGBATZt2iQqEJLV/fv38fPPPwuxrq4uDh06JOlOcfPnz0f79u2F2N/fX1RAIa9MTU1x+fJljB8/Xu66KiEhAa9evcL9+/fx/PlzufNhftDV1cXhw4fx888/y92lLSUlBW/evMH9+/fh7e0tuSBNpkaNGuHmzZuoXLmyaHlqaipev36Nhw8fKvyS4ujRo/Hff/9BX19f7e3JT4sWLcIvv/wi975lHg/Pnj0THQ8zZswQChzlF11dXZw4cQJVqlTJ1/Vk0tLSwp49ezB37ly5u7KGh4crPS9YW1vj8uXL6Ny58xfJMzdkMhmCgoLg5eWFe/fu4fnz53JfNi9RogROnDiB7t275zheSkqKqBDK4MGD81wkrLCpW7cubt++jRYtWkhq6+bmhmrVqonObZq+zlPEyckJ1apVEy3T0tLCuHHjcj3m13SNpy5tbW2F1xfjxo2TOy/kRkxMDF6+fIl79+7B09NT4Ws4YcKEfL1GS0xMhI+PD9zd3eHu7o6AgAC5v3M0b94ct27dkrt+ISIiIiIiIiIi0rQLFy7ILcupqEGzZs2EgjRaWlqoWLEimjVrhsaNG6NMmTJyN4aZPXu2wv/zVqhQAc2bN0fDhg1hYWEhei4lJQVOTk5wc3PLcRsWLVqESZMmyRWkMTY2Rq1atdCqVSs0btwYNjY2cv+vUiY0NBS2trZy/2syNTVFnTp10Lp1azRq1Eiu+EVISAiqVasmVygiq1atWin8H4GDgwNat26Nli1bok6dOrCwsJD7v5EinTt3litIY2RkhBo1aqBly5Zo3bo1GjRooPC90bTAwEAMHz5cKEhjYmKCevXqoXXr1qhRo4ZcMfcrV65g7dq1OY7r4OAgV5BGV1cXNWrUQKtWrVCnTh2h4H96ejqGDRv2RW9OI1V6ejoaNmwoFHDR19cXtqF+/fpy/xcIDw9Ht27dNLLu3PyNPac++bU9+Xn8fU0iIiKEm6BlyvpdBCKiwo5FaYiIiIiIiIiIiIgK2N69e0UTWmrVqoXatWtrZOzsxW0OHjyI1NRUjYz9NRg9ejRev36NdevWoXXr1ihbtiz09PRQvHhxODg4YMaMGfDy8sKmTZugp6cnuus7IH3SnLGxMfbt24fHjx9jyJAhkiZB16hRAz/88APu3Lkj3KG7sCgK29OpUye4u7ujZ8+eStvo6upiwIAB8PLywqBBg3K1Hh0dHezbtw9ubm6YOnUqmjRpAktLSxgYGOQ29Ry1adMGPj4+WLhwIcqWLau0XbFixdCnTx88evQo10U0SB5f/9wzNzfH7t274e7ujh9++AG1a9dGiRIloKurCysrK7Rr1w7r1q2Dr6+vUFQtt+dddSxatAhnzpxBrVq1lLapW7cuzp8/j1WrVkn6Ipky33//PXx9fTFjxgyULl1aZVtzc3MMGTJEuAOVKuXLl4e7uzt27Nihcju0tLTQtGlTnD59GufPn89x4uyyZctw+PBhDB8+HBUqVFDZFgCKFy+O4cOH49GjR1i6dGmO7TN17twZfn5++Ouvv+Dg4JDja1y8eHF0794df//9N0JCQjT2xa7sihUrhkmTJgnxiRMn1C7EUVDGjh0riqtXry75zoQ5MTMzw7Vr1/D7778rvWuZvr4+vvvuOzx//lyuSGJOvvvuO/j4+GDq1KlyX2jNysTEBCNHjsTLly8xdepUhW1iY2MxZMgQ0fXvsmXL0Lx5c0m5aGtrY//+/aLCR0ePHsW2bdskbk3O9PX1sX37dnh6eqJfv34wNjZW2d7GxgY//PADLl68qLEcstLS0sKvv/6KFy9eYPTo0Tle55UuXRrjx4/HgwcPchy7du3aeP78OVatWiVXnCYrXV1dtG/fHrdu3YKzs7PkLxx/aUuXLsWtW7dU7k/29vY4ceIENmzY8EVyqlWrFp48eQJnZ2cMGDAA1apVg5mZmdyXhjVFS0sLK1euhKenJ3r16qXyGrhs2bJYtGgR3rx5IypsVdDOnDmDTZs2oXv37pJ+rylbtizmzp2L169fo3fv3pLW8e+//yIiIkKIf/jhh9ymW6jVqFEDbm5uOH36NIYOHQo7OzsYGxtDX18fFStWRL9+/XD48GF4eHigevXqAMTXel+iKA0AjBkzRhR37Ngxx2utnBS1azxNyv566ujoyF2H5GTSpEk4ffo0xo8fDzs7uxzbGxgYoG/fvrh16xa2bdumkYkZDg4OuH37Nv73v/+hYcOGksZs0aIF9u7di9u3b+f4vhMREREREREREWlC1hsxAMjx71Lp6el49uwZgM9Fu2NiYhAQEIC7d+/iwYMHeP/+PUJCQoT2d+/elSs6UqNGDQQHByMwMBB37tzBw4cPERkZicuXL4v+r5WRkYFu3bqpLDBx4cIF/Pbbb6Jl1tbWuHr1Kj59+gRvb2/cvHkTDx48gL+/P1JSUnDx4kU0b95c7sZUWTVt2hTx8fFCXKVKFbi7uyM2NhZPnjzBjRs34O7ujvDwcHh5eaFcuXJC2+joaHTp0kXhuBs3bhT9f7hUqVLw9vZGVFQUvLy8cOPGDbi5ueHJkyeIjIxESkoKtm/fjho1aij8O21UVJToO1N6eno4fPgw4uPj8eLFC7i5ueHGjRvw8PDA+/fvkZqaisuXL6Nt27b58jdIFxcXZGRkwMjICCdPnkRcXBwePXqEGzdu4MWLF4iLi0Pjxo1FfZYsWaJyzEWLFuHVq1eiZWPGjEFqaipevHiBmzdv4smTJ0hISMDvv/8ObW1tpKWlwdPTU9Obl2f+/v6Ijo6GlpYWfv75ZyQnJwvb4Onpifj4eMycOVPU58aNG6IbpeXWtWvXIJPJIJPJRP/nq1KlirA8+0/279d8qe3Jr+Pva5P9/4NWVlYwNTUtoGyIiNSnJVN0i1IJGjZsiNDQUFhbW8PDw0PTeX2VvtRrVmdPnXwbO789HfVU7T6ywDX5kAlR0aFVcXbuO8fLV8gl+qYY/9/dz1PW5fzlNqKvmf4sn1z3nVQt/yZaEhUFW19nrVj9vsDyICoc/m9StuzKt/FPAiJltDrkzwRRKnibNm3C9OnThXj37t0YNWqU2uNkZGTA09MTr1+/RkREBOLi4mBkZARzc3NUqVIFNWvWRKlSpTSZer4q7NsTEhKCW7duISgoCAkJCTA1NYWdnR1atGghaeJpYff48WM8e/YMYWFhSE5ORqlSpVChQgU4OjrK3cWGNI+vf/5q1KiR8H81fX19fPz4Efr6+vm2vhcvXuDBgwcICwtDWloaypQpgyZNmqBmzZoaX5dMJsPjx4/x4sULhIeH4+PHjzA2Noa1tTVq1qyJ2rVr57qIQGBgIO7fv48PHz4gLi4OJUqUQJkyZdCiRQtRcQ11BQcH4+XLl3j79i2io6ORnJwMIyMjlCxZErVq1UKdOnU0UpQrPDwc9+/fR2hoKCIjI5GRkQFTU1NYW1vD3t4eVatW/WJFKj58+IDKlSsjISEBALB27VrMmjXri6w7L3755Rf8+uuvQrxq1SrMmTNH7XFcXV3h5OQkxIsXLxZ90TAtLQ13797F06dPER0dDVNTU5QvXx5OTk4a+YxNT0/HgwcP8ObNG4SFhSEjIwOlSpVC5cqV0aJFi0JbrCQvkpOTcffuXQQEBCA8PBwpKSkwMTFBxYoVUbt2bVSpUuWL5pP5Hvj6+iI8PBwJCQkoXrw4ypUrh1q1aqFGjRq5Ltb18uVLPHr0CGFhYUhISEDJkiVRrlw5ODo6frHiFJri5+eHe/fuCV8Qtra2RqNGjVCnTtH9/kZuJCQkwM3NDYGBgYiIiICBgQGsrKxQq1Yt1KtXr6DTy5FMJsPr16/x5s0bBAYGIi4uDunp6TAxMYG1tTUcHBxQrVo1te9O2a5dO1y/fh3A5zseXrlyJT/SL3Iyr1Ey74bbtm1b4XXKTyNHjsS+ffuE+NixYxgwYIDGxi+K13h5ce3aNdGdPLt3746zZ8/maczw8HA8f/4cfn5+iIqKQkJCAoyMjFCiRAnUqFEDdevWzbGIW17Fx8fj2bNn8PX1xYcPHxAfHw9dXV2YmZmhcuXKqF+/fpH6uw0RERERERERUWH15MkTnDx5sqDTUKpfv35wcHDQ+Lhubm5o1aqVEJuZmeV4c4yffvpJ7oY406ZNw8aNG0XL7Ozs4OvrK1o2YMAAHDt2LMe8qlSpAj8/PyGuU6cOnjx5orT9+/fvUaVKFSQlJQnLJk2ahH/++UdhewsLC0RHRwuxg4MDHj16JOnv7hkZGQrbLVy4EL///rsQS/07fMWKFfHu3Tsh9vf3h42NjahN69atcevWLSEOCQmBtbV1jmMrs2bNGtH/bDdv3ozvv/8+1+NlMjc3R2xsLADV+1L2/Q4ADA0N4e/vr/JvzCVLlhQVPLl+/Tratm2rsK2BgYGoMNHChQuxfPlypWOfOnUK/fr1k1uey2nvCrVt2xY3btyQNLauri7S09NFy86dO6fyRjmdOnWCi4uLEI8ZMwa7du3KQ8bKc6pSpQp8fKTP/cnv7cnP4y+3FJ0D1VW2bFkEBwcrfT7r/8d1dHSQlpamcryjR4/K3TBvy5YtmDJlSp7yJCL6kliU5gtiUZqcsSgNkfpYlIYoD1iUhkjAojREuceiNERZsSgNUSYWpfl69e3bF//++68Qe3t7q7xbORER5V5ERATKli2L1NRUAJ8L1Li7uxdwVvQtmjdvHlatWgXg85ejfH19oaurW8BZKZeeng5bW1vhjmX6+voICgrK1cTpnIrSEBGRag8fPhTdRfTOnTto3rx5AWZUeJw+fRp9+vQR4jlz5gift/klJiYGZcuWRWJiIoDPd6EMCgr6KoucfSlDhw7FoUOHhPjff/9F7969CzAjIiIiIiIiIiIqSliU5jNlhUTev3+PkydP4q+//sKbN29EzxkaGiIuLk7u/5bZCzKYmJggLi4ux5yePXuG2rVrC7Guri4+fPgACwsLlf3279+PESNGCLGRkRHi4+Pl2jk7O2Ps2LFCbGxsjIiICBQrVizH3FQxMTHBp0+fAACmpqaIjo6WVOTmxYsXopvTDBw4EEePHhW1sbe3x8uXL4V8M9eTW9OnT8emTZuEODo6WiM3+shLUZrDhw/LFcvIbunSpaL/EX///ffYvHmzXLsNGzZg5syZQlyhQgUEBgbmmH+HDh1w9epV0bLCUpSmb9++OZ6jAgICYGtrK8Q1atTAixcvcp+wipzyWpRG09uTn8dfbmmiKI21tTVCQkKUPi+lKM379+/h4uKCLVu24MGDB6LnvtSNGoiINEm9W/cQEREREREREREREX2j/P39cebMGSG2sLCAvb19AWZERPR1+/vvv4WCNADQsmXLAsyGvmXz5s2DqakpACAwMFA08bowOn/+vFCQBvj8xbLcFKQhIqK8+/PPP4XH3bp1Y0GaLLLfvfdLXOvt27dPKEgDfL67JwvS5F5ERIToy+vlypVD9+7dCzAjIiIiIiIiIiKioik2NhZaWlpyP+XKlcP06dPlCtLo6Ojgxo0bkm6kMXToUEk5bNiwQRR37Ngxx4I0ADB8+HCUKFFCiBMSEvDw4UO5dtn/Jjxnzpw8F6RxcXERFYoZN26cpIIYwOeCM9bW1kJ8584duTbGxsbC44SEBEREROQhW8gVoNFUEY7cMjExybEgDQBMmDBBFD958kRhuwMHDoji2bNnS8oj+75XmGzZsiXHNjY2NjAyMhLi9+8L701uNbk9+X38FRXp6elKz9+jR48WFaTR0tLCqFGjWJCGiIokFqUhIiIiIiIiIiIiom+SOndUSU1NxahRo0R3Dhk1apTkf6QSEX3r1L2LlZeXF1asWCFalvWuaURfkqWlJRYvXizES5cuFRVMKmxWrlwpiqdOnVpAmRARfdsePXqEEydOAAD09PSwZs2aAs4o/6h7rbd161bRXU9Lly6d78VMUlNTsW7dOiHW1tbG5MmT83WdX7v169cjOTlZiCdNmiRpEgwRERERERERERHlXvny5fHs2TM0btxYUvsxY8ZIanf79m1RPH36dMk5derUSRQfPHhQrs3Lly9F8c8//yx5fGX2798vitX9TkG5cuWExx8+fJB7PmsxdZlMhpo1a8LT01PNLP9P//79RfHUqVPltuFLql27tqR2ZcuWhZaWlhDHxsYqbPf69WtRPGXKFEnj16pVS1QEpbAoXry4qHCKKlkLM2X9u3lhountye/jT1PWrVsHmUym1k9ISIjG8zAwMICnpyd2796t8bGJiL4EflueiIiIiIiIiIiIiL5JDRo0wNGjR5GSkqKynZ+fHzp06ICbN28KywwMDPD999/nd4pERF+NAwcOYOTIkfDy8sqx7YkTJ9C2bVskJSUJy9q1awcHB4f8TJFIpenTp8Pe3h4A4Ovri23bthVwRort3r0bbm5uQtyoUSO0atWqADMiIvp2LViwQCjWMm3aNNSoUaOAM8o/s2bNwi+//JLj3T+TkpLw66+/yv0+PXXqVOjp6eVnili+fDnevn0rxP369YOtrW2+rvNr9uLFC6xdu1aIjYyMMGnSpALMiIiIiIiIiIiI6Ouko6MDS0tLODk54cqVK3j37h2qV68uuX/Tpk0ltcv+992OHTtKXkeHDh1EsaLvBSQmJgqPLSwsNFLg2sPDQxTXqVMHWlpakn+y9s96o7JMv/76qyjP8PBwNGzYEJaWlujZsyd27NiBT58+Sc63Xr16sLGxEeK0tDSMGDECRkZGaNWqFX7//XcEBgaq8xLkSfny5SW3zXrjtqzvZVZZXwsjIyPo6+tLHj9rgZLCwszMTHJbAwMD4bGifakw0PT25Pfx97VJTk5Go0aNcO3atYJOhYgoV3hrEiIiIiIiIiIiIiL6Jj1+/BiDBg2Cubk5OnfujMaNG8PGxgbFixfHx48fERgYCFdXV1y4cEHuH5+//fYb7OzsCihzIqKiJy0tDfv27cO+fftQs2ZNtGvXDnXr1oWVlRV0dXURFRWFp0+f4ty5c3j27Jmor4mJCXbs2FFAmRN9pqenh+fPnxd0GiIhISHC8RIWFoYrV65g7969ojbLli0riNSIiAjAxYsXCzqFLyYmJgYbNmzA8uXL4ejoiJYtW6JmzZqwsLBAWloawsPD8eDBA5w+fVrubpf169fH/PnzNZqPn58f/Pz8IJPJ8P79e5w+fRqnTp0SntfR0cHixYs1us6vWVJSklD0Li4uDp6enti0aZNo4sG0adNgZWVVUCkSEREREREREREVaWZmZoiJidH4uFkLieQkOTlZeKyrq6tW0ZjsRdmzb0tQUJAoLlGihOSxVYmNjdXIOACEIvNZmZqa4sCBAxgyZAgyMjKE5ZGRkTh79izOnj2LCRMmwNzcHI0bN8bChQvRtm1bleu5efMmateujY8fPwrLEhMT4ebmBjc3NyxcuBCGhoaoWbMmJv8/9u47Oqpyffv4NamkkJCQQAgk9N6lNwkEUNCDgqBSpBxBBUUpx4YNUMCjSLOh6KEKiCBFBCkiRJCOFJFOCCWEJCSBFEhIef/wZX7s1JkUJsD3sxZrzf3M8zz73jOzJ+eYzDUvvKAhQ4YU2jlm5uHhka912T1WkjFYpESJElbtaU1gyp1izTmYTCbz7ZweH1sr7PMp6uvvbmFvb6/U1FTDWHR0tHbv3q05c+Zo5cqV5vvT0tLUpUsXHTlyxKpwMQAoDgilAQAAAAAAAADc1+Li4vT999/r+++/t2j+yy+/rNGjRxdxVwBw7/r7778tDvfw9PTU8uXLVbly5SLuCrj7rF+/XoMHD87x/l69eqlr1653sCMAwP0uPT1dISEhCgkJsWh+3bp1tXLlSjk6OhZqH/Pnz9f48eNzvH/UqFGqV69eoR7zXhYREZHrtyJXqlRJ77777h3sCAAAAAAAAIAlbg+WyMvNmzfNt+3t7a06jq+vr6FOTEw01BEREYba1dXVqv1zkpSUVCj75ObJJ59UkyZN1LdvX+3Zsyfb8Iy4uDht3LhRGzdulL+/v5YuXao2bdpku19gYKAiIyM1ePBgLV++3PC433L9+nXt27dPQ4cO1SuvvKJJkybplVdeKfRzK0rWhBpJkrOzcxF1gqJyJ66/u5WPj4+6deumbt26KTIyUjVq1DCH+KSlpSkoKEiXLl2ycZcAYB3Low4BAAAAAAAAALiHlC9f3qr5AQEBmjNnjmbMmGHVNwkBAP75gwtr/4ioS5cu2rFjh4KDg4uoK+De1aJFC3377be2bgMAcJ/w9/e36sMNjo6Oev7557V9+3YFBgYWYWdZde/eXZMmTbqjx7yXlSlTRqtXr5abm5utWwEAAAAAAABQALeHh6elpVm1NioqylBn/u+F/v7+hrqwwiycnJwMdWxsrDIyMvL9LydVq1bVrl27lJCQoGnTpikoKEilS5fOdm54eLjatWuX65ejlShRQosXL1ZKSop++OEH9ejRQwEBAdn+PVpSUpJGjhyZ65eVFBe3/54gOTnZqrXx8fGF3Q6K2J26/u52ZcqU0Z49ewzXd0REBGH/AO46/NU8AAAAAAAAAOC+dP78eW3fvl0ffPCBunfvrrp168rLy0uOjo5ydnaWn5+fGjRooOeee07ff/+9Tp06pUGDBtm6bQC4Kz366KOKjIzU0qVL9fLLLysoKEgVK1aUu7u77O3tVbJkSVWsWFHt27fXu+++qz179mj9+vWqXbu2rVsH7gomk0menp5q06aNPv30U/3+++/y8PCwdVsAgPvEpEmTFBYWpq+//lqDBg1SixYt5OfnJxcXF9nb28vLy0vVqlVT9+7dNXXqVJ06dUqzZs2Sp6dnkfdmZ2cnb29vBQcHa8GCBVq5cqXhwxWwnqurq+rVq6c33nhDf/31l+rXr2/rlgAAAAAAAAAU0O1fMpOamqr09HSL1x47dsxQlypVylBnDqWJjY21vsFsZP596MmTJwtl35y4urpq5MiR+u233xQdHa2bN2/qf//7n1q0aGEIZMnIyNCAAQOUkpKS5569evXSjz/+qHPnziktLU2bNm3SY489luVLf+bOnastW7YU9ikVqtv/23tCQoJVayMjIwu7HRSxO3393c2qV6+u4cOHG8Y++ugjpaam2qgjALCeg60bAAAAAAAAAADAFkwmk1q3bq3WrVvbuhUAuC94eHiod+/e6t27t61bAe4JgwYNKvLAvKCgoHv6W8kAAIUnICBAQ4cO1dChQ23disaNG6dx48bZuo17RqVKlfjfAwAAAAAAAMA9zt/fX3FxceZ6/fr16tq1q0VrN23aZKgbNmyYZY6Li4uuX78uSYqJiVFqaqocHAr28eZatWrpxIkT5nrFihVq1qxZgfa0hoODgwYPHqzBgwfr3Llzqlmzpm7cuCFJSklJ0WeffabRo0dbtWdwcLCCg4OVmpqqOnXqGII+3nvvPW3durVQz6Ew+fn56dy5c5KktLQ0HTlyRHXr1s1zXXp6OqE0dyFbX393mxkzZujbb781vw8mJydr9OjRmjlzpo07AwDL2Nm6AQAAAAAAAAAAAAAAAAAAAAAAAAAAAAB3Xrt27Qz1p59+avHajRs3Guq+fftmmVOnTh1DPWHCBCu6y16/fv0M9Q8//FDgPfMrMDBQr7/+umHs999/z/d+Dg4O2rJli2Hs9gCQ4qh58+aG+r333rNo3cyZM5Wenl4ULd0T7Oz+LwagOD1Oxen6uxvY2dlp1KhRhrGvv/66WD2nAJAbQmkAAAAAAAAAAAAAAAAAAAAAAAAAAACA+9Arr7xiqDdu3KiYmJg81y1evNgwz83NTU2bNs0yb+TIkYZ66tSpSklJyV+z/1+vXr3k7Oxsrk+dOqV169YVaM+CyHzeycnJBdrP399fJpPJXKelpRVov6KWOWhoxYoVioiIyHVNenq6xeE19ysHBwfz7cTERBt2YlTcrr+7wcSJE+Xi4mKuk5OT9eqrr9qwIwCwHKE0AAAAAAAAAAAAAAAAAAAAAAAAAAAAwH2odu3aqlq1qrlOTU1VUFBQrmsiIiL07LPPGsYGDBiQ7dz+/fvL29vbXCcmJqp58+ZKT0+3qL/s5tnZ2WnYsGGGsccff1x//fWXRXvesmzZMh09ejTL+MKFC5WammrxPosXLzbUNWvWNNRr1qzJM6TldmvXrlVGRoa5LlOmjMVrbaF27dqqUaOGuU5PT1fDhg117dq1bOenp6erWbNmOd6Pf5QqVcp8Ozo62qrXZFEq6uvvXjVixAhD/cUXX1j8PggAtkQoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAHCfWrBggaE+fPiw6tSpk22QyubNm1W9enVdv37dPFayZElNnz49x/2XLl1qqA8ePKgKFSpo8+bNOa759ddf1aZNmywBL7d88sknKl++vLlOSUlRw4YNNWLECKWkpOS4b3h4uF588UX5+Piod+/eOnToUJY5L730klxcXPTwww9r48aNOe4lSa+//roWLVpkGBszZoyhnjJlivz9/fXAAw/o66+/zjWIYvny5erRo4dhLKfAn+JkzZo1srP7v4+tR0ZGytfXV8OGDdOhQ4eUmpqq06dP65133pG3t7f2798vSXJ3d7dVy8Ve48aNzbdvBf2EhITYsKP/U5TX371q8uTJKlGihLm+ceOG3n77bRt2BACWcbB1AwAAAAAAAAAAAAAAAAAAAAAAAAAAAABso1WrVho9erSmTp1qHjt69KjKlSungIAABQQEKCUlRaGhobpy5YphrZ2dndauXSsnJ6cc9w8ODtbbb7+tDz74wDx26dIlBQcHy83NTVWqVJGXl5eSk5MVGRmpixcvmoMt/Pz8st3Tzs5O+/fvV9WqVZWQkCDpn+COzz77TJ9//rnKlSuncuXKyd3dXQkJCYqNjdWlS5cMYTq5SU1N1fr167V+/XrZ29urTJkyKlu2rDw9PZWWlqaIiAiFhYXp5s2bhnWPP/64KlSokGW/jIwM/fnnn3r++ef1wgsvyMvLS/7+/vL09JS9vb2uXLmis2fPKjEx0bDOz89Pr732mkU921L16tX15Zdf6vnnnzePpaSkaNasWZo1a1a2a5o0aSJ3d3dt3br1TrV5V5k4caLWrl1rrv/++2+1b98+yzwfHx9FRUXdydaK/PorDKNGjdKoUaOsXle9enWdOHGi0Puxs7PT8OHDDe+zM2bM0KRJkwr9WABQmAilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAO5jn3zyiZycnPThhx8axs+fP6/z589nu8bR0VEbNmxQ27Zt89z//fffV7ly5TRixAilp6ebxxMTE3X48OF89VymTBldvHhRLVu21NGjR83jGRkZCg8PV3h4eJ57uLi45DknLS1Nly5d0qVLl3KdFxQUpBUrVuS5X0ZGhmJiYhQTE5PrvLJly+b7sbGF5557TiVKlNBzzz2n5OTkXOd26dJF69atU8uWLc1jJpOpqFu8qzRq1Egvv/yyZs6cmeu8zMFId8qduv7utNvfnwrbxx9/rM8//9x8fSQlJWncuHEaN25ckR0TAArKztYNAAAAAAAAAAAAAAAAAAAAAAAAAAAAALCtyZMna8+ePapZs2au8+zs7NS1a1dFRkYqKCjI4v2HDx+u8+fPq2PHjrK3t891rslkUq1atfTpp5/mOs/Dw0N///23lixZosDAQIv6cHNzU3BwsLZt26bu3btnuf+rr75S69at5erqatF+5cqV08KFC/Xbb79le/+UKVP02GOPycvLy6L9SpYsqdGjRysiIkI+Pj4WrSkuBgwYoJiYGA0ZMkRlypQxP88mk0kuLi5q0aKFfvnlF61fv152dnaKi4szr83rNXE/mjFjhn777Te1bt1aHh4exS64pyiuv3uZnZ2dnn/+ecPYlClTbNQNAFjGlJGRkZGfhU2aNFFERIT8/Py0b9++wu7rnnSnHrP68+oX2d5F7fBA6xMbM859UgSdAHcPU+CY/C9OXFd4jQB3I7eu5psp06rZsBHA9pxGncr32udrOBdiJ8Dd56sTtye4553kDdzb/M23MjY9bMM+ANszdfrF1i0AAAAAAAAAAAAAAAAAQLF16NAh/fjjj7ZuI0c9e/ZUgwYNbN2GTSUkJOibb77RgQMHFBERIUdHR5UvX16dOnVSr169CuUYq1ev1vr163Xp0iVdu3ZN7u7uqlixooKCgvTYY4/Jzs7O6j1v3LihhQsXavfu3YqMjFRCQoLc3d1VpkwZNW/eXN26dZO/v3/eG/1/kZGRWr16tfbv36+LFy8qMTFRzs7OKl26tJo3b65evXrJz8/Pqv7WrFmjXbt26cyZM4qPj5fJZJKXl5fq1Kmjnj17ql69elaf993K1dVV169flySVLl1a0dHRNu4IBVHY1x8AwPYcbN0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAgOLD3d1dI0eOLNJjdO/eXd27dy/UPUuUKKEhQ4ZoyJAhhbJfmTJlCm0v6Z/+evXqVWjBPnezzZs3mwNpJKlWrVo27AaFobCvPwCA7VkfEQgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQD4NHjzYUA8aNMg2jQAAgBwRSgMAAAAAAAAAAAAAAAAAAAAAAAAAAIA7plSpUrZuIVfFvT+gOFq6dKnWrVuX57z09HS1adNG586dM4+5uLhoyJAhRdkeAADIBwdbNwAAAAAAAAAAAAAAAAAAAAAAAAAAAID7R2BgoP79738rLi7O1q1kUapUKQUGBtq6DeCu8/PPP2v+/Pny9vZWly5d9Oijj6p9+/by9/dXTEyM9uzZo8WLF+uHH37QjRs3DGunTJlio64BAEBuCKUBAAAAAAAAAAAAAAAAAAAAAAAAAADAHRUYGEj4C3APiomJ0ZIlS7RkyRKL5vfo0UPDhw8v4q4AAEB+2Nm6AQAAAAAAAAAAAAAAAAAAAAAAAAAAAADA3cvNzc2q+Q4ODho7dqx+/PHHIuoIAAAUFKE0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAIB8++KLL7Rp0yb16NFDAQEBKlGihEwmk/l+k8kkZ2dn1axZUyNHjlR8fLwmTpxow44BAEBeHGzdAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADg7hYcHKzg4GBbtwEAAAqJna0bAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAUH4TSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADMCKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAJgRSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMCOUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgRigNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMCMUBoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgBmhNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAM0JpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABmhNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMwcbN3A/eKnn35SVFTUHTnW4YGH78hxigtT4BhbtwDcvdy62roDoNhwGnXK1i0Ad62vTiTbugWgGPG3dQNAsWHq9IutWwAAAAAAAAAAAAAAAAAAAAAAAACQT3a2buB+MWXKFKWlpdm6DQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADIFaE0d0hCQoKtWwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAPDnYugEUgXGetu4g/8ZdtXpJRsTsImgEuHuY/Ibmf3Hy9sJrBLgbObcx30yfVduGjQC2Z/fC0Xyvfb6GcyF2Atx9vjqRfFsVbrM+gOLB33wrY0MXG/YB2J6pywZbtwAAAAAAAAAAAAAAAAAAAAAAAJBvhNIAAAAAAAAAAAAAAAAAAAAAAAAAAADgjrp69U8lXT9n6zaycHUJlKdnY1u3AQAAANgcoTQAAAAAAAAAAAAAAAAAAAAAAAAAAAC4Y65e/VN79/WydRs5atpkGcE0AAAAuO/Z2boBAAAAAAAAAAAAAAAAAAAAAAAAAAAA3D+Srp+zdQu5Ku79AQAAAHcCoTQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADNCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAZg62bgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYLlSpUrp6tWrkiRPT0/FxcXZtqFiJigoSFu3bjXXGRkZNuzmznJwcFBaWpokqWrVqjp16pSNO7K9lJQULV26VIcPH9bFixcVFRUlR0dHlS5dWv7+/urcubOCgoJkZ2dn61YNTCaT+Xb79u21ZcsWm/VSrVo1nT59WpJkb2+v1NRUm/UC4M4hlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAoYtu2bVO7du0smuvg4CBvb281aNBA7733ntq2bVvE3QHAvSU9PV2vv/66vvvuO0VEROQaTPThhx9Kkvz8/NSjRw+NHTtWFSpUuFOtAkCxVbyiugAAAAAAAAAAAAAAAAAAAAAAAAAAAID7XGpqqiIjI7Vp0ya1a9dOVapUUWhoqK3bAnCH9OjRQ0FBQQoKCtKbb75p63buOpMnT5a7u7umTJmiS5cu5RpIc7uIiAh9+eWXCggIUNu2bXX69Oki7hQAijcHWzcAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIGehoaGqUaOG9uzZo0aNGtm6HQBF7KefflJaWpok6cKFC5o8ebKNO7p7dO7cWZs2bcr2vtKlS6t06dLy8vKSnZ2doqKiFBsbqytXrmSZu337dnXu3Flnzpwp6pYBoNiys3UDAAAAAAAAAAAAAAAAAAAAAAAAAAAAwP3G09NTGRkZWf6dP39eM2fOVLVq1QzzU1NT1bZtW6Wnp9uoYxQncXFx5tdMXFycrdspdrZs2WK4rnB/qF27dpZAmpIlS+qDDz5QfHy8oqOjdfz4ce3cuVN//PGHTp48qejoaMXHx2vSpEkKCAgwrOW1839OnTplvp5SU1Nt3Q6AO4RQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAKCYqFChgkaMGKGTJ0/q1VdfNdyXmJioMWPG2KgzACi++vXrp2PHjhnGBgwYoGvXrumtt96Su7t7jmvd3d315ptv6ty5c1q2bJlKly5d1O0CwF2BUBoAAAAAAAAAAAAAAAAAAAAAAAAAAACgGProo4/UuHFjw9iSJUts1A0AFE+rVq3SokWLDGNvvvmm5s2bZ/VeTzzxhCIjI9W1a1c5ODgUVosAcFcilAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAopiZOnGioIyIibNQJABRPI0aMMNQNGzbUpEmT8r2fnZ2d1q5dq19++aWgrQHAXY1oLgAAAAAAAAAAAAAAAAAAAAAAAAAAAKCY6tq1a5axiIgI+fn5WbzH0qVLtW3bNp07d043btxQs2bN9P777+c4PykpSbNnz9aBAwcUEREhJycn+fv7q1OnTnriiSfydR6ZhYSEaOXKlQoPD9eVK1fk6uqqChUqqH379urZs6ccHKz/GHRYWJjmzJmjM2fO6PLly3J3d1f58uX11FNPqU2bNvnqMz09XatWrdKmTZt08eJFJSQkyMnJSaVKlVLNmjXVvn17BQUFWbXnjh07tHr1ap08eVLXrl2TnZ2d3N3dVaVKFTVv3lzdu3eXk5NTvvq11oULF/TVV1/p5MmTiouLU9myZdW5c2f17dtXdnZ2+d534cKF2rx5s8LDw+Xl5aVq1appyJAhqlixYiF2X/Ru3LihL774QocOHdKlS5fk7e2txo0b64UXXpCHh4et27NaUZ5PUVx/lggJCdH58+cNYxs2bCiUvatWrWr1mqNHj2ru3LkKCwtTTEyMvL29VblyZQ0aNEg1a9YslL4ssX37di1atEhhYWFydHRUlSpV1KNHD7Vt2/aO9QDg7mfKyMjIyM/CJk2amP8H6759+wq7r3vOrcdLUtE/ZuM8i27vojbuqtVLMiJmF0EjwN3D5Dc0/4uTtxdeI8DdyPn//o9s+qzaNmwEsD27F47me+3zNZwLsRPg7vPVieTbqnCb9QEUD/7mWxkbutiwD8D2TF0K5xdZAAAAAAAAAAAAAAAAAHAvuhSxSn//PdrWbeSoTp2pKuf3WKHvu23bNrVr185ce3p6Ki4uzqK19vb2Sk9PN9d79uxR06ZNzXW1atV0+vRp89zU1FRJ0qBBg7R48WKlpKQY9itRooSuX7+e5Tj79+9Xv379dOzYsVx7efjhh7Vo0SKrQyyio6M1aNAgrV+/3txjdkwmk6pUqaJx48apf//+ee778ccfa9KkSbk+niVKlNALL7ygTz75xKKwlWvXrql379769ddflZaWlutck8mkqlWr6quvvlLHjh1znDdq1Ch9/fXXSkpKyvP4vr6+evXVV/Xqq69me3+pUqV09eo/n8vN7bWU+XX37LPP6ptvvtHp06fVtWtXnTx5Mtt1Dg4OGjFihKZOnZpnr7cbMWKEZs2alePzW7lyZf3888+qXbu2goKCtHXrVvN9+fzYe7as2dvBwcH8HFetWlWnTp1SSkqKHnnkEW3evNlw7d0uODhYq1evlqura5H0bInsnvs7fT5Fcf1ZI/PjVr9+fR06dKhQj2GJjz76SB988IHi4+NznOPh4aEJEybolVdesWhPk8lkvt2+fXtt2bIlzzXz58/XSy+9lGMfpUqV0meffaZ+/fpp+vTpGjVqlPm+adOmaeTIkdmuy+nnDIB7W+G+YwMAAAAAAAAAAAAAAAAAAAAAAAAAAAAoVJlDJCpUqJDr/ISEBJUvX17z5s3LEkiTk7Fjx6pJkya5BtJIUlpamn7++Wf5+voqJCTEor0l6euvv1bZsmX1888/5xlmkJGRodOnT+cYyHJLdHS0KlSooNdeey3PgJ8bN25o+vTpqlChgqKjo3Od+9dff6ls2bLasGFDnoE0t/o9deqUVq5cmeOxq1SpounTp1sUSCNJUVFRmjNnjkVzrbV48WLVrFkzx0AaSUpNTdW0adPUuXNni/ZMSUlR1apV9dlnn+X6/IaGhqp+/fpaunSp1X3fKaGhofL19dWmTZtyDHCRpF9//VWVKlWy+Dm1laI6n6K6/qx18OBBQ/3CCy8U6v55uXHjhqpXr67XX38910Aa6Z+wq5EjR6pu3boWvzdbo2/fvho4cGCufcTFxal///4aPnx4oR8fwL3HwdYNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMjeunXrsoz5+fnluqZly5YKDw+XJJlMJgUEBMjf319paWm6cOFClsCCMWPGaOrUqVn2CQgIUIUKFZSSkqLQ0FDFxMSY70tJSVGHDh20detWtW3bNtd+3nnnHX3wwQdZxt3c3FSpUiV5e3vrxo0bioyMVHh4uG7evJnrfpIUERGhatWqKTEx0TDu4eGhihUrysvLS0lJSTp79qwhBOPSpUuqUaOGIiIi5OTklO3e7dq1040bNwxjXl5eCggIUKlSpZSWlqZr167p4sWLio2NVUZGRq69PvTQQwoNDTWMubq6KjAwUKVLl5a9vb0SEhJ06dIlRUVF5RnaUxDnzp1T//79zeEkJUuWVNWqVeXh4aHIyEidPHnSEMSzadMmTZ06VaNHj8513wYNGujMmTOGMQcHB1WrVk2+vr6Ki4vTqVOndP36daWlpalfv35q0KBB4Z9gAaWlpalJkya6du2aJMnJyUlVqlSRr6+vEhISdPz4cUNoS1RUlLp166YtW7YU+Nh2dnaFvqaozqcorz9rJCUlZQnEefLJJwu8r6XS09NVpUoVXbp0yTDu6Oio6tWrq3Tp0rpy5YpOnjxpeF/7+++/Va1aNZ07d67Qehk2bJgWL15sGDOZTAoMDFT58uWVnJxseB//8ssv1b1790I7PoB7k/U/mQAAAAAAAAAAAAAAAAAAAAAAAAAAAADcEW+//bahLlu2bK7z09LSdOTIEUlShw4dFBcXp7CwMO3YsUO7d+9WeHi4IUBhx44dWQJpatWqpYsXL+rcuXP6448/tHfvXl25ckUbNmyQm5ubeV56erq6deumlJSUHPtZt25dlkAaPz8//frrr0pISNBff/2lkJAQ7d69W2fPnlVKSop++eUXtWrVSu7u7jnu26JFC0MgRtWqVbVnzx5dvXpVhw4d0tatW7Vnzx5FRUXp4MGDKl++vHlubGysHn744Wz3/fTTTw0hF76+vvrrr78UExOjgwcPauvWrdq2bZsOHTqkK1euKCUlRbNnz1atWrVUsmTJLPvFxMQoJCTEXDs6OmrJkiVKTEzU0aNHtW3bNm3dulX79u0zB/Js2LBBQUFBeT7X+bFx40alp6fL1dVVP/74o65du6Y///xTW7du1dGjR3Xt2jU1a9bMsGbcuHG57vnOO+/o+PHjhrHBgwfr5s2bOnr0qEJCQnTo0CElJSVp8uTJsrOzU2pqqvbv31/Yp1dgZ8+eVWxsrEwmk95++20lJyebz2H//v1KTEzUyJEjDWu2bt2qCxcuFPjYmzdvVkZGhjIyMmRvb28er1q1qnk887/bg6Lu5PkU1fVnrc2bNxtqZ2dn+fj4FMrelujVq1eWQJqXX35ZKSkpOnLkiEJCQnTkyBGlpKRo+PDhhnnnz59X3759C6WPvXv3atasWYaxxo0bKzo6WmfPntX27dvN7+ObNm0yv4+vXr26UI4P4N5FKA0AAAAAAAAAAAAAAAAAAAAAAAAAAABQDL311ltZgjt69+5t0dpevXpp8+bN8vDwyHLf7WEv/fv3N9xXv359HT16VP7+/lnWde7cWSdOnFCJEiXMY/Hx8Xr55Zdz7KNfv36GukGDBrp48aI6duyY45qHHnpIf/zxR5agk1vGjh2rc+fOmevg4GCdOnVKTZs2zXZ+gwYNdOHCBQUEBJjHfvvtN4WFhWWZ+8MPPxjqQ4cOqW7dujn26uDgoCFDhujo0aOaOHFilvvnzJljqKdPn66nnnoqx/2kfx7n3377Tb/99luu8/LLxcVFoaGh6tGjR5b7XF1dtXv3bnl7e5vH4uPjtWXLlhz3++ijjwz12LFj9b///S/buW+88YaWLVuWv8bvoDVr1uj999/P9r5p06apc+fOhrF33333TrSVb4V5PkV5/Vnr9j6kf16/d8qFCxe0YsUKw9isWbM0Y8aMbOd//vnn+uSTTwxjixcvVkRERIF7GThwoKFu0qSJ9u/fb7iObwkODtaxY8fk7Oxc4OMCuPcRSgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAUE+Hh4frss89Uo0YNTZo0yXCfi4uLpk2bluceJUuWzBKukp0jR47ozJkz5trBwSHX8BFJ8vf31+zZsw1jCxYsyHbunDlzFBsba67d3Ny0a9cu2dlZ9hHnnOZ9+umn5tseHh7asGGDRfutX7/eUL/66qtZ5kRFRZlvu7m5yc/Pz6K9c3L27FlD3bdv3wLtVxjmzJmjMmXK5Donc9BQTq+nGTNmKCUlxVwHBARkG85zux49eig4ONjCbu+8Hj16qFu3brnOyXwN7NixoyhbKpDCPp+ivP6sdfHiRUN9e2BWUct8jbRo0ULPP/98rmtGjx6thg0bGsZGjBhRoD4iIiL0999/m2t7e3tt2rQp1zUVKlTQ1KlTC3RcAPcHQmkAAAAAAAAAAAAAAAAAAAAAAAAAAACAO+zq1asymUxZ/pUvX14jRozQyZMnDfPt7e21detWOTg45Lm3pcEnM2bMMNSdO3eWt7d3nuv69+8vLy8vc52UlKS9e/dmmXd7eIUk/ec//ylwaMTGjRuVkJBgrp999lmLQ25q165tCJn5448/ssxxc3Mz305KSlJ0dHQBupVKlSplqJcuXVqg/QqqZMmSeuqpp/KcN3ToUEN96NChbOd99913hnrMmDEW9ZH5tVecfPHFF3nOqVixolxdXc11eHh4UbZUIIV5PkV9/Vnr9tArybpQmsDAwGzfgzP/e+6557Jd/9tvvxnqWbNmWXTcL7/80lBv3rzZ4p6z89VXXxnqdu3aZXnfyc7w4cPvaIgPgLsToTQAAAAAAAAAAAAAAAAAAAAAAAAAAABAMVahQgUdOXJEzZo1s2j+4MGDLZq3fft2Qz1ixAiLe+rSpYuhXrRoUZY5x44dM9Rvv/22xfvnZOHChYb63//+t1Xry5cvb759+fLlLPe3adPGfDsjI0N16tTR/v37rezy/zzxxBOG+sUXX8xyDndSvXr1LJrn7+8vk8lkrq9evZrtvBMnThjqYcOGWbR/3bp1DSEoxYW7u7shOCU3twczJScnF1VLBVLY51PU15+1Moev3Lhxw+K16enp+Z6XkpKiuLg4c+3u7q5GjRpZtF+rVq0M4VcxMTFKTU21aG12Mofa9OnTx+K1lr4fALh/EUoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAFCP29vby8fFRhw4dtGnTJp0/f141a9a0eH2LFi0smhceHm6oO3fubPExOnXqZKgPHjyYZc7169fNt729veXg4GDx/jnZt2+foa5fv75MJpPF/25fn5aWlmX/999/39BnVFSUmjRpIh8fH/3rX//SN998o4SEBIv7bdSokSpWrGiuU1NT9cwzz8jV1VXt2rXT5MmTde7cOWseggKpUKGCxXPt7P7vo+i3P5e3u/2xcHV1lZOTk8X73x5QUlx4enpaPNfZ2dl8O7vXUnFQ2OdT1Neftfz9/Q21NaE0BZH5cbj9GrdEYGBgrvtZ4/z584a6e/fuFq9t3Lhxvo8L4P5AKA0AAAAAAAAAAAAAAAAAAAAAAAAAAABwh3l6eiojIyPbf6mpqYqKitLmzZsVHBxs1b63B4nkJTk52XzbwcHBqtCYWrVqGeq4uDhDfeHCBUPt5eVl8d65uXr1aqHsI0kZGRlZxjw8PPTdd99leRyvXLmiNWvWaOjQoSpZsqS8vLzUpUsXbdmyJc/jhISEqGTJkoax69eva9u2bRo7dqwqVqwoV1dXNW3aVN98802BzikvHh4e+VqX3WMlGYNFSpQoYdWe1gSm3CnWnIPJZDLfzunxsbXCPp+ivv6slTncJSkpyeK1Fy5cyPb999lnn81zbeYgmDJlylh83OzmZ97PGomJiYbaz8/P4rWZQ30AIDNCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAIB7xO3BEnm5efOm+ba9vb1Vx/H19TXUmYMRIiIiDLWrq6tV++fEmtCJ/HryySd14sQJNW/ePMfHMy4uThs3blSHDh1Uvnx5bd++Pcf9AgMDFRkZqaefflqOjo7Zzrl+/br27dunoUOHys3NTTNmzCiUc7mTrAk1kiRnZ+ci6gRF5U5cf9bo2LGjoU5OTlZ0dHSRHzfzMdzc3Kxa7+7ubqijoqLy3Utqamq+1xbW+zKAexehNAAAAAAAAAAAAAAAAAAAAAAAAAAAAMB96PaAlLS0NKvWZg5RyBzK4O/vb6gLK8zCycnJUMfGxiojIyPf/3JStWpV7dq1SwkJCZo2bZqCgoJUunTpbOeGh4erXbt2+v7773Pcr0SJElq8eLFSUlL0ww8/qEePHgoICJCdXdaPeyclJWnkyJEaPHiwhY+K7dwe2pOcnGzV2vj4+MJuB0XsTl1/lnJ3d5enp6dhbMmSJQXeNy8+Pj6GOnMoV14SEhIMdeaQL2u4uLgYamtCau5EgA+AuxuhNAAAAAAAAAAAAAAAAAAAAAAAAAAAAMB9yNnZ2Xw7NTVV6enpFq89duyYoS5VqpShzhxKExsba32D2fDw8DDUJ0+eLJR9c+Lq6qqRI0fqt99+U3R0tG7evKn//e9/atGihSGQJSMjQwMGDFBKSkqee/bq1Us//vijzp07p7S0NG3atEmPPfaY4fmQpLlz52rLli2FfUqF6vZgo8xBG3mJjIws7HZQxO709WeJhg0bGuqvvvqqyI8ZEBBgqK19LWeen3k/a2R+Tnbu3Gnx2hMnTuT7uADuD4TSAAAAAAAAAAAMKlWqJJPJJJPJpEqVKtm6HaDYOXv2rPkaMZlMGjRokK1bAoA7Zu7cuYb3wLlz59q6JQAAAAAAAAAAAABAAWQOjlm/fr3Fazdt2mSoMwdDSJKLi4v5dkxMjFJTU63sMKtatWoZ6hUrVhR4T2s4ODho8ODB2rlzp86ePasSJUqY70tJSdFnn31m9Z7BwcFauXKlEhISVL16dcN97733XoF7Lkp+fn7m22lpaTpy5IhF69LT0wmluQvZ+vrLzvjx4w31X3/9VeSvrSZNmhjqsLAwq9afO3cu1/2s0bhxY0P9448/Wrz2zz//zPdxAdwfCKUBAAAAAAAAgLvQ5cuXtWHDBs2ePVsff/yxPvjgA02fPl1z587Vzz//rAsXLti6RQAAAAAAAAAAAAAAAABAMdeuXTtD/emnn1q8duPGjYa6b9++WebUqVPHUE+YMMGK7rLXr18/Q/3DDz8UeM/8CgwM1Ouvv24Y+/333/O9n4ODg7Zs2WIYO3HiRL73uxOaN29uqC0N0Zk5c6bS09OLoqV7gp3d/8UAFKfHqThdf7cEBQWpQoUKhrHOnTsX6TGdnJzk5eVlrhMSEnTgwAGL1u7atUuJiYnm2tvbWw4ODvnupVevXob6+++/t2hdZGRklnAcAMiMUBoAAAAAAAAAuEucPHlSr732mqpXry4/Pz899NBDeu655/Taa6/pnXfe0ahRozR48GA9+uijCggIUJkyZdSrVy/98MMPun79uq3bBwAAAAAAAAAAAAAAAAAUM6+88oqh3rhxo2JiYvJct3jxYsM8Nzc3NW3aNMu8kSNHGuqpU6cqJSUlf83+f7169ZKzs7O5PnXqlNatW1egPQsi83knJycXaD9/f3+ZTCZznZaWVqD9ilrmoKEVK1YoIiIi1zXp6ekWh9fcr24PKbk9wMTWitv1d8v06dMN9aFDhzR27NgiPWaHDh0M9QsvvGDRuuHDhxvqggbo9OjRQy4uLuY6PDxcCxcuzHNdz549C3RcAPcHQmkAAAAAAAAAoJi7ePGi+vXrp1q1aunjjz/WqVOnLFoXFRWl5cuX68knn1TZsmX1zjvvKC4urmibBf6/s2fPymQymf8NGjTI1i0hn7Zs2WJ4LseNG2frlrI1d+5cQ59z5861dUsAAAAAAAAAAAAAAADFXu3atVW1alVznZqaqqCgoFzXRERE6NlnnzWMDRgwINu5/fv3l7e3t7lOTExU8+bNlZ6eblF/2c2zs7PTsGHDDGOPP/64/vrrL4v2vGXZsmU6evRolvGFCxcqNTXV4n0WL15sqGvWrGmo16xZk2dIy+3Wrl2rjIwMc12mTBmL19pC7dq1VaNGDXOdnp6uhg0b6tq1a9nOT09PV7NmzXK8H/8oVaqU+XZ0dLRVr8miVNTXX3498cQTeuqppwxjkydP1sCBAwvtGJnNmDHDUO/atUtfffVVnmv2799vGJs5c2aBe8l8noMHD9auXbtynD9mzBht3769wMcFcO8jlAYAAAAAAAAAirGff/5ZDRo00KJFi7L95bqbm5sqVqyoJk2aqF69evL19ZWdXdb/9BsfH68PPvhAlStX5he5AAAAAAAAAAAAAAAAAACzBQsWGOrDhw+rTp062QapbN68WdWrV9f169fNYyVLltT06dNz3H/p0qWG+uDBg6pQoYI2b96c45pff/1Vbdq0yRLwcssnn3yi8uXLm+uUlBQ1bNhQI0aMUEpKSo77hoeH68UXX5SPj4969+6tQ4cOZZnz0ksvycXFRQ8//LA2btyY416S9Prrr2vRokWGsTFjxhjqKVOmyN/fXw888IC+/vrrXAN5li9frh49ehjGcgr8KU7WrFlj+NvFyMhI+fr6atiwYTp06JBSU1N1+vRpvfPOO/L29jaHcri7u9uq5WKvcePG5tu3gn5CQkJs2NH/KcrrryCWLFmi6tWrG8bmz58vDw8PTZw4UUlJSbmu379/vx599FHNmTPHouNVqFAhy/U6bNgwjR49Otv5L7/8skaOHGkY69OnT6EET3355ZeGALDU1FS1atVK3bp106+//qqkpCRFRETo66+/Vo0aNTR16lRJXIMA8uZg6wYAAAAAAAAAANlbsGCBBg8erLS0NMN43bp1NWTIEAUHB6t+/fpZ1qWkpCgkJETr1q3T8uXLFRYWZr4vLi4u11/4AQAA5GbQoEEaNGiQrdsAAAAAAAAAAAAAABSiVq1aafTo0eaQAkk6evSoypUrp4CAAAUEBCglJUWhoaG6cuWKYa2dnZ3Wrl0rJyenHPcPDg7W22+/rQ8++MA8dunSJQUHB8vNzU1VqlSRl5eXkpOTFRkZqYsXL5r/zs3Pzy/bPe3s7LR//35VrVpVCQkJkv4J7vjss8/0+eefq1y5cipXrpzc3d2VkJCg2NhYXbp0yRCmk5vU1FStX79e69evl729vcqUKaOyZcvK09NTaWlpioiIUFhYmG7evGlY9/jjj6tChQpZ9svIyNCff/6p559/Xi+88IK8vLzk7+8vT09P2dvb68qVKzp79qwSExMN6/z8/PTaa69Z1LMtVa9eXV9++aWef/5581hKSopmzZqlWbNmZbumSZMmcnd319atW+9Um3eViRMnau3ateb677//Vvv27bPM8/HxUVRU1J1srcivv4I4ceKEOnTooC1btpjH4uPj9fbbb+vtt99W6dKl5ePjIy8vLzk5OSkxMVFRUVG6fPmykpOTsz3XVq1a5Xi8ZcuWqXz58uYQr4yMDE2bNk2ff/65qlevLm9vb8XExOjkyZNZ/n43ICAgS6hVQYSEhKhJkybm88jIyNC6deu0bt26bOe7u7vrrbfe0ptvvmkey+29HMD9iVAaAAAAAAAAACiGdu/erWeffdYQSFOqVCnNnDlT/fr1M3yjSGZOTk7q1KmTOnXqpA8//FBz587VxIkTDeE0AAAAAAAAAAAAAAAAAADc8sknn8jJyUkffvihYfz8+fM6f/58tmscHR21YcMGtW3bNs/933//fZUrV04jRoxQenq6eTwxMVGHDx/OV89lypTRxYsX1bJlSx09etQ8npGRofDwcIWHh+e5h4uLS55z0tLSdOnSJV26dCnXeUFBQVqxYkWe+2VkZCgmJkYxMTG5zitbtmy+HxtbeO6551SiRAk999xz2YZ73K5Lly5at26dWrZsaR4zmUxF3eJdpVGjRnr55Zc1c+bMXOdlDka6U+7U9Zcfv/32myZMmKDJkyfrxo0bhvuuXLmSJVwrJ61atdKyZcvk7++f4xw7OzuFhoaqXr16On36tHk8JSVFR44cyXFdrVq1dPDgQYv6sFTdunW1b98+dezYUZGRkbnODQgI0M6dOzV79mzDeJkyZQq1JwB3v5w/tQAAAAAAAAAAsImYmBg9+eSThl8UVqhQQdu2bdMzzzyTayBNZo6Ojho6dKiOHz+uYcOGFUW7AAAAAAAAAAAAAAAAAIB7wOTJk7Vnzx7VrFkz13l2dnbq2rWrIiMjFRQUZPH+w4cP1/nz59WxY0fZ29vnOtdkMqlWrVr69NNPc53n4eGhv//+W0uWLFFgYKBFfbi5uSk4OFjbtm1T9+7ds9z/1VdfqXXr1nJ1dbVov3LlymnhwoX67bffsr1/ypQpeuyxx+Tl5WXRfiVLltTo0aMVEREhHx8fi9YUFwMGDFBMTIyGDBmiMmXKmJ9nk8kkFxcXtWjRQr/88ovWr18vOzs7xcXFmdfm9Zq4H82YMUO//fabWrduLQ8Pj2IX3FMU119heffdd5WYmKhRo0bJz8/PojUmk0llypTRsGHDFBUVpT/++CPXQJpbSpQooVOnTmny5MkqWbJkrnM9PDw0bdo0HT16VE5OThb1ZY26devq8uXLmjx5sqpWrSpHR0fzfc7OzqpWrZqmT5+uc+fOyd/fXxcvXjSsL1euXKH3BODu5mDrBgAAAAAAAAAARuPGjVNYWJi5dnR01OrVq1W3bt187+ns7KwvvvhCwcHBRfJLLAAAAAAAAAAAAAAAAABA7tq2bauMjIwi2fvUqVOFsk/Tpk117NgxJSQk6JtvvtGBAwcUEREhR0dHlS9fXp06dVKvXr3yvb+/v79+/fVXSdLq1au1fv16Xbp0SdeuXZO7u7sqVqyooKAgPfbYY1Z9gdtTTz2lp556Sjdu3NDChQu1e/duRUZGKiEhQe7u7ipTpoyaN2+ubt265RkycWsvSYqMjNTq1au1f/9+Xbx4UYmJiXJ2dlbp0qXVvHlz9erVK8/Ai6ZNm2rlypWSpBs3bmjNmjXatWuXzpw5o/j4eJlMJnl5ealOnTrq2bOn6tWrZ9E53x7okpuCvO5SU1OtXuPq6qrZs2dr9uzZec69cOGC+banp6fVx8rNli1bLJ6bn/OUCu+6y01QUJC2b99u1Zo7fT6Fef0VJjs7O02dOlVTp05VSkqKFi9erL/++kvh4eGKjo6Wo6OjfHx8VLZsWXXq1EnBwcFWve9k9sYbb+iNN97QkSNHNHfuXIWFhSk2NlZeXl6qVKmSBg8erNq1a1u1Z36v3Vu95GXv3r2GulmzZjnOvROvdwDFD6E0AAAAAAAAAFCMXLlyRd9++61hbOzYsWrcuHGh7P/EE08Uyj7FyeXLl7Vr1y5FRkYqOjpadnZ2KlWqlGrUqKFGjRqpVKlS+do3Li5O27dvN//y8dYvRhs3bqwaNWoUqOf09HT9/fffOnTokKKiohQfHy8nJye5u7srICBA1apVU82aNQv0y81z585p7969unz5smJjY+Xp6Sk/Pz+1adPG4m/9KI7S09O1e/dunTx5UpGRkUpLS1OZMmVUuXJltW7d2vCtHsVVQkKCjhw5omPHjunKlSu6fv26PD095ePjowceeKDAr6/Mbr0WoqKidOXKFTk5Ocnb21s1a9ZUo0aN5ObmVqjHs0RkZKT++usvnT59WnFxcUpNTZW3t7f8/PzUokWLYvcaLc7XU1E9v5GRkfrjjz8UERGhmJgYeXp6qmzZsmrRooUCAgIK+Sz+OY8//vhD586dU0ZGhnx9fdWoUSM1bty42H3DU04iIyO1c+dORURE6MqVK4Y/qKlcuXKhH2/v3r06deqULl26pBs3bqhixYrq27dvjvPj4+P1559/6vjx44qLi1NycrJcXV3Nf/RSp04dlS1bttD7zE5KSop27Nih0NBQRUZGyt7eXmXKlFH16tXVvHnzAv38y87169cVEhJi/qPJW+fcvn17m7wHAgAAAAAAAAAAAMDdwN3dXSNHjizSY3Tv3l3du3cv1D1LlCihIUOGaMiQIYWyX5kyZQptL+mf/nr16lWgYJ97xebNm3X9+nVzXatWLRt2g8JQ2NdfYXJyctLAgQPvyLHq1q2rjz/++I4cqyBSU1N1+PBhc12yZEm++BJAFoTSAAAAAAAAAEAx8uWXXyopKclcu7q6atSoUTbsKHdBQUHaunWrubbmGxnmzp2rwYMHm+s5c+Zo0KBBFq1NSUnRN998o6+++kqHDx/O8bj29vZq2bKlBg0apL59+8rV1TXPvUNCQjR+/HiFhITk+G0h1apV0/Dhw/Xiiy9a9Qu4a9eu6cMPP9TcuXN16dKlXOd6eHjowQcfVP/+/c3fupOXlJQUffnll/r666/1999/ZzvHZDKpSZMmeueddwr9DzokqVKlSgoLC8syPm/ePM2bNy/HdXk9/5GRkZowYYKWLFmiK1euZDunZMmS6tGjhyZMmKCKFSta3XtROnHihBYvXqxffvlFe/fuzfWbaPz8/DRs2DCNGDFCXl5e+TpeQkKCZs6cqblz5+rkyZM5znN2dtaDDz6oZ599Vj179jSE+uQUBDJ+/HiNHz8+xz1/++03BQUFGcYyMjK0bds2LV26VBs3btTx48dz7b9+/foaM2aM+vXrJweHnH+ddfbs2RzDPgYPHmx4j8ksNDRUlSpVyvH+4nA95aQwnt+crFy5Uh9++KF2796d43vrrefnmWeesTg85PbXU/v27c3fRLVnzx698cYb2rx5c7brAgICNGHChFzfH1JSUlSuXDnFxMRI+udnZ0REhEqWLGlRb7ecPXtWVapUMZ933bp19ddff+W6Jj09XQsXLtSnn36qffv25fiY1a5dW6+99poGDBhg0WOW08/I69eva8qUKZozZ45CQ0MNazw9PbMNpdm/f78++OAD/fzzz0pJScn1uJUrV9YjjzyiYcOGqU6dOlb1ZonQ0FC9++67WrVqleLj47Od4+Pjoz59+ujdd9+Vj4+PRfsOGjTI8DPm1vV99epVjR8/Xl9//bUSExOzrHNyctKzzz6rCRMmWHwsAAAAAAAAAAAAAABQODL/bY+lf38AoHAMHDjQ8LeMrVu3tmE3AIqrwv2KOQAAAAAAAABAgaxatcpQ9+7dW56enjbqpnjasWOHatSooRdffFGHDh3KNQgnLS1N27dv19ChQ7V06dJc901JSdGAAQPUvn17bd68OdfQkFOnTmn06NGqV6+ejh07ZlHfBw8eVO3atTV58uQ8A2mkfwJs1qxZozFjxli0/65du1SrVi2NHDkyxwAN6Z9wkL179+qxxx5T9+7ds/2QfnGzdOlSVatWTZ9//nmOgTSSFB8fr/nz56tmzZr6/PPP72CHuVuzZo1q1qypcePGaefOnbm+tiQpIiJC7733nurXr689e/ZYfbxVq1apcuXKeuutt3INLJGk5ORkbdy4UU8//bS2b99u9bEs9eqrr+rBBx/UZ599lmcgjSQdPnxYgwYNUocOHRQZGVlkfeWkOF9PRfX8Xr16VQ899JB69OihXbt25freeuv5admypUXvZzmZOXOmWrVqlWMgjSSdP39egwcP1nPPPaf09PRs5zg5Oenpp58210lJSVq2bJnV/cyfP99w3nl9M9LJkyf1wAMPaODAgdq7d2+uj9nRo0c1ePBgtWnTRlFRUVb3JklhYWFq2rSp3n333SyBNDn58MMP1axZM61YsSLPQBrpnzCXzz77TIsWLcpXj7mZPn26atWqpYULF+YYSCNJ0dHR+vTTT1W1alX9+OOP+T7e33//rYYNG2ratGk5Xpu3wqdatmyps2fP5vtYAAAAAAAAAAAAAADgn7/zWrduXZ7z0tPT1aZNG507d8485uLioiFDhhRle8A97+TJk/r4448tmvvmm29m+RuhTz75pCjaAnCXI5QGAAAAAAAAAIqJhIQE/fnnn4axRx55xEbdFE9LlixRhw4dFBYWluW+cuXKqXHjxnrggQcUGBho1b7Jycl65JFHtGDBgmz3bdq0qWrUqCFHR0fDfSdPnlTbtm2zPG+ZXb58WcHBwQoPDzeMOzg4qGrVqmrWrJmaNWumGjVqqESJElb1Lkk//fSTOnTokCWkwMnJSTVr1lTz5s1Vq1YtOTg4ZFnXsWNH3bhxw+pj3imzZ89Wnz59sgQYuLu7q27dumrUqJFKlSpluC85OVkvvfSSJkyYcAc7zVl2j6+Li4tq1KihJk2aqGnTpqpcubLs7Iy/trl48aKCgoJ09OhRi481depU9ezZU9HR0YZxk8mkgIAANWnSRI0aNZK/v3/+TiafsnsMvLy8VLt2bbVo0UKNGjVSuXLlsszZtm2bOnbsqOvXr9+JNiUV7+upqJ7fmJgYBQUFacOGDVnuCwwMVNOmTVWlSpUsr9E9e/aoTZs22b4n52XWrFl65ZVXlJaWJumfa6J27dpq1qxZtv3Pnj1b06dPz3G/zAEy8+fPt7qn29fY29urf//+Oc7dtWuXWrdurYMHDxrG7e3tVa1aNTVv3lx16tTJ8p6+c+dOtWrVyupgmmvXrqlLly6GkKQyZcqocePGqlOnjtzc3LKs+fbbb/Xmm29mCfMpWbKk6tevr5YtW6phw4YKCAiQyWSyqh9rvfPOOxo1alSWYJxSpUqpYcOGqlevntzd3Q33Xbt2TU8++aTmzJlj9fHOnj2r4OBgw2uzYsWKatasmWrXri17e3vD/NOnT6tnz555hoYBAAAAAAAAAAAAKDyuLtb9fc+dVtz7A4qjn3/+Wd26dVPp0qXVp08ffffdd7pw4YLS09MVHR2tdevWacCAAXJzc9Mff/xhWDtlyhQbdQ3cO86dO6fXXntNLi4u6tSpkz799FMdP35cKSkpSkpK0t69e/Xqq6/Kz89PH374oWFtly5dVLduXRt1DqA4c8h7CgAAAAAAAADgTtixY4f5w/m3NG3a1EbdFD979uzRwIEDDR9o9/Dw0Kuvvqr+/furUqVKhvmxsbEKCQnR0qVLtWzZslz3Hjt2rDZt2mQYe/zxxzV+/Hg1aNDAPBYTE6Nvv/1W7733njkk48qVK+rdu7cOHDiQ5QP1t0yYMEFXrlwx11WqVNHEiRP1r3/9K0uQQFpamo4fP67169dr2bJlhm+Dyc6RI0f01FNPGUI72rVrp9dff13BwcGGQIT4+HgtWbJE77zzji5fvixJ2r17t0aNGqUvv/wy1+NY6rvvvtP169d1+fJlQ6BDly5d9Oqrr+a4LrtfZu7fv18vvviiIVAhMDBQU6ZMUffu3eXs7Czpn8ds06ZNGjNmjI4cOWKe+95776lZs2bq2rVrYZxagdjb2+vhhx9W9+7d1bFjx2wDPuLj47V69WpNmDBBJ06ckCQlJSWpb9++2r9/f56hEStXrtSYMWMMY2XLltVbb72l3r17y8/Pz3BfZGSkfv31Vy1ZskRr1qzJst/GjRslSQcPHtR//vMf8/gzzzyjAQMG5NhHw4YNsx0vWbKkevXqpUceeUStW7fONoTm4sWLWrhwoT788EPFxcVJ+uc1/sYbb2jGjBlZ5vv5+Zn73LBhg+FbXl599VV16dIlxz4zPx63jlWcrqfbFfbze7vnnntOBw4cMIwNHTpUr7/+uqpWrWoeCw8P16effqopU6aYwztCQ0PVt29fhYSEZAn6yMmpU6f0yiuvSJJq166tDz74QN26dTM8vvv379dLL72kHTt2mMfeeecdDRw4UKVLl86y562woGPHjkmStm7dqrCwMFWsWNGinrZv367Tp0+b686dO2f7GpWkiIgIde/e3RAO1KBBA7355pt69NFHDT8Lbty4odWrV2vs2LHm/U+fPq1BgwZpzZo1FofBTJo0yfw6e+qppzR27FjDz6ebN28afo4lJyfrtddeM+zxxBNP6M0339QDDzyQ5bjx8fHas2eP1q5dq4ULF1rUk6V++uknffDBB4axevXqacqUKerUqZP5dZOSkqJVq1ZpzJgxOn/+vKR/3t9feOEFNWnSxHC+eXn22WcVEREhFxcXvfrqq3r++ecNYUexsbH673//q48++kgZGRmSpD///FOzZ8/WsGHDCnrKAAAAAAAAAAAAACzg6dlYTZssU9L13P82xhZcXQLl6dnY1m0Ad62YmBgtWbJES5YssWh+jx49NHz48CLuCrh/3LhxQ7/++qt+/fVXi+ZXqFBBP//8cxF3BeBuZcq49Vd2VmrSpIkiIiLk5+enffv2FXZf95xbj5ekon/MxnkW3d5FbdxVq5dkRMwugkaAu4fJb2j+FydvL7xGgLuRcxvzzfRZtW3YCGB7di8czffa52s4F2InwN3nqxPJt1XhNusDKB7+70OeGRty/gA4cD8wddmQr3Wff/65XnrpJXPt4eGhq1et/29mBVWpUiWFhYVJkipWrKizZ8/mODcoKEhbt24119b8J+e5c+dq8ODB5nrOnDkaNGhQtnOTk5NVq1YtQy916tTRL7/8ooCAgDyPdfHiRSUkJKhmzZpZ7tuzZ49atGhh6P3dd9/V+PHjc9xv37596tixo65du2Yee+WVVzR9+vRs55ctW1aRkZGSJF9fX/31118qU6ZMnn1L0tGjR1W7dvb/vzU1NVWNGzfWX3/9ZR4bP3683nnnnVyDDi5evKgOHTro5MmT5rH9+/ercePC+2Oas2fPqnLlyuZ64MCBmjt3rsXrMzIy1LBhQx0+fNg81rBhQ23ZskWlSpXKdk1ycrK6deumzZs3m8fKlSun06dPy8XFxepzyIm15xYaGio7OzuLwzFu3LihJ554QmvXrjWPrV27NtdwncuXL6tWrVrmIBfpnzCV1atX5/h43e7EiRNydXVVhQoVsty3ZcsWdejQwVy/9957GjdunEXncsu+fftUvXp1eXh4WDQ/LCxMHTp0UGhoqCTJxcVFFy5ckLe3d45rrHlPyU5xvp6K8vldvny5evXqZRj75ptv9Oyzz+a439q1a/X444/r5s2b5rFp06Zp5MiROa7J7jF86KGH9OOPP8rV1TXbNUlJSWrdurUOHjxoHps+fbo50CazDz/8UG+++aa5fv/99/X222/n2NPtnnvuOc2e/X+/c1q8eLGefvrpbOd27dpVv/zyi2HtZ599JkdHxxz3j4uL0yOPPGL4lq8ff/xRPXr0yHZ+5tfzLbmd/+3Wrl2rRx55xFwPGDBA8+bNy3Od9E84zIULF1SlShWLesvtWktKSlKVKlXMgTrSP4E/P/30kzlYLLPY2Fi1b9/e8P7fqFEj/fnnnzn2PGjQoCzn5+3trV9++UXNmjXLcd3EiRMNr5G8jgMAAAAAAAAAAAAAAHI2fPhwq77MycHBQa+99pomTpxYhF0B94/t27erbdu2Vq3p3LmzfvnllyxfsgcAt/DuAAAAAAAAAADFRExMjKH28fGxUSfFz7x58wyBNKVLl9amTZssCqSRpPLly2cbSCP9E6RweyDNo48+mmsgjfRPEPnXX39tGPvmm2+yDRGKi4szB9JI0hNPPGFxII2kHANpJGnZsmWGAI3nn39e7777bq4BGtI/j8fy5csNv0T85JNPLO7pTti4caMhkMDV1TXPAA5nZ2f9+OOP8vPzM49dunRJixYtKspW81S5cmWLA2kkqUSJElqwYIE8Pf8vhH7OnDm5rpkxY4YhsKR69epat26dRYElklSjRo1sA0sKS5MmTSwOpJH+CcS6PSDk+vXrFn9zUn4V5+upKJ/fzL2+9NJLuQbSSFK3bt30/vvvG8amT5+utLQ0i/qRpMDAQH3//fc5BtJI/1z3H374oWFs3bp1Oc7v37+/4XlYsGCBRb3cuHFDP/zwg7n29PTU448/nu3cnTt3GgJpunbtqlmzZuUaSCNJpUqV0vLly1WyZEnzmLWvk6efftqiQBrpnyCi21nzbWJOTk45BtJYa+HChYZAGn9/fy1btizHQBpJ8vLy0urVqw1hYgcOHLD4m5tu+fbbb3MNpJGk119/3XBtHDhwwNAvAAAAAAAAAAAAAACw3BdffKFNmzapR48eCggIUIkSJQx/e2MymeTs7KyaNWtq5MiRio+PJ5AGKERt2rTRiRMnNHToUNWsWVPu7u6Gv6cymUxydHRU+fLl1bt3b509e1YbNmwgkAZArniHAAAAAAAAAIBiInMoze2BFJYICQnRpk2b8vy3ffv2wmz7jpg+fbqh/uijj1SuXLkC7xsXF6fly5eba5PJZHFIwFNPPaWWLVua68TExGzDT65fv26o8wousMbtj4urq6smT55s8dr69evrscceM9erVq2yKlCiqH3zzTeGetSoUQoMDMxznaenp8aNG2cYyxwgdDfw9vZW165dzfUff/yR49yUlJQs3zA0a9Ysubm5FVl/d0JwcLDhOs/tMSgMxfV6Ksrn9++//9aOHTvMtZubW5awmZyMHj3aEAwWFhamDRs2WHzsN954w6Kfc507d5aXl5e53r9/f45zK1SooODgYHN94sQJ7dy5M89jrFq1yhD68+STT6pEiRLZzs3882jatGl5Bhfd4ufnpyFDhpjr7du3WxWAYulzIxXtzx5rZH4vHz9+vEUBVZUqVcoSwGPNe3nz5s1zDBa6nYODg3r27GkY27dvn8XHAQAAAAAAAAAAAAAARsHBwfrxxx917tw5Xb9+Xenp6crIyFBGRobS09N148YNHTt2TNOmTcvx7zMA5F/16tX19ddf69ixY4qPj1daWprhGkxJSdGFCxe0dOlSq75sD8D9i1AaAAAAAAAAACgm4uPjDbW1gQM9e/ZU586d8/zXr1+/wmy7yF24cEFHjx4116VLly60c9ixY4dSUlLMddu2bVWjRg2L1//73/821CEhIVnm+Pj4yMnJyVyvWbNGV69ezUe3RleuXNHu3bvN9aOPPmoIbrBEly5dzLcTEhL0559/FrivwpL5sRw8eLDFa/v06SMXFxdzvW/fPiUlJRVab3dK5cqVzbcvXryoqKiobOft3r3bEKhRr149dezYsajbuyMqVapkvl2Ur8/ifD0V5fO7detWQ92zZ0+VKlXKorWOjo565plnDGPZvQdmx2Qy6cknn7Rorr29verXr2+uo6KilJycnOP8gQMHGup58+bleYz58+fnusct6enp+uWXX8x18+bNVbNmzTz3v93trxNJ+v333y1a16xZM1WrVs3i4/j7+xvqhQsXWry2sCQkJBhChFxdXfX0009bvD7zz1hLHyvpn+A4SzVq1MhQnz9/3uK1AAAAAAAAAAAAAAAAAADcywilAQAAAAAAAIBiomTJkoY6MTHRRp0UL5k/hN6xY0c5OzsXyt67du3Ksrc1goODDfXOnTuzzHF0dFT79u3NdWhoqDp06KD169crPT3dquPdbtu2bcrIyDDXTZs2tXqPwMBAQ317+I8tnT17VpcvXzbXFStWVNWqVS1e7+HhYXg80tLStGfPnkLtMb/i4uL07bffavDgwXrggQdUrlw5ubm5yWQyZfk3efJkw9ro6Ohs98x8jXTt2rXI+i8MZ8+e1UcffaTevXurdu3a8vX1lbOzc7aPwY4dO8zrcjr/wlCcr6eifH7vxHtgdipVqqTSpUtbfJwyZcoY6tyCvXr27CkPDw9z/f333xvCxzK7fPmy1q9fb66rVaumNm3aZDv38OHDhmPfyddJ8+bNrTpOx44dZW9vb66nTZum4cOH68yZM1btUxB79+5VWlqauW7WrJnc3d0tXl+9enUFBASY60uXLiksLMyitdY8N9a8vgAAAAAAAAAAAAAAAAAAuJ842LqB+1FkZKSaNGlSoD327dtXSN0AAAAAAAAAKC68vb0NNR+K/sfp06cNdX5CAHKS+cPtDRo0sGp9lSpVVLJkScXHx0uSzp8/r4yMDJlMJsO8d955R7/++qs5hObPP//Uww8/rLJly+rhhx9WUFCQWrVqpZo1a1p87MxBBq+99ppee+01q/rPLCYmpkDrC0tBnxdJatiwoSHM49y5cwXuqyASExM1fvx4zZw5U8nJyfnaIy4uLtvxorxGClNYWJheeeUVrV692hAAY6mczr8wFOfrqTi/BzZs2NBQW3qdZQ4ByYubm5uhvn79eo5zXVxc1Lt3b3377beSpNjYWP3000964oknsp3/3XffGYJTBgwYkOPemV8nX3zxhb744os8+8+Npa+TypUrW7VvQECA/v3vf2v27NnmsS+//FJffvmlmjRpok6dOunBBx9Uy5Yts/zvj8JSWO/l58+fN9fnzp1TxYoV81xnzWvMmtcXAAAAAAAAAAAAAAAAAAD3E0JpbCA9PV0RERG2bgMAAAAAAABAMZP5Q+FXrlyxan10dHS241u2bFGHDh3y3ZetZf7AvrVhBrmJjY011D4+PlbvUbp0aXMoTVpamuLj4+Xh4WGY065dO33zzTd64YUXlJKSYh6/fPmy5s2bp3nz5kmS/Pz8FBwcrKeeekoPP/ywHB0dczyuta8PSxSXIKTCeF4yr8m8550UHR2tjh076vDhwwXaJ6cwm6K8RgrL7t271aVLlwK9xm6/dgpbcb6eivN7oLe3t+zs7MyBW5ZeZyVKlLDqOJnlFWo0cOBAcyiNJM2bNy/HUJpb77+SZDKZ9Mwzz+S4ry1fJ5l/rlhi5syZioiI0E8//WQY37dvn/bt26f//ve/MplMatiwobp27ap+/fqpbt26Vh8nJ7Z8Ly/Iayw/oVkAAAAAAAAAAAAAAAAAANyLCKW5w+zt7eXr62vrNgAAAAAAAAAUQzVq1DDUV69e1dmzZ1WpUiXbNFRM3Ap8ucXd3b3Q9k5ISDDUbm5uVu+ReU12oTSSNHjwYLVq1Urjx4/Xjz/+mG3ARkREhL777jt99913qlSpkiZNmqQ+ffpke9y4uDire83LrWAJWyuq58VWevfunSWQJiAgQB06dFCdOnVUoUIFubu7y8XFRXZ2duY58+fP14IFC/LcvyivkcJw5coVdevWLUv4RoMGDdSuXTtVq1ZN/v7+cnFxUYkSJWQymcxzxowZo0OHDhV5j8X5eirO74Emk0kuLi5KTEyUZNvr7HZt27ZVlSpVdObMGUnSL7/8oqioqCy/ozt48KDh9dW+fftcf+ba8nWSW0hZTkqUKKFVq1ZpyZIl+uijj3TgwIEsczIyMnTgwAEdOHBAkydP1iOPPKLp06erWrVqVh8vs3vtvRwAAAAAAAAAAAAAAAAAgPsNoTR3mK+vr/bt22frNgAAAAAAAAAUQ61atZK9vb3S0tLMY3v37r3vQ2lKlixpqDN/yL0gMoc73ApWsEbmNZn7vV2tWrW0ePFixcbGasOGDdqyZYtCQkJ09OhRZWRkGOaePXtWffv21e7duzVt2rQse7m6uhrqkSNH6pFHHrG6/9tVqVKlQOsLy51+XorS6tWrtWXLFkMfX375pfr06WMIoMnOr7/+atExivIaKQwTJ07UlStXzHX16tW1cOFCNW/ePM+1mV/nRaU4X093+j2wdOnSFq/PyMjQ9evXzbWtrrPMTCaTBgwYoHHjxkmSbt68qcWLF+vll182zJs3b56hHjhwYK77Zn6d9OnTR//+978L1Ku/v3+B1ufFZDKpT58+6tOnj/7++29t3LhRW7Zs0bZt2xQdHZ1l/s8//6yQkBD9/PPPateuXYGOfS+9lwMAAAAAAAAAAAAAAAAAcD8ilAYAAAAAAAAAigl3d3c1btxYe/fuNY+tXbtWvXr1smFXuTOZTPlem5SUZNE8b29vQx0ZGZnvY2bm5eVlqG8PzrDU7Wvs7e0t+sC8l5eXnnrqKT311FOSpOjoaG3dulWrV6/WsmXLDI/N9OnT1apVKz355JOGPXx8fAx1uXLl1KlTJ6v7L44K43nJHLaQec87ZcmSJYb6q6++Up8+fSxaGxMTY9G8orxGCsP3339vvl2iRAn98ssvFge2WPoYFFRxvp7u9HtgYGCgxetjYmKUnp6e4362NGDAAI0fP94c+DVv3jxDKE1qaqoWLVpkrt3c3PL8eZv5dVKqVKli8zqxRJ06dVSnTh298sorysjI0LFjx7RhwwYtW7ZM27ZtM8+Lj49Xr169dPr06SzBMta4l97LAQAAAAAAAAAAAAAAAAC4H+X+FZwAAAAAAAAAgDvqscceM9RLly7VtWvXbNRN3kqUKGGor1+/bvHaqKgoi+ZVr17dUN8e2lNQFStWNNQHDx60av2ZM2cUHx9vrgMDA/MV1OPj46MnnnhC8+bNU1hYmLp162a4/5NPPsmypnLlyob61KlTVh+3uCro85Ldmsx73ik7d+403y5dunSWcKHcHDlyxKJ5RXmNFNS5c+cUHh5urh9++GGLA2muX7+u0NDQomrNoDhfT8X5PbC4XGfZqVy5stq1a2eu9+/fb7im1q9fr8uXL5vrnj175hnAUpxfJ9YymUyqXbu2XnnlFf3+++8KCQkxhO5ERkZqwYIFBTrGvfReDgAAAAAAAAAAAAAAAADA/YhQGgAAAAAAAAAoRoYNGyZXV1dznZiYqBkzZtiwo9x5eHgY6ts/4J+XPXv2WDTv9lABSdq8ebOSk5MtPk5uWrZsmWVva2Sen3m//PDx8dGiRYvk5uZmHtu7d2+Wc+7QoUOuvdianZ3xVxAZGRkWr61UqZLKli1rrsPCwnTmzBmL18fHxxuCOxwcHNS0aVOL1xem26+JatWqyd7e3qJ1165d0759+yyam/kaWbduneUNWqAgz2Xm94SaNWtavPb333/XzZs3LZ5fkD6L8/VUlM9vcXwPLEwDBw401PPnz8/2dnZzs9O8eXPDz+g//vhDN27cKGCXxUO7du304YcfGsa2bdtWoD2bNm1qeM/bs2ePEhISLF5/6tQpnT9/3lyXK1dOgYGBBeoJAAAAAAAAAAAAAAAAAABYjlAaAAAAAAAAAChGSpcurX//+9+GsQ8++ECHDh2yUUe5q1ixoqH+888/LVoXHR1tcfiBv7+/6tevb66vXLmi7777zvImc9GyZUs5OTmZ623btunUqVMWr//f//5nqNu3b18ofXl6eqpevXrmOj09XTExMYY55cuXN8w5ffp0oYeRFMTtoTqSlJSUZNX6zI/l3LlzLV67ePFiXb9+3Vw3bdrUECRxJ90ejJKSkmLxuv/9738Wh100a9ZM3t7e5vqvv/4q1FCVgjyXmYNhrHkMvvjiC4vnSgXrszhfT0X5/Ga+zlasWKGrV69atPbmzZtasGBBrvvZWu/evQ3X/sKFC5Wenq64uDitXr3aPB4QEJAlmCg7Tk5O6tixo7lOTEzUnDlzCrdpG2rTpo2hjo6OLtB+7u7uatKkiblOSkrS0qVLLV5fVD9jAQAAAAAAAAAAAAAAAACAZQilAQAAAAAAAIBiZty4cQoICDDXKSkp6t69u44dO2bDrrL3wAMPGGpLP2w+YcIEQ2hIXl555RVD/dprr+nSpUsWr89JqVKl1KtXL3OdkZGh//znPxatXbZsmXbs2GGu3d3d1adPnwL3dEvmMAAvL68sc1599VVDPXLkSIsDJYqah4eH7O3tzXVoaKhV64cMGWKop06dqgsXLuS57tq1axo3bpxhbOjQoVYduzD5+fmZbx85ckRxcXF5rrl48aLGjx9v8TEcHR01fPhww9gLL7ygxMREi/fIze2BKJJ1z+Xt5y/9E/xkibVr12rVqlUWH0cqWJ9S8b2eivL5rV27tlq3bm2uExIS9N5771m0dsaMGTp37py5rlSpkjp37lzgngpTyZIl1bNnT3MdHh6uTZs2aenSpYbQp2eeeUZ2dpb92jTz6+S9994zPA53M0t+7lgr83v5u+++q4SEhDzXhYWFacaMGYYxW76XAwAAAAAAAAAAAAAAAABwPyKUBgAAAAAAAACKmdKlS+v777+Xo6OjeSwsLExt2rTRkiVLlJGRYdV+J06cKOwWzTp16mToc+nSpXmGTnzzzTf67LPPrDrOM888o6pVq5rrK1euqFOnThaFlEj/hHwcP3482/tGjRplCCNYtWqVPvjgg1z3O3DgQJYP2g8ZMkQeHh5Z5m7evFlvvPGGwsPDLepVklasWKHTp0+b6zp16qhEiRJZ5vXr109169Y11ydOnFDXrl2tOtbNmzc1b948/fe//7V4jSUcHR1Vo0YNc33gwAHDOeWlU6dOatCggblOTEzUY489lmtISEpKinr37m0ILCpXrpz69u1rZfeF5/bAj5SUFL355pu5zo+KitKjjz5qUXjN7V5++WVDKMvJkyfVrVs3i/c5fvx4jtdTxYoV5e7ubq5//fVXxcbGWrRvYGCgypcvb6737Nmj77//Ptc1u3fvVv/+/S3a/3a3XwuStHr1at28edPi9cX5eirK53fMmDGGeubMmZo/f36u+61fv15vvfWWYWzkyJEWB7vcSQMHDjTU8+fP17x583Kdk5sHH3xQDz30kLmOiopSly5drAqOS09P18qVK/X6669bvMZa77zzjhYuXKjU1FSL5mdkZOiTTz4xjDVp0qTAffTr109ly5Y11xcvXtSTTz6plJSUHNfExcXpscceU1JSknmscePG6tixY4H7AQAAAAAAAAAAAAAAAAAAlit+fxkKAAAAAAAAAFCrVq309ddfGz7gHxMToz59+qhx48b69NNPdfTo0WzXZmRkKDQ0VLNmzVLbtm31/PPPF1mfPj4+6tGjh7lOT0/Xo48+qrlz52b5wPnhw4fVv39/DR06VBkZGYaQmbw4OTnp+++/l7Ozs3ns77//Vr169TRx4kSFhYVlWRMXF6dVq1apb9++qlKlinbs2JHt3k2bNtWoUaMMY++884569eqlv/76yzAeGxurKVOmqE2bNoZwlKpVq+YYZHPt2jX997//VaVKldStWzd9++23OnHiRLbhQufPn9e7776rp556yjA+dOjQbPe2t7fX8uXL5enpaR7bsWOH6tWrp/feey/HQKLLly9rzZo1ev7551W+fHkNGjQox9dTQXTp0sV8Oy0tTQ8++KDGjx+vFStWaOPGjdq0aZP53+1BMpJkMpk0Z84cQ+jR/v371ahRIy1fvtzw+kpPT9fGjRvVtGlTbdiwwbDP//73v2wDfe6UAQMGGOpZs2Zp4MCBWV6z8fHx+vbbb9WgQQMdOHBAklS7dm2Lj+Pr66u5c+fKZDKZx0JCQlS7dm199tlnunz5cpY1kZGRWrx4sbp37646dero1KlT2e5tZ2en4OBgcx0XF6eWLVvqv//9r1atWmV4Hjdt2pQlsCbzYzBgwABNnjxZ165dM4xfuHBBb7/9th588EHFxsaqRIkSqlSpksWPQZkyZdSwYUNzffLkSbVq1UozZszQmjVrsvR548YNw/rifD0V5fPbs2dPPfHEE+Y6IyNDgwYN0gsvvKAzZ84Y5l66dEljx47Vo48+argGW7durZdeeqmgp1kkOnbsqAoVKpjrZcuW6Y8//jDXLVu2NARoWWL+/PkKCAgw18ePH1eTJk00cuRIHTx4MNv399jYWG3atEmjRo1SpUqV1KNHD+3atSsfZ2SZw4cP65lnnlH58uU1bNgw/fLLL7py5UqWeenp6dq2bZu6dOmilStXmsddXV0LJdDL1dVVs2fPNoytW7dOzZs318aNG5Wenm4eT0lJ0fLly9WoUSMdPHjQPO7k5KS5c+cWuBcAAAAAAAAAAAAAAAAAAGAdB1s3AAAAAAAAAADI3qBBg+Tl5aVBgwYpLi7OPH7w4EG9/PLLkiR3d3f5+vrKx8dHGRkZio+P14ULF5SYmJjtnj4+Pnr33XcLtc+PPvpIa9euVUJCgiTp6tWrGjx4sEaMGKGqVavK3t5eFy5cUGRkpHnNgw8+qP79++u5556z+DhNmjTR/PnzNWDAACUnJ5uP9fbbb+vtt99W+fLlVbZsWZlMJkVFRen8+fPZBgNkZ+LEiTp48KA2bdpkHlu+fLmWL18uf39/+fv7Kz4+XmfOnNHNmzcNa0uXLq2lS5fKzc0t12PcvHlT69at07p16yRJJUuWVLly5VSqVCmlp6crPDxc4eHhWda1bdtWI0aMyHHfmjVrasWKFXriiSfMYSCxsbGaMGGCJkyYIB8fH/n5+cnNzU3Xrl1TdHS0oqKiLHpcCmr48OH66quvzOEf4eHhGjduXLZz58yZo0GDBhnGHnjgAX3++ed64YUXzMEFZ8+eVa9evVSyZElVqlRJ9vb2CgsLyxKEIknjx4/Xww8/XKjnZK0uXbqoW7duWrt2rXls/vz5mj9/vqpUqSJfX1/FxcUpNDTUEPLRt29fVa9eXePHj7f4WP/61780depUjR492vzaj4iI0IgRI/Tyyy8rMDBQvr6+SktL0+XLl7N9veVk1KhRWr16tXnfEydO6I033sh27m+//aagoCBz/Z///EcLFizQhQsXJP0TPDF27Fi9++67qlmzptzc3BQVFaWzZ88artmZM2fqu+++09mzZy3uc8yYMYYQnH379mnfvn3Zzg0NDc0SelOcr6eifH6//vprnTp1yhwEkpGRoa+++kpfffWVKlWqJF9fX8XExCg0NNQQIiJJlStX1qJFi2Rvb194J1uI7Ozs9Mwzz2jy5MmSZP75ccvAgQOt3rNMmTL6+eef9cgjj+j8+fOSpKSkJM2YMUMzZsyQp6enypcvr5IlSyohIUExMTFZgrfulMjISM2aNUuzZs2SJJUrV04+Pj5yc3NTYmKiQkNDzT+/b/fJJ5+ofPnyhdLDv/71L7399tuG8LaDBw+qS5cu8vLyUsWKFZWWlqazZ88qPj7esNbOzk6zZs1SgwYNCqUXAAAAAAAAAAAAAAAAAABgOUJpAAAAAAAAAKAYe+yxx3To0CG9+uqrWrp0aZaQlYSEBCUkJCg0NDTXfby8vPTss8/qrbfeUqlSpQq1x4oVK2rZsmXq2bOnkpKSDL3dCji4XceOHfXjjz9qxYoVVh/rySeflL+/v/r06WMOuLjl4sWLunjxovUnIMnZ2Vk///yznn32WS1cuNBwX05hMZJUvXp1rV69WrVq1bL6mPHx8Vk+fJ9Z9+7d9d133+UZ9tChQwft2bNHffr00Z49ewz3RUdHKzo6Otf1JpNJAQEBljVuhRo1amjBggUaPHhwtqEHlhg6dKg8PT01ZMgQw+MVHx+vw4cPZ7vG2dlZn3zyiV588cV8HbOwfffdd3rooYe0e/duw/iZM2d05syZLPOffvppzZkzR5MmTbL6WCNHjlRgYKCGDh2qmJgY83hGRobCwsIUFhZm/QlIat++vWbMmKExY8ZkCWbKi7e3t1avXq2uXbvq8uXL5vHU1FQdOXIky3w7OztNmTJFQ4cO1XfffWfVsZ555hn99ddf+vjjjy0OpcqsuF5PUtE9v97e3tq6dat69+6tjRs3Gu47e/ZsjsFAzZo106pVq1SuXLl8HfdOGThwoDmU5nbOzs566qmn8rVn/fr1tW/fPj3zzDNav3694b6rV6/q6tWree4RGBiYr2MXxKVLl3INyHFxcdG0adP0/PPPF+px33//fZUuXVqvv/66IYArNjY221AxSfLw8NCcOXPUs2fPQu0FAAAAAAAAAAAAQPGx92qiwq4n5z3xDqvo4qymnrl/QRUAAABwPyCUBgAAAAAAAACKuYCAAC1ZskTjx4/X7NmztWLFimyDLDIrW7asWrVqpaefflqPP/64nJ2di6zHW4Eb//nPf7R+/fpswyD8/Pz0xhtv6KWXXsozZCU3bdu21cmTJ/XFF1/o66+/1vHjx3Oc6+TkpAcffFADBw5U7969c93XyclJCxYs0JAhQzRhwgSFhIQoNTU127lVq1bV8OHD9dJLL8nJySnXfR955BFt3LhRP/30kzZv3qwjR47kGpZhb2+vjh076uWXX9ajjz6a696Ze9q9e7d++uknTZ8+Xdu2bTN88D+74zRv3lxdu3ZVv379VKVKFYuPZY1evXqpTZs2mj9/vn777Tf9/fffio2NVWJiosWhIU8++aSCgoI0YcIELV682BDGcbuSJUuqR48eGj9+vCpVqlSIZ1EwpUqVUkhIiCZOnKiZM2fmGFZRt25dvfXWW+rTp0+BjtezZ08FBwfrk08+0fz583MNKnFzc1NwcLD+/e9/q127drnuO2LECHXr1k3z58/Xtm3bdOzYMcXFxen69et5PpeNGzfWvn379MYbb2jx4sVKS0vLMsdkMqlTp06aOHGimjVrZtnJZuO///2v+vXrpwULFmjnzp06ceKErl27phs3bli8R3G9nqSie349PT21YcMGrVixQh9++KH27NmT4/Nar149jRkzRgMGDJCdnV2BzudOqFmzplq0aKFdu3YZxv/1r3/Jy8sr3/v6+vrql19+0e+//66PP/5YmzdvVmJiYo7zTSaTGjVqpIceekh9+/ZV/fr1833svMyePVuPPfaYfv75Z23bts0QCJUdb29vPfnkk3rjjTdUsWLFIulp5MiR6t69u9577z2tXLkyx7Cy0qVLq2/fvnrnnXfk6+tbJL0AAAAAAAAAAAAAsL29VxP16P6Ttm4jR2seqE4wDQAAAO57pox8fk1kkyZNFBERIT8/P+3bt6+w+7rn3NHHa5xn0e5flMbl/a2RmWVEzC6CRoC7h8lvaP4XJ28vvEaAu5FzG/PN9Fm1bdgIYHt2LxzN99rnaxTdB5uBu8FXJ27/doZwm/UBFA/+5lsZG7rYsA/A9kxdNhT5MS5duqRDhw4pLCxMsbGxSklJUcmSJeXl5aXSpUurfv36Rfah8rxcvnxZW7duVXh4uBITE+Xt7a0GDRqoZcuWBQqjyUlYWJj27NmjyMhIxcbGytnZWd7e3qpZs6YaNWokN7f8/XFIXFyctm3bpvDwcF25ckVubm4qW7asGjVqpJo1a+a736tXr+rIkSM6ffq0oqKilJSUJGdnZ5UqVUrVq1dXo0aNVKpUqXzvf0tSUpJ27typ8+fP68qVK7p+/brc3d3l4+OjmjVrqnbt2vl+bGwpLS1Nu3fv1smTJxUZGan09HT5+vqqSpUqat26tRwdHW3dYq5u3LihHTt26OjRo4qNjZWTk5P8/f3VrFkz1ahRo0iOefToUR06dEhRUVGKi4uTq6urfH19VatWLTVo0KBIA6uyExMTo5CQEIWFhSk+Pl5ubm6qXLmyWrdurTJlytzRXixVnK+nonp+L1++rD/++EMRERGKjY2Vh4eHypYtqxYtWigwMLCQz+LecPPmTe3evVuhoaGKjo5WYmKi3Nzc5OXlpRo1aqhOnTry9LTN7/FCQ0N1/PhxhYWF6erVq0pJSZG7u7t8fX1Vv3591alTRw4Od+77TFJSUvTHH38oNDRUUVFRsrOzU5kyZVSjRg01b978rgg7AgAAAAAAAAAAAFAwyyNi9OLRc7ZuI0ef1w7UE37etm4DAAAAsClCae4QQmksRCgNYDVCaYACIJQGMCOUBsg/QmmA2xFKA9xyJ0JpAAAAAAAAAAAAAAAAAOBuRSgNAAAAUPzxFXMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADNCaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAZoTSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACKjenTp8tkMpn/TZ8+3dYtAQBw3yGUBgAAAAAAAAAAAAAAAAAAAAAAAAAAALgDtm3bZgjayO2fo6OjypYtq86dO2vbtm22bh0AAAD3GQdbNwAAAAAAAAAAAAAAAAAAAAAAAAAAAADAKDU1VZGRkdq0aZM2bdqkypUr69dff1XlypVt3ZpV3nzzTe3YsUOS5OXlpRUrVti4o7vX9OnTtXLlSnO9ZcsWm/UCAADufYTSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMVcaGioatSooT179qhRo0a2bsdiP/zwg06fPi1Jsre3t3E3d7eVK1dq69attm4DAADcJ+xs3QAAAAAAAAAAAAAAAAAAAAAAAAAAAABwP/L09FRGRkaWf+fPn9fMmTNVrVo1w/zU1FS1bdtW6enpNuoYuDNGjhxpuCZGjhxp65YAALjvEEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAFCMVKlTQiBEjdPLkSb366quG+xITEzVmzBgbdQYAAID7BaE0AAAAAAAAAAAAAAAAAAAAAAAAAAAAQDH10UcfqXHjxoaxJUuW2KgbAAAA3C8IpQEAAAAAAAAAAAAAAAAAAAAAAAAAAACKsYkTJxrqiIgIG3UCAACA+4WDrRsAAAAAAAAAAAAAAAAAAAAAAAAAAAAAkLOuXbtmGYuIiJCfn59F65cuXapt27bp3LlzunHjhpo1a6b3338/x/lJSUmaPXu2Dhw4oIiICDk5Ocnf31+dOnXSE088ke/zKCw7duzQsmXLdP78ecXFxal06dKqUqWKhgwZosqVKxdo7+joaM2ePVvHjh3T5cuX5erqqvLly+vxxx9XcHBwvvcNDQ3VDz/8oD///FOxsbFKS0uTm5ubypcvr8aNG+vxxx+Xj49PgXq3xtGjR7Vu3TodOHBA0dHRSktLU+nSpVW5cmX1799ftWvXzvfecXFxWrZsmbZv366oqCjduHFDLi4u8vX1Vb169dS1a9cC7V9cjw0AwL2GUBoAAAAAAAAAAAAAAAAAAAAAAAAAAACgmLOzs1N6erq5vnDhgjmUplq1ajp9+rQkyd7eXqmpqZKkQYMGafHixUpJSTHstXXr1mxDafbv369+/frp2LFj2fYwa9Ys2dvb6+GHH9aiRYvk4eGR7bzp06dr1KhRWcbT0tJkMplyPMeMjIwc77tx44aee+45LV26VMnJydnOmTRpkkqXLq1PPvlEAwcOzHGv7Kxfv15Dhw7V+fPns73/s88+k6Ojo/r166fZs2fLwcGyj2l/9913Gj16tCIjI3OdN3ToULm5ual79+5atGiReXzbtm1q165dtmtyeyx///13tW3b1jA2f/58ffrppzp8+HCOj6H0z+Po7u6ul19+WRMnTsy179sdOnRITz/9tI4ePZrrvDFjxsjR0VGNGjXShg0bVKpUqSxzMr+Gpk2bppEjR96RYwMAgH/Y2boBAAAAAAAAAAAAAAAAAAAAAAAAAAAAALm7PZBGkipUqJDj3ISEBJUvX17z5s3LEkiTk7Fjx6pJkyY5BtLckpaWpp9//lm+vr4KCQmxaO+C2rJli0qVKqUFCxbkGqYiSVeuXNGgQYPUpUsXi/fv06ePHn744RwDaW65efOm5s6dKy8vLx0/fjzPfZ9++mn1798/z0CaWxITE7V8+XKL5lorLCxMAwcO1N69e/N8DKV/XkOTJk1SjRo1lJCQkOf8r7/+Wo0aNcozFOaWmzdvas+ePTp37pxF84vrsQEAuJdZFsEHAAAAAAAAAAAAAAAAAAAAAAAAAAAAwCbWrVuXZczPzy/H+S1btlR4eLgkyWQyKSAgQP7+/kpLS9OFCxcUHx9vmD9mzBhNnTo1yz4BAQGqUKGCUlJSFBoaqpiYGPN9KSkp6tChg7Zu3aq2bdsa1plMJqvOLzfLly9X7969lZGRYRj39fVVYGCg3NzcFBcXp9OnTysxMdF8/8aNG9W+fZ1R47gAAQAASURBVHtt3bo11/0ff/xxrVq1Kkv/lStXVrly5ZSYmKjTp08bHrOEhAQ1aNBAJ06cUMWKFbPdd+rUqfr+++8NYw4ODgoICFDZsmVVokQJJSQkKDo6WhEREbpx40a2++T3scxrnbOzs8qUKSNfX1+5u7srNTVV0dHROnfunKGXkydPqm3btjpw4ECOex0/flwvvPCC4TkymUzy8/OTv7+/SpYsqevXrysuLk7h4eFZXn8FYctjAwBwryOUBgAAAAAAAAAAAAAAAAAAAAAAAAAAACjG3n77bUNdtmzZHOempaXpyJEjkqQOHTpo5cqV8vDwMMxJSEgw396xY0eWQJpatWrp119/lb+/v2F848aN6tGjhzn8JT09Xd26dVN0dLScnJzM81555RW98sorkqRq1arp9OnTkiR7e3ulpqZadM6SFB4erj59+hgCRx566CHNnTs321CexYsXa/DgwUpOTpYkhYSEaPr06Ro5cmS2+8+fPz9LIE27du20du1aubu7Z5k7ZMgQ3bx5U9I/oTzt2rXTuXPnst37/fffN9SjRo3KNvjnlqNHj2ry5Mn67bffDONt2rQxn39QUJAhZCdzUE9uTCaTvLy81KdPH40ePVpVq1bNce7y5cs1ePBgc4DLwYMH9e233+rZZ5/Ndv6wYcMMvbRp00Zr1qxRqVKlsp0fExOjadOm6auvvpK9vb3F51Dcjg0AwL3OztYNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMjeW2+9pf379xvGevfunee6Xr16afPmzVkCaSQZAlf69+9vuK9+/fo6evRolkAaSercubNOnDihEiVKmMfi4+P18ssv59lPfjz66KPmEBhJmjJlin755ZdsA2kkqU+fPjp+/LgcHR3NY++9916O+2fu+5FHHlFISEiWQBpJGjBggHbu3GkIMjl//rw+++yzLHOTkpIUFxdnroODg3MNpJGk2rVra/78+Tp//nyu8/IrMDBQMTEx+vzzz3MNpJGkJ554QhcuXJCLi4t5bMKECTnO37dvn/m2l5eXtm3blmMojCR5e3vr/fffV2RkpOrWrWv5SRSzYwMAcK8jlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAoRsLDw/XZZ5+pRo0amjRpkuE+FxcXTZs2Ldf1JUuW1A8//JDncY4cOaIzZ86YawcHB23ZsiXXNf7+/po9e7ZhbMGCBXkey1qnT5/Wn3/+aa7btm2rMWPG5LmuYsWKevvtt831tWvXtGrVqizzli5dqqtXr5prd3d3rV69Ote9H3jggSw9fPjhh1nmnThxwlAHBQXl2Xdx4+HhoWHDhpnrc+fOKT09Pdu5169fN9+uUaNGkfdWXI4NAMC9jlAaAAAAAAAAAAAAAAAAAAAAAAAAAAAAwAauXr0qk8mU5V/58uU1YsQInTx50jDf3t5eW7dulYODQ6779u3b16Ljz5gxw1B37txZ3t7eea7r37+/vLy8zHVSUpL27t1r0TEtNX78eEP9+eefW7x27NixMplM5nrhwoVZ5syaNctQDxkyRHZ2eX/0+r///a8cHR3N9cWLF5WUlGSY4+PjY6h///13i/oubjp16mSo16xZk+2821+Pp06dKtKeitOxAQC41xFKAwAAAAAAAAAAAAAAAAAAAAAAAAAAABRzFSpU0JEjR9SsWbM85w4ePNiiPbdv326oR4wYYXE/Xbp0MdSLFi2yeK0lbg9ycXJyUoMGDSxe6+DgIBcXF3N96NChLHMOHz5sqN966y2L92/UqJGh/uGHHwx1hQoV5OTkZK43bNigUaNGKTU11eJjFKXTp0+rX79+qly5slxcXGRvb59tOFK3bt0M644cOZLtfpUrVzbfvnLlitq0aaPo6OgiPYficGwAAO51hNIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAxYy9vb18fHzUoUMHbdq0SefPn1fNmjUtWtuiRQuL5oWHhxvqzp07W9xfp06dDPXBgwctXmuJ23tLSUnJNjQlt39JSUnm9fHx8Vn2j4uLM992cnKSj4+Pxb21bNnSUO/YsSPLnN69exvq6dOnq0SJEmrQoIFGjx6tvXv3Wny8whIdHa2WLVuqWrVqWrRokc6ePasbN24oPT3dovURERHZjk+aNMlQ//HHH/L19VWlSpU0cOBArVmzxuJjWMuWxwYA4F5HKA0AAAAAAAAAAAAAAAAAAAAAAAAAAABgA56ensrIyMj2X2pqqqKiorR582YFBwdbvKedneUfH05OTjbfdnBwkIODg8Vra9WqZahvD3kpDDdv3iy0va5fv55lLC0tzXzb1dXVqv2qVKliqKOiorLMWbhwoerXr5/lmIcPH9a0adPUrFkzOTo6qnr16ho7dqwSEhKs6sFakZGRqlq1qnbt2pXvPW4P+rndY489pmHDhmUZDwsL0/z58/Wvf/1LDg4O8vf314ABAxQaGprvHorTsQEAuNcRSgMAAAAAAAAAAAAAAAAAAAAAAAAAAADcI0wmk8Vzbw9+sbe3t+o4vr6+hjoxMdGq9XnJyMgo0r1uH3NycrJqP29vb0N97dq1bOcdOnRIH3zwgTw9PbO9PzU1VadOndLkyZPl6empAQMGWNWHNTp27Jilz3r16unVV1/VypUrdebMGcXGxhqCkX7//XfD/Nyeky+++EI//fSTKlasmO39GRkZunTpkhYsWKAqVaqoWbNmio6OLviJ2fjYAADcywilAQAAAAAAAAAAAAAAAAAAAAAAAAAAAO5Djo6O5ttpaWlWrY2KijLUbm5uhdJTdtzc3AxhKdb+i4uLy7Ln7eE9KSkpVvUTExNjqD08PHKc+9ZbbykuLk47d+7Us88+qxo1asjZ2TnLvPT0dC1YsEA1atSwqhdLHDhwQEeOHDHXzs7O2rdvnw4fPqyPPvpIjz32mCpXrqxSpUoZ1mV+jvPy6KOP6uzZswoLC9Mbb7yhJk2ayN3dPdu5e/fuVUBAgM6dO2f1+RS3YwMAcK8ilAYAAAAAAAAAAAAAAAAAAAAAAAAAAAC4D90ejpKamqr09HSL1x47dsxQZw40KSgHBwfz7eTk5ELdW5Ls7e3Nt5OSkqxae+bMGUPt6+ub55oWLVrom2++0fHjx3Xjxg0dO3ZMzz//vLy9vQ3zTp48qWHDhlnVT16mT59uqD/++GM98MADea47depUvo4XGBioyZMna+/evYqPj9eVK1c0fvx4Va1a1TDvxo0b6ty5c76OURyPDQDAvYZQGgAAAAAAAAAAAAAAAAAAAAAAAAAAAOA+5O/vb6jXr19v8dpNmzYZ6oYNGxZKT7eULl3afDs1NVWhoaGFuv/tITopKSmKjo62eO3OnTsNdatWraw+fs2aNTVr1ixduXJFzz77rOG+xYsXW71fbv7++29D/eKLL1q0bvPmzYVyfG9vb7377rs6deqUfvjhB8N9J06cUExMTKEcp7gdGwCAux2hNAAAAAAAAAAAAAAAAAAAAAAAAAAAAMB9qF27dob6008/tXjtxo0bDXXfvn2znefg4GB9Y5Jat25tqCdNmpSvfXLSoEEDQz1x4kSL1x44cMBQP/XUUwXq5ZtvvpGrq6u5vnr1arbzHB0dDXVKSopF+8fHx5tv29nZyc7Oso+Yh4SEWDTPGr169VLTpk0NY5lfS0XFlscGAOBuRCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAcB965ZVXDPXGjRsVExOT57rFixcb5rm5uWUJ+7j9vlvS0tIs7u3NN9801PPnz1dcXJzF6/MybNgwQ/3NN98oPT09z3VvvfWWbt68aa4rVKigEiVKFLif0qVL5znHw8PDUIeGhlq09+3PQXp6uqKjo/NcM2HCBCUlJVm0v7WqVatmqIvqOMXt2AAA3G0IpQGA/8fefUdHUbb/H/9sGqEm9BA6oTepAgoSEFBQOkrvHX0EVHxUVBAFBURQsCEIqHSQB6UJKC30hN6lBAIhhBZCIBBSfn/wY76Z7CbZDQkb5P06Z8/Z6567XLM72eGws9cAAAAAAAAAAAAAAAAAAAAAAAAAAPAEqlChgvz8/Iw4NjZW/v7+KY4JCwtT3759TW09evRItn+JEiVM8YoVK+zKrXbt2qpUqZIRx8TEqGrVqg4VEYmPj9fEiRNtbuvQoYO8vLyMOCoqSq1atUpxvn379mn8+PGmtvfff9+q34EDB7Rz506784yKilJoaKgRe3h42OxXvnx5U7xw4UK75q9SpYopfv3111Psv3XrVn388cd2zS1Js2bNsruvJG3bts0U165d26HxmWVtAAD+7ShKAwAAAAAAAAAAAAAAAAAAAAAAAAAAADyhfvnlF1N88OBBVaxYUWFhYVZ9//77b5UpU0bR0dFGW86cOTVlypRk509a6KVr166aO3euYmJiUs3tjz/+kLu7uxGHhISoYMGC+u6771Ict3v3brVr107ZsmXTO++8k2y/r7/+2hSvXLlS/v7+NgvfzJ07V3Xq1FFcXJzRVrRoUQ0ePNiq799//626deuqWLFiGjlypCIiIpLN4ejRoypbtqxp3uQKpXTs2NEUjxs3Tt9++62ioqKSnV+Shg8fbooXLlyY7Ovy+eefq2HDhoqPj09xzsT69OkjLy8v9erVS8ePH0+2X1RUlJ5//nmdO3fOaMuZM6cqV65s91qZaW0AAP7t3JydAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADnqFevnt588019+eWXRtvRo0dVqFAhFS1aVEWLFlVMTIzOnDmjq1evmsa6uLho1apV8vDwSHb+nj17avDgwUYhm8jISHXr1k3dunWz6puQkGCKS5Ysqd9++02tW7c2iqRERUVpyJAhGjp0qIoWLar8+fMrS5YsioyM1OXLl3Xp0iXFxsbate89evTQb7/9puXLlxttmzZtUo4cOVSyZEn5+vrq9u3bOnnypCIjI01jPTw8tGXLlhTnDwkJ0bhx4zRu3Dhlz55dhQsXVu7cuZU1a1ZFRkbq7NmzVq+pq6ur5s2bZ3O+qlWrqlChQrp48aIk6e7du3rttdf02muvWfXdtm2b6tWrZ4yrUaOG9uzZY2yfOHGipk2bpnLlyilXrly6du2a/vnnH929e9fo86CAkD0iIyM1Z84czZkzR1myZFGhQoWUN29e5cyZU9HR0bpw4YIuXLhg9R5PnTrVrvkz69oAAPybUZQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAeIJNmjRJHh4e+vzzz03tISEhCgkJsTnG3d1da9euVf369VOdf/78+WrXrp1RWMYRL7/8snbv3i1/f3/dvHnTaL93755Onz6t06dPpzjeYrGkuP1///ufOnfurAULFhhtCQkJKc6dI0cOBQYGqnjx4nbvx61bt3TixIkU+7i7u2vNmjUqVqxYsn1WrVqlOnXqKCYmJsW54uLiTPGmTZtUrFgxXb9+3WiLjo7Wvn37bI4fOHCgunXrZndRmsTu3r2r4OBgBQcHp9hv9OjR6tmzp8PzZ9a1AQD4t3FxdgIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAnOuzzz7T7t27Va5cuRT7ubi4qHnz5goPD5e/v79dc7du3VqnT59W27ZtlS9fPrm6ujqUW40aNRQREaH3339fXl5edo3Jly+funfvnmpxEul+0Zw1a9aoSJEiKfZzd3dXjx49dPXq1RRfpy5duqh///4qUqRIqkVxHsz74DVt3Lhxin2rVaumS5cuqWfPnvLx8ZGbm1uq80v3C+mEhoaqUaNGKebk4+OjBQsW6Pvvv7drXkkaNWqUKlSoYFcuFotFVapU0f79+zVq1Ci718iMawMA8G9nSUhISEjLwJo1ayosLEw+Pj4KCgpK77z+dR7p6zXavn9MZ0qjbzg8JCHsxwxIBHh8WHz6p33w3a3plwjwOMryrPE0/vsKTkwEcD6XQUfTPHZg2SzpmAnw+PnhxN1EUajT8gAyB1/jWcLaZk7MA3A+S7O1zk4BAAAAAAAAAAAAAAAAADKtpWHX9NrRc85OI1nfVCim9j55nJ2GU0VFRWnGjBnat2+fwsLC5O7ursKFC6tJkybq0KGDs9NTWFiYZs2apRMnTig8PFz37t1Tzpw5Vbx4cT3zzDNq0aKFsmXLlqa5r1y5oh9++EFHjx7V5cuXlS1bNvn6+qpVq1Z64YUX0jTnxo0btWnTJh07dkzXr19XXFyccuXKpRIlSujFF19U06ZN0zRvWl27dk0zZszQrl27FBkZqfz586tUqVLq1auX/Pz8Hmruo0ePavXq1Tpw4IAuX76su3fvKnv27CpUqJAaNmyotm3bytPTM532JPOsDQDAv5F9pe8AAAAAAAAAAAAAAAAAAAAAAAAAAAAAPBFy5MihYcOGOTuNZPn4+Oi9997LkLnz5cunkSNHpuuc/v7+8vf3T9c5H0aePHn0zjvvZMjcFSpUUIUKzrmBuDPXBgDg34iiNBnkjz/+0BdffKGoqChJUnh4+KNbfPSNR7dWJmDx6e/sFIDHV5ZnnZ0BkGm4DDrq7BSAx9YPJ+46OwUgE/F1dgJApmFpttbZKQAAAAAAAAAAAAAAAAAAAAAAAABII4rSZJAvvvhCJ0+edHYaAAAAAAAAAAAAAAAAAAAAAAAAAAAAmUrxrFmcnUKKMnt+AAAAwKNAUZoMEhUVJUlycXFRgQIFFB4ervj4eCdnBQAAAAAAAAAAAAAAAAAAAAAAAAAA4Fy1vLJrRY0yOht919mpWCmeNYtqeWV3dhoAAACA01GUJoMVKFBAQUFBqlmzpsLCwh7JmpM6vvxI1skIby1c4fCYhNBvMyAT4PFh8R2S9sF3N6VfIsDjKEtD42nCj5WcmAjgfJb+h9M8dmBZKsDjyfbDicRfhIU6LQ8gc/A1niXs7OTEPADns9RZ4OwUAAAAAAAAAAAAAAAAACBTq+WVneIvAAAAQCbm4uwEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACZB0VpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGitIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwUpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGChKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwUJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGCgKA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEBRGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgaI0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADRWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAaK0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADBQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYKAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFEaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAIDBzdkJAAAAAAAAAAAAAAAAAAAAAAAAAAAA4Mly+Zp067azs7CWPZuUP4+zswAAAACcj6I0AAAAAAAAAAAAAAAAAAAAAAAAAAAAeGQuX5PWb3V2Fslr8iyFaQAAAAAXZycAAAAAAAAAAAAAAAAAAAAAAAAAAACAJ8et287OIGWZPT8AAADgUaAoDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFEaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBojQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANFaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAA8sby9vWWxWGSxWOTt7e3sdJ4obm5uxmtfunRpZ6cDAAASoSgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAkAECAgKMghsZ8XBzc3P2LgIAAOBfin9pAgAAAAAAAAAAAAAAAAAAAAAAAAAAAADSxN/f33jepk0bDRs2zGm5AACA9ENRGgAAAAAAAAAAAAAAAAAAAAAAAAAAACADWCyWx3p+wB6bNm0yxRSlAQDg38HF2QkAAAAAAAAAAAAAAAAAAAAAAAAAAAAA/0bPPvusEhISUn1s2bLFNM7Ly8uucffu3XPSngHpIzY21jieT5486ex0AABAIhSlAQAAAAAAAAA8FoKDg2WxWIxHr169nJ0SkG78/f1Nx/fjrFevXqZ9CQ4OdnZKkv5drzFs27hxo+k9Hj16tLNTAh5bo0ePNv09bdy40dkpZUqZ9ZwHAAAAAAAAAAAAAAAAAOnBzdkJAAAAAAAAAAAAAMC/TWRkpPbu3avTp0/r6tWrunPnjnLmzKncuXOrcOHCqlWrlry8vJydJgAAAAAAAAAAAAAAAAAAgE0UpQEAAAAAAACATKZEiRI6e/asEW/YsEH+/v5OnwsAAKQsMjJSP/30kxYsWKDdu3crPj4+2b4Wi0UVKlRQhw4d1KNHD/n5+T3CTNPm+++/1+DBg01tvXr10qxZs5yUEQAAAAAAAAAAAAAAAAAAyCgUpQEAAAAAAAAAAHhIFovFeN6wYUNt3LjRecngsZK4eFjx4sUVHBzs3ISQJrGxsZoyZYo++eQTRUZG2jUmISFBR44c0ZgxYzRmzBi1bdtW48ePV5kyZTI427SbPXu2VduSJUs0bdo0Zc+e/dEnhGT5+/tr06ZNRpyQkODEbAAAAAAAAAAAAAA8an/++af+/PNPnT17Vjdv3lSZMmX0zTffJNv/6NGjWr16tfbt26crV64oLi5OefPmVcmSJdWtWzdVqFAhXfObP3++Nm/erODgYGXNmlWlS5fWwIED03xDl+3bt+v333/XP//8o8jISLm4uChHjhwqVaqUnn76abVq1UoeHh7pug9J/fnnn1qxYoUuXLig27dvq2DBgqpQoYIGDBigPHnyPPT8YWFhmjlzpv755x9dunRJCQkJypcvn2rVqqUOHTqoSJEids8VExOjP/74Q1u3blVwcLBu3ryp7NmzK3/+/PL399crr7yS4a9Xelq0aJE2bNig0NBQxcTEqFChQqpevbr69u2rbNmypds6q1at0po1a3Tq1Cm5u7urWLFi6tOnj6pVq5ZuawAAkNlQlAYAAAAAAAAAAAAA0igsLEwdOnTQ1q1bbW739PRUgQIFlD9/ft25c0cXL17UtWvXrPotW7ZMK1as0M6dO1W9evWMTtthx48f186dO63ao6KitHTpUvXo0cMJWQEAAAAAAAAAAADAkym5G1W8//77+uqrr3T79m1T/3Xr1lkVpfn55581depUHTx4UHfv3k12rXHjxilHjhx64403NHbsWLvyK126tE6dOiVJcnV1VWxsrCRpxIgRmjp1qs31Jk6cKD8/P/3vf/9T5cqV7Vpn+PDhmj59utX+2pI/f36NGDFCI0aMsGtue8TExKh3795atGiRsY9JvffeeypevLhmz54tf39/h9eYMGGCPv/8c12/ft3m9rlz52r48OHy8vJSu3btNH36dLm5Wf98/OzZs/rkk0+0YsUKXbp0Kdn1ZsyYoW7duqlmzZqaPXt2iu9F4vc5sU2bNplu8pWYn5+fTp48aWpzc3NTXFxcstttuXbtmrp06aJ169YpPj7eavusWbP0xhtvqFKlSlqwYIFdx1Ryf1dfffWVPvjgA0VFRVmNmTp1qnx8fDR37lw1btw41TUAAHjcuDg7AQAAAAAAAAAAAAB4HJ0/f14NGjSwKkjj7e2td999VwEBAYqKitLZs2cVGBioQ4cO6erVq4qIiNC8efPUqVMnubu7G+Pu3bunGzduPOrdsMvs2bOT3TZnzpxHlwiQicyePVsJCQnGo0SJEs5OCQAAAAAAAAAAAE+wqlWr6rPPPrOrQMvZs2fVs2dPBQYGpliQ5oGoqCiNGzdOZcuWtVmYIzXx8fF66qmn9MUXX6S43qlTp1S9enVt3749xfnu3LmjUqVKacqUKXbtryRdvnxZs2bNcijvlBw6dEi5c+fWvHnzki1I88DZs2fVqFEj9evXz+75Q0NDVahQIf33v/9NtiBNYjdu3NCsWbN04cIFm9vbtGmjmTNnpliQJrGgoCA99dRT+u677+zO+VH5888/VbBgQf355582C9IkdvjwYVWpUkWffvppmtZq3ry5hg0bluJxHxYWpiZNmmjRokVpWgMAgMzMutQdAAAAAAAAAAAAACBF9+7dU4cOHazuztW/f3999tlnyps3b7Jjvby81LlzZ3Xu3FmnTp3SyJEjtWjRIuMOW5lNfHy8fvnlFyPOnj27ypQpo3379kmSNmzYoHPnzqlYsWJOyhAAAAAAAAAAAAAAnmwtWrTQwYMHjbhQoUIqUqSIXF1dFRoammyhkgeyZMmiAgUKKH/+/MqRI4diY2N15coVnTt3Tnfu3DH6/fPPP6pfv77xfbG9GjVqpAMHDkiSXFxcVKxYMfn6+io2NlZnzpzR5cuXjb6xsbFq0aKFrl69KhcXF5vzvfDCCzpz5oypLVu2bCpWrJjy5s0rV1dXRUVF6eLFi7p8+XKqRWMc9c8//6h69epW8+bMmVN+fn7Knj27Ll68qDNnzpiuBZg5c6Zu3bql+fPnpzj/mTNnVKFCBasCPq6uripWrJjy588vDw8PXbt2TSEhIbp586ZD+VssFnl5ealQoULy8vKSp6enbt68qfPnz5uK1sTHx2vIkCGqVKmSnnvuOat5knt/UpKWMYmtW7dOzZs3t7rGIk+ePCpZsqQ8PDx0/vx5hYSEmLZ/+OGHunv3rj755BO71+rTp4/WrFkj6f5r5uvrq8KFC8vNzU1nzpzRxYsXjb4JCQnq3r27mjVrJm9v77TvIAAAmczDnbkBAAAAAAAAAAAA4An03nvvaefOnaa2sWPHavr06SkWpEnKz89PCxYs0LJly5QzZ870TjNdrF+/3nSBYtu2bdW3b18jTkhI0M8//+yM1AAAAAAAAAAAAAAAklavXi1JqlKlikJCQhQaGqpdu3Zp+/btOnv2rCIiIkz9LRaLcufOrSFDhujkyZO6c+eOzp07p6CgIG3atElbt27V8ePHFR0drSVLlpi+z96/f79mzpxpd25xcXHavHmzpPvFc27cuKEzZ85o69at2rlzp8LDw7VkyRK5uroaYyIiIvTFF1/YnO/atWvGfJLk7u6uBQsW6NatWzp69KgCAgK0adMmBQUFKTQ0VPfu3dPatWvl7++vggUL2p13Sho2bGgqSJMlSxb9+uuvioyM1N69exUQEKBTp04pIiJCdevWNY1dsGCBFi1alOzcsbGxqlOnjqkgjaurq0aMGKHY2FidPn1aO3fu1JYtW3T48GFFRkYqJCRE/fv3l6enZ7LzFihQQE8//bR+/fVXxcTE6Pr16zpy5Ii2b9+uDRs2KDAwUGFhYbpx44ZatWplGtu+fXubc544cUIJCQlWBWIaNmxotCd9nDhxItkcU3Pnzh21bdvWtF7OnDn1119/6erVqwoMDNS2bdt07tw5hYSEqHTp0qbxY8eOVWBgoN3rzZo1S5JUs2ZNhYaG6vz589q5c6e2bt2q0NBQbdu2zfSax8TEaOjQoWnePwAAMiOK0gAAAAAAAAAAAACAA/bv368vv/zS1DZo0CC9//77aZ6zdevW2rFjhwoVKvSw6aW72bNnm+Ju3bqpU6dOcnNzM9rmzJnziLMCAAAAAAAAAAAAACRWs2ZNHThwQEWKFLHaliNHDlNcrFgxXbt2Td988438/PxSnLd9+/Y6f/68smbNarSNGTPG4fwGDx6slStXWuXyYI1vvvnG1PbDDz/YnOdBoZAHpkyZoo4dO6a4dtOmTbVhwwZt2LDBwaytffnll7p48aIRu7m5KSgoSF27drXqmytXLm3fvl2NGjUytQ8aNCjZ+f/zn//o8uXLRuzu7q5du3ZpwoQJyY4pUqSIpk+frujoaBUtWtRmnz///FM7d+5U165dTd/328p5+fLlGj58uNF25coVrVq1Ktkxj8qQIUN069YtI86ePbtOnz6txo0bW/UtUqSI/vnnH5UpU8ZoS0hIUJcuXRxas2nTpgoMDJSPj4/Vtnr16hkFoR5Yvny5Q/MDAJDZJf+vBgAAAAAAAAAAHoH4+Hht27ZNJ06cUFhYmDw9PVWqVCk1aNBAefPmzbB1r169qh07dig0NFSXL19Wjhw59OKLL6ps2bIpjjt+/Lj27t2r8PBw3bp1S/ny5ZOvr6/q168vLy+vDMv3cbV79279888/unDhglxcXOTn56dGjRql+lrduXNHAQEBOnr0qG7evKncuXOrfPnyatCgQYoXRdjr9u3b2rp1qy5cuKDw8HC5urqqQIECqlixomrUqCGLxfLQa6SnGzduaPPmzTp58qSio6OVN29elS5dWvXr11eWLFmcnV66io2N1datW3Xy5EldunRJnp6e8vPzU4MGDZQnTx5np5fp3blzR0eOHNHRo0d1+fJl3bp1Szlz5lTevHlVpUoVVa5cWS4u6X/filOnTunAgQMKDQ1VRESE8uXLp06dOv1rPxcnTpxouutW8eLFNXHixIeet2LFig6P2bdvn44cOaLw8HDduXNHBQoUUNGiRVW/fn3TBYFpFRkZqf/9739G7OPjoyZNmsjV1VUvvPCCVq5cKUk6efKktm7dqmefffah13TU2bNntXfvXp0/f16RkZGyWCzKnj27ChUqpFKlSqlSpUop3g0uM9m/f78CAwMVHh6uLFmyyMfHR88884xKlCjh7NRM7t27p23btunQoUOKiIhQrly5VLRoUTVs2FC5c+dOlzXOnTunwMBAXbp0SdevX5eXl5d8fHz07LPP2rzYzxFhYWHas2ePgoODFRkZqfj4eGXLlk0FChRQqVKlVLlyZZsXwqanmzdvau/evTp+/LgiIiJ09+5dZcuWTblz51aJEiVUsWLFdLtLIwAAAAAAAAAAAP793Nzc9Pfff2fY/Lly5dLgwYONG7icO3dO8fHxdl+DUKhQIX377bcp9hk4cKDeeusto+hISEiIzX7BwcGm2NEiIw/riy++MMWjRo1SpUqVUhyzZs0aeXt7Kzo6WpJ0/fp1LVu2TG3btjX1i4+Ptyq6M2fOHNWoUcPu/NLrupAvv/xS3377re7evStJ+u6779SiRYt0mTutFixYYIp/+eUX5cuXL8UxmzdvVuHChRUfHy9J+ueff3T8+HGVK1cu1fWyZs2qNWvWpNjH399fRYoU0fnz5yXdv84rJiZGHh4eqc4PAMDjgKI0AAAAAAAAAPCE27Fjh+rVq2fETZs21dq1ax2e5+uvv9bQoUON+J133tH48eOT7R8XF6fJkydr8uTJCg0Ntdru6uqqdu3aacKECWn6IXaJEiV09uxZSfeLBTy4GGH79u367LPPtGbNGt27d880ZvLkyTaL0ty9e1dTp07V999/r1OnTtlcz83NTQ0bNtTo0aNVv379ZPO6ceOGqlevrjNnzhhtY8eO1fvvv2/XfgUGBurZZ59VTEyMpPuv04YNG9SgQQNJj/b93Lhxo+kuPqNGjdLo0aMVFxenb775RtOmTdM///xjNXe2bNn02muvacyYMVZFAm7evKlPP/1U33//vSIjI63G5s+fX5999pn69u3r8D5J0tatWzV27Fj9/fffxgUTSRUoUEADBw7UiBEjlDNnzmTn8vf316ZNm6zaN23alGJRmwevkz0uXryokSNHau7cucZ7nlj27Nk1bNgwvffee8qePXuy87z44ov6888/jXjr1q165pln7MrhgXv37qlIkSIKDw+XJHl6eio0NDTdig/cvXtX48aN07fffqsrV65YbXd1dVWHDh00YcIEFStWTMHBwSpZsqSxvWfPnpo9e3a65NKrVy/NmTPHiM+cOWP351Byfxcpzf/A2bNnUzx2ktvH8+fPa8GCBVq5cqW2b9+e7LEtSblz51bv3r311ltvydfXN/UdklJ8rX/99VdNnTpVu3btshpXp04dHTt2TJ07dzba+vfvr+nTp9u1bmJvvvmmJk+ebMTffvutBg8e7PA86eH8+fNatGiRqe2tt97K8AIWid28eVPjx4/XrFmzbJ5Dpft/oy+++KI++eQTVa5cOc1rLVy40LgoTpI6deokV1dXSVK3bt2MojSSNHv27EdWlCY+Pl4zZszQN998owMHDqTY18PDQzVr1lT79u01ZMgQU7GezHDelKT58+fr448/1vHjx23OX6dOHX3xxRcp/htj9OjR+vjjj21uS+mzpWHDhtq4cWOy2xO7e/euJkyYoClTpujatWtW2x98Vo8fP17Fixe3a87EYmJi9N1332n69Ok6cuSIzT4Wi0U1a9bUhx9+qFatWjk0/+LFizV58mRt3749xX6urq6qWrWq2rRpo9dffz3ZomhpOV/s2bNHn376qVauXGnz3J5YyZIl9dJLL2nw4MFpKloFAAAAAAAAAACAJ8dzzz2nXLlyZegaTZo0MYrSSNKKFSvs/s5u2LBhdvUrX768goKCJN2/VuX27dvKli2bqY+3t7cpXrRokQYMGGDX/A8rKipKFy9eNGIPDw998MEHqY7z8PBQz5499f333xtt3377rVVRmiVLlpiu+yhcuLDpuotHrWDBgjp37pyk+zfNcabt27ebrl/Inz+/1etni4+Pjxo1aqS//vrLaPvqq69SLZIkSa+88opdRX7q1q2rJUuWGHFAQIAaN26c6jgAAB4H6X8bRAAAAAAAAADAY6Vu3bqqWrWqEa9fv97qbjL2+PHHH43nFotF/fv3T7bvtWvX9Mwzz2jEiBHJ/pg+Li5Oixcv1lNPPaUNGzY4nI8tEyZM0LPPPqs//vjDqiBNcg4fPqyKFStqxIgRyRakkaTY2Fj99ddfatCggfr06ZPs/F5eXpo/f77c3d2NtlGjRmnr1q2p5hIZGalOnTqZfsA8atQooyCN5Jz3M7Fbt26pefPmGjp0qM2CNJJ0+/ZtTZw4Uc2aNTNdKHDq1CnVrFlTEyZMsFmQRpIuX76sfv36afjw4Q7szf28Xn31VdWvX1+rV69OsWhHeHi4PvnkE5UtW1a7d+92aJ30tGXLFlWuXFmzZs1K9kfrt27d0tixY9WoUSObxQEeGDhwoCmeMWOGw/n8/vvvRkEaSerQoUO6FaQ5d+6cqlWrpjFjxtgsSCPd/0xYuHChqlatarMY0JPqwIEDKlasmEaMGKGNGzemeGxL9++09eWXX6pixYpavXp1mte9e/eu2rdvr+7du9ssSPNAu3btlD9/fiNesGCBcUczR9b6+eefjThbtmzq2rWr40mnk6TnEE9PT3Xr1u2Rrb9p0yaVLl1aY8eOTfYcKkl37tzR//73P1WrVk0jR45M83pJCyEl3tfWrVubinctWrTI9LmeUa5fv67nnntOAwcOTLUgjXS/0Mn27dv19ttv68KFC6Ztzj5vxsTEqFu3burSpUuyBWkkaefOnfL390+34ltpceHCBdWtW1cfffRRsuecB5/VtWrV0t69ex2af+fOnSpfvryGDRuWbEEaSUpISFBgYKBat26tVq1a2fWZcvfuXbVr106vvvpqqgVpHuzH3r17NWrUKO3Zs8eh/UjJ559/rtq1a2vZsmWpFqSR7he5mTZtmubNm5duOQAAAAAAAAAAAODfyZ7iHMk5deqUunbtqpIlSypr1qxydXWVxWKxerRo0cI07vDhw3av0a9fP7v6Jb5pjiSr73glqX379qb4tdde06+//mp3Lg9j4cKFprhGjRp2j/3www9N8f79+636LFiwwBR36dLFgezs98MPP6h27drKmzev3N3d5eLiYvM9f1CQRpIiIiIyJBd7JX1tmjZtavfYN954wxRv2bLFrnH2vv4VKlQwxSldzwEAwOOGojQAAAAAAAAAAA0aNMh4npCQoJkzZzo0fseOHTp06JAR+/v7q3Tp0jb73rx5U82aNbMqYmCxWFSiRAnVqlVLJUqUMNojIyPVunVrHT161KGckvrhhx/03//+VwkJCZLu332mbNmyql27tgoXLiyLxWI1JjAwUA0aNNDp06dN7e7u7ipTpoxq1aolX19fq3GzZs1Sq1atkv2xcZ06dTRu3Dgjjo2NVefOnVMsKiLdLyySuDBO48aNbRYaeJTvZ2IJCQnq1KmT1q1bZ7T5+vqqVq1aqlixolxdXU39t2zZoqFDh0q6XwimcePGRiEbi8WiUqVKqXbt2ipVqpTVWlOmTNHcuXPt2p/w8HA1bNhQixcvttpWpEgR1axZU9WqVbMqsBIWFiZ/f38FBATYtU56CgoKUvPmzY1jwsXFRX5+fnr66adVpkwZq+N19+7d6tWrV7LztWzZ0nSsLlq0KNnCP8lJXHBBkt0FF1ITFhamRo0a6dixY6b2pMfAg32+ceOGWrZsadX/SRUTE2N8rj3g4eEhPz8/Va9e3Thm3NzcTH1u3Lihl19+Oc1Fv3r16qXffvvNiHPmzKlKlSqpevXqypcvnymX3r17G/HNmzetLhJKzbJly3T16lUj7tixY4bf3S0lSS9Meu6559KtQFNqVq5cqRdffNFUIEq6XxinfPnyqlGjhqkIkHS/uMa4cePUt29fh9f7559/tG3bNiMuX768atasacRZs2Y1XdgYGRmpZcuWObyOIxISEtS6dWurYm4Wi0W+vr6qUaOG6tSpo4oVK1rdHS85zjpvSlLPnj1N57PcuXOratWqqlGjhlX+cXFx6tevn1MKpkVERKhJkyamO98VLlxYtWrVUuXKlZUlSxZT/ytXrqhVq1Z2n2v++OMPNWrUSGfOnDG1e3h4qFy5cnr66adVvnx5q8+yP/74Q40bN9adO3dSnL9fv342j80CBQqoevXqqlu3ripVqmT195OeZs6cqffee0/x8fGm9pw5c6pKlSqqW7eunnrqKRUtWtTmv0sBAAAAAAAAAACAlDz//PMOj7ly5Yrq1q2r0qVLa968eQoODtadO3esvtNKTlhYmF39LBaL8uTJY1ffxDdGkWT1/bgkVatWTcWLFzfi2NhYde/eXdmyZVODBg302WefmYqppKedO3ea4rp169o91tfXVx4eHkZ8/fp1qz5Jb+DRqVMnBzNM2Q8//KCsWbNq0KBBCgwM1LVr1xQbG2t17Ykt9tx4IyMdPHjQFDdp0sTusUkLKtkqdmRL9erV7eqX9Pv9xNe5AADwuKMoDQAAAAAAAABAXbt2VY4cOYx41qxZiouLs3u8I8UqRowYoaCgICO2WCx6/fXXFRwcrDNnzmj37t06c+aMTp8+rYEDB0q6X8RgyJAhdueT1LVr1zR8+HBJUqFChTRz5kxduXJFx48f165du3T+/HmdOnXK9EX1zZs39corr5i+/M+WLZsmTJigsLAwnThxQrt379aFCxe0f/9+tWnTxrTmmjVr9NFHHyWb01tvvaXmzZsbcUhIiPr06ZNs/xkzZpgKOeTPn1+//vqrXFys/6v/Ub6fif38889asWKFJKlz5846cuSILly4oN27d+vw4cO6dOmS1fs4Y8YMHTx4UD169NC5c+fk6empjz76SKGhoTp16pR27dqlU6dO6dixY3ruuedMY99++23du3cvxZzi4+PVqVMn0zGXP39+TZw4URcvXlRISIgCAwO1d+9eXblyRQEBAWrcuLHR9/bt2+rcubPNCwUmTZqkdevWmYrwSFLVqlWNdluPHj16pPpavvrqq7p165by5MmjL7/8UuHh4Tp58qR27typEydO6Pz581ZFaP744w+tXr3a5nxubm6mghS3bt3SvHnzUs3jgbNnz5r2s1y5clbvR1oNHDjQVPjJzc1N7777rvF3+eAYCAkJ0TvvvCM3N7eH/kxwtnfeecc4HgoWLGi0FyxYMMVj55133kl2zoYNG2ry5Mk6dOiQbt26pZMnT2rPnj3GMXPz5k3973//09NPP22MiY+PV7du3RQVFeVQ/mvWrDE+j2rVqqU///xT165d06FDh7Rnzx5dvnxZ27dvV+HChSVJAwYMMBVYSPoZk5qMKoiUVkmL0tSqVeuRrBsSEqJu3bqZim/kzZtXP/74oy5fvqyjR48qKChI4eHh2rZtm+rXr28a/9NPP+n77793aM05c+aY4q5du1r16datW4pj0tuSJUtM70H27NmNz/QLFy4oKChIO3bs0OHDh3X9+nWdO3dOc+bMUbt27awKmjzgrPPmL7/8Yvwtvfjii9q+fbuuXr2q/fv3KygoSFeuXNGyZctMRcXi4uL0+uuv25yvR48exudF1apVTdtS+myZNGlSqrm+9dZbOnbsmNzc3PT666/r5MmTOn/+vHbv3q2DBw/q6tWr+uKLL+Tu7m6MOX/+vMaOHZvq3IcPH1bHjh0VHR1ttDVo0EArVqzQjRs3dOzYMe3cuVNHjx7VtWvXNH36dNNn565du4x/59mye/du010R3dzc9N577yk4OFiXLl3Snj17tH37dh06dEjh4eEKCwvT4sWL1aNHD2XNmjXV/O1x9+5dq8/w9u3bKzAwUDdu3NCBAwe0fft27du3T+fOndONGzf0119/6a233jLtKwAAAAAAAAAAAJCcxEVa7BEeHi4/Pz+rIiuOuH37tl39bF3fZG/f5IqlbN682aqATXR0tAICAvT++++rePHiypYtm2rVqqUZM2bYvX5qrly5Yopt3WwrJYm/g7T1vXTSG38k/e73YYwYMUKDBg1K9aYfybG3WFFGiYiIMMXly5e3e6ybm5vphmr2vgYFChSwq5+9xy0AAI8j21fdAQAAAAAAAAAyjaCgIMXGxqZprL1fnubKlUudO3c2flR94cIFrVq1Si1btkx17M2bN7Vw4UIjzps3r9q1a2ez744dOzR9+nQjtlgs+vnnn61+0C5JJUuW1Pfff6+aNWtqwIABCg4OtmtfkstRkipUqKC///5bPj4+NtdL7P333zet6eXlpQ0bNti8+0nVqlW1bNkyffDBB6YfX0+cOFGvvvqqatSoYTXGYrFozpw5qlatmkJDQyVJy5cv19dff6033njD1PfIkSMaOnSoaezPP/+sQoUK2dzfR/V+JvXg9friiy/01ltvWW3PmzevvvnmG0VHR2vWrFmS7n8B37FjRx09elQ5cuTQqlWr1KBBA6ux5cqV0+rVq1WrVi0dPXpU0v27La1cudKqIFBiEydO1IYNG4y4Tp06+v33321eMODi4qJnn31W69at09ChQzVt2jRJ939UP2bMGH311Vem/jVr1rS5Zu7cuR26E48tp0+fVokSJbR+/Xr5+flZbff19dWsWbPk7u5uKoYwffp0U7GjxPr3769x48YZF7T8+OOPGjRokF35/PTTT6YLS/r16+fI7iTrjz/+0O+//27E7u7uWrZsmV566SWrvoULF9b48eNVv359tWvXTmfOnEmXHJyhYsWKqlixoiTJ09PTaPf09HT42ClWrJgOHTqkSpUqpdjP09NTrVu3VsuWLTVw4EDjgqvQ0FD98ssvGjx4sN1rXrp0SZL0yiuvaN68eTYLfSS+E5ifn5+aNGliFDbauXOnDh48qCpVqqS61unTp01/w5UqVVK9evXszjW93bp1S+fPnze12XtXrIc1ZMgQ0wVWRYsW1ZYtW2xeWFivXj1t2rRJvXr10i+//GK0v/XWW2rVqpWpyElyEhISTGMtFovNojTPP/+8ChUqpIsXL0qS1q9frwsXLhhFidLb4sWLTfEff/yhRo0aJdu/aNGi6tGjh3r06KGQkBDlzp3bqo+zzpsPCnJ9+OGHGjNmjNV2V1dXtWnTRhUrVlT16tWNCzp37dql/fv366mnnjL1L1WqlHHBY9L9TI/zUpYsWfTbb79Z3UFOul8c6K233pKPj4/p33WzZ8/Wp59+aipWk1hsbKw6depkKkjz8ccf68MPPzQVs3ogZ86c6t+/v1q0aKFGjRrpn3/+kSR9//33GjBggM2/x6THzPTp09W7d+9k97VgwYLq0KGDOnTooC+//DJdLqz866+/dO3aNSPu0aNHigWccubMqcaNG6tx48YaN26c1ecOAAAAAAAAAAAAkFS2bNkc6t+4cWOrAiiVK1dW8+bN9eyzz6pq1arKnTu3vL29je0BAQGm63qcWXijWLFiCg8PV+/evbV06VKbN7aKjo5WUFCQ+vfvr6FDh2rcuHGma7DSIulrZus76JRkyZLFeG7r9bt7964pTu7mK47aunWrvvjiC1Nbjhw51KRJEzVr1ky1atVS0aJFlSdPHnl4eBh9SpcurVOnTiWb76N069YtU5wvXz6Hxru5uRnXTaX1ekwAAJ5E9pcXBAAAAAAAAAA4xdtvv62mTZum6fGgcIA9Bg4caIoTF7pIyfz5801f+Hbv3t305XliX331lenL6ddee81mQZrE+vfvny4FMNzd3bVo0SKbBWmSioiI0E8//WRqmzlzZqqFBz799FNTQZD4+HhNnjw52f758+fX3LlzTXdKGTFihPbs2WPE0dHR6tixo+nOQm+99ZZefPHFFHN5FO+nLR07drRZkCaxTz/91LTPD4rMfPnllzYL0jyQLVs2ffjhh6a21atXJ9v/9u3bmjBhghEXKlRIq1atSvUONi4uLpoyZYqpqMZPP/1kdbedjOTq6qrFixfbLEiT2Pjx401FTdauXZvsj+eLFi1qOj737NljOtaSExcXZ/p78PDwUM+ePVMdZ4+vv/7aFI8cOdJmQZrEWrZsqXfffTdd1v83KFCgQKoFaRJzcXHRN998Yzq2HhSJckSpUqU0Z84cuy9+Sutn0owZM0znjf79+9ufZAZIXFTiAXvvivUwjh8/rpUrVxqxi4uLlixZkuKd7lxcXPTTTz+Ziv/cvn1b3333nV1r/v333zp37pwRP/PMM1YF3B6s07lzZyOOj483FbNJbydOnDCely9fPsWCNEkVLVpUOXLksLnNWefN1q1b2yxIk1jZsmX1n//8x9SW0vkvo3z++ec2C9Ik1rVrV9WpU8eIw8PDFRQUlGz/JUuW6NChQ0Y8cOBAffTRRzYL0iRWuHBhLV261PTviUmTJtnsm/iYyZEjh3r06JHi3InlzZtX+fPnt7t/chLnIN0vMmUvDw8Ph++uCAAAAAAAAAAAAKRk3759Onz4sBFnyZJFQUFBOnjwoCZMmKDWrVurZMmSpoI0knT58uVHnGnKPD09NX/+fMXExGjx4sVq27atihYtavoe8YHbt29r2LBhKd7Awh65cuUyxdevX3dofOKiM7a+F018HZCUfsVTkn4n3rZtW928eVPLli3T4MGDVbt2bfn4+JgK0kj23xTvUciePbspvnLlikPjE7+W6VXsBwCAJwFFaQAAAAAAAAAAkqSaNWuqVq1aRrxq1SpduHAh1XFJf7SdXMGAiIgILVu2zIg9PT318ccf25XbuHHjrL7wdlTHjh1VuXJlu/rOmzfPVATm2WefVfv27e0a++WXX5riRYsW6caNG8n29/f3NxVaiYmJUceOHXXz5k1J0tChQ00/1n766ac1bty4VPPI6PfTFovFkuoP6yXJ19fXlJskFS9eXH369El1bMuWLU0XjuzduzfZvj///LOpgMTo0aOVJ0+eVNeQ7heFee+994w4KipKf/75p11j00P79u2tXiNbcufOrWbNmhnx7du3dezYsWT7Dxo0yBTbU3RhzZo1On/+vBG3bt06XX6gf+HCBf31119G7O3trf/+9792jX3vvffk5eX10Dk8qTw8PPTKK68Y8d69exUdHe3QHO+//76yZs1qd//WrVurUKFCRvzrr7+meuFSbGysZs+ebcRZsmRR9+7dHcozvdkqSpP0IryMMHPmTFNxns6dO+vpp59OdZybm5smTpxoavvxxx/tuntZ4tdeUopF5JJumzNnTqrzp1XiY9Xd3T3d5nXGeVOSXed06f6/YxKzp6hYeipcuLBee+01u/o6kuuUKVOM59myZdNnn31md05VqlRR69atjXj58uXGXe0SS3zMuLi42LwANaMl/YxNz2MXAAAAAAAAAAAAcFTi7+kkaeLEiapRo0aq406ePJlBGT28Dh066LffftO5c+cUFxen9evXq3Xr1lY3FZk9e7Y2btyY5nXy5ctnik+fPu3Q+MTfHbq6ulptT1r0Zt++fQ7Nn5zE1xN5e3vrt99+s2vcg2vYMoOk12ekdI1UUrGxsabvk5MW/wEAAMmjKA0AAAAAAAAAwJD4jihxcXGaNWtWiv3379+vwMBAI37mmWdUsWJFm323b99uutPLSy+9ZHeBkPz586tFixZ29U1O586d7e67adMmU2xPsZQHypcvr2eeecaIY2JitGPHjhTHfPjhh2rYsKERnzx5UoMGDdLChQtNP3b38vLSggUL7P4hc0a+n7ZUrVpVZcuWtatv0gJBbdu2tXmhRVI5cuRQiRIljPjcuXPJ9l21apXx3M3NTZ06dbIrtweef/550w/Xt2zZ4tD4h5H0B/0pqVatmikOCQlJtm/z5s1VrFgxI05agMmWGTNmmGJHCy4kZ9u2babCGO3bt7f7go9s2bKpXbt26ZLHk6pkyZLG89jYWFPxq9S4urrq1VdfdWg9Nzc302fp9evXtWTJkhTHrFy5UhcvXjTi9u3b233eyCi2LrZKeieujPAw56WmTZuqSJEiRnzp0iWdOHEixTE3b940XYDm7u6e4ntevXp10/ni2LFj2rlzp905OsLX19d4fuTIkRSLkznqUZ83q1SpYnf/ypUrm+7UltJnfUZo166d3f/+sPe8dPXqVe3atcuIX375ZeXOnduhvBIXZouKirJ5PCQ+ZiIjI/XHH384tEZ6SJyDdL8wFwAAAAAAAAAAAOAsR44cMcX23qDi77//zoh0MsTzzz+v//3vf4qKilKZMmVM20aNGpXmeevUqWOKU7suLLGwsDDFxMQYsa3vRytVqmSKFy1a5GCG1q5cuWIqyFKvXj27xsXExCgyMvKh108vVapUMcXr16+3e2zi68ik+zdmAQAA9qEoDQAAAAAAAABkchs2bFBCQkKaHsWLF3dorc6dO5vutjJz5kxT0YikEhdMkVIuVpH4R8eS5O/v71BujvZP6umnn7a7b9If0jdu3NihtZ5//nlTnNrFB66urpo7d67pTjrz5s1Tjx49TP2mT59uKiSRmox8P22pWbOm3X3z5s1riu2525Ktscld+JCQkKCtW7cacdmyZa3uJJSa7Nmzm9Y6evSoQ+MfRq1atezuW6BAAVN848aNZPu6uLiY3tfIyEgtXLgw2f5hYWFasWKFEZcsWVJNmjSxO7eUJC7kIMlUzMkejvZ/Ety+fVsLFizQwIEDVbduXfn6+ipnzpxycXGRxWIxPRIX35DuX4Bkr/LlyytnzpwO5zdgwABToaeknzlJPexnUkawtd+3bt3K0DXv3r1ruvOZu7u76tevb/d4FxcXNWrUyNSW2nlp8eLFpoJVzZs3T7UgUNeuXU3x7Nmz7c7REU2bNjWex8XF6cUXX9SMGTNSLbBlj0d93nTks97d3d1017eUPuszQkaclwICAkyvryNrPJC40Jpk+1yd+JiR7h+rkyZNUkREhMPrpVXjxo1NxfcmT56sIUOGOHzXRAAAAAAAAAAAACA9JL4hi4uLi+m7/JRs3rw5o1LKMG5ubtq4caOpLbUbuaQk6Y3R9uzZY/fYMWPGmOKnnnoq1fnnzZvnQHa2Jb3pV+Jr1FLyySefpHnNxEVw0kuXLl1M8bp16+weO3XqVFPcoEGDdMkJAIAnAUVpnOCPP/5Qw4YNVbNmzTQ/AAAAAAAAACAjZM+eXd27dzfi4ODgZL+8jY6O1ty5c404V65cevXVV5Od+8yZM6a4cuXKDuWW9E4njsiRI4fdX6YnJCQoJCTEiHPlyqUSJUo4tF7SCwaSfrFvS+HChTVnzhxZLBajLfGdcQYMGJDi62tLRr6ftuTPn9/uvtmyZUuXsdHR0Tb7XLp0SdeuXTPiI0eOWBXlsOdx+fJlY47E82W0pD/oT0n27NlNcXKvyQN9+/aVm5ubEadUGGTWrFmKjY01jU18jD6MCxcumOIKFSo4NN7R/v9m9+7d0/jx4+Xj46POnTtr+vTp2rlzpy5evKioqKgUi2o84EhxBkeKYyVWrFgxNW/e3Ig3b96c7IVe58+f15o1a4y4TJkyD12c7IEjR45o/fr1dj0uXrxoGmurMEtGFwdJeqe08uXLy8PDw6E5HD0vJS0o061bt1TX6Nq1q+nzYeHChbp79679Sdpp0KBBKliwoBGHh4erf//+yp8/v1q3bq0pU6Zo9+7dunfvnsNzP+rzpiOf9Q/yS7z+o5QR56WkBWTeeecdh8/TL730kmkOW+fqV155RRUrVjTiqKgovf322ypYsKCaNWumzz//XAEBAbpz547d++iookWLqk+fPqa27777Tn5+fqpVq5beffddrVq16pH+WwMAAAAAAAAAAABPrsTf6cXHx9t1I5sxY8aky81CnMHX19f0ffbDFEzJli2bChUqZMQxMTH69NNPUx0XGxurOXPmmNpef/11q37t27eXp6enEV+4cEHz589Pc76S5OPjY4pPnTqV6pjY2FhNmTLFoXUSv8YZcS1FnTp1lDVrViO+fPmyli1bluq48PBwbdiwwdQ2fPjwdM8PAIB/K4rSOMEXX3yhkydPKiwsLM0PAAAAAAAAAMgoAwcONMUzZsyw2W/JkiWmIgZdu3a1KjSSWNKCB3nz5nUoL0f7J5YrVy67+964cUPx8fEPtW7SAjjXr1+3a1yLFi305ptvWrVXrlzZ4S/5H8io99OWxBdEOOphxtpy9erVdJ1PyvjCE4k9zOuRWgGSQoUKqVWrVka8fft2HTlyxOY8M2fONGJXV1f17t07zXkllfQzwcvLy6Hx3t7e6ZbL4yw6Olovvvii3n33XdOdzBzlSPEQRz5Tk7L3M2nWrFmmi8D69euX5jWTmjBhgpo2bWrX488//zSNtVWUJjw8PN1ysyXpOcTeImspjUnpvHT69GkFBAQYsZeXl1q2bJnqGsWLF1f9+vVNayxfvtzhXFOTJ08erVixwnSRnyTdvn1bv//+u4YPH66nn35aefLkUYsWLTRjxgyHii49LudNe4pNpaeMyPVRnavd3d31xx9/qFy5cqb2mJgYrVu3Tu+9954aNGggb29vNWrUSFOmTNGlS5fSPbevv/7a5t9SUFCQxo8fr5deekn58uVT9erV9f777+vw4cPpngMAAAAAAAAAAAAgWd8czFZxlMS2bt2qjz/+OCNTcsiKFSsc+p3tqlWrTN9bOnoDkaTeeecdU/zxxx9b3ZQjqebNm5uK+uTJk0etW7e22TfpDS969uypffv22Z1f4mvfpPtFeVxdXY14586dqd4ww9/fX1FRUXavKZm/Vz59+rRDY+3VpUsXU9y9e/dUiyo1bNjQdA1KuXLlVKZMmQzJDwCAfyOK0jjBg3+Iubi4yMfHJ00PAAAAAAAAAMgoVapU0TPPPGPEy5cv1+XLl636/fjjj6a4f//+Kc6b9EtqR3+4nfgOPY5yd3e3u2/SPNOybtIxjhSKsPVl/tNPP226y4sjMur9zOwcKUBgr6QXbDzOBg0aZIqTvv+StGHDBtOdkV566SX5+vqmWw5Ji6B4eHg4ND5LlizplsvjbMiQIfr7779Nbfnz59crr7yiTz/9VHPmzNGyZcu0Zs0arVu3zniMGDEizWs68pmaVIsWLVS0aFEjnjNnju7du2fqEx8fbyqI5O7url69eqV5zfSUPXt2FSlSxNS2d+/eDF3zUZ+X5syZY7oYr1q1agoICND69etTfVSqVMlqroxQq1YtHTp0SCNGjLBZKEi6/7qtXr1a/fv3V7FixTR69GirY82WJ/W86QyP8lxdqlQp7dmzR2PHjlXhwoVt9rl79642btyo4cOHq1ixYnrjjTceqthXUp6enlq+fLnmzZunatWq2eyTkJCgffv26bPPPlPlypX18ssv6+TJk+mWAwAAAAAAAAAAACBJw4cPN8ULFy60KrTywOeff66GDRtmqutmvvjiC/n6+qpGjRqaPn16irktXbpUbdu2NbX16NHjodYfNmyY6UYqsbGxql69uhYuXGjVNyoqSvXr19f69etN7d99912y80+dOtVUOOfevXuqVauW3nvvvWTHhIWFafDgwcqWLZtCQkKstleuXNl4HhcXp/Lly5uuC3rg3LlzqlSpkrZu3ZrsWsnx8/Mznt+6dUuNGjVyqJiOPaZNm2a6BuLWrVvy8/PT5s2brfqGhoaqXLlyOnbsmNFmsVj066+/pmtOAAD827k5O4EnWYECBRQUFOTsNAAAAAAAAADAysCBA7Vt2zZJUkxMjObMmaO3337b2H78+HFt2bLFiGvWrKnq1aunOGfSH8QnvvOLPW7duuVQ/7TKkSPHQ6+bdEzOnDntGrdkyRL98MMPVu0//fSTXnrpJbVr187hXKSMeT8zu6RFjypWrKivvvrqoeZMa2GgzKhJkyby8/MzLi755Zdf9Pnnn5sKvWR0wQUvLy9T7OjdlSIjI9MzncfSvn37TEU/3N3dNWHCBA0ZMiTVIj+2Lix6FFxdXdWvXz+NGjVKkhQeHq7ly5erQ4cORp9169bp7NmzRtyqVauHvktZemrQoIHmz59vxIGBgRm63qM8LyUkJOjnn382tW3atEmbNm1yeE1J+vPPPxUWFpYhN93IkyePJkyYoE8//VQbN27Uhg0btGnTJgUGBloVn7l586Y+/vhjrV+/XmvXrk21MN6TeN50hqTvw7Bhw/TSSy891JylSpVKcb33339f7777rrZu3aq///5bGzdu1M6dOxUdHW3qGxMTo6lTp2rt2rXavHlzun0GWSwWde7cWZ07d9aRI0e0bt06bdy4UQEBATbv3rdy5Upt3rxZK1euVIMGDdIlBwAAAAAAAAAAAKBq1aqqUaOG9uzZY7RNnDhR06ZNU7ly5ZQrVy5du3ZN//zzj+mmQ127dtXcuXOdkbKVhIQE7d27VwMHDtSgQYOUO3du+fr6ysvLS66urrp69aqCg4Otvi/38fFJtgCPIzZt2qSKFSsqNjZW0v2bYHTq1EkDBgyQn5+fsmfPrtDQUJ05c8Z0YxhJ6tSpk1599dVk53ZxcdG2bdtUqVIl4/WPi4vT559/rokTJ6p48eIqUKCA3N3ddf36dZ07dy7V62h+/PFHPf3000Z8+fJllS5dWkWLFlXx4sUVExOjkJAQXbx40ejj5eWlvHnz6vTp03a9Ju+++666detmxBs3brT5XXqFChV05MgRu+ZMytPTU8uWLdMLL7xgvK6RkZFq2LCh8ubNq5IlS8rDw0MhISE2i/OMHDlStWrVStPaAAA8qShKAwAAAAAAAACw8uqrr2rYsGG6fv26JGnGjBmmH2PPmDHD1N+eYhXe3t6m2NYPb1Ny9epVh/qnlZeXl1xcXIw76KRl3aT7ljt37lTHBAcHq1+/fslu79evn2rVqqVixYo5nE9GvJ+ZXb58+UxxQkKCmjRp4qRsMh+LxaIBAwbov//9r6T7x/lvv/2mzp07G/GyZcuM/kWKFFHz5s3TNYc8efKY4tDQUFWrVs3u8aGhoemaT2IWiyXNYx0tuPUwFi1aZLpw6eOPP9awYcPsGnvt2rUMyip1/fr10yeffGJcmPXjjz+aitJk9GfS7NmzNXv27DSPT1qUZvPmzYqIiLA6z6WXpOeQjDwvbdq0ScHBwQ7Pn5y4uDj9+uuvpnNOevPw8FCzZs3UrFkzSVJ0dLR27NihVatWad68eabPiq1bt+rtt9/Wt99+m+KcT+J50xmSnqsLFSr0SM7VLi4uatCggRo0aKBRo0bp3r17CgwM1Jo1azRv3jydPHnS6Hv8+HH16tVLq1atSvc8KlasqIoVK2ro0KFKSEjQsWPHtHbtWi1ZskQBAQFGv5s3b6pDhw46deqUVZEqAAAAAAAAAAAAIK02bdqkYsWKGd+LSve/b923b5/N/gMHDlS3bt0yTVGaxBISEnTt2rVUr4UoWLCgDh48mC5rlilTRkFBQapbt67pJhiRkZHau3dvsuN69eqlWbNmpTq/n5+fTpw4oVq1auny5ctGe1xcnE6fPm13oZgHateurdGjR2v06NGm9uSKt+TIkUN79+5V06ZN7V6ja9euWrRokX7//fcU+8XExNg9py1NmzbVypUr1apVK+PaE+n+9RQpXVPx8ccf66OPPnqotQEAeBK5ODsBAAAAAAAAAEDm4+npqZ49exrx8ePHtXnzZkn3vxSeM2eOsS179uzq0qVLqnOWKlXKFB86dMihnA4cOOBQ/7SyWCwqWrSoEUdGRjr8A/39+/eb4uLFi6fYPzY2Vp07d9aNGzeMtoEDB6pHjx5GfP36dXXp0sX0Rbq9MuL9zOx8fHyUNWtWIz579qzu3bvnxIwyn969e8vDw8OIf/zxR+P5L7/8YrrTVZ8+feTq6pqu61esWNEUp3RBji3JXQSVHjw9PU1x4ouHUpP4QqCMtmPHDuO5i4uLBg0aZPfYw4cPZ0RKdvH19dXLL79sxOvWrTM+Z8PDw7V8+XJjW4kSJYxiI5lFy5Yt5eb2f/f+iI6OztCL7goVKmT6Wz127JjDF2jZe156mGI9yUl8jnkUsmbNqkaNGmnixIkKDg7WyJEjTdtnzJiR6h3insTzpjOULFnSFCcuBvMoubu7q169evr444914sQJffPNN3Jx+b9LKVavXq2jR49maA4Wi0UVKlTQ0KFDtWXLFm3evNlUtCc8PFy//PJLhuYAAAAAAAAAAACAJ0uOHDkUGhqqRo0apXjzHh8fHy1YsEDff//9I8wuZV988YVat25t143CJClnzpx68803FRYWZnXzjIdRtWpVXbt2TZ07dzZdR2BLsWLF9Ndff9lVkCbxmPDwcI0aNUo5c+ZMtX/u3Lk1ePBgFS5c2Ob2UaNGad68efLy8kp2Djc3N7344ou6ePGi1Xe69li+fLkWL16s6tWrK3v27A91Y6iUNG/eXJcuXVLTpk1N3+/aUrFiRe3fv5+CNAAApBFFaQAAAAAAAAAANg0cONAUPyhYsXz5clPRhU6dOtn1pXft2rVN8aZNmxzKx9H+D6Nu3bqm+O+//3ZofNL+SedLauTIkabiEpUrV9aUKVP0zTffqGzZskb71q1bre5WY6/0fj8zO3d3dz377LNGfPv2be3cuTPD1kt8AUVCQkKGrZOe8ufPr3bt2hnxxo0bjYIAM2bMMNpdXFzUp0+fdF//6aefNsUrVqxwaHxqd1V6GLly5TLFly5dsnvs7t27HVor8YUxjh47ifPKnz+/3RdbxcfHP9LPVFsSF9BJSEjQzJkzJd0vYJK4gFTfvn0z7AKltCpSpIheffVVU9ukSZN069atDFnPw8ND1atXN+KYmBgFBATYPT4hIUEbN240tdk6L926dUtLly41tZ05c0YJCQkOP0qXLm3McejQIQUFBdmdb3pyd3fXp59+qvr16xtt9+7ds+t88LifN5NedJcZz02NGjUyxY7+eysjWCwWDRkyxKrQkCN/c+mhQYMG+vzzz52aAwAAAAAAAAAAAB69+vXrm757jYiISLbvxo0bTX3TwtPTU3///beuXLmi8ePHq3379mratKm6dOmiDz74QCdPntTFixfVsWNHm/klvr4kqZMnTxr9HLkJ14wZM0xrJP6+94FatWrpf//7n65du6bo6GgtXrxYb7/9ttq1a6emTZuqWbNm6tixoz7++GMdPHhQkZGRmjRpUqprR0RE2PXaJ+bp6al58+bp3r17WrVqlYYMGaI2bdqoWbNm6t69u8aNG6erV6/q7Nmzaty4sd2vQ2KjR49WZGSkTp48qffff19dunRR06ZN1bx5c/Xo0UNff/21Ll68qGvXrunbb79NsUBO586dFRERob/++kv9+/dX8+bN9dJLL2nw4MGaPXu27t27p9WrVytHjhyS0vY+dujQQXv27FFUVJTi4+OtrimwdcOS2NjYFLfbkidPHq1du1ZxcXFasGCB+vfvr5dfflkvvPCCevbsqSlTpujWrVs6fPiwqlatatecaf27GjZsmGncsGHD7B4LAEBml3LpPQAAAAAAAADAE6t8+fJq2LChUbhgyZIl+vrrr60uJujfv79d8z3zzDPy8PBQTEyMJGnlypW6du2a8uTJk+rY8PBwrVq1ysE9SLuGDRtq4cKFRjx79my7i3IcP35cW7duNeIsWbKoTp06yfZfu3atJk6caMTZsmXTwoUL5enpKUlauHCh6tatq7t370qSPvvsMzVu3NjhixTS+/18HLz44otav369EU+dOtXmhSrpIXv27IqKipJ0vwDO42LgwIFasGCBJBkXC7Vu3VqHDx82+jRr1kzFixdP97Vr166tggULGoVVduzYoaCgINWsWTPVsbt27XK4+Isjku7v3r175e/vn+q4e/fuadmyZQ6tlT17duO5o8dO4otfHny22uP333/X+fPnHVorvTVr1kwlS5bUmTNnJEmzZs3SqFGjjOI0kuTq6qrevXs7K8UUvf3225o3b54RnzlzRv/97381bdq0h5r31KlTiouLMxUkk+6flxIXUpk9e7bd54F169YpJCTEiAsVKmQ1v3T/vPDgc0y6X7imRIkSDu7BfZ06ddKnn35qyteev+2M8uyzz5oKely5ciXVMY/7eTPxZ4t0//MlaZuzFS5cWJUrV9ahQ4ck3T/+V69erebNmzs5s/vHzK+//mrE9hwzGZFDYs7IAQAAAAAAAAAAAE+GPHny6J133nF2Gmni6empDh06qEOHDs5ORc2bN8/Q7zv9/Pw0duzYdJkrLdefZWYdO3Y0iicBAID05ZJ6FwAAAAAAAADAk2rQoEHG8zt37ujTTz/VunXrjLYqVaqkWHAlMW9vb7Vt29Y036hRo+waO3LkSIcKLjyszp07m364vWXLFv3vf/+za+xbb71lil999VV5eXnZ7BsWFqbu3bubCkt89dVXqlixohFXq1bNVLQmPj5e3bp10+XLl+3KJ7H0fD8fB/369ZO3t7cRL1myRCtXrsyQtRIXVwoODs6QNTKCv7+/ypcvb8SzZ8/Wt99+a+qTUQUX3N3drQqODB48WHfu3ElxXHR0tOlYzgg1atQwxYsWLbJr3NSpUxUaGurQWomPnatXr+rmzZt2j/Xx8TGeX79+XUeOHEl1TFRUlNXnlDNYLBYNGDDAiC9cuKB3331Xx48fN9patGihwoULOyO9VFWvXt3qrlbffPON6fPaUWvXrlW9evVsHkN9+/aVxWIx4rlz5yooKCjVOePi4qwuHOzXr5/NvnPmzDHFnTt3tidtmzp16mSK58+f/0jP40klLeaRO3duu8Y9zufNpEX/HhSAymxGjBhhiocNG6YbN244KZv/k9Zj5t+WAwAAAAAAAAAAAAAAAIAnG0VpAAAAAAAAAADJateunfLnz2/EX375pamAiqPFKoYOHWr6Uf0333yjuXPnpjhmxowZmjFjhkPrPCxvb2/16dPH1NanTx8dOHAgxXGjRo0yFT1xcXHR8OHDbfaNj49X9+7dFR4ebrR17NjRZrGA//znP2rVqpURX7x4UT179jS9F/ZI7/czs/Py8tJ///tfI46Pj1fnzp31+++/OzRPUFBQqnfSqVSpkvH8ypUr2rhxo0NrONPAgQON55cuXdKvv/5qxAULFlTLli0zbO3hw4ebCifs3r1brVu3Nv1dJHbp0iW1bNlSe/fuNX2WpLeaNWuqQIECRrxjxw7NmzcvxTGrVq3S+++/7/BaiY+dhIQELV261O6xzzzzjCl+5513FB8fn2z/27dvq127djp9+rTDeWaEPn36yN3d3YgnTZpk2p7ZP5PGjx+vWrVqmdreeecdDR48WNevX7d7nkuXLmnAgAF68cUXky04VrZsWb388stGHB8fr/bt2+v8+fPJzpuQkKB+/fpp//79Rlv27NltFnU6e/as6XPLxcVFr776qt37kFSlSpVUpUoVI7569apWrFiR5vkeiIyMVNeuXbV37167xwQHB2vJkiVG7OLiomrVqtk19nE+byb+bJFkeg0yk65du5pyPXHihJo3b+5Qga979+5pzpw5Gj9+fLJrbNq0ye75rl+/bvVvz5o1a9o93pYPP/xQv/76q2JjY+3qn5CQYPWZ+LA5AAAAAAAAAAAAZDbZszk7g5Rl9vwAAACAR4GiNAAAAAAAAACAZHl4eKh37942t3l6eqpbt24OzVevXj317dvXiBMSEtS9e3e98cYbCgkJMfUNDg7W4MGDNWDAAElSiRIlHEv+IY0dO9a05vXr1/XMM89o0qRJVsUGDh06pPbt22vMmDGm9hEjRqh69eo25//888+1fv16Iy5ZsqR++OGHZPOZNWuWihQpYsSrV6/Wl19+6cgupfv7+Th455131KJFCyO+efOm2rRpo3bt2unvv//W3bt3rcbcuXNHu3bt0rhx41SzZk3VqlVLixYtSnGdZs2ameK2bdvq3Xff1eLFi7V27VqtX7/eeGSWgiAP9OzZU56enja39erVy1Q0JL0VKFBAX331lalt7dq1KleunAYPHqy5c+dq9erV+vXXXzVo0CCVK1dOf/31lyRzMZ305u7url69epnaevfurUmTJunWrVum9lOnTumNN95Qq1atdPfuXfn5+Tm0VtJjZ/DgwfrPf/6j+fPn688//zQdO0eOHDH17datm1xc/u/rvpUrV6ply5ZW/e7cuaMlS5boqaee0rp16yRJFSpUcCjPjFCgQAG1bdvW5rbChQub/nYzIw8PDy1dulSlSpUytX///ffy8/PTyJEjtX37dsXFxVmNjYyM1OLFi9W9e3eVKFFCP/74Y6qFxr799lt5e3sb8dmzZ1W9enX99NNPVsfljh075O/vr9mzZ5vav/jiC/n6+lrNPWfOHNP6/v7+8vHxSTGf1HTq1MlqjYcVHx+vefPmqUaNGqpdu7bGjx+vPXv2KCYmxqpvZGSkZs6cqXr16unGjRtGe8uWLe3et8f5vNm0aVNT/Mknn6h379765ZdftGbNGtNnS1BQkJOylFxdXbV06VJ5eXkZbdu3b1flypU1atQonThxwua4S5cuacWKFRo4cKAKFy6sXr166ejRozb7rly5Uv7+/qpYsaJGjRql7du36/bt21b9oqOjtXjxYtWpU0dnz5412p966inVrl37ofbz4MGD6t69uwoXLqzBgwdrzZo1unr1qlW/+Ph4BQQEqFmzZvrf//5ntGfLlk1dunR5qBwAAAAAAAAAAAAym/x5pCbPSvWqZ75Hk2fv5wcAAAA86dycnQAAAAAAAAAAIHMbMGCAJk6caPVj+Q4dOih37twOzzdp0iQFBQVp7969ku4Xppk6daqmTZumkiVLKm/evLpy5YrOnDljjMmZM6e+/fbbR1qgIGfOnFq8eLGaNWtmFKG5deuW3n77bb333nsqWbKkcuXKpYsXL+rChQtW41988UWrIjUPbNu2TaNGjTJid3d3LViwwPSD7KTy5MmjefPmqVGjRkaBg/fee08NGzZUrVq17N6v9H4/MzsXFxfNmzdPbdq00caNGyXdP+aWLVumZcuWKUuWLCpevLhy586tO3fuKCIiQufPn7dZRCIlPXr00NixY3XlyhVJUkREhMaPH2+z76hRozR69OiH2a10lTt3br366qv6+eefTe0Wi0X9+vXL8PW7deum4OBgffjhh0ZbRESEvv/+e33//fc2x7Rp00YjRowwbXdzS9+vvUaOHKm5c+caf98xMTF6++23NXLkSJUpU0aenp4KCwvT+fPnjTHly5fXZ599lmyhFVtefvlllStXTsePH5d0v4DMtGnTNG3aNKu+PXv2NBUZKV++vAYNGqRvv/3WaFu1apVWrVqlokWLqlChQoqKilJwcLCpAMNzzz2n7t27q3///nbnmVEGDRpks+hT79695erq6oSMHFOsWDFt2bJF7du3144dO4z269eva9y4cRo3bpyyZs2qAgUKKH/+/Lpz547CwsJ09epVm0VoPD09k/0sLlKkiH799Ve1b9/eKKh15coV9e3bV6+//rpKliyprFmzKiQkROHh4Vbj+/Tpo0GDBtmcO+nff9KCMmnRqVMnjRw50ohXr16ty5cvK3/+/A89tyQFBgYqMDBQ7777rjw8PFSkSBHlzp1brq6uunr1qoKDg60+y/Pnz6+pU6c6tM7jet6sXr26GjdurL///lvS/WIns2fPtipUJEkNGzY0zpHOUK5cOS1btkzt27c3/s11/fp1jRkzRmPGjFG+fPnk4+Oj7NmzKzIyUleuXNHly5cdXufo0aPGnK6uripSpIjy5MkjDw8PRURE6PTp07p3755pTLZs2fTTTz+ly35KUnh4uOn8VqhQIeXLl0/Zs2fXrVu3dObMGUVFRVmNmzRpkgoXLpxueQAAAAAAAAAAAGQW+fNQ/AUAAADIzFxS7wIAAAAAAAAAeJL5+fmpSZMmVu1pLWaQK1curV271qqQSkJCgk6fPq3du3ebCtLkypVLv//+uypUqJCm9R5GrVq1tHnzZpUqVcrUfu/ePZ04cUKBgYE2C9L06tVLv//+uzw8PKy2RUREqEuXLoqNjTXaxo4dq6effjrVfBo0aKCPPvrIlEenTp108+ZNu/cpvd/Px4GXl5fWrVunN99806pwyd27d3XixAnt3LlT+/fv19mzZ20WpClatGiKa+TJk0dLly5VgQIF0jX3R2XgwIFWbf7+/ipduvQjWf+DDz7QrFmzUi3wYLFY9Nprr2nRokWmIiuSUizqlBYPPnvy5ctnar97964OHTqkwMBAU0Gap556SuvXr5e3t7dD67i5uWnp0qXy8/NLU56TJ0/Wyy+/bNUeEhKiXbt26ciRI6bXqlGjRlq+fHm6F/FJq0aNGqls2bKmNovFor59+zopI8f5+vpq8+bN+uyzz5QzZ06r7dHR0Tp79qwCAwN16NAhXblyxarAiYuLi7p27arjx4/rqaeeSnatl156SX/++afVZ010dLSOHDmioKAgq4I0rq6ueu+99zRz5kybc27ZskWnTp0yYnd3d7Vv3z7V/U5NqVKlTOe2e/fuae7cuQ89ry0xMTE6ffq0goKCtGvXLp06dcrqs7x8+fIKCAhI9fM8qcf5vPnLL7+oRo0azk7DLo0aNdLu3btVu3Ztq21XrlzRoUOHtHPnTh09etRmQRqLxeLQexsXF6ezZ89q79692rlzp44fP25VkKZw4cJav359hr6GFy9e1MGDB7Vjxw4dPHjQqiBN1qxZ9f333ydbUAoAAAAAAAAAAAAAAAAAMhJFaQAAAAAAAAAAqerTp48pLleunJ577rk0z5cvXz5t375d48ePV6FChWz2cXV1VYcOHbR//375+/unea2HVblyZR05ckQTJ060Kk6TmJubm55//nlt2bJFs2bNkru7u81+ffv21dmzZ434hRde0Ntvv213Ph988IHp9Th16pTDP1RO7/fzceDm5qZJkybp+PHjGjBggF3FY0qUKKEBAwZo7dq1Cg4OTrX/c889p2PHjmnatGlq2bKlSpYsqZw5c8rFJfN/HfPMM8+oXLlyprZHXXChV69eOnHihCZPnqznnntOvr6+cnd3V44cOVS1alW98cYb2r9/v6ZNmyZ3d3ddu3bNND69i9JIUo0aNRQUFKTOnTvL1dXVZh9vb2999NFH2rFjhwoXLpymdSpVqqQDBw5o1qxZ6tChg8qWLSsvL69k10zMw8NDy5cv1+TJk+Xj45NsvxIlSmjatGlpKpyT0Xr37m2KmzZtqhIlSjgnmTRyd3fXu+++q3PnzmnSpEmqVauWLBZLimNcXFxUuXJlffLJJwoODtavv/6qYsWKpbpWw4YNdfLkSb3//vvy9fVNtp+np6fatGmjvXv3aty4ccn2mzNnjilu1qyZ8uRJn1sRdu7c2RTPnj37oebz9vbW3r17NXr0aD377LPKkiVLqmOqVq2qr7/+WgcOHLAqgGSvx/W86evrqx07dmjx4sXq0qWLKlWqJG9v70xTlCopPz8/7dq1S7///rsaN25ss7hfYq6urqpXr57GjBmjkydP6pNPPrHZb9euXZowYYKef/55Zc+ePdU8SpcurU8++UTHjx9XvXr10rQvSf3444/66aef1L59exUsWDDV/nny5NGgQYN09OhRm4XjAAAAAAAAAAAAAAAAAOBRsCQkvQ2fnWrWrKmwsDD5+PgoKCgovfN67CV9fRLHkjL0tZvU0fpumI+LtxaucHhMQui3GZAJ8Piw+A5J++C7m9IvEeBxlKWh8TThx0pOTARwPkv/w2keO7Bs6j/+Af7NfjhxN1EU6rQ8gMzh/34Qm7CzkxPzAJzPUmeBs1NIdx999JHph74TJ050qJBKSuLj4xUQEKATJ04oPDxcWbJkUalSpdSgQQPly5cvXdZIT8eOHdPevXsVHh6u27dvK2/evCpcuLDq16+fIUUxMkJGvp+Pi4SEBB0+fFiHDx/WlStXFBERoSxZssjLy0slS5ZUxYoVUyz28G9z8+ZNFSpUSLdu3ZIk5c2bVxcuXLCr4IOzTJs2Tf/5z3+MePbs2erZs2eGrRcREaGNGzcqJCREN27ckJeXlypVqmR3YYxHITY2Vrt379aBAwd09epVubq6ysfHR9WqVdNTTz3l7PSS1aNHD/3yyy9GvHjxYnXo0MGJGaWPiIgI7d27V2fOnNGVK1d09+5d5cyZU7lz51bRokVVq1Yt5cqV66HX2bdvnw4fPqzw8HDdvXtX+fPnV9GiRVW/fn1ly5YtHfYk87p7966OHDmikydP6uLFi4qKipLFYlGuXLlUokQJVatWLc3FohLjvOkct2/f1o4dOxQSEqKrV68qOjpaOXLkUL58+VSuXDlVqFDBriIzicXFxeno0aP6559/dOHCBd28eVOSlDNnThUuXFjVqlVTyZIlM2J3TM6cOaPjx4/r7NmzunHjhmJiYpQjRw7lz59fVapUUcWKFTNt8SAAAAAAAAAAAAAAAAAATw6uYgIAAAAAAAAApCguLk6zZs0yYg8Pj3Qt/ODi4qLnnntOzz33XLrNmZHKly+v8uXLOzuNNMvo9/NxYbFYVLlyZVWuXNnZqWQK8+fPNwrSSFL37t0zTaGV5Pz111+muFatWhm6nre3t9q0aZOhazwsNzc31atXT/Xq1XN2KnaLiIjQkiVLjLhAgQJq3bq1EzNKP97e3mrUqJEaNWqUoetUq1ZN1apVy9A1MqssWbKoevXqql69eoatwXnTebJly6bGjRun65yurq6Z4vxfsmTJR1L8BgAAAAAAAAAAAAAAAAAehouzEwAAAAAAAAAAZG6rVq3S+fPnjbht27bKnz+/EzPCw+D9hC0//PCDKR4wYICTMrFPcHCw/vjjDyPOkyePKlSo4MSMkFa//PKLoqOjjbh3795yd3d3YkaAGedNAAAAAAAAAAAAAAAAAMCTiqI0AAAAAAAAAIAUTZgwwRS/9tprTsoE6YH3E0mtX79ee/bsMeLGjRs/8gIvCQkJdve9d++eevbsqbi4OKOtZ8+ecnHha6/Hzb179zR58mQjdnFx0aBBg5yYEWCN8yYAAAAAAAAAAAAAAAAA4EnF1bkAAAAAAAAAgGTNnj1bAQEBRlyrVi01aNDAiRnhYfB+Iqlbt25p2LBhprY333zzkedRo0YNLVq0SDExMSn2O336tJo0aaLNmzcbbVmyZNGQIUMyOkVkgLFjx+rMmTNG3K5dO5UoUcJ5CQFJcN4EAAAAAAAAAAAAAAAAADzJ3JydAAAAAAAAAAAgc7h48aIOHz4sSQoPD9f69ev1888/m/qMGTPGGakhDXg/YcvWrVsVHR2tu3fv6sSJE/rmm2906tQpY3udOnX00ksvPfK89u3bp44dO8rb21svvPCCateureLFiytHjhy6efOmzp07p40bN2r16tWKi4szjf30009VunTpR54zHHP69GmdPn1aCQkJCg0N1fLly7Vs2TJju6urq0aNGuXEDPGk47wJAAAAAAAAAAAAAAAAAIAZRWkAAAAAAAAAAJKkP//8U7179052e4cOHdS8efNHmBEeBu8nbOnatavOnj1rc5u7u7u+//77R5yRWUREhBYuXKiFCxfa1f+NN97Qm2++mcFZIT38/PPP+vjjj5PdPnz4cFWuXPkRZgSYcd4EAAAAAAAAAAAAAAAAAMDMxdkJAAAAAAAAAAAyvzp16mjmzJnOTgPphPcTSbm7u+unn35StWrVnLJ+4cKFHepftGhRzZo1S1999ZVcXPi663HXqlUrjRs3ztlpAMnivAkAAAAAAAAAAAAAAAAAeBK5OTsBAAAAAAAAAEDmY7FYlCtXLlWuXFmdOnXSwIED5e7u7uy0kEa8n7DFw8NDvr6+atSokYYPH64qVao4LZeQkBBt375dGzZs0K5du3Tq1CmFhoYqKipKLi4uyp07twoUKKC6devq+eefV5s2beTh4eG0fPFwXFxc5O3trerVq6tXr17q2rWrLBaLs9MCDJw3AQAAAAAAAAAAAAAAAACgKA0AAAAAAAAA4P/r1auXevXq5ew0kE54P2FLcHCws1OwyWKx6JlnntEzzzzj7FSQQUaPHq3Ro0c7Ow0gWZw3AQAAAAAAAAAAAAAAAAAwc3F2AgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAzIOiNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA0VpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGitIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAxuzk4AAAAAAAAAAAAAAAAAAAAAAAAAAAAAT5bbV8N1LyrS2WlYcc+RS9nyFnB2GgAAAIDTUZQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAj8ztq+E69/dKZ6eRrGKNX6IwDQAAAJ54Ls5OAAAAAAAAAAAAAAAAAAAAAAAAAAAAAE+Oe1GRzk4hRZk9PwAAAOBRoCgNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAURoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIGiNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA0VpAAAAAAAAAAAAAAAAAAAAAAAAAAAAADxS/v7+slgsxuNR69evn2n9gICAR54DUjdlyhT5+fnJw8PD9H5ZLBZVr17d5pg7d+5o0KBBKlSokNzc3KzGvffee0Zfb29vo93b2/sR7dX/yaj1AwICTPvcr1+/dJsbAPDkcHN2AgAAAAAAAAAAAAAAAAAAAAAAAAAAAMC/kb+/vzZt2mSKN2zY8FBzxsbGKkuWLIqPjzfa9u/fr6pVqz7UvEBmcufOHVWuXFmnTp1yaNyBAwdUr1493b59O4MyAwDgyeHi7AQAAAAAAAAAAAAAAAAAAAAAAAAAAACAf6PRo0eb4oCAgIeec+LEiaaCNAUKFKAgzRMmICBA/v7+xiM9jqvMpnnz5qkWpHF1dbVqa9iwYaoFaVxc+Ik9AAD24IwJAAAAAAAAAAAAAAAAAAAAAAAAAAAAZAB/f3/lypXLiGNjY/Xdd9891JzTp083xV27dn2o+fD4CQwM1KZNm4xHYGCgs1NKV2FhYdq4caMRu7m5ady4cbp8+bISEhKMR9L9njp1qiIiIow4b968WrBggaKjo03jxo4d+4j2BACAx5ubsxMAAAAAAAAAAAAAAAAAAAAAAAAAAAAA/q3atWun2bNnG/HXX3+twYMHp2mu0NBQBQcHG7HFYtHo0aMfLkEnSVx0BEhs6tSppnjixIkaNmxYquMS/51J0oEDB+Tr65vimMRFbP5N6tevr4SEBGenAQB4zLk4OwEAAAAAAAAAAAAAAAAAAAAAAAAAAADg32rs2LGm+Pjx44qMjEzTXB988IEprlixonLlypXm3IDMaPv27aZ4yJAhdo07ffq08TxPnjypFqQBAAApoygNAAAAAAAAAAAAAAAAAAAAAAAAAAAAkEF8fX1VsmRJI05ISNDo0aPTNNeyZctM8fDhwx8mNSBTunLlivHcxcVFHh4edo2Ljo42nlOsCQCAh0dRGgAAAAAAAAAAAAAAAAAAAAAAAAAAACADDRo0yBTPnTvX4Tm2bt2qiIgII/bw8FDfvn0fNjUg00lcXMZisdg9Li4uznju6uqarjkBAPAkcnN2AgAAAAAAAAAAAAAAAAAAAAAAAAAAAMC/2Ztvvqn333/fKJoRHh6uo0ePqkKFCnbPMWrUKFPcqFGjVMfEx8dr8eLF2rRpky5evKjo6GgVLFhQTz31lAYMGKAcOXI4tiP/35kzZ7R48WLt3btX169fV1xcnLJnz67ChQurevXqatOmjfLly5emuR2xYsUK/fnnn7p48aJu3Lih7Nmzq1SpUmrSpIlatGiR4euvXr1aq1evVmhoqG7evKn8+fOrQoUK6t+/vwoUKJDh62eE48ePa86cOQoODtbly5eVP39+ff755ypWrJjN/uHh4frjjz+0c+dOXbp0Sbdv35aXl5d8fX3Vpk0bNW7c2OEcEhIS0pR7Wsc9jDt37ujnn39WYGCgLl26pOjoaHl5ealy5cpq2bKlatSo8chzSm+rVq3SmjVrdOrUKbm7u6tYsWLq06ePqlWrluY5r1y5om+++UZHjx5VRESEihYtqho1amjgwIFycXFJv+QBAA+FojQAAAAAAAAAAAAAAAAAAAAAAAAAAABABnJzc1OdOnW0bds2o23kyJH67bff7J5j8+bNpviTTz5Jtu/Zs2fVqVMn7dq1S/Hx8Tb7vPXWWypfvrx++eUX1apVy64c5s6dqzfffFPh4eEp9uvfv7+yZ8+uVq1aad68eTb7+Pv7a9OmTUZsb0GRU6dOqWfPntq+fXuy+zZ58mS5uLioUqVKmjhxol544QW75rbHtWvX1K1bN61bt06xsbE2+3zwwQcqWrSofvjhBzVv3jzd1nZzczMKGyU2fPhwDR8+3OaYvn37asaMGUY8ZcoUU9/Jkydr2LBh+v3339WvXz9dvnzZao7WrVubitJs3bpVY8eOVUBAgG7evJlsvlOnTpWbm5tatWqlWbNmKVeuXMn2tVgsNtvj4uJsbvPz89Prr7+e7H6fOnXK5rikr4e3t7du3LghSfLy8lJERESyOSa2evVqvf766zp9+rTN7UuWLNHo0aOVJUsWNWjQQNOnT1fJkiXtmjs1nTp10sKFC43Y1dVVS5cuVevWrY22gIAANWjQwIiT7ndiyf0tfvXVV/rggw8UFRVlNWbq1Kny8fHR3LlzHSo8dP78ebVo0UIHDx60uf0///mP2rRpoyVLlkgyH/N+fn46efKk3WsBAB4eZcIAAAAAAAAAAAAAAAAAAAAAAAAAAACADPbRRx+Z4jVr1tg9dvr06bp3754R58mTR7Vr17bZd+zYsSpZsqR27NiRbNGWB44dO6batWtr7NixqebQqVMndevWLdWCNA/cunVLS5cutauvvUaOHKnSpUtr69atqe5bfHy8Dh48qHfffTfd1v/1119VoEABrV69OtmCNA+EhISoRYsW6t+/f7qtn1EGDx6s1q1b2yxIY0uTJk20evXqFAvSPBAbG6vffvtNvr6+2rNnz8Om6nTx8fHy9/dXixYtki1Ik9jdu3e1fv16TZ8+PV3Wrl27tqkgTdasWRUYGGgqSJMemjdvrmHDhtksSPNAWFiYmjRpokWLFtk1519//aUSJUokW5BGul+EaOnSpSpcuLBu377tcN4AgPTl5uwEAAAAAAAAAAAAAAAAAAAAAAAAAAAAgH+7F154QTly5DCKPERHR2v58uV2FZP46quvTHGnTp1s9hsyZIi+++47U5uLi4uKFCkiHx8fZcmSReHh4Tp9+rSpyM0HH3wgd3d3vfPOOzbn/fLLL02FMCTJzc1NRYsWVcGCBeXp6amoqChduXJFYWFhunPnTqr75KguXbpo/vz5Vu1eXl4qVqyYcufOrVu3biksLExhYWGKi4tL1/UnTZqkt99+29RmsVjk4+OjwoULK1u2bLp69apOnTpl2v8ZM2YoLi5OP/3000PnYLFYHB7j4uKS4vaVK1dq/fr1Ruzt7a0SJUooR44cCg8P16lTp1Ic7+rqqrx588rHx0c5c+aUi4uLIiIidO7cOd24ccPod+vWLTVo0ECXLl1Sjhw5HN6PpFxcXDLk9UhJfHy8SpUqpbNnz1pt8/Hxka+vr3LkyKHIyEiFhITo2rVrSkhISPN6iUVERKhy5cq6cOGC0ZY/f34dOHBAPj4+6bLGA3369DGKZlksFvn6+qpw4cJyc3PTmTNndPHiRaNvQkKCunfvrmbNmsnb2zvZOQ8dOqQXXnjB6u/S29tbpUqVUtasWRUaGqrg4GAlJCQoNDRUDRs2TNf9AgA4Lu1nTQAAAAAAAAAAAAAAAAAAAAAAAAAAAAB2a9WqlSkeN25cqmMiIyN19OhRU9snn3xi1W/RokWmgjQuLi568803dffuXZ09e1Y7d+7U5s2bdezYMd25c0fDhw83FfV49913dfz4cZs5JF1v+PDhunfvnk6fPq3t27drw4YN2r17t86cOaPo6GgdOXJE3bt3V4ECBVLdP3tMmzbNqiBN2bJltX//fkVEROjAgQPatGmTAgMDdf78ecXGxmrevHmqUqWKsmbN+tDrBwYGasSIEaa27t27KzIyUqGhodq9e7c2bdqkQ4cOKTo6Wl988YVcXV2NvrNmzdLq1asfOo979+4pISFBkydPNrVPnjxZCQkJNh/Tp09Pcc4HBWm8vb21fv16Xb9+XXv37tWWLVt0/PhxxcTEqGXLlqYxBQsWVJs2bbRp0ybFxsbq0qVL2r9/vwICArR582YdOHBAEREROnTokMqUKWOMu337tl555RWbeSTO2c/Pz2h3dXW1uV8nTpzQ0KFDTW2JX3M/P780vR4pad68uVVBmpdeeknXr1/XxYsXFRQUpE2bNmnv3r26cuWKbt++rXHjxqlAgQIPVQzn+PHjKlq0qKkgTfny5XX+/Pl0L0gj3T9eJalmzZoKDQ3V+fPntXPnTm3dulWhoaHatm2bPD09jf4xMTEaOnRoinO++OKLpoI0WbNm1fLly3X9+nUFBQUpICBAp0+fVkREhOrVqyfp/t9deheXAgA4hqI0AAAAAAAAAAAAAAAAAAAAAAAAAAAAwCOQtAhNYGCgYmNjUxzz8ccfKyEhwYjLli2rPHnyWPXr1auX8dzV1VU7duzQpEmT5ObmZtXXxcVFX375pX755RejLSEhQf3797fqe/v2bUVERBjx888/ry+//DLFnCtUqKCff/5ZISEhKfazR2xsrN58801T28svv6zjx4+ratWqyY7r3LmzDhw4oICAgIfOoVWrVsZ7YLFYtGTJEv3888/KkSOHzf5vvfWWNm3aZCr68/rrrz90HhklZ86cOnv2rJ5//nmrbS4uLsqWLZupLTg4WMuWLdNzzz2X4ryVKlXSiRMnVLlyZaNt3bp1iomJSZ/EH6HNmzdr7dq1prZvvvlGK1askLe3t80xnp6eeu+993Tp0iWbhaTssW7dOlWpUkVRUVFG2wsvvKCjR4/Kw8MjTXPao2nTpgoMDLRZ9KZevXpWRZaWL1+e7FwzZ840FdRxd3fXwYMHrYp0SVKuXLm0bdu2VI8tAMCjQVEaAAAAAAAAAAAAAAAAAAAAAAAAAAAA4BEoXry4ihYtasTx8fH64osvUhwzd+5cUzx06FCrPmPHjlV0dLQRf/bZZ6pdu3aq+XTt2lVPP/20EQcEBCg+Pt7U58SJE6bY398/1XnT04gRI3Tv3j0j9vX11R9//GH3eBeXh/s59apVq3Tx4kUj7tGjh9q3b5/quGeffVYdO3Y04tOnT+vs2bMPlUtGWbBggXLlypVh88+fP994HhcXp0WLFmXYWhllyJAhprh9+/ZWbSlJy3H43Xff6YUXXjAd/6+//rrWrFnj8FyOyJo1a6pr+Pv7q0iRIkZ848aNZIsNff7556b4gw8+kJ+fX4rz//nnnxladAcAYB+K0gAAAAAAAAAAAAAAAAAAAAAAAAAAAACPyIABA0zx9OnTk+179OhRXbp0yYjd3NxsFsL48ccfjedZsmTRiBEj7M5n2LBhxvOEhAQtX77ctD1fvnymeMuWLXbPnR4WLlxoir/99ttHuv7YsWON5xaLRdOmTbN77JgxY0zxzJkz0y2v9JI7d261aNEiQ9eoXLmyqSjLihUrMnS99BYfH68jR44YsYuLi37++ecMXfPNN9/UkCFDlJCQIOn+sff1119r6tSpGbquJL3yyit2FdGpW7euKQ4ICLDZ79SpU8ZzNzc3ffTRR6nO7enpqebNm6faDwCQsdycncCTJjw8PMPXeGvh4/UPsYdl8bW/iiCAJLI0dHYGQKZh6X/Y2SkAj60fTtx1dgpAJuLr7ASATMNSZ4GzUwAAAAAAAAAAAAAAAAAAAMiU3nnnHY0aNUrx8fGSpDNnzigsLEw+Pj5WfUeOHGmKGzRoYHPOkJAQ47mfn59D+TRq1MgUr127Vm3btjXiIkWKyMPDQzExMcb24cOHa+LEiXJzy9ifKsfHxyssLMyIPT091bp16wxdM6mDBw8az/PmzascOXLYPbZMmTKyWCxGYZFt27ale34Pq3r16mkeGxUVpU8++UR//PGHQkJCdPv2beO4Tsm5c+fSvKYzLFmyxHgPpftFdrJly5Zh67300ktatWqVEbu5uWn58uUZXjzogS5dutjVr0KFCqY4NDTUqs/mzZtNr13p0qXtzqN3795WRbIAAI9W6iXKkC4e/AMzPj7ern9MAQAAAAAAAAAAAAAAAAAAAAAAAAAA4N/Hw8NDtWrVMrV9+OGHNvuuWbPGFI8ZM8aqz+HDh02/XT1y5IgsFovdj0KFCpnmu3TpktUar7zyiimeMmWKPD09VbVqVb355psKDAxMeafTaM+ePaaCFiVKlMiQdVJy8+ZN4/mVK1ccem0TF6SRpGvXrj3y/FPz1FNPpWlcnz59lCtXLk2YMEFHjx5VVFSU3b+hvnHjRprWdJaNGzea4vr162fIOgkJCapUqZKpIE327Nm1d+/eR1aQRrK/UJG3t7cpvnr1qlWfnTt3muLKlSvbnUfz5s3t7gsAyBgUpXlERowYodKlS8vHx0cuLrzsAAAAAAAAAAAAAAAAAAAAAAAAAAAAT6qRI0ea4qVLl1r1Wb58uaKjo43Y29vbZjGMU6dOpWtuERERVm2//vqrqlSpYmqLi4vTwYMHNXnyZNWuXVvu7u4qU6aM3n//fUVFRaVLLsHBwaa4YMGC6TKvva5cuZKu86XX65KeChQo4PCYmjVratasWaaCO46IiYlJ0zhnSVqoqUyZMhmyTmRkpI4cOWLEefPmVXBwsEOFXNKDvcdE0t/M2zoewsLC0jS3dL+Al8Visbs/ACD9uTk7gSfFyy+/rJdfflnS/X9oJT2BpqvRXhk3d0Yb7Xhlw4Fls2RAIsDj44cTd9M8NuHwm+mYCfD4sVT60nj+ZgXOJ3iyfXn0Ic4nQX3SMRPg8WOp+VOiKNRpeQCZg+//PY077Lw0gMzAtZKzMwAAAAAAAAAAAAAAAAAAZGKtWrVS9uzZdevWLUnS9evXtXPnTtWpU8foM3bsWNOY9u3b25zr4sWL6ZpbfHy8zfYDBw5o7Nixmjhxom7csP49aGxsrE6ePKnPPvtM48ePV9euXfXzzz8/VC6XL182xTly5Hio+Rx17ty5dJ0vudfWmTw9PR3qP3z4cO3Zs8fUVrhwYTVr1kzPP/+8qlWrpoIFCypPnjymoiVubm6Ki4uTZLt4SWYWGRlpivPkyfNI1r1+/bq2bdumVq1aPZL1MkLiwlqS48ebxWJ57I4XAPg3cUm9CwAAAAAAAAAAAAAAAAAAAAAAAAAAAID01KJFC1P80UcfGc9jY2MVFBRk2v7pp5/anMfb29sU16lTRwkJCWl+bNy4MdmcR44cqYiICO3YsUN9+/ZV2bJllSWL9c2i4+Pj9csvv6hs2bKpvAopK1CggCmOiop6qPkclTdvXlNctGjRh3ptT548+UjzzwjffvutVXz+/Hn99NNP6tq1qypVqqR8+fKZCtJImbMgj728vLxM8bVr1zJsnebNmxtxfHy82rRpo/nz52fIeo9Cvnz5TPH169cdGv84HzcA8G9AURoAAAAAAAAAAAAAAAAAAAAAAAAAAADgEfvss89MceJiMBMnTjQVYyhVqpR8fHxszlOqVClT7GjRh7SoU6eOZsyYoePHj+vOnTs6duyYBg4cqDx58pj6/fPPPxo8eHCa1ylZsqQpDgsLS/NcaVG8eHFT/KiL4mQ2f//9t2JiYoy4UaNGdr2/UVFRSkhIyMjUMlTBggVN8YkTJzJsrVWrVqljx45GnJCQoC5duuiHH37IsDUzUrFixUzx6dOn7R575syZ9E4HAOAgitIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAj5ifn58KFy5sxDExMZozZ44kWRWgGDJkSLLzPPXUU7JYLEYcEhKSzpmmrly5cvr+++919epV9e3b17Rt/vz5aZ63WrVqpn07e/ZsmudKK09PT+N5RESEqVjQk2bLli2muHv37naNW7hwYUak88g0btzYFG/dujVD11uwYIH69+9vahs0aJAmTpyYoetmhDZt2pjiI0eO2D123rx56ZwNAMBRFKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAnKB3796meNKkSQoNDTUVX3F1ddXw4cOTncPDw0M+Pj5GHB0drc2bN6d/snaaMWOGsmXLZsQ3btxI81wuLi4qVKiQEd+5c0fLli17qPwcVaFCBeN5QkKCpk6d+kjXtyVLliym+N69e49k3fDwcFNcpEgRu8Z99913GZHOI9O2bVtTcaRDhw7p9u3bGbrm9OnT9fbbb5va3nnnHX344YcZum56y5cvn+nz4PLly/rnn3/sGjtr1qyMSgsAYCeK0gAAAAAAAAAAAAAAAAAAAAAAAAAAAABOMHLkSKtiF++8846pT7169eTikvJPgjt16mSKkxa7edTy5s2bbnN17drVFL/++uvpNrc9hg4daopHjx6t2NjYR5pDUvnz5zfFYWFhj2TdPHnymOKgoKBUx+zbt8+ufpmZi4uLqlSpYsTx8fHq0aNHhq87ceJEffzxx6a2Tz/91OqYzOxefPFFU/zqq6+mOub333/XqVOnMiolAICdKEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAOIGnp6eqV69uxAkJCZo3b56pz0cffZTqPF988YWyZMlixKdPn1b79u0dyiUiIkLfffedVfuBAwe0c+dOu+eJiopSaGioEXt4eDiUR1Ljxo0zzREaGqqWLVvaPT4+Pv6h1u/Zs6epCExERITq1q3r0Lx37tzRlClTHiqPxJ5++mlTHBAQkG5zp+T55583xd98802K/SMjI9W4ceOMTOmR+fbbb03x0qVLrdpSktbj8KOPPrI6dr7++munF55yxOTJk03Ft/bt26dBgwYl2//QoUN65ZVXHkVqAIBUUJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAcJL33nvPFCckJBjPc+bMqaZNm6Y6h4uLi3744QdT22+//SY/Pz9t3rw5xbGLFi3Ss88+qzx58mjMmDFW2//++2/VrVtXxYoV08iRIxUREZHsXEePHlXZsmUVFxdntNWuXTvV/FPi5uZmVZRjxYoVqlChgg4cOJDsuCVLlqhq1aqqX7/+Q63/YK7ERTWCgoLk4+OjpUuXpjhu3bp1atasmXLmzGn1Pj+MYsWKmYoQ7dq1S2+88YbOnz+fbmvY4u/vL09PTyM+f/686tevrzt37lj1XbdunYoVK6br169naE6PyrPPPqvmzZub2l577TW1bNlSkZGRNsfExMRowoQJKlSokD788MM0rz106FD9P/buOyqK6+0D+Hdp0ougItjBgiIqVqzYe+9RwW6Mxq6JsWvU2E0sUew9iuVn74rYCyo2bKioKFKkKAgo8P7BYd6dbezCLkW/n3M4hzt7Z+bZ3Tuz0+5zN27cKGqDmzdv1jjxVG4pUaKEXBKatWvXonTp0li9ejXCwsKQmJgIf39/dO7cGW5ubkhOToaBgYHoPRMRUc4zyO0AiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIh+VN26dYOJiQm+fPki91qnTp3UXo63tzdu376Nf/75R5j24sULNGrUCKampihVqhRsbGwAAHFxcQgLC0NUVBRSU1PVWv6bN28wb948zJs3D2ZmZnB0dISNjQ1MTEwQFxeHkJAQREVFiebR19fHzp071X4PygwfPhwXL17Erl27hGmPHz9GlSpVYG1tjRIlSsDGxgYJCQkICwvD+/fv8e3bNwBA1apVs73+hg0bYtmyZRgzZowwLSIiAt26dUOBAgVQsmRJ2NrawsDAALGxsQgPD0dERIQoOY+BgXa7dbdp0wYHDhwQyitWrMCKFSvk6g0fPhyrV6/W2nrHjh2L+fPnC+XLly/D3Nwczs7OKFKkCD59+oQXL14gNjZWqFO/fn1cvXpV9HnkR0eOHEGZMmUQEhIimmZlZYWiRYvCwcEB5ubmiIuLw9u3bxEZGSkkmVJ3O1NmwIABsLS0RI8ePYRl7d+/Hy1atMCpU6eyteycsHr1aty8eRO3bt0Spr169QojRozAiBEjFM6zadMm9O/fX2g3+vr6ORIrERH9PyalISIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiLKRa1atRIlGMnw559/arScv//+GxUrVsSIESNECUASEhLw6NGjTOc3NDRUaz3x8fF4+vRppss6ceIESpQoodYyM7Nz504UK1YMixYtEk2PiYlBTEyMVtahyujRo+Hk5IRu3bohKSlJmJ6UlJTpZwFoP6HGf//9hzJlyiA0NFRlveTkZK2ud968ebh06RIuXrwoTEtJScGTJ0/w5MkTufqVKlXChQsXYGRkpNU4coOenh5evHghJNmR9v79e7x//16n6+/atSuOHTuGtm3bCtv36dOnUbduXVy5ckWn69aGmzdvon379jhy5IjKevr6+tiwYQP69u0LLy8vYbq5ubmuQyQiIhl6uR0AEREREREREREREREREREREREREREREREREREREdGPbO7cuXLTSpQokaWELsOGDUNkZCS6deuGAgUKZFpfT08PJUuWxB9//KEwucpPP/2EIUOGoFixYpBIJJkuz9DQEK1bt0Z4eDiaNGmicfyqLFy4EI8ePUL16tUzjUVfXx/u7u5YtmyZ1tbfrl07xMTEYNiwYTA1Nc20vkQigYODA3755Re8fv1aa3EAgJGREd6+fYtZs2ahTJkyan3X2uLv748xY8bAwMBAaR0zMzOMGzcODx48gJ7e99OlXU9PD1euXMH+/ftRrFixTOsbGxujTZs2GD58uFbW37JlS1y4cEGUQOrq1atwc3NDamqqVtahS4cPH8alS5fQsGFDmJqaCtuxvr4+ChYsCG9vb7x9+xbe3t5ITk5GWlqaMK+lpWVuhU1E9MOSpEnviTVQvXp1hIWFwd7eHgEBAdqOK99T9fno/LObaaX9ZeaUmbEazzKsXM4dJBPlRWufJmVeSYm0h+O0GAlR/iOptFT4f5wLf0/ox7Y0KBu/JwEDtRgJUf4jqb5RqvQu1+Igyhsc/v/flIe5FwZRXqBfKbcjICIiIiIiIiIiIiIiIiIiIiIiyrNiQ57j/Y2LuR2GUkVrNYBVSefcDkNr7t27h127duH169eIjIyEnp4erKysUK5cOXh6emqcOMbPzw8XLlzA48ePER0djZSUFFhaWqJUqVJo1aoVmjdvrqN3Ipaamopdu3bh4sWLeP/+PRISEmBhYQFnZ2c0bdoULVu21HkMwcHB2LZtG168eIHw8HCkpaXB0tISZcqUQYMGDdCqVSuViVvyu2/fvmHnzp04deoUwsPDYWFhgVKlSqFt27ZaT0iUV8XExGDDhg148OABPnz4gJSUFNjY2MDV1RUdOnSAm5tbboeYb+3Zswc9e/YUyiNHjsSKFStyMSIioh/P93sUQ0RERERERERERERERERERERERERERERERERERET0g3Nzc9NqYgxPT094enpqbXlZpaenhz59+qBPnz65FoOTkxNmzpyZa+vPbQYGBvDy8oKXl1duh5JrrK2tMX78+NwO47u0atUqUblTp065EwgR0Q9ML7cDICIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiICgNu3b+PixYtC2cjICE2bNs3FiIiIfkxMSkNEREREREREREREREREREREREREREREREREREREOcbQ3DK3Q1Apr8dHRJQfzZw5E+Hh4ZnWu337Nho0aIC0tDRhWtu2bXUZGhERKWGQ2wEQERERERERERERERERERERERERERERERERERER0Y/D1LYwSjRpi6+f43I7FDmG5pYwtS2c22EQEX13li5ditmzZ6NcuXLo2LEj2rZtCzc3N1haWuLNmzc4evQotm7diuvXr4vmMzY2xubNm3MnaCKiHxyT0hARERERERERERERERERERERERERERERERERERFRjjK1LQww+QsR0Q8lLS0NT548wcKFC7Fw4cJM6+vp6WHfvn2wtLTMgeiIiEgWk9IQERERERERERERERERERERERERERERERERERERERERkc4YGhpqVL9w4cI4fvw43N3ddRQRERFlRi+3AyAiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiKi79f79++xcOFC1K1bF7a2tjAwMBC9rq+vD0tLS9SrVw///fcfPnz4wIQ0RES5zCDzKkREREREREREREREREREREREREREREREREREREREREREWWNkZISJEydi4sSJuR0KERGpSS+3AyAiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiKivINJaYiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIhIwKQ0RERERERERERERERERERERERERERERERERERERERERERERCRgUhoiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiEjApDREREREREREREREREREREREREREREREREREREREREREREREJmJSGiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiARMSkNEREREREREREREREREREREREREREREREREREREREREREREAialISIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIBk9IQERERERERERER0Q9v8+bNkEgkwt/mzZtzO6Q8aebMmaLPyc/PL7dDytMCAwMxYsQIuLm5oWDBgtDX1xd9fvl9fUSUs/r37y/apl+9epXbIf3QPD09uY/Np4KDgzFx4kTUrFkTdnZ2MDAw4LZF3z3us4jk8fw2b+Gxbv729etXVKxYUfj+Vq1aldshUR7FY5Lc899//wmfe/HixZGQkJDbIRERERERERERERERUT5hkNsBEBEREREREREREVHmvnz5gtu3b+PZs2eIjo5GfHw8TExMYGlpiRIlSsDJyQllypSBnh5zkRPlttTUVEyaNAlLliz5LtdHlFekpKQgICAAjx49Qnh4OL5+/Qpzc3MUK1YMLi4uqFChAn8XiUhk2bJlmDhxIlJSUnI7FCIiIqLvxooVKxAUFAQAcHJywtChQ3M5IiKS1bNnTyxYsAB3797F27dv8ddff2H27Nm5HRYREREREREREREREeUDTEpDRERERERERERElEelpaXh0KFDWLNmDc6cOYNv376prG9hYYHq1aujUaNGaN26NWrWrMnO+ES54K+//srRBDE5vT6i3Pby5UssWrQIu3btQkxMjNJ6lpaWaNy4MYYOHYo2bdrkXIBElCft3LkT48aNy+0wiIjyvNTUVDRo0ABXrlwRTW/UqBH8/PxyJygiyrMiIyNFiS1mzJgBQ0NDlfOUKlUKISEhQvn8+fPw9PTM0vo1XdbMmTMxa9Ysuel16tTB1atXsxTD58+fYW9vj/j4eLnXshqPLDMzM1hbW6NYsWKoWbMmmjZtinbt2sHAgI8Ak3okEgnmzJmD9u3bAwAWLVqEIUOGoHjx4rkcGRERERERERERERER5XXsjUBERERERERERESUB4WEhKBZs2bo1KkTTpw4kWlCGgD49OkT/Pz8MGvWLNSpUweHDx/OgUjzhv79+0MikQh/r169yu2Q6Af18eNHzJkzRyhLJBJ4eXlh586dOHXqFE6fPi385cf1fW9mzpwp2newk23elpqaivnz58PFxQX//vuvyoQ0ABAXF4eDBw9i69atORMgUT7m6ekp2h9+b759+4YJEyaIprVv3x6bN2/GyZMnRb+X9vb2uRQlkXLS22dWO+0TqWvlypVyCWmISDM/0nWqhQsXIjY2FgBQokQJ9O7dO5cjyppr167h2bNnWZp33759ChPSaFN8fDxCQ0Nx/fp1rFy5Ep07d0apUqWwbds2na6X8g5tXMNq27YtXF1dAQCJiYmYO3eulqMkIiIiIiIiIiIiIqLvEYdJICIiIiIiIiIiIspjXrx4gYYNGyI0NFTuNSMjI5QuXRpWVlZISkrCx48fERoaitTUVLm6aWlpOREuEUnZs2cPEhMThfK0adPUGvE6v6yPKLd8/foVffr0ga+vr9xrVlZWKFq0KCwtLfHp0yeEhIQgISEhF6IkorzqzJkzeP/+vVD28vLCli1bcjEiIqK8KSQkBFOmTMntMIgon/jw4QNWrVollMeOHQsDg/z7SOrWrVtFiX/VlVvHlaGhofDy8oKfnx/Wr1//XSaXJO2SSCSYMGEC+vfvDwDYuHEjfv/9d5QqVSpX4yIiIiIiIiIiIiIiorwt/94BJCIiIiIiIiIiIvoOff36Fe3btxclpJFIJOjTpw+GDRuGOnXqyHXu+Pz5MwICAnD8+HHs3bsXwcHBOR02/SBmzpyJmTNn5nYYedrVq1dF5WHDhn1X6yPKLYMGDRIlpDEwMMCwYcPg7e2NGjVqiDrfpaam4unTpzh58iT27NnDjnlExN9LIiI1DR06FJ8/fwYAmJmZIT4+PpcjIqK8bMWKFUJCUBMTEyHRRX6ip6cnJPvevn07Zs+erdE5ZEhICPz8/BQuLytatGiBiRMnyk2Pi4tDcHAwjhw5An9/f9FrGzduhIODQ5YS6tCPp2fPnhg7diyio6Px9etXLF++HMuXL8/tsIiIiIiI6Af37tVVxH58kdthyLEqWAYOpTxyOwwiIiIiolzHpDREREREREREREREeciaNWvw6NEjoWxsbIx9+/ahTZs2SucxNzdHo0aN0KhRI/z111+4cOECli1bBn19/ZwImYikPH36VPjf2toaDg4O39X6iHLD9u3bsW3bNqHs4OCA48ePw83NTWF9PT09VKhQARUqVMDo0aMRHR2dU6ESUR4l/XsJAJUqVcqlSIiI8q7Nmzfj1KlTANKvM0ycOBEzZszI5aiIKK9KTEzE2rVrhXK3bt1gbW2dewFlUePGjXH27FkAwKtXr+Dv749GjRqpPf+2bduQlpYGADAyMkLt2rVx8eLFLMdTtGhRNGvWTOnrEydOxIkTJ/DTTz+JznUXLFiAgQMHonTp0lleN/0YjI2N0adPH6xcuRIAsGnTJsyZMwcWFha5HBkREREREf2o3r26it2r6+Z2GEr1/OUKE9MQERER0Q9PL7cDICIiIiIiIiIiIqL/t2XLFlF5xowZKhPSKNKoUSP873//Q/v27bUZGhGpISYmRvjf0tLyu1sfUU6LjIzE2LFjhbKVlRUuXLigNCGNIjY2NroIjYjyEenfS4C/mUREsj58+IBx48YJ5Tlz5qBEiRK5GBER5XW+vr6IjIwUyv369cvFaLKuVatWKFy4sFCWvTabma1btwr/t2vXDgULFtRabMq0atUKe/bsEU37+vUrVq1apfN10/fBy8tL+D8uLg47duzIxWiIiIiIiOhHF/vxRW6HoFJej4+IiIiIKCcwKQ0RERERERERERFRHvHx40cEBAQIZT09PQwZMiQXIyIiTSUmJgr/SySS7259RDlt7ty5oo6O8+bNg7Ozcy5GRET5kfTvJcDfTCIiWSNGjEB0dDQAoEaNGhg1alQuR0REed327duF/21sbNC4ceNcjCbrDAwM8NNPPwnlvXv34suXL2rNe+XKFTx79kwoe3t7az0+ZZo1a4bmzZuLpp06dSrH1k/5W40aNVCsWDGhLL09ExERERERERERERERyTLI7QCIiIiIiIiIiIiIKF1oaKiobGdnB1tb21yKJmc8efIE169fx7t376Cvr4/ChQujVq1acHFxye3QRFJTU3Hjxg0EBgYiKioKZmZmKFq0KBo2bAh7e3utrCM8PBzXrl1DWFgYoqKiYG5uLnwepUuXztayP378iNu3b+P58+eIjY3Ft2/fYGpqCjs7O5QuXRqVKlWCjY2NVt6HMl++fEFgYCAePXqE6OhofPnyBSYmJrC0tESpUqVQoUIFFC9eXKcxyEpISMClS5fw5s0bREREwNjYGIULF0alSpVQpUqVLC0zLS1Ny1HmzvpCQ0MREBCAd+/eISoqCtbW1ujcuTMcHBx0sr6sSEtLw7179/D48WNEREQgNjYWpqamsLe3h4uLCypXrgx9ff1ciS0mJgaXL1/Gu3fvEBkZKWzP1apVQ7ly5bK17NTUVDx69Aj37t1DREQEPn36BCMjI5ibm6N48eJwdnZG+fLloaeX9bEpXr9+jVu3buHDhw+Ijo6GlZUV7O3tUa9ePa3t89SRlJQkGnXe3t4ew4YNy7H1a8uDBw9w69YthIWFwdDQEI6OjqhTpw5KlSql1fV8+fIF/v7+ePz4MT5//gwbGxuUKlUKjRo1gpmZWbaXn9/bNZC+3wgICMDdu3cREREBU1NTODo6okGDBihSpEi2li0rNjYW/v7+eP78Ob58+QJbW1s4Ozujfv36KFCgQJaXm5qaimfPnuHhw4d49+4d4uLiUKBAARQsWBDOzs6oVatWtpavbSkpKbh16xaeP3+O8PBwJCUloVChQihdujTq1auXY7Hq6vcyKioK165dw7t37xAREQFzc3O0atUq023iyZMnuHPnDsLDwxEfHw87Ozs4ODigfv36sLKy0nqcN2/exLNnzxAaGgo9PT04OTmhcePGma4rMTERly5dQlBQED59+gQbGxtUqFABDRo0gIFB7jxuktNtKqvfcX6jq32WIhnf3/v375GYmIiSJUuKkgEooovjdmlPnz5FYGAg3r9/j0+fPsHAwABmZmZwdHSEk5MTKlasmK02r8tzPW04cOAA9u3bByA9OcO6deuy/ZurS8+fP8f169eFayiOjo5wd3fX+nWEr1+/4sqVK3jw4AFiYmJgaWmJ4sWLo1GjRlo5f87v7TrD93Cs+/LlSzx8+BCvX79GbGwsDAwMULBgQZQsWRJ16tSBubm5lt5F9mWcBwcFBYmOI4oVK4YGDRrkWKzh4eE4e/asUG7Tpk2uHRtog7e3N5YvXw4A+PTpEw4cOJDpbxMAbNmyRfi/UKFCaN26NTZu3KirMOW0a9cOp0+fFsoPHz5EamqqTvbhYWFhuH37Nl69eoW4uDikpqbC1NQUhQsXRpkyZeDq6qqV9peTxyQ55fXr17hy5Qpev36NtLQ0FCpUCFWrVkW1atVyLVmmRCJBhw4dsHr1agDpCZZCQkJQsmTJXImHiIiIiIiIiIiIiIjytvx7J5CIiIiIiIiIiIjoO/Pp0ydROSUlRevrePv2LUqVKiUsu1y5cnjy5InGyzl06BA6duwolHv06IHdu3eL6rx69UrUwc7b2xubN28GkD5y75QpU3Dr1i2Fy3dxccGCBQvQvn17pTFs3rwZAwYMUPiaqo59JUuWxKtXr5S+Li01NRX//vsv/vrrL7x9+1budYlEghYtWmDx4sVwdXVVa5myy9++fTtWrFiBgIAApR2mXVxcMGnSJHh5eWnUseTs2bNYsGABzp49i9TUVKX1JBIJypcvj44dO2LEiBFKk8PMnDkTs2bNEsrnz5+Hp6enyhieP3+O2bNnY//+/YiPj1dZ18HBAS1btsTQoUNRp04dlXWzIzAwEDNmzMDJkyeRmJiosI6joyMGDhyISZMmqexUI/uZSAsJCVHauSSrneO1tT7peo0aNYKfnx8A4OjRo1i6dCn8/Pzk2oyjoyM6deoEQPX2rQ5PT09cuHBBaXyqvH79GvPmzcOBAwcQHh6utJ6NjQ3atGmDYcOGoUGDBsJ0Pz8/pSO4Zzaye2Zx+vv7Y9asWfD398e3b98U1nF2dsYvv/yCESNGwMjISOXypMXFxeGvv/7C5s2b8f79e5V1LS0t0bBhQ/Tt2xc9e/ZUa/nJycn4999/4ePjg0ePHimsI5FIUL16dUybNg0dOnRQO/asOnDgAD5+/CiUe/XqlWuJhlRRtj3t378f06ZNU/h5SiQS1K1bF4sWLYKHh0e21h8bG4tZs2bBx8dH4X7WyMgIgwYNwuzZs2FnZ6fx8vNTu1a2b0pLS8P69esxd+5chISEyM2nr6+PFi1aYMmSJdnu0P7+/XtMmTIFO3bsQHJystzrZmZmGDNmDCZPnqx2B+qMjrH/+9//4Ofnh+joaKV1CxQogPbt22Py5Mlwd3dXuVxVvymqOkdKt3NlXr16hTlz5uB///ufaDuWZmpqii5dumD27NlaT8qg6jgRUP7+Xr58KepEX6pUKaHNSB9DXr16FfPnz8eJEyfw9etX0TKWLVumMGFJUlISVqxYgTVr1iA4OFjh+g0MDNCoUSPMnDkT9evXV/UWBbK/azNmzMDMmTORkpKCVatWYeXKlXj27JncfKamphgxYgRmz54NY2Nj0WufPn3Cn3/+iTVr1iAuLk5u3kKFCmH+/PkYNGiQWjFqg67aVFa/Y1NTU52fV8keL2W4cOGCym00ow2oQ9v7LNltb9OmTejfvz++fPmCxYsXY9OmTXj58qVoHisrK6Ud/7V53C4rKSkJy5cvx7p165RukxlMTEzg4eGB7t274+eff1Zr+bo+19OWmJgYjBgxQiiPHTsWVatWzfE4pCnbLv38/DB58mRcu3ZN4XxVqlTB3Llz0bZt22ytPykpCQsXLsTy5csV7m/09fXRrVs3LFiwIEud9/Nbu/4ej3UTExNx9OhR7N+/H+fOnUNYWJjSuvr6+mjatCkmT56c6fUPXV6nioiIwPz58/Hff/8pPW42MjJCq1atMGfOHLi5ualcXnYdP35cdN0ys3PpvK5q1aqoXLky7t+/DwDYunVrpklpEhMTsWfPHqHcu3dvGBoa6jROWWXKlBGVU1NTER0drdUk576+vli2bBmuXr2qsp6+vj7c3NzQqVMnjBw5EgULFtRoPdo+Junfv78oaZDscb4qyo6vlVG2n7x58yZ+//13nDt3TuF8xYsXx+zZs9G/f3+1Y5GW3WtYjRs3FpLSpKWl4ejRo/jll19UzkNERERERERERERERD+mvDusDREREREREREREdEPxtraWlSOiorC8+fPtbqOYsWKiTpoPX36FP7+/hovZ926daLy0KFD1Z530qRJaNmypdKENAAQFBSEDh06YM6cORrHpi1xcXFo0aIFRo4cqTAhDZD+wP7JkydRu3ZtnDx5UqPlP3v2DO7u7vD29satW7dUdhQICgrCgAEDUK9ePURERGS67LS0NPz6669o1qwZTp8+rTIhTUb9x48fY8GCBRq/D1W2bdsGV1dXbNu2LdOENADw7t07bNq0CStXrtRaDNLS0tLw22+/wd3dHQcPHlTaARAAQkNDMWfOHJQtWxaXLl3SSTx5SUabadeuHc6dO5dpm8kNaWlpmDVrFsqWLYu1a9eqTEgDANHR0dixYwcaNmyo89iSk5Ph5eWFRo0a4dy5c0oTdwDpiZrGjRsHV1dXPH78WK3lBwYGwsXFBfPnz880cQeQvv86cuQIxo8fr9byr1+/jgoVKmDMmDFKE9IA6d/BrVu30LFjR3To0EGt7To7jh49Kirnl46OGdtT165dlX6eaWlpuHz5MurXr5+t37pHjx6hSpUqWLZsmdLvIyPhUJ06ddROypYxX35u1xmSk5PRrVs3DB06VGFCGiA9EeDx48dRrVo1bNq0SaPlS7t48SJcXV2xadMmhR0pASA+Ph5z585F48aNlSbWkFW6dGl4e3vjwIEDKhPSAOmd0ffu3YsaNWpg3rx5Gr8Hbfjzzz9Rvnx5bNy4UeV7TEhIwPbt21GhQgVs2LAhByPMnoULF6JevXo4fPiwXLISZR4+fIiKFSti4sSJKpMEfPv2DWfPnkWDBg0wcOBAtZcvKz4+Hq1bt8bo0aMVJqQB0j//RYsWoUWLFvjy5YswPTg4GNWrV8fChQsVJqQB0jvGDx48GGPHjs1SfJrK6TalznecF86rsktX+yxZISEhqFGjBqZPny6XkEYZXR+3v379GlWrVsXvv/+eaeIOAPjy5QvOnTuH4cOHq/w9zKDLcz1tGzdunPA7XLp0abUTGuW0RYsWoUmTJkoT0gDpxxbt2rXDzz//nOUkoKGhoahTpw6mT5+utM2npKRg9+7dqFGjBu7cuaP2svN7u5Z+H/n9WLd+/fro1q0bdu7cqTIhDZD+fZ86dQqNGzfGyJEjNfqstGXDhg1wcnLCsmXLVB43Jycn49ChQ6hWrRpmz56t05hkrx01atRIp+vLCd7e3sL/Z86cyfQc5eDBg4iJiVE4f05RlARHW200KSkJXbp0QY8ePTJNSAOkbyt37tzBjBkzcPv2bY3WlVPHJDnpn3/+gYeHh9KENADw5s0bDBgwAEOHDs2V63Cy18tOnDiR4zEQERERERGR7jk7O0MikUAikcDAwCC3w/muXLp0SfhsJRIJBg8enNshac29e/cwfvx4dO7cGc2bN0fXrl3xxx9//BDPT+lCZGQkevToATs7O+jr64vajUQikRsIkIi0x9raWtjWZJ/Pzg+WL18u2l8sX75caV1PT09RXSJtY1IaIiIiIiIiIiIiojyiTJkyMDY2Fk377bffstyZShnZEbBlO0JmJjQ0FMePHxfKZcqUQZMmTdSa9/fff8eiRYuEsoWFBSpVqoQaNWqgUKFCcvWnT5+O/fv3axSfNnz9+hXt2rXD2bNnhWmFCxeGu7s73Nzc5EbkTUhIQLdu3ZR2dpd1/fp11K1bF4GBgaLp+vr6cHZ2Rq1atVCxYkW59nDt2jV4eHhk2llx+vTpChO7FCxYEFWqVEGdOnVQuXJlFC1aVK14s+L06dPw9vZGUlKSaLqpqSkqVqyIOnXqoFq1aihVqhT09HR/uyItLQ39+/fHwoUL5Tp62NnZwd3dHS4uLnKfeVhYGFq0aKHVZD150R9//CFqMyYmJqhQoQKqV68Oe3v7XIws3devX9GjRw/MnDlTroOSkZERnJycUKtWLVSqVCnHbyAnJSWhbdu22LZtm9xrRYsWRY0aNVCuXDm5zmLPnj1D/fr1M+3I+uHDBzRt2hTv3r0TTTcwMICTkxNq1qyJmjVroly5cnLtVx2HDx9G48aN5TqJGxkZoXz58qhVqxYqVKgg94Da4cOH0aRJE5WdabPr5s2bonKVKlUA/H8CkV69eqF8+fIwMzODtbU1ypYtix49emDTpk1ISEjQWVyZ+fPPP0Xbk6mpKSpVqoSqVavKtc/U1FRMnz4df/31l8brefXqFZo2bSr67SlZsiRq1qwJFxcX6Ovri+oHBwejS5cuanVSzO/tWtrw4cNFxxKWlpZwc3ODq6srzM3NRXWTkpIwePBg7NixQ+P1BAQEoHXr1kIHST09PWHfVLZsWbkHPm7evIn+/furtWzZ7UwikaB48eJwc3NDnTp1UKlSJZiamorqpKWlYcqUKTrvkCwtJSUF/fv3x7Rp0+T21ba2tnBzc0ONGjXg6Ogoei05ORmDBw/GsmXLcizWrFq7dq3o+NzIyAjlypVDzZo14ejoqPDBnlu3bqFBgwZ48eKFaLqhoSHKli2LGjVqwMHBQW6+TZs2oUOHDko75iqTlpaGXr164fTp08I0BwcH1KhRAxUrVpTbN1y8eBGjR48GAISHh6NJkyZCIhuJRIIyZcqgZs2aKFOmjNy6li9fnqXtRV250aY0+Y5z87wqu3S5z5KWkehTOnFF4cKFUa1aNVSsWFHuvArQ/XH7ly9f0KxZM7kkanp6eihZsiSqV68uHP/I/k6oQ9fnetp05swZUTK2NWvWyP2e5AXbtm3DpEmThO2yQIECKF++PNzd3RVeR1i7dq3c9qmOmJgYNGvWDHfv3hWmOTo6okaNGnB1dUWBAgVE9SMjI9GhQwelCbyk5fd2Le17ONZVdA7j4OAAV1dX4XqJlZWVXJ1Vq1blaPIwAJg2bRoGDx6MT58+iaZbWlqiUqVKqFWrFkqVKiV6LTU1FTNmzBB+33Xh4sWLwv/W1tZwdnbW2bpySp8+fYR2lZKSgu3bt6usv2XLFuH/SpUqwd3dXafxKSJ7PgWkX/vThsGDB+PAgQNy0zN+xzPOQxTthzWRU8ckOWnNmjUYPXo0UlJSAKRfY3NxcUHNmjUVHvevW7dOZUcCXSlcuDCKFy8ulKW3ayIiIiIiItIN2c7i2hiU5du3b3JJLu7du6eFaOlHdODAARQpUgRVqlTB0qVL8b///Q9nzpzB/v37MX/+fDRo0ACGhoZwd3fHsWPHcjvcfOH48eMoWrQofH19ERUVlScHCSMiIlIHk9IQERERERERERER5RHGxsZo2rSpaNr+/fvRtGlTXL58WWvradmypajDyt69exEdHa32/Bs3bhQeqgfSOymok1nf398fCxYsAADUrl0bp06dwsePH/HgwQPcvHkTHz58wPnz51GhQgXRfKNGjVLYqahly5Y4ffo0Tp8+jRYtWohe2759u/Ca7J86HWf/+usv4UH8Pn364N69e/jw4QMCAgIQGBiIqKgobNy4EZaWlsI8nz9/xqRJkzJddlhYGDp06IDIyEhhmpubG3bt2oWYmBg8e/YM169fx8OHDxEdHY3du3fDyclJqBscHIz+/fsrTVYUGhoqfM4Zhg0bhkePHiEqKgp3797F1atXce/ePbx79w4fP37EkSNH8PPPPyvsdJVVY8aMEcXYuHFj+Pn5IS4uDg8fPsTVq1dx+/ZtvHz5Ep8/f8aVK1cwdepUhR2etWHlypXYunWraFqDBg1w+fJlhIeHIyAgAI8ePUJERATWrVsn6sTz5csX/PTTTwo7+3h5eYnaV5EiRYTXihQporQdZpUu1vfo0SMsXLgQAFC2bFn4+vri48ePCAoKwq1bt/D+/Xvcv38fbm5uWY47uyZNmoS9e/eKplWtWhX79u3Dx48f8fz5c1y/fh0PHjxAdHQ0nj17huXLl6NmzZpyy6pSpYrwufTr10/02uLFi5V+hso+xz/++ANnzpwRTevUqRMCAwPx7t073Lx5E0+ePEFYWBgWLlwIExMToV5UVBS6d++Oz58/K33vs2fPRlRUlFAuU6aMsL94/vw5bty4gRs3buDJkyf4/PkzHj58iKVLl6Ju3bqZ7psfPnyInj174suXL8K0Bg0a4MiRI4iNjcXjx49x/fp1BAUF4ePHj/Dx8RG1uRs3bmDs2LEq15FVsbGxePr0qVDW19dHyZIl8eLFCzRo0ABt2rTB7t278fTpUyQkJCA2NhbPnz+Hr68vBg4ciLJlyypMqKJrz58/F5KAFClSBFu3bkVUVBQePHiAO3fuIDIyEidOnICLi4tovilTpqg1+ry0QYMGISwsDCYmJpg+fTpCQ0Px6tUr3LhxQ9if/fbbb6J2cOfOHbWSJuTndi3Nz88PGzduBAA4OTnh4MGDiIqKQmBgIO7fv4+oqCjs3r1blNAiNTUVQ4cOlUvUlJkePXogPj4eBQsWxNKlSxEeHi7sm54+fYq3b9/KdZ48fPiwKCGFKhUqVMC0adNw5coVfP78Ga9fv0ZgYCCuXr2KBw8e4NOnT7h69Sp69eolmm/27NlyCZ4ySP+myO7jVe0LlyxZonB5s2fPFnXONTQ0xOjRo/Hw4UNERkYiMDAQN2/exNu3b/H8+XMMGzZM9H1OmjQJV65cUevzyIz0caIm709VIraPHz8K+7yiRYtiw4YNiIyMxJMnT3Djxg28ffsWwcHBaNasmTDPp0+f0L17d9GxtqmpKRYuXIiwsDA8ffoUN2/eRGhoKAIDA9GpUyfROk+cOIHp06dr9N63bt2KI0eOAAB69+6NR48eITQ0FDdv3sTDhw/x4cMH/PLLL6J51q9fj/v378PLywuvX7+GsbExpk+fjnfv3iE4OBg3btxAcHAwHj9+jIYNG4rmnTBhAr5+/apRjOrK6Tal6Xes6/OqJUuWKDwGcXNzU7mNenl5ZbpuXe+zMsybN0/4Pe/ZsycCAwPx4cMH3L59Wzjf2bVrl2geXR23Sy8/I/ESABQqVAg+Pj6IjIzEq1evcOvWLeH4Jy4uDs+fP8eaNWvQokWLTH+DdH2up03x8fGi5Bp9+vSRO6/OC2JjY/Hrr78CSE9q+/fffyM8PByPHz9GQEAAwsPDcfnyZXh4eIjm8/Hx0Xhk0fHjx+Px48cwMDDAyJEj8fz5c7x9+xY3b94UjhsWL14sSsr39u1bzJ07N9Nl5+d2Le17OdYFgBIlSmD8+PE4e/YsYmNjERoaivv37wvXS6KjoxEYGIjhw4eLEuBs2rRJYaIOQPvXqTZt2oQ///xTKEskEnh5eeHmzZuIjo7GgwcPcP36dbx8+RKhoaGYPHmyqH3+888/2LNnj1qfhybCw8Px9u1boVypUiWtryM32Nvbi743VeeTYWFhOHXqlFD29vbWaWzK+Pn5icrFihWTSxyaFTdv3hQl5TEwMMDkyZPx6tUr4Xc84zwkPDwcYWFh8PX1hZeXl+jcUB05dUySU54/fy4khHJxcRGuWz169Ag3btxAaGgoAgIC5H63pk2bJjpHzqDta1iyKleuLPyfcT5OREREREREujNz5kxR+dKlS9le5qJFi0RJLgoXLpzlZzs6d+4MT09PeHp6YvLkydmOjfKXiRMnokuXLggPD1dZ79u3b7hz5w5GjRqVQ5HlX3FxcejUqVOmydQ1uU6fF02ePFnYd3Tu3Dm3w6FcwDZA9GMwyLwKEREREREREREREeWU33//HUePHhVNO3/+POrXr4+SJUuiRYsW8PDwQK1ateDi4gI9Pc1zj+vp6WHIkCGYMmUKgPQRordv3y509FIlLS1N6NwNpHdKGDBggFrrzejc3b9/f6xfv15uVGuJRAJPT0/4+/ujatWqQoer0NBQHD16FB07dhTVL1q0KIoWLQoAcqMX16tXT26kaE28ePECEokEPj4+GDx4sNzrBQoUwIABA+Ds7AxPT0/hAY8DBw4gIiJC5SjBAwYMEN3AHzp0KFauXKmw04ixsTF69OiBFi1aoG3btkKn2mPHjuF///ufwpt4Bw8eFHUMnj59OmbNmqU0HhsbG7Rt2xZt27bFokWLNOpIq8yjR4/w6NEjody4cWOcOXNGaXs1MTGBh4cHPDw8MGvWLK13gHj79i1+++030TRvb29s2rRJ7sa+ubk5Bg8ejGbNmqF+/foIDQ0FkN5J+ZdffsH//vc/Uf0yZcqIEulIj2xvbGws6piuDbpYX0REBID0TpHHjh2Dubm5XB1XV9csRKsdJ0+elBsxetSoUVi2bJnSNuXs7IzRo0dj9OjROHv2rOg1Gxsb4XOSfcirevXq8PT0VDu2mzdvYtmyZaJpyra5ggULYuLEiWjSpAmaNGmCuLg4AOmdj6dOnap0VGzpZDyFChXC1atXUbhwYYV19fX1UbFiRVSsWBFjx45FUFCQ0ti/ffuGXr16iRLSzJo1C9OmTVP4wIuFhQWGDBmCNm3aoHHjxkKn1zVr1mDo0KGoVq2a0nVlxYsXL0Qdsi0sLPDo0SPUrVsXsbGxmc7/7t07eHl54eHDh/jrr7+0GpsqGfuMkiVL4vLly6JkJ0D6d9SyZUvcunULLVq0EJLOZSRCuXfvntoPHL148QIFCxbEiRMnFCZgsrGxwV9//QULCwtMnTpVmO7j44Phw4crXW5+bteyQkJCAADu7u44f/68KJkcABgZGaFHjx5o3Lgx6tevLyROSEhIwMiRI+WOyVR58eIFSpUqhTNnzogSDGRwcHDApk2bYGhoKOos7ePjg9atW6tc9pEjRzLdN+np6aFOnTqoU6cOWrduLXSMTUlJweLFixV2zJf+TbGxsRG9punvyZUrV0Sdpu3s7HD8+HHUqFFDYX0nJyesWbMGjRs3xk8//YTU1FR8+/YNw4cPR2BgoEbrVkT6OBHI/vsD0hPMAOmdS8+dO6cwgU3p0qVF5T/++AOvXr0SylZWVjh//rzCfaabmxsOHDiAqVOnihIcLFq0CD169IC7u7tacWasb/HixRg/frzc67a2tli1ahW+fPmCTZs2AUg/xu/ZsyeCgoJgbm6OY8eOoUGDBnLzli9fXvheM7bFsLAwHD16VC6hTnblRpvS9DvW9XlV9erVFc4rfSyTVbrcZ0n78OEDAGD58uVCB21phoaGouXp8rg9g6+vr/B/gQIF4O/vL5cYNYNEIoGTkxOcnJwwbNgwPH78WO48Vpquz/W0acqUKcI5uq2trdxvf14RExMDIL3d+/v7KzwvqVu3Li5evIiePXti3759wvTRo0ejbdu2Cs9vFHnx4gUKFCiA/fv3o02bNnKvm5mZYfz48bC3t0ffvn2F6Zs3b8aff/6pNBFEfm/X0r6HY10A+Pfff1G3bl2V71sikcDNzQ2rV69G586d0b59eyQlJQFIT2SsaBvV5nWqFy9eYOTIkULZxMQE+/btU7oPdnBwwLx589CqVSu0atVKOM/79ddf0aFDB9G1g+ySHWm7fPny2V5mQEBAph0zlElMTMz2+jN4e3sLyU7u37+PO3fuKDxu2759u5BYTl9fX7RPyCkhISFyx/hNmjTRyrKl9ylA+nal6vpvkSJF0K1bN3Tr1g1Lly7VaLTnnDomySkZ+8mWLVti//79MDU1lavj7u6OM2fOoG7dusJxakJCArZv3y53vKTNa1iKVKhQQTSqeWBgIJydnbO1TCIiIiIiIlLO09MTlpaWwj3Vb9++4d9//830epYqPj4+onKfPn2yvKzDhw8L1zzevn2L+fPnZ3lZlL9s374dixcvFk3LuJZctGhRfP78GSEhIfj48WMuRZg/TZgwAcnJyUK5VKlSWL16NZo3bw4Dg++na7+vry+Cg4MBQO37DfR9YRsg+jFo3luBiIiIiIiIiIiIiHSmfv36mDZtmsLXQkJCsG7dOgwcOBCurq6wsrJC06ZNMWfOHNy6dUuj9QwcOFDUYUrdkaRPnz4t6ljbrl07hZ01lalRowZ8fHxU3nwqVKiQ3GeQG6Pfjho1SmFCGmkNGjRA9+7dhfLXr1/lEmBIu3btGk6cOCGUW7dujTVr1mQ6irG1tTX27dsHCwsLYdqSJUsU1s3oUJ/hl19+Ublsaebm5ihevLja9ZWRjWHYsGFqJ1DS09NDuXLlsh2DtNWrV4sSb1SpUgXr169X2RmuVKlS8PX1FdU5dOiQaPT574mNjQ327NmjdofNnCSbCOOnn37C33//rXabatq0qS7CAgAsW7ZMlDilXbt2KpNAAemdhmQfDFu/fr3CRCsxMTGijs1du3ZVmrhDERcXF6Wv7d27Fw8ePBDKw4YNw/Tp0zPtJOro6Ih9+/aJPn9l+6PsyOiAnEEikaBdu3bC52RqaorBgwdj69atOHr0KLZu3YpBgwbJjcq+YMECrFixQuvxqSKRSODr6yvXSVeaqakp9u/fDysrK2HagwcP1B7NPMOGDRsUdtKV9ttvv6FYsWJC+e7du0KSAEXyc7tWxNTUFAcOHJBLSCOtUKFC2Ldvn+jBr2PHjsn9nqmir68PX19fhR0ppS1YsEDUKfjUqVOZdtrUtKOhl5eXqGPs/v371UrmlB2zZ88W3oeenh4OHjyoNHmItJ49e4oSp9y7dw9nzpzRWZzZZWhoiD179qh1/BsTEyNKOgKkb7OZJfH6888/RR1sU1NTNU4WIfu5KluP9L48I8nM0qVLFSakyWBqapojx+q51aY0+Y6B3Duvyi5d7rNk9erVS2FCGkVy4rhdet/euHFjpYk7FKlQoYLSWHLiXE9brl27Jjo+Wrx4scrEqnnBunXrVCbK1NfXx/bt20VJPz58+ICdO3dqtJ6//vpLYUIaaX369EHt2rWFcnh4OAICApTWz8/tWpH8fqwLpF/L0eSh6ObNm2PixIlC+caNG6JEvLqwcOFCJCQkCOWNGzeqlYCjYcOGos4j4eHhcglysisjoVUG6c8/qyZMmIDmzZtn6S+z71sTHTt2FLXbLVu2KKwnPb1Zs2aiZIg5ISwsDJ06dRISJWXw8vLSyvKl9ynm5uYaLdfW1laj35ScPCbJKSVKlMDu3bsVJqTJYGpqKpdANzeufzs4OIjK0senREREREREpBtdunQRlf/5558sL+vdu3eiczmJRIKZM2eK6jx//hxpaWlIS0vLclJgUqx+/frCZ5uWlob169fndkhZJp0gG0i/bvzx40c8e/YM/v7+uH37NqKiovD+/Xv069cPRkZGuRRp/nLkyBHhfwMDAzx58gStW7f+rhLSEFHe4efnJ/pdItI2JqUhIiIiIiIiIiIiymNmz56Nv//+O9NRjD9//oxz585h+vTpqFmzJlxdXbFx40a1Hsi3t7dHx44dhfL9+/dx/fr1TOeT7WQ5ZMiQTOeRNnv27Ew75QFAjx49ROXbt29rtJ7sMjExUZocSFbPnj1FZVWxLl++XFRetmyZ2p2/7O3tRUlyLl++rLDjjXRnNwBqfd7alhdiyJCWloYNGzaIpi1evFitG/weHh6i7ze/P0SiyogRI3K0I7S6bty4gatXrwplCwsLrFq1Khcj+n8xMTHYt2+fUJZIJGp3IO7Zsyfq1KkjlOPj4xV2mNXltiS9PzI1NdVolLPKlSuLfkMOHjwojJimLbJJaaKjo4XOj9WrV0dQUBDWrVuHfv36oU2bNujXrx/Wr1+PR48ewc3NTTTvxIkTNUoukl3dunXLtPMsABQuXFguaYRsAgtVatWqhU6dOmVaz8DAQO7hRmUdp/N7u1ZkxIgRKFGiRKb1XF1d5TpbavJ9dO3aVa2EGTY2NmjRooVQTkhIwOPHj9Vej7qkk9J8+/YNN2/e1Po6MgQFBeHkyZNCuWfPnqhbt67a8//222+i32XpNpjX9OzZU2VSBGk7d+4UdSavV68eunbtqta8S5cuFZX37NmjdmIhiUSC2bNnZ1rPwcFBrs2WLFkSAwcOzHTe9u3bixLa3LlzR63Y1JWbbUqT7xjIvfOq7MrJfdacOXPUqpdTx+3Sv0O6OrYCdHOupw3JyckYNGiQcN2gSZMm6N+/v07WpS01a9ZUa/9pbGwst//T5Lfc0dERI0aMUKuuutcB8nu7ViQ/H+tmh/SxFQBcuXJF6+vI8PHjR2zdulUoe3h4oFevXmrPP2TIEFHSR20fW71580ZUzumELLpkbGwsuia5a9cuuc5at2/fFiV49fb2zpHYPn/+jLt37+LPP/+Em5sb7t69K3q9efPmWkvKK71P0dPTUzspcFbktfMobfj9999FyY2Uad68OWxsbIRyTl//BuS3X9ntm4iIiIiIiLRv7ty5ovKTJ08QFxeXpWVNnTpVVK5YsaLKgTqIFLl69aroPqSVlRUePnwIa2trubr29vbYunUrPn36JJdwl+RFREQI/zs5OTGZDxER5WtMSkNERERERERERESUB40aNQrPnj3DL7/8otZD7ADw8OFDDBo0CLVq1UJISEim9YcNGyYqy3aMlBUREYFDhw4J5eLFi6NVq1ZqxQak37Ru2bKlWnULFiwo6kCe0w/EN2vWDLa2tmrVrVq1qqisLNbU1FScOHFCKNeqVQvly5fXKC7pzhcAcPHiRbk6siPcantEbHXIxrBjx44cjyHD48ePER4eLpRLlCihUScd2Y7Z/v7+WostL+ndu3duh6DQqVOnRGUvLy+FD77khqtXryI5OVko169fH+XKlVN7fnXalp2dneihlCNHjqidlECVqKgo3LhxQyi3a9dO1BFKHdL7o8+fP2s9IcHnz58VTi9WrBhOnz6tNMlIqVKlcPbsWVGSpaSkJCxevFir8amiySjyXl5eog7rFy5cUHte2c7Qqqj7W5Wf27Uymnwfsh1K/fz81J5XF99HdpQuXVpU1vY2Ku348eOicr9+/TSa39bWFtWrVxfKio5v8gpNfi9lt2d1kr1kqFChgigJS3JyMq5du6bWvG5ubmpvt7LJVzp37gx9ff1M5zM3N0epUqWE8uvXr9Van7pys01l5Zgop8+rtCGn9lk1a9aEs7OzWnVz6rhd+lzF399fK+03p871tGHOnDl49OgRgPTkC2vXrtXJerRJk9/yrl27wtzcXCjfunUL8fHxas3bpUsXtRO6qLtd5Od2rUx+PtbNjpw8tvLz8xMlBdH0d9DQ0BCNGzcWyleuXFErgbW6ZI/dpbe574H0OUF4eLho/w4AW7ZsEf63tLRUK3mSJrZs2QKJRCL3Z2FhgWrVqmHatGmiziwAUK5cOezatUtrMUjvU+Li4nD48GGtLVtWXtvWs0sikcglW1dGX18flStXFsoRERFISkrSVWgKyW6/ujw3JyIiIiIionQODg6ia01paWmYOXNmlpZ14MABUXns2LHZCY1+UL6+vqJyjx49Mk1SbGRkhG7duukyrO+CdMJrOzu7XIyEiIgo+5iUhoiIiIiIiIiIiCiPKlasGFatWoUPHz7g0KFDGDt2LGrUqJHpqBkBAQGoVasWgoODVdZr2rQpypYtK5T/++8/fPr0SWn9LVu2iDqqDxw4UKORct3d3TWqLz2qc04/EK/OCL0ZpOMElMd6//590WuarCODbBKGoKAguTrNmzcXlcePH4+pU6ciLCxM4/VlVe3atUWjL+3fvx89evTA/fv3cyyGDNevXxeVGzduLOoUl5mGDRuKRrG/c+eOaDv4HlhYWMDFxSW3w1BItjNumzZtcikSebJtq0mTJhrNL9sZVVGyAUNDQzRq1Egov3z5Eo0bN8bJkyez1anw0qVLSEtLE8q62h9lh7GxscLpixYtyjSBjp2dndyoXNu2bRN17NQViUQi+s4yU7JkSVFih7CwMLU7Eevityo/t2tFbG1t5ZJuqOLh4SE6zrp79y6+fv2q1ry6+D5kpaam4ty5cxg/fjyaNWuGkiVLwtraGvr6+nKdVmWTIURGRqodn6Zk99XZ3ac8fvxYtI/KS2rVqqV23ZzYnhSRTsaSGdkkjO7u7lmaN6sjeCqTm21Kk+84Q06fV2lDTuyzgOxtM7o6bpc+X4qNjUXjxo3h6+ur9v5ekZw618uue/fuYcGCBUJ52rRpaicNyk2enp5q1zU1NUXNmjWFckpKCgICAtSaNyeOrfJTu1Ykvx/rKnLjxg388ccfaNOmDcqUKYOCBQvC0NBQ7tiqQIECovny07FVXFwcQkNDsx1XhoSEBFHZxMQk28s8f/480tLSsvRXsmTJbK9fWr169eDk5CSUt27dKvz/9etX7Ny5Uyh3795dK+8/O7p164YrV66onVxbHbLXFvv06YMlS5YgJiZGa+vIkFPbek4pVaqURt9Fbr8nU1NTUVndRG5ERERERESUPT///LOonJWBni5fviw6VzcyMsKgQYOyGxr9gN6/fy8qa/t6G6WTvtZPRESUH2X7lyw8PFyjh5p+FNKj3Kiqk9XPTt0HJoiIiIiIiIiIiCj/K1CgANq3b4/27dsDAJKTk3H//n1cvnwZZ86cwcmTJ+U6JYWHh6Nr164ICAiAvr6+wuVKJBIMHToUEydOBJD+0PmuXbswdOhQhfXXr18v/K+npyc3YndmZB+yz4yZmZnwf04kEpCmSazScQLKY5XtVLh69WqsXr1a8+CkfPz4UW5a3bp10bx5c5w+fRpA+qgrc+fOxfz581G3bl00bdoUDRo0QK1atWBhYZGt9StjbGyM3377DVOmTBGm+fr6wtfXFy4uLmjRogUaNWoEDw8P2Nvb6ySGDCEhIaKym5ubRvMXKFAAFSpUwIMHDwAASUlJ+PDhA4oXL661GHNbyZIlNeoYmZNkk2tlpTOermS3bZUpUwYWFhZCp/U3b94gLS1N7ruYNm0azp49KyTruHPnDlq1aoUiRYqgVatW8PT0hIeHh1wCClVk90eTJk3CpEmTNIpfluz+KDo6Wu37WTY2NnL3zGRH6waAggULomvXrmots2fPnhg9erTQoSsxMRE3btyQ60QbEBCA6OhotZZZvXr1TBPilChRQuN9q6urK16+fCmUX758KdcxXRFd/Fbl53atiCYJaYD0hDnly5cXkqglJSXh3bt3aj14p4vvQ9rBgwcxduxYUVvRhC46kGaQ3adoeswnKyUlBXFxcbCyssrWcrTN3Nxc7RHs0tLS8ObNG6FsaWkp6pSvjipVqojK6nbiL1SokNrrkO2EmtV5tX2snlttSpPvWFpOn1dpg673WRmkR3vNTE4dt0+cOBE7duwQkiq8ePECPXr0gLW1NVq2bAlPT0/UrVsXrq6uaicLyolzvcTERFy6dEmteU1MTFCvXj3RtJSUFAwaNEhIUlK5cmWhzWrTo0eP8O7dO7XqVqpUCUWLFlVZR19fHxUqVNAoBldXV5w/f14ov3z5Eg0bNsx0vrx4bJWb7VqR/H6sK+3ixYsYOXIk7t27p/Z6pOXksVVWEqbJ+vjxo86uY+TVZILZ4eXlhRkzZgAADh06hJiYGFhbW+PYsWOihETe3t45HptEIoGTkxOaNm2KIUOG6OTZ3e7du2Pu3Ll49OgRAODz58+YMGEC/vjjDzRq1AhNmjRB/fr1UaNGDaUJZdWVU8ckOSU717+BnH9P3+P2S0RERERElB+MGzcOf/zxB1JSUgCkP+MVFBSk0WBCGdcuMjRu3FirMWZHamoqTp48CX9/fzx79gxxcXEoUKAA7OzsUKdOHfTp00fh8wBZ9e7dO6xZswbPnz9HZGQk7O3t0bVrV3Ts2FHlfJcvX8bOnTvx8uVLSCQSlCpVCsOHD9f4HrM2HDt2DCdOnEBwcDAMDQ1RokQJDBw4EFWrVtX5uq2trUXlmzdv6nyd2nLkyBGcPHkS79+/R2xsLMzMzFCmTBk0a9YsS4NeJSQkYN26dbh79y7CwsJgZGQEBwcHNGvWTO1nVbQpOTkZhw8fxuXLl/Hq1St8+vQJZmZmKFSoEDw9PdG9e/dMBzdUR2JiIrZu3Ypbt27hw4cP+PLlC6ysrODq6or27dtrNLCJNkRGRmLdunV4/PgxPnz4AFNTUzg6OqJTp05yA7tkR2JiItatW4f79+/jzZs3MDAwQK9evdCnTx+trUPWnj174Ofnh1evXkFPTw/VqlXDr7/+qvK6YkJCAnx8fHD58mXExsbC1tYWDRo0wNChQ7Od7Oj48eM4fvw43r17h0+fPqFQoUJwcXHBkCFDsn2PPqtu3bqF06dP4/79+8L9Ujs7O1SoUAFeXl5q3WNSV2JiIlavXo379+/j/fv3sLOzQ7Vq1TBixIhsX/s+efIkjhw5gtDQUCQkJKBIkSJwcXHB0KFDUbBgQS29A+14+fIlfH19cefOHURHRyMlJQVmZmZwdHREtWrV0KlTpyw9R5EhNTUVGzduxI0bN/D69WsYGxujdu3a+PXXX1UeD0RGRmLt2rUICAjA58+fUahQIbRu3Rp9+/bNciyUPVne42R80ampqTk6uur3gJ8dERERERERERERZYeRkRGqV6+O6tWrY9SoUYiKisKCBQuwdOlS4YEFAAgMDMR///2n8kZZ//79MXXqVCQlJQFI7yCpqPPkxYsX8eTJE6HcsmVLjW/wZPdGTU7KTqzKHuaPiorK8jKVUTZ67s6dO9G+fXtcu3ZNmJaamopLly4JHRkNDAxQo0YNtGvXDn369NG4o3RmJk+ejJCQEPj4+IimBwUFISgoCH///TcAoHz58mjZsiV++ukn1K5dW6sxAJBLNpGVG2Sy80RHR39XSWksLS1zOwSlpDvjSiSSbN3g1DZttC1bW1sheUdKSgo+ffok9300aNAA69evx88//yxKQPbhwwds2bIFW7ZsAQDY29ujadOm6NmzJ1q1agVDQ0Ol682J/VFgYKDc6OrKNGrUCH5+fqJpim46e3h4qHxf0oyNjVGrVi0hQReQ/uCCbFKa8ePH48KFC2ot8/z58/D09FRZJyuj08vOo24HV138VuXndq0slqzELy0mJkatpDS6+D4y/PHHH5g/f36Wlw9AONbTBV3tU/JaUhpNfi9jY2OFpEtA1tqiouMPdWSnLeaV4/XcalPZOSbKyfMqbdDlPkuaJp9pTh23ly1bFnv37kWvXr0QFxcnTI+JicHu3buxe/duAOlJ8xo3boxu3bqhU6dOMDExUbrenDi2CgsLU/vYqmTJknj16pVo2tKlS3Hr1i0A6cmQfHx8NP5dVcfChQuF3/HMbNq0Cf3791dZx8rKSuMHar+3Y6vcateK5Pdj3Qxr167F8OHDs5WMIT8eW2mLbFK7xMRErS07r+jXrx9mzpyJtLQ0JCUlYffu3Rg2bJho/1a6dGnUr19f6+tu0aKFXNIwiUQCU1NTWFlZwcHBQa6jkLYZGhri8OHDaNOmjei4JTk5GadPnxbOswsUKAAPDw907NgRvXv3RpEiRTReV04dk+SU7B5P5/R7kk2CI5skh4iIiIiIiHTDwMAAtWvXxpUrV4RpU6ZMwf79+9Vehr+/v6g8Z84chfWcnZ2FwYD09fXx7ds30euenp4K75cHBwcrHdzIyspK7jpfZGQk5s2bh7179+Lt27dKz3E3b96Mn3/+GeXLl8eaNWsyvf+eQTqWjOcLHjx4gC5duuDZs2dy9bdt2wZLS0vs2rVLLjnJvn37MHToUIUDga1evRpOTk44efIknJycVMZ06dIlNGjQQCgPGjRINDCBNNnPOePz+fvvvzF16lR8/vxZbp4VK1bA3t4eO3bsQJMmTVTGkh3t27fHmjVrhPLx48eRmJiYZ+4bygoODoa3tzeuXr0quh8rbdmyZdDT00OlSpWwaNEitGzZUuUyb9++jT59+uDx48cKX1+zZg309fXRqlUr7Ny5U+n9r8GDB2PDhg0KX7tw4YLCbWrZsmUYM2aMUA4JCcGcOXNw5MgRfPjwQWnM69evR9++fVG9enVs3rw5S8mUjh8/jpEjR+LFixcKX9+7dy9mzpyJAgUKoEGDBvDx8REGpFi+fDnGjh0rN09KSorKgdFUXf86efIkhgwZIhr8RdrKlSthaGiIPn36YN26dWrdPzIwMBCep3VycsLz588RExODVq1a4caNG3LxvH37NltJaZRt66NGjcK6devkrqcfPXoUf/75J1q1aoWjR4+KBhZITExEly5dcOLECbk4//vvP4waNQqTJk3CvHnzNIrx48eP6Nu3L06fPi33m5Bh6tSpKF68ONauXYvWrVsrrKOtNvDt2zesWrUK69atw5MnT5TGBKQPgGVra4sZM2bg119/VVovM4mJiWjTpg38/Pzk4tmxYwcmTpyI5s2b4+DBgxrtC5OTkzFgwADs2bNH6fuYPHkySpYsic2bN6v9G6guZe1PmR07dmDcuHEIDw9XWW/IkCEwMzNDhw4dsHPnToV1FB1vfPv2DX379sW+ffvkPo+DBw9iypQp6N+/PzZu3Ch6LTIyEu3atcP169fl1rNz504MHjwYS5cuxS+//KIybtK+LA99MnHiRDg7O8Pe3p5/Cv5UjSqjjc+OiIiIiIiIiIiIKIOtrS0WLlyI//3vf9DX1xe9tn37dpXz2tnZiUbyuHnzJgIDA+XqrVu3TlQeMmRINiL+MeliFG1lN9ft7Ozg7++P1atXw9nZWWGdb9++4dq1a5g6dSqcnJzQr18/lTezNSWRSLB27VqcOHFCZSedJ0+e4J9//kGdOnVQv359oaOktsg+uJGVzhWy82QkW/he6KIzqrZIf9ampqYq77/ktJxsWwMGDEBgYCB69eqldJSjsLAw7NixAx06dEC5cuWwa9cupevNyf1RVinqzFauXDmNllG+fHlRObOb6Nog20FTHbLtQNEDZzklP7drRfL79wEAW7ZskUtIY2JighYtWmDq1Knw8fHB/v37cfToUaGD6OnTpzM9BtSm/LBP0QZNfi95/JE9udWmsnNMxPMqxfLqdtO6dWs8fPgQQ4cOVTr6WnR0NPbv34+ffvoJJUuWxIoVK5Q+uJfX94NfvnwRjZo7fPhw1KlTR2vL16X8/luen9u1Ivn9+wDSE03KJqQxMDCAp6cnfvvtN6xevRp79+7FkSNHRMdW0skudS2v71NkE6J8j8copUuXFnUo2rp1K6KionD06FFhmpeXl8oH67OqaNGiaNasmeivadOm8PDwQMWKFXWekCZDmTJlcPv2bcydOxeOjo4K6yQlJcHPzw9jx45FiRIlMGrUqO+yPXzPZPfJeS0xKBERERER0fds+vTpovKJEyfUntfHxwdfv34VygULFkTNmjW1FltWDBkyBMuWLcObN2/Uuub45MkTNG7cGJMnT87S+g4cOIAqVaooTEiTIS4uDu3atROSdgPAuHHj0K1bN4UJaTIEBwfD1dUVL1++zFJs6mrdujXGjBmj8pppWFgYmjVrhj179ugsjjZt2oiu/SYnJ8sN9pNXTJkyBc7Ozrh8+XKm1zxTU1Nx//59/P777yrr/fHHH6hevbrShDQZUlJScPToURQqVEguKZQ2derUCRs2bFD7Gb6AgABUqVIF//77r9rrSE1NhaenJ9q0aaM0IY20pKQknDlzRm5gOm3q3bs3WrVqpTQhTYavX79i8+bNsLGxESXUVtfly5dhb2+P69ev51iC6CpVqmDFihUqE7yfOHFC9IzTu3fvYG9vj+PHjyuNMyUlBfPnz890AAhp27dvR+HChXH8+HGVyV8A4M2bN2jTpo3O72fv27cPY8aMwcOHDzONCUhP7D9q1CjRNXxNvH37FkWKFMH58+eVfrZpaWk4deoU7Ozs8Pr1a7WW++DBA9jY2GDnzp2Zvo+QkBA0btwYgwcP1jh+benVqxf69u2r9rN08fHx2Ldvn9rLT0hIQMmSJbF7926ln0daWho2bdqExo0bC9Pu3r2LYsWKKUxIkyEpKQkjRozA7Nmz1Y6HtEOzoWSktGvXDu3atdNmLN+V6tWrIywsTOFr/OyIiIiIiIiIiIhIF9q1awdvb29R5vhLly5lOt+wYcNEGezXrVuHlStXCuWYmBjs3btXKNvb26N9+/ZaivrHIdt5q3fv3hg4cGC2lung4KD0NUNDQwwfPhzDhw/HrVu3cPbsWfj5+eHKlSuikdOB9JvN27dvx5kzZ+Dn5yeXyCE7WrZsiZYtW+Lly5c4deoU/Pz84O/vj3fv3snVvXz5MurVq4ft27eje/fuWlm/bGe8+Ph4jZchO4+FhUW2YiL1WVhYIDo6GkD6zcrU1NQ8k5gmp9tWhQoVsGvXLkRHR4u2paCgILmb5K9evcJPP/2EGzduYNmyZXLLkt0fjRkzBm3bttU4fmllypTJ1vyynJycYGRkhOTkZGGastGmlJGtn9GWdCkhIUHjeWTbgbJOxDkhP7drRfL795GcnIzffvtNNG3gwIFYuHAhbG1tVc6blQewssrU1FR0bHH8+HG1RiVTJb8P1MLjj+zJr22K51XZk9PbTbFixbB27VosXbpUOA/y9/dHYGCgMGJihoiICIwaNQoXLlzA7t275ZLB5vS5nqaSkpLw5csXobxq1SqsWrVK4+UoGsUzOjpapwkS8vtveX5u14rk9+8DAMaPHy86zmzbti3WrFmDYsWKqZwvKSlJ16EJZPcpmzZtyjS+zFSpUiVb80srXry4qPz+/XutLTsv8fb2FjqXXLlyBXPmzBHOTyUSCby8vHIzvBxhamqKP/74A7///jsuX76Mc+fOwc/PD9evXxf9rgDp5y4rVqzAqVOn4O/vj8KFC+dS1KQJ2euzJUqUyKVIiIiIiIiIfjwtW7aEubm5kJDky5cvOHjwIDp27JjpvH///beo3KtXryzHkZXnL9SZx9LSEvb29rC2toapqSni4+MRGhqK9+/fi67P/fXXXyhXrhwGDBig9vo/fvyIbt26CUlJLCws4OzsDHNzc4SGhuLly5fCOtLS0tCvXz+0bdsW69evF91nLlKkCEqUKAFDQ0O8fPlSdJ0rMTERzZs3x/Pnz9WOSxMDBw4UEhFJJBI4ODjA0dERBgYGcrFkvIcWLVro7Hr8pEmTMHPmTKF848YNDB8+XKNEJ7r2008/KRzExsrKCiVKlICNjQ3i4+MRFhaGsLAwuWviiowfPx5Lly6Vm168eHEUK1YMycnJePnypSiJUXJyMho3bowLFy7IDdKWle1JVeJriUQCKysrFC1aFFZWVjA2NsanT5/w9u1bUdKa1NRU/PLLL6hUqRIaNmyocn2pqakoU6YMQkJC5F6zt7eHg4MDzM3NERcXhzdv3uDjx48KE3doM2F3p06dcPDgQbnlly5dGkWLFkV8fDyCg4NFCbE/f/4MNzc3PH36FCVLllRrPcnJyWjevLlwvd/Q0BBlypRBoUKFEBcXh5CQEK0/k9a6dWvcu3dP7j0lJSXhyZMnovf0/PlzDBw4ED4+PqhcuTJiY2MBAEZGRnBycoKdnR1iYmLw+PFjUWKyLVu2oGfPnmjdurXKWJYsWYIJEyaIpkkkEtjb28PR0RGmpqaIiopCcHCwKIHO+vXrkZKSInomOWNeXTAxMUGRIkVga2sLc3NzJCUlITw8HG/evBG970uXLqFTp0743//+p9HyPTw8hGcR9PT0hO8kLi4Oz58/F90Li4+PR8WKFREWFqby/tazZ89QrVo1ueQrFhYWcHJygpmZGd6/fy/6fQKADRs2ID4+XuMBurJr6dKlooRtQPoADsWLF0eRIkVgbGyMz58/IzIyEmFhYSoTKilTs2ZN4fqzvr4+nJycULhwYcTHxyMoKEi0TD8/PyxduhReXl7w8PAQtlETExM4OzvDxsYG4eHhePbsmWjfPnPmTPTs2VOrzzqTatl7aoeIiIiIiIiIiIiI8pQePXqIbgB9/vwZsbGxKkc5bdiwIVxcXBAUFAQA2LFjBxYtWgQTExOhLN3RoH///tnuFPojsrOzE5Wtra3RrFmzHFl3jRo1UKNGDfz2229ITU1FYGAgTpw4gd27dyMwMFCoFxYWhm7duiEwMFDrN1lLly6NYcOGYdiwYQCAFy9e4OzZs9i/fz9OnTolPCySnJwMLy8v1K5dWysdIWxsbETlqKgojZcRGRmpcpk/ouzeWFa3M2PBggWFRCJpaWmIjIzMMx2btNG2pOfR19dXK+GAjY0NevbsiZ49ewJIb58XLlzAoUOHsHfvXtFnu3z5cnh4eKBHjx6iZcjujzJGYtcmT0/PbI0qpK+vj3LlyuHBgwfCNE07gsreFJft3Amk39jWJtn9hTpk244uO3ZnJj+3a0Xy+/fh5+cnepisRYsW2LBhg1rzqhrdT9vs7OxECUTc3d3zzL46t1hZWUFPT084vuHxh2bya5vieVX25NZxu5mZGTp27Cg86B8XF4dLly7h6NGjQuK0DPv27cOSJUswadIk0TJy4lyvVKlSOTZiY3Zs3rwZmzdv1tryYmNj8fXrVxgaGqo9T176Lc/P7VqdWNSRl76Pp0+f4s6dO0LZ1dUV+/fvh5GRUabz5vSxlbSKFSuiVq1aObb+zJQqVUpUfvv2be4EomPdu3fHyJEjhd9u6c5e9erV03pi1rxMT08PDRo0QIMGDTBjxgx8/foVt27dwokTJ7Bz505R56wnT56gf//+OHbsWC5GnL3rVllJwJVfySalkd2+iYiIiIiISLc6dOggSnY/b968TJPSxMXFCfchMsyZMyfLMZw7d07438DAQOjs7eTkpFFCFhsbG1SqVAkjRozAgAEDYGxsrLBecnIyRo4ciXXr1gnThg8fDm9vb7WfU7p//z4AoECBAti6davcfeOQkBC4u7sL1/S+fv2Kvn374vDhwwAAR0dHnDhxAq6urqL5jh07hg4dOgifQXBwMM6ePYumTZuqFZcmNm3aBACoXr06jhw5Ije4wtWrV9GkSRPhmYfk5GSMHj0aW7Zs0XosADBjxgz8/fffomvHGcm8p0yZopN1amLlypVySRvKlSsHX19fuLm5KZxn165dmD9/vnCfTtbVq1flEtJUqFABZ8+elRs44PTp0+jcubOQhD01NRVt2rRBZGSk6Pqyj48PfHx8hLL0NapGjRqp9XxK4cKFUatWLYwaNQo9e/ZUei8xLi4O/fr1w6FDh4RpXbt2RUREhMrlt27dWi4hTdu2bbF9+3aF1+8TExOxbNkyLF++XLSNjh49GqNHjwYAODs7Izg4GED6MyKyiTlU2bp1q1xCmgYNGuDYsWNySUC2bt2KwYMHC4lJkpOT0aBBA7x+/Vqtdb158wZA+vcyduxYLFmyRK5ORqIwbclIPtWkSRMcPHhQ7j1NmjQJixYtEspbt27Fhw8fhP3XhAkTRK8D6e+7YcOGuH79ujDtl19+wcuXL5XGcevWLUycOFE0rV+/fli9erXCZCtLlizBb7/9JuwPN23ahO7du4sS32irDRgZGaFo0aLo378/Ro0apXKwmZUrV2L8+PFCAvmDBw/i5s2bqFmzplrrio2NFZL9eHp64ujRo3LPki1duhQTJ04UnvWIj49H27ZtceHCBaXLbdSokeg9FyhQABs2bECfPn1E9eLi4tCyZUtcu3ZNmPbff/+hc+fOaj0DpS2yxw1jx45VmKArQ1BQEObPn4/z58+rtfyUlBQ8evQIQHpCsS1btsjty7p37y4a0GfmzJnYsmULEhMToa+vj8WLF2PMmDGieWJiYuDu7i609bS0NAwaNEitgVtJO/LGUJJEREREREREREREpBWKHhxX50H6jEQhQPrFe19fX6Es/SCARCLBkCFDshfkD6p06dKisq5G08mMnp4eqlWrhsmTJ+Pu3bvYt2+f6Ab8gwcPcPLkSZ3HUaZMGQwZMgTHjx9HYGCgqDNPYmIiVq1apZX1yI6GIp2ERx0ZI5NkKFCgAIoUKaKV2PIz2QeIZEfIzkxmD0FkKFu2rKh869YtjdajS9ltWy9evBCNeFOiRIksdZqys7ND165dsWXLFoSEhKBNmzai1xU9RJFX9keZcXd3F5Wlk3OoIzw8XFS2tbXNdkyZefPmjSiRgjoyHprLIPv95KT83K4VkU5qpI6vX7/K7fNlHzjLSdIPogDpDxKp6+HDh9oOR6n8sk/JSRKJBMWLFxfKcXFxePXqlUbLkN3+1B3h7XuQn9sUz6uyLq8ct1taWqJNmzZYtWoV3rx5Izc667Jly+SSw+TnNpvXpaSk4PHjxxrN8z0dW+Vmu1Ykvx/ryh5bDR48WK2ENACPraTJdvKQbqPfEwsLC3Tu3Fnha97e3jkcTd5iaGgIDw8PzJo1C0+fPsWqVatEHVKOHz8u1zkup2XnupW616y+B7K/sco6cREREREREZFuzJs3T1S+detWph35Z82aJbqWV65cORQsWFAn8Wli48aNePDgAYYPH640IQ2QnnzAx8cHa9euFaYlJSVh/vz5Gq3PwMAAQUFBCjvxlyxZEleuXBFNO3jwIFJTU+Hg4IBXr17JJaQBgDZt2sh11M9Owp/MNG/eHLdu3VKYgMHDwwPHjx8XTZNN3KFNjRo1EiWkyTB16lTRd5Ubvn37hnHjxommtWvXDk+ePFF5LaN37964d++e0mQFffv2FZUrV66MoKAghc8HNG/eHE+fPhW17U+fPmHUqFGavBW1nDx5EtevX0efPn1UDm5haWmJgwcPYuzYscK0yMhIlcmi/f39cerUKdG0VatW4ciRI0oTyhsbG2Py5Mn48OGDTrYH2c+wbdu28Pf3V5goxcvLC9euXYO+vr4w7c2bN1i5cqVG69y6davSZ04UrTe7OnfujLNnzypc9sKFC+Hh4SGUU1JShO9w8+bNcglpgPT96LVr10TLe/XqlcqBBTp06CD8dkgkEuzduxdbt25V+n7Hjx+PCxcuiJ73GTlyZCbvNGs6d+6Md+/eYd68eSoT0mTE8OjRI1EbyMp22KpVK5w/f17h4Gbjxo2T29/6+/vj3r17Cpe1dOlSvH//XigbGBggICBALiENkL7dXr16FY0bNxZN//nnnzV+D1mVkJCAmJgYody0aVOVCWkAwMXFBVu3bhUSO6nr999/x44dOxTuy3x9fUX3UT99+oR79+5BIpHg3LlzcglpgPSBL+7evStanuy9P9ItJqUhIiIiIiIiIiIi+o5kjMghTZ1O+N7e3qLEJBkdJm/evCnqNNWkSZM8ORKw7Gg5eXEE+Vq1aoluZF25ckUY0SY3denSBePHjxdNy+nRA1xdXUUjxWgzhjp16ojKfn5+GrWPixcvCiOsAOlJMtTttPY9s7S0FJU1SRYSGRmpdsf8Bg0aiMraHmk7O/sO2bYlPYKYOmTryy4vK+zs7LBz506YmZkJ027duoWkpCRRPdmb25rGnlM6dOggKgcEBGg0v2z98uXLZzumzKSlpcHf31/t+iEhIaLtwd7eHiVKlNBBZOrJz+1akY8fP2qUmObq1avCiEoAULVqVRgaGmoWsBbJ7ls1acOafnfZ2R/ml31KTsuL21N+kZ/blK7Oq6Qf+MuL5zrakBeP283MzODj4yNK/hoWFiaXICKvnutlMDc3x+nTpzX+kx0x0c3NTa6OLh7OlaVq5EFZCQkJokSW+vr6qF69ui7CUkt+bteK5PdjXR5baUfhwoVRrFgxoaxpIsj8xMvLS26asbExunfvngvR5E0SiQS//PILfvrpJ9H03B6ZNDvXrW7evKntcLRCF9e/pROHWVtbw9nZOdvLJCIiIiIiIvWVLFlSNMhBamoqFi9erHKeHTt2iMqjR4/WSWy6NnToUBQuXFgo7969W6P5p06dqjIBdvny5RWe5547d05loo/JkyeLXtfVtS8TExOcOHFCZR1PT0/RdbjY2FjRvWxtqVy5sui6r+ygM8OHD8eePXu0vl51TZw4UXSd3MHBAYcPH1Z7ftlrKkB6EvIXL14IZQMDA/j5+alcjoODg2gACgDYtm2b2nHoytKlS1GgQAGh/O+//yqtKzsITteuXTUaGEfRZ5kde/bsQWxsrFA2NzfHoUOHVM7j7u4u95zjX3/9pfY6PTw85BIS6ZKZmRn279+vso6ixDO1atXKNDl6z549RWVl6zl27JgoaYqXlxe6du2qctkAUK9ePdE6Xrx4gZCQkEzn0zUnJye0atVKKN+5c0ej+U1MTDLdh7Rr1w7t2rUTTZNtdxlkf7dnzJiBSpUqqVz+iRMnRM8UREdH48CBAyrn0ZanT5+Kyp6enjpZT7FixTJNOCedVCtDz5490bBhQ6XzWFpaol69ekI5JSUFd+/ezXKcpBkmpSEiIiIiIiIiIiL6jsg+NG9vb69WhyVra2vR6DGXLl3C48ePsX79elG9IUOGaCdQLZPuqA6kd0TLa4yMjNCkSROhHB8fj02bNuViRP9P+kYNAJUjZ+S3GMqXLy8aST4kJATnz59Xe/6NGzeKyo0aNdJKXPmdiYkJChUqJJTv3buH1NRUtebV5GEi6ZvIQPoDHdKjdWRXdvYdderUEe1fL126pNEI9rpqW1ZWVqJRvVJTU/Hx40dRHUdHR1Gd4OBguVG28oJWrVqJRpq6d+8enj17pta8Dx8+lBuZXVc30mVt3bo1y3VV3VjPCfm5XSujyfexZcsWUTm39/mynQzVfcjww4cPmT7YJCs7+0PZfbWPj4/owcAflWz72bx5s9rzPnnyBJcvXxbKBQoUQO3atbUVWp6Xn9uUrs6rpLfRvHiuow159bjdwMBAbvuTPVfJy+d6QPp7aNasmcZ/FStWFC3HxsZGro6qh/e1RZPf8n379uHz589CuXr16nK/cTkpP7drZfLzsW5Wj62SkpLkvovMZOfYSnbb+u+//xAVFaXR+nVNOoFsXFyc2udp+U2zZs3kRkbu1KkTrKyscimivCsvXFuUJj3CKqB+x4SvX7/m2MP/mtL29e8PHz7g7du3Qlk2MTQRERERERHljKFDh4rKsgM6SQsKChIlXjUwMNAooUReI528XzpBSGb09fUxbdq0TOvVrFlTbn3qJKouWrSo8L82n0+R1r17d7USfMgmPtd2IuAWLVqIEu+UKVMGwcHBokT8aWlp6N27N06fPp3p8vT09CCRSCCRSODk5KSVGGWfMVq9enW2l/n333+Lys2bN0fBggUzna9v376wsbERyrKJ8nOL9H0IZckZUlNT8ejRI6Gsp6en0fV+XVizZo2oPHjwYLW2iwULFogGFwoNDVX7WtmSJUs0CzKb1ElwXq9ePbn3rShRjawuXbqIyleuXFFYb+7cucL/EokEK1euzHTZGWbPni0qb9iwQe15dcnDw0P4PykpCWFhYWrP26tXL7Xub65du1ZUln5+I8Pnz59FCX+MjIwwderUTJdtZGQkl3RIG/s2ddjZ2YnKFy9e1Ml6xowZk2mdPn36yE1bsWJFpvM1a9ZMVFbn94m0g0lpiIiIiIiIiIiIiPKIyMhI7Ny5U+3EDrKSk5PlLsq3bNlS7fl//vlnUXnZsmXYtWuXULazs0Pnzp2zFJuuyd4YfvnyZS5FoprsSPMzZszA69evcyma/yfbUUT6Bnp+j0EikWDQoEGiaRMnTkRKSkqm8964cQP//fefaFmDBw/WSlzfA3d3d+H/6OhotW7wxcbGYsGCBRqto379+kI5Li4OI0aM0CxQFbKz77C2tka3bt2EclpaGiZMmKDWvHv37sXVq1eFsrm5OXr37q32ujOjzvYkuz8aM2aMaASivMDMzExuhKI///xTrXllH4xo1KiRaKQ1Xdq7d69aI6uHh4fLPfAycOBAXYWllvzerhVZtWoV3rx5k2m9hw8fyj10ldvfh729vais7kOGv/76K5KSkjRaV3b2h9WrV0fjxo2F8ps3b9R60OZ717t3b1HH0YsXL+J///ufWvPKjrLVo0ePH6rzc35vU7o4r5LeRl+9epWt+PKqvHzcnpVjq7xyrvc9uHnzJvbt25dpvcTERMyYMUM0Lbd/y/N7u1YkPx/rZvXYatq0aaLOPurIzrFVkSJF0K9fP6EcHx+v1fNgbZC93ig9kvL3RF9fH8HBwfj06ZPwl9sdNfKqvHBtUZr0NSsAao/mvWLFCrx7904XIWWbtq9/y263mtxHICIiIiIiIu2ZNGmSKBnBy5cvlXaunzJliqicVxOM7tmzBw0aNEDhwoVhaGgoSlQi/Xft2jVhni9fvqi9/GLFiqmVuMLZ2VlUlk2qq4z0cwXqXMvNip9++kmtei4uLqKyNq9bbNq0SfScj7W1NYKCglC6dGncu3dPNIBQamoqWrdurfLacGpqqigxuDoJgDKTmpoq2h6MjY3RsWPHbC9XNrnEr7/+qva8LVq0EJV37tyZ7XiUWbt2LWrWrAlbW1uV25L0/ShliZT27t0r+n5cXV1FyYdyw/3790Vl2X2cKlWrVhWVfX19M53HwMBAlMwkJ6i7rUtvb3p6emol+Ze9Bqosub3052xrawtzc3O1YgKAsmXLQiKRCGVliW+05fbt2+jUqRNKlCgBY2NjpW1e9tmBwMBAtdfx22+/qVXPwcFBNGDfly9f5AbPkk2aJfudqCKbXE2T95AdxYoVEw1WdurUKYwdOxbfvn3T6noGDBiQaR07OztR+7KwsJBLmqNIpUqVROXw8HDNA6QsYVIaIiIiIiIiIiIiojzi8+fP6NOnDypXrozt27drdMM9MTERffv2xcOHD0XTvby81F5GnTp1UKVKFaHs4+ODT58+iZYlfUMiL5G90bB3795cikS1hg0bih7wj4iIQIsWLfD48WO1l5Gamor//e9/Sm+QjRgxAocPH5YbAVyZpKQk/PPPP6Jp1atXVzseRf7++2+sWrVKoxF7ZUf4yG4M0oYPHw4TExOhfPv2bfz8888qP6PXr1+jW7duojodO3bU2kg+34M2bdqIyr/99pvK7zw+Ph49e/ZUKzmEtBkzZohuQO7cuRNjxoxRu42fPXtW6Wuy+w51OrtKGzt2rOhhp4MHD2aaNOXu3btynUkHDx4MS0tLubrnzp3D77//rtGDRQcOHEBwcLBQrlixoujhhQx9+vQRvf+nT5+idevWGq3r69ev2LJli0aJhjQ1Y8YMUfxbt27Fxo0bVc6zevVquQ5nkydP1kl8iqSlpaFHjx4qP8svX76gS5cuokRAFStWlHuAKTfk53atSEJCAjp37iw6ppEVERGBLl26iB6yaNWqlVYelMuOunXrisp//fWXXGdPWVOnTlXrgStZ2T2WmjNnjqjdLFy4ELNnz1Z7Xw0Ab9++xcSJE9Xq6J4fWFtby3W+HzhwIO7du6dyvhkzZuDo0aNCWU9PD2PHjtVJjHlZfm5Tujivkt5GIyMj4efnl+048yJdH7cHBQVh+PDheP78udox3bx5U/R5W1tbi0ZvzZAT53o/siFDhohGa5WVmpqKfv36iTroFy5cWO2HfXUpP7drRfLzsa7ssdWaNWsy/dzWrl2LxYsXa7yu7B5bTZ06VdQhYPfu3Rg2bBiSk5PVXsbHjx/x559/4vDhwxqtWx2tWrWCvr6+UD5//rzW15FXGBsbw9zcXPiTHoH3e9WnTx9cuHBB7frR0dFYv369aJo2r+tlRfXq1UUdyK5du5ZpB6Fjx47hjz/+0HVoWZbda1iyZI8nZa/zERERERERUc4wMjJCjRo1RNNkO6pnOHHihKgsO1BLbjt06BCsra3Rs2dPXLp0CREREfj27Zta93Y0GcBN3cForK2tRWV1n/eRHnRCV6pVq6ZWPdn3oCzpRFbI3n88fPiwcP/KyckJt27dEt3PSklJQf369REUFKRwebLXGjw9PbMd4+3bt0Xtp1SpUtleJiCf3Kd58+Zqz9usWTNRWReJJNauXQsTExP8/PPPuHXrFj5+/Kj2tqTsGrLs9yM9QFhukU6gY2RkpFYyigx16tQRlaUHUFImNxJpq7utS193lr6npIrsvvDz588K60nfp46MjFSY5EXVn3S7k03Koi3BwcGoUKECqlevjoMHD+LNmzdISkpS+9mAt2/fqlVPT09Po+eAZJObnTt3TlS+fv26qCzbLlVxcHAQ7WOjo6PVnje7unfvLiovX74cxsbGcHNzw7hx43Dr1q1sLV8ikcglWVdG+pkQdQdqsrW1FZWVJeMi7WNSGiIiIiIiIiIiIqI85tGjR+jXrx/s7e0xZMgQ+Pr64v379wrrvnv3DqtXr4aLi4tcB+QuXbqgSZMmGq172LBhSl/T5ojc2ubp6Sm6ObdlyxZ06dIFGzduxPHjx3HmzBnhT3a0k5y2detWFC9eXCg/efIE1atXx5gxYxAYGKjwZlp0dDTOnDmDsWPHolSpUujcubPcTa0Mly9fRocOHVC6dGlMmDABfn5+iIuLk6v39etXnDhxAvXq1RN11LW3t0e7du2y9R5fvnyJkSNHwsHBAd7e3jhw4IDSNnz37l306tVLlBhHT09PqyOoFytWDAsXLhRNW79+PZo0aSIaeQlIT5yyYcMGVK9eXZQ8pWDBgli1apXWYvoe9OnTR9RJLjAwEM2aNcOdO3dE9RITE7F//37UrFkTJ0+ehJGREYoVK6b2epo1a4Zx48aJpv3999+oXr069u/fj/j4eLl5nj9/jr///hs1atSQeyBEWrVq1UQPNfj5+aFJkyZYs2YNjh07Jtp3nDlzRm7+GjVqyD2oM23aNHTr1k2u02x0dDQWL16MevXqiTpnOjk5KU34ERcXhwULFqBUqVJo06YNNmzYgKdPnyrcT7x58wbTp09Hz549RdOHDBmicNn6+vrYt2+f6Ibu1atX4erqihkzZuDp06cK5/vw4QOOHDmCYcOGwdHREf3791f60JE2FCtWTK5j9uDBgzFy5Ei5BEevX7/G8OHDMXLkSNH03r1759iI346OjjAwMMCrV6/g7u6O7du3IzExUXg9NTUVJ0+eRI0aNUS/RxKJBD4+PqIETLklP7drWSVLlgQABAQEoFq1ajh8+LAo8UxycjJ8fX3h7u4uavMmJiZYuXKlWuvQpUaNGgnvAUj/POrVq4fTp0+LPq+0tDRcuXIFzZs3x9y5cwHIj5qXGdkH7ebMmYMBAwZg27ZtOHHihGhfGBAQIDd/vXr1hHVnmDFjBmrWrIn//vtP4QM0KSkpCAoKgo+PD1q2bInSpUtj8eLFCvfr+dXcuXNFD0dGR0ejbt26WLJkidxn8uDBA3Tt2lXuAeKJEyeq/bDa9yS/tyltn1fJJnLo3Lkzfv/9d/j6+uLUqVOibfTFixcaLz+v0PVxe1JSEtasWYPy5cujUaNGWLFiBR48eKBwpNOIiAgsXboUTZs2Fb3u7e2tNKmQrs/1fkTW1tawtLQU9p8rVqyQO7+9evUqGjRoIJf0Y/ny5bCwsMjJcBXK7+1aWn4/1nV2dhaNgvrp0yc0bNgQvr6+ciNABgYGomfPnkICIU2PrbJ7napMmTLYsGGDaJqPjw8qV66MdevW4cOHD3LzpKWlITg4GNu2bUOXLl1QrFgxTJs2TasdVTIUKVJEdM3x2LFj+Pr1q9bXQ7nj6NGj8PT0RMWKFTFjxgxcvXpVYRLiL1++wNfXF7Vr10ZISIgwvUqVKqhZs2ZOhizH0NAQ/fv3F00bMGAAlixZIndsGBwcjFGjRqFDhw5ISkrKswmps3sNS1paWhoOHToklD08PFC6dGmdxU5ERERERESqTZkyRVRWlIj04MGDosHNrK2t80RSiQz//vsvOnbsKLpnrCvSz6moIt3RHVC/s3tOXMdUN7GO7HvQZPAGVY4cOSL6ripUqCDXnipVqoSrV6/CwMBAmJacnIzq1asrTACxbds24X+JRCL33EZWvHr1SlQuUqRItpcJpF9Xz2BgYCB6j5mpUKGCqKztZAgTJ07Ezz//LLr2rgllCZ5krymXLVs2S8vXJun7FOpu1xlkE+1HRERkOo+m69AGTRLtZFC3Paqzf8hs0CNNKUt8kx0PHjxAxYoV8eTJkywvQ924ChQooNFy7e3tReXQ0FBRWfbzVXcAiAzSCYgU3dfTle3bt6Ny5cqiaSkpKbh//z6WLVuGmjVrwtDQEGXLlsUff/yh8fcu2zbVpe73o6vfRsqc+r+WRERERERERETb/V37AAEAAElEQVRERJSj4uLisH79emGkWVtbW9jZ2cHa2hqJiYl4//49wsPDFc5bu3ZtbNq0SeN19u3bF5MmTZK7kVC/fn2NO+DkpMKFC6Nfv37YuHGjMO3AgQM4cOCAXN2SJUvK3bTOSYULF8bRo0fRtm1boZNZQkIC/v77b/z999+wsrKCo6MjLCws8PnzZ3z8+FFpQhdVQkJCsGTJEixZsgQSiQSOjo6wtbWFiYkJ4uLi8OLFC7kb2Pr6+li/fr3aI25kJjY2Flu3bsXWrVsBAIUKFULhwoVhYWGBxMREvHr1SuHN+UmTJml9ROURI0bgxo0boocg/Pz84OHhgUKFCqF48eJITEzEy5cvRQ/yAOk3AHfu3AkHBwetxpTf2draYvbs2ZgwYYIw7erVq3B3d4ejoyMcHBwQHx8v95n+888/2LVrl9qjlADAX3/9hTdv3mDPnj3CtDt37qBr164wMjJCyZIlUbBgQcTHx+Pt27dqP/RhaGiIUaNGYfr06cK08+fPKx3lXdFNzLlz5yIwMFDU4Wffvn3Yt28fHBwc4ODggE+fPuHFixdynfRsbW2xZ8+eTEfa+vr1K44fP47jx48DACwsLFC0aFFYW1sjNTUV7969kxvJCUjfd//6669Kl1u+fHkcOHAAXbt2FTr3R0dHY/bs2Zg9ezbs7Oxgb28PMzMzxMXFITIyUq0HObRt2rRpCAgIwJEjRwCkfw+rVq3C6tWrUbp0adja2iIqKkphJ3x3d3f4+PjkWKzOzs74+eefMW3aNHz48AH9+vXDzz//jNKlS8PQ0BCvXr1SmEhh9uzZqFevXo7FmZn83K6leXp6wsDAABs2bEBwcDA6dOgAKysrlCpVCmlpaXj58qVoVCgg/QG5NWvW5ImOiIaGhli0aBF69OghTHv69ClatGgBGxsblClTBikpKXj9+rVoRKoiRYpg7dq1aNiwodrrqlatGpo0aSKM7JSamorNmzdj8+bNcnUbNWokN5IaAPz+++8IDw/HsmXLhGkBAQHo3bs39PT0UKJECWHEopiYGLx//15h59bviYWFBXx9fdGiRQth24+Pj8eECRMwefJklC5dGpaWlnj//r3cA0wA0KpVqzw3ymVOys9tStvnVV5eXpg7d67wYFtMTAwWLFigsO6MGTMwc+ZMjdeRV+TEcXtqair8/f3h7+8vzOfo6CiMkBgeHo7Xr1/LHfuVLVsWc+bMUbrcnDrX+5FYWVlhzpw58PLywqdPnzBq1ChMnDgRZcqUgampKd68eaPwusigQYPQu3fvXIhYsfzcrqV9D8e6ixcvhqenp3AM+/79e/To0QPm5uYoW7Ys9PT08PbtW9ED+mZmZtixYwfc3d3VXo82rlP16tUL7969w8SJE4WOBE+fPsXQoUMxdOhQFC9eHHZ2djAwMEBMTAzCwsLkjm11qV+/fjh9+jSA9PPI8+fPyyVRo/wtKChIuD6gr6+PYsWKoWDBgjAyMkJMTIzC80FTU1NRu89NU6ZMwY4dO4Tj7OTkZEyYMAFTpkxB2bJlYWxsjLCwMNH1qQoVKmD+/Pno3LlzboWtlDauYWW4deuW6H337dtXe4ESERERERGRxjp06AAzMzMhkWp0dDSuX7+O2rVrC3VkE/l37do1R2NU5d27d3IJSAoUKABPT0+0atUKNWrUgLOzMywtLUVJITw9PXHhwoWcDpcAues3AwYMUFjP3d0dfn5+aNSokZAw4cuXL3B1dcWLFy9QsGBBoe6xY8eE/zOu4WeX7PMp5ubm2V4mANE1LX19fY3mLVSokKiszcExLl++jMWLF4ummZubo1mzZmjRogVq1KiB4sWLC9foMjg7OyM4OBiA8mtCsgn/pb+73CIdqzqJ86XJxq9owD5ZmiQf+l68fv1aq8tTlvQoO5o0aYLk5GShLJFIUKNGDbRq1Qp16tSBq6urMIhFhuXLl4sG21I3KYn0YALqkB38QvYemGy7y7gnpy7pJCw5nVjl3r17mDt3LhYtWqQwody3b9/w/PlzzJ8/HwsWLECfPn2EZ4Dpx/Xj7UWJiIiIiIiIiIiI8ihzc3NUqVIFgYGBCl+PiorKdHRjPT09DB06FAsXLszSiOAWFhbo3bs31q1bJ5o+ZMgQjZeV05YvX443b94IHWLyssqVKyMgIAD9+vXDyZMnRa/FxsaqNXJQiRIl1F5fWloa3r59qzIJiI2NDTZt2oS2bduqvVxNRUREqExooa+vjylTpmDWrFlaX7dEIsGWLVtgb2+PJUuWiG6SqorL3t4evr6+eWqEqbxk7NixePLkidw+IzQ0VK5zvZ6eHpYuXYphw4Zh165dGq3HwMAA//33H8qVK4cFCxaIHhBJTk7Gs2fPsvwe/vjjDzx9+hTbt2/P0vwFChTA0aNHMWjQILllKEuqAaR3/jx06JDcKErq+PTpU6adDTt06IAdO3Zk+gBN48aNcfPmTfTu3Rs3b94UvRYZGZnpqDkSiQTFixdXL/As0tfXx969ezFs2DBs2bJFmJ6WloYXL14oTEYD/P9noK0Hk9Q1depURERE4J9//gGQ/hDSgwcPFNbV09PDtGnTMHXq1JwMMVP5vV1LW716NWJiYoQRBWNjY5UeaxkZGWHlypXw8vJSP3Ad6969O+bOnYupU6eKHkKJjo5GQECAXP3ixYvj2LFjWWr327ZtQ/v27XH79u0sx7t06VJUrVoVo0ePFiUIS01NxatXrzJNDGhhYQFra+ssrz8vqlGjBvz9/dGxY0fR/urr1694+vSp0vn69+8PHx8fjR+M+t7k1zal7fOqggULYt++fejevbvSxKTfi9w4bv/y5QueP3+uso6HhwcOHDiQ6Xl2Tp/r/Qj69euHDx8+YNKkSUhLS0NSUhKCgoKU1h80aFCOJiVUR35v19Ly+7Fu3bp1sW7dOgwZMkR0Xvn582fcuXNHrr6NjQ3279+PatWqabwubVynGjduHCpVqoQBAwbIJbF68+aNkABLmQIFCqg96rKmunfvjnHjxgnnjNu2bWNSmu9YSkoKQkJCEBISorSOo6MjfH19NUrgpEuWlpY4dOgQWrZsKbq2kZSUpHC/VaVKFRw9ejRb15h0LbvXsDJIJ0mzsLBgUhoiIiIiIqI8oE2bNvD19RXK06dPF64xf/v2Te6+4J9//pmj8akyePBg0TVPd3d33Lx5E3p6eirnk03sTzlH9tpx3bp1ldatV68eTp06hebNmwvfc2xsLCpUqIBXr17B1NQU/v7+ovtX2ho8QfbaprbajKGhIb59+wYAQrIddclez89ssB5NDBs2TFTu3Lkz9u/fn+l8soPSKWJlZSUqSw96k1skEonwDIR0UhJ1yMYvnbCE/l/GAC8ZihcvrvVENdmxadMm0TZlZWWFBw8eoFixYirny2r7lU2ynhnZZ5hkk87ItjtFAzeokpSUJPwvkUg0mlcbpkyZgilTpuD69etYt24dLl68iJCQEFFcQPqzGdu2bcO1a9dUPuNC3z/VR3ZERERERERERERElGPs7Oxw9+5dBAcHY+nSpWjfvr3a2fOLFi2K0aNH4+7du/j333+zlJAmw8CBA0Vla2trdO/ePcvLyykWFhY4efIkTpw4gYEDB6Jq1aooWLBgnu3IW6hQIZw4cQL+/v5o3759pjepJRIJqlWrht9//x337t1TOvLA4cOHsXLlSrRt21atDrgODg6YOHEinj59io4dO2blrciZPXs2/vvvP/Tt21etZBXm5ubo27cv7ty5o5OENBkkEgkWLlyI27dvo0OHDqLRJmQ5ODhg2rRpePbsGRPSqKCnpwcfHx9s3rxZZefZBg0a4MqVKxg9enSW1yWRSDBnzhwEBQWhf//+mbbvIkWKYPDgwbhx44bKevr6+ti2bRsuXbqEESNGoFatWrCzs1PZPmQZGRlh27Zt8PPzQ5MmTVSOruPk5IQlS5bgwYMHmSbuaNu2LU6fPo1Ro0bB1dU10xvQ+vr6aN68OQ4fPoyDBw+qnZTCyckJN27cwKFDh9CkSZNMRyDS19eHh4cHZs+ejefPn2POnDlqrSc7ChQogM2bN+P48eOoV6+e0noSiQS1a9fW+DPQtr///hv79u2Di4uL0jp169bFxYsXtfZAlrbl93Yt/T58fX3h4+OjdD+lr6+Pli1b4s6dO3kyEd8ff/yBo0ePokqVKkrrWFpaYvz48bh//z5cXV2ztB4HBwdcu3YNvr6++Omnn1CpUiVYW1trPGKYl5cXXr16hTlz5qBcuXKZ1rexsUG3bt2wdetWhIWFoWrVqlmKPy9zdXXFo0ePsGjRIpQpU0ZpPQMDAzRt2hQXL17Epk2b8uxxbE7Lr21K2+dVDRs2xOPHj7Fy5Uq0b98epUuXhoWFRaYPdedHujpud3Nzw+XLl/Hbb7+hevXqau3f6tati61bt+Ly5csoUqSIWvHr6lzvRzZhwgScP38etWrVUlqncuXKOHToENavX58nt4v83q6l5fdjXW9vb/j7+6Nhw4ZK6xgbG2PgwIF4+PAhPD09s7QebV2natmyJV68eIF//vkHbm5umR4/m5ubo23btvj333/x/v17tGnTJkvxZ8bY2FjUSWLfvn2iBHKUf924cQMLFy5E06ZN1epU4+zsjDlz5uDJkyfw8PDIgQjV5+7ujoCAAPTu3VtpclNra2tMnz4d165dg6OjYw5HqBltXMNKSkrCjh07hPKAAQPYaYeIiIiIiCgPmD9/vqjs5+cn/L9o0SJR0pcyZcrA3t4+p0LL1OXLl4X/DQ0NcfXqVbWu0eaFpBg/qoyELBlMTU1V1m/SpAkOHjwoujYZEREBFxcXfPv2TTToS6FChbSWALd06dKiclhYmFaWK30t5du3b6LtKzOPHz8WlbU5OIb0sq2trdVKSAPIJ85QRPZeQF5ILCF9vS4hIUGjeWUHrypUqJBWYvrelCxZUlTOa8nANm7cKCofOXIk04Q0ALKcWEc22UpmZPc5steP7ezsRGVlg6op8+XLF+F/TQbn0rbatWtj/fr1ePLkCRITE/H48WMMGzYMBQsWFNV79uwZhg8fnktRUl4gSZMeTo20pnr16ggLC4O9vb3CEep0aqZV5nXyqpmZjwgla1g59W+oEX2P1j7V7GBIWtrDcVqMhCj/kVRaKvw/zoW/J/RjWxqUjd+TgIGZVyL6jkmqS18MUzxSPNGPw+H//015mHthEOUF+pW0tqi0tDSEhITg6dOneP36NWJjY/HlyxeYmprCwsICDg4OqFKlilo3Y9S1ceNGDBo0SCiPGDECK1eu1NrySbGvX7/ixo0bePnyJSIjIxEfHw8zMzPY2NigXLlyqFixotyoJZlJS0vD06dP8ezZM7x+/RpxcXFISUmBhYUF7O3t4ebmhnLlyum8w15oaCgeP36Mly9fIjo6GklJSTA1NYWtrS0qVaqEypUra9R5QlsSEhJw6dIlvH79GpGRkcII4pUqVfouO8TrWlpaGu7cuYM7d+4gMjISaWlpKF68OOrWrSv3oIg2pKSk4MaNGwgODkZERAQSEhJgbm4OR0dHVKpUCRUqVMiVUUQAICYmBpcuXcK7d+8QFRUFMzMzFClSBFWrVkX58uWzvNzY2Fg8fPhQ9J4LFCgAa2trlC1bFlWrVtXKAy8JCQm4du0a3rx5g6ioKHz58gXm5uaws7ND+fLl4eLiotXRnrIiNDQUV69eRUhICBITE2FjY4OiRYuiXr16cqNl6Zp0O2vUqJHoAUEAuH//Pm7duoWwsDAYGRmhaNGi8PDw0Ml2oUv5oV2/evVK9Ll6e3tj8+bNQjk1NRUBAQHCfsrExASOjo5o2LBhnnpwU5WgoCBcv34d4eHh+PbtG2xtbeHi4oI6depkmlAqt4SGhuLmzZsIDw9HVFQU9PT0YGlpCUdHR7i4uMDJySlPJg/QpcePH+POnTsIDw9HQkICbG1t4ejoiPr162t8vPcjyi9tiudV2qOr4/b4+HjhN+jDhw+Ij4+HgYEBrKysUKZMGVSrVk0rD7Hq4lzve1aqVCmEhIQASH9Y9tWrV6LXnz9/jmvXriE0NBQSiQRFixaFu7s7KlXS3rWYnJBf2vX3fqz76tUrXL58Ge/fv0dSUhKsra1Rvnx51K1bN9OOGLklIiIC169fR1hYGKKiopCamgpLS0vY29vDxcUFZcuWzbHEdh8+fECZMmWEDgNLly7F2LFjc2TdlDNSUlIQFBSEZ8+eITQ0VOjgYmFhAUdHR1StWjXfbO8xMTHw8/PDmzdvEBsbCysrK1SqVAn16tXLleuSuWXr1q3w9vYGkN5R8MmTJ/nmOyQiIiIiou9T0O0dOPGfdhJY6EKrXtvh4t4nR9ZVrFgxhIaGCuXNmzfD29tbdM0UABYvXozx48ertUxnZ2cEBwcDSO/wLpuMRJqBgQFSUlIApA9K8vz5c7XWoaenh4wuypUqVcKDBw/Umq9AgQJITk4Wyqq6OWd2nVKR5cuXi65VLVu2DGPGjMl0Pk9PT1y4cCHTuC5duoQGDRoI5UGDBmH9+vXZWqasrL6HzDRs2BAXL14UyqtWrcIvv/yS6Xx79+5Fjx49RPHb2toiKipKKO/cuRO9e/fOdoxA+v19AwMDYX3GxsaiJA5ZValSJTx69EgoHzt2DK1bt1Zr3l69emH37t1CeezYsVi6dKnCupq028jISNG1+9atW+PYsWOZxpOcnCy6tqVsO9+3bx+6desmlN3c3BAYGJjp8tWhyX5GWqFChRAZGSmUIyIi5JJ8KFOrVi3cvHlTKGfsL2Vldb+WVVnd1q2trREbm9633srKSu0E8Oq0MRMTEyQmJgr1v337pvV76VltA8WLF8fbt28BpA84pW7SGNltWNW+UfqzBdKf1VD3GafChQsjIiJCKEdFRYkStWzYsAGDBw8WynXq1MHVq1fVWnZYWBiKFi0qlAsVKoTw8HC5epr8DmS1/akyePBgbNiwQSgra59ZbQNZ2UY1+f0l7dJsSDMiIiIiIiIiIiIiylESiQSlSpVCqVKlcmydshfohwwZkmPr/pEZGhqiXr16qFevntaWKZFIUL58+WwlC9AGR0fHPDnSsKmpKVq0aJHbYXw3JBIJ3N3d4e7uniPr09fXh4eHR54bhRtIv6Hdrl07rS/XysoKdevWRd26dbW+bGmmpqZo0qSJTteRXY6OjqIHZvKyypUro3LlyrkdRrbl93YNpD8QWbNmTdSsWVPn69IVFxcXuLi45HYYGsmrxwG5qUKFCqhQoUJuh5Fv5Zc2xfMq7dHVcbuZmRlq1aqFWrVqaX3Z0nRxrvcjc3Z2hrOzc26HkW35vV1nyO/Hujl9zU0bChUqpJPj8qwoUqQIRowYgUWLFgFIfzj6119/hYEBH0v9Xujr68PV1RWurq65HUq2WVtbo1OnTrkdRq5bvHix8P+AAQOYkIaIiIiIiCgPGTBgAP7880+hvGTJEjRv3lyUkEZfX19nSYH19PSEjuGpqalqzyfd6V3dBOz79u0TJaShnFWtWjVRUppNmzaplZSmW7ducsk/pBPS1K1bV2sJaYD0Nlm0aFG8e5c+aGxiYiIOHDiAzp07Z2u5DRo0ECW0WLFihdpJaU6fPi0q//TTT9mKJcPr169FZXWTs8yZM0etep07d4ZEIhG21wcPHiAhIUErydmzej3Yzc0N586dE8pz587FsmXL1Jr37t27onLPnj2zFMOPwMXFBXfu3AGQvr9esWIFRo8erdV1ZLUNZCS8B6B24vCEhAQ8fvw4S+sDgEWLFqmVwOTdu3eihDQmJiaihDQA0Lt3b1FSmtu3b6sdx+zZs0XlKlWqqD1vTlq/fj127dolfFfSCX7ox5P7Q0MRERERERERERERUZ5x7949Ubb+2rVr59kbHkRERERERHkRz6uIiOhHMWnSJFhaWgJI7zixa9euXI6IiJQ5evQo7t+/DyB9ZPGpU6fmckREREREREQkbcqUKZBIJEL5wYMHmDRpkqiOh4cH9PR00yVYOqlAfHy82vNJxxwaGqrWPLpKrEPqGT9+vKh869Yt0X0tVby8vLBmzRq56RKJBMePH9dKfNL69OkjKo8cOTLby5RNyHH69Gl8/Pgx0/l27dolqmdmZoYaNWpkOx4AsLe3F5WDg4Mznefbt29Yvny5WsvX09MTJZhPTU2Fl5eXRjEqY2ZmJvyfkdhKHcOHDxeV169fr1ZCrClTpuDr169CuVixYjA2NlZ7vT8a2fY+c+ZMfPv2TavryGobkP7e1P3dGTRokEaJ02Tt2rVLrfll26eiwUdMTU1RtGhRoZycnCxKLqfMt2/fsGXLFtE0bezbdMXW1ja3Q6A8gklpiIiIiIiIiIiIiEiwcOFCUXnEiBG5FAkREREREVH+xPMqIiL6UdjZ2WHGjBlCedasWaIOAUSUN6SlpWHatGlCecKECShevHguRkRERERERESyjI2NUa1aNaGclpaGnTt3iupMnz5dZ+u3trYW/o+MjFQ7aYGFhYXwf0hICB48eKCyvre3N968eZOlGEk7SpQoIUpQAgCNGzfGw4cP1ZrfxMQE+vr6omlpaWlo2LCh1mLMMG/ePBgZGQnld+/eoX379mrPryj5hIuLC5ycnITyt2/f4OnpqXI5YWFhGDRokGiatpK6AICDg4PoM71+/XqmiXI8PT3x+fNntdexevVqUXnfvn1y01RRlsijVKlSovKRI0fUWl63bt1gZWUllD9//owOHTqonOfu3btYsGCBaNoff/yh1vp+VN7e3ihUqJBQjomJQZ06dTRK7JKYmKgyAVJW24D0dpiamopZs2aprL979278999/ai1bmYSEhEzb2fHjx3Ho0CHRtCVLliisK5s8btasWQgKClK5/NatWyMhIUEoFyxYEB07dlQ5j7bcu3cP169fV7v+58+f8e7dO6EsvT+mHw+T0hARERERERERERERAOD8+fOihxkcHR3Ro0ePXIyIiIiIiIgof+F5FRER/Wh+/fVXuLi4AEgfwdfHxyeXIyIiWbt378adO3cApI8ePXny5FyOiIiIiIiIiBSRPV9LS0sT/rewsEDz5s11tm7phDipqamoUqUK/P39M52vcePGonLt2rUVdniPiYlBo0aNsHXr1uwHS9l24MABURKUpKQkuLm5YezYsUoTEl29ehVubm7w9vZGSkqK3OuBgYGidqQNBgYGcskwjhw5AhcXF9y7d0/pfHv37oWbmxvq16+v8PVt27aJyvfv30fFihURFhYmV/fcuXMoW7Ysvnz5IkyzsLBQmaQjK1xdXYX/U1JSUKFCBQQHB8vVe/36NSpVqoTLly9rtPx69eqhdevWomkjRoxA+/btERcXp3Ce5ORkLFy4EEWLFhUlPJYmm+CjT58+2LFjB5KTkzON6Z9//hGVjx49Ck9PT1HCjgw7duxA7dq1RW2vePHiGD58eKbr+dHt3bsXEolEKAcEBMDe3h779u1TOd/p06fRokULWFhYqLyemNU2MGzYMFF51qxZWLlypcK6o0aNQu/evVUuT11Hjx5Fs2bNkJiYKPfaihUr5BJfNWzYEG5ubgqXNWbMGBQtWlQof/v2DdWqVcPu3bvl6n7+/Bn169fHmTNnRNP//fffrLyNLDl37hzq1KmDEiVKYMqUKYiJiVFaNygoCOXKlRNtczVr1syBKCmvMsjtAIiIiIiIiIiIiIgo50VHRyMgIAAA8PHjR1y+fBk+Pj6ihxmmTJmCAgUK5FaIREREREREeRrPq4iIiABDQ0M8evQot8MgIhV69eqFXr165XYYRERERERElIlu3brBxMRElPwiQ6dOnXS67rlz5+LYsWNC+dGjR2jUqJFcPTs7O0RERAhlHx8fHDlyROi0npCQgDp16sDe3h6lS5cGALx79w6vX78W7p8YGRnBzc0Nt27d0uVbIhWcnJywf/9+dOrUSfheUlNTsXz5cvzzzz8oXrw47O3tYWBggKioKISEhMi1Sz09PbRq1UrUbu7evQt3d3fcvn1ba7EOHz4cFy9exK5du4Rpjx8/RpUqVWBtbY0SJUrAxsYGCQkJCAsLw/v374XEOlWrVlW4TA8PD4wbNw5Lly4VpgUFBaFo0aIoXrw4ihcvjuTkZLx8+RJRUVFy7/vYsWMwMjLS2nsEgHXr1qFWrVpCOSIiAs7OzihevDhKliyJ5ORkvHnzBu/fvxfqWFlZwdbWFi9evFBrHUeOHEGZMmUQEhIimmZlZYWiRYvCwcEB5ubmiIuLw9u3bxEZGSlqH4p4e3tj+PDhQvuIi4tD37590bdvX7m60vdQAcDLywv79+/HwYMHhWkXLlyAubk5SpcuDQcHByQkJOD58+dyiXOMjIxw8eJFtd73j65hw4ZYtmwZxowZI0yLiIhAt27dUKBAAZQsWRK2trYwMDBAbGwswsPDERERIUpGYmCgPB1FVttAnz59MHHiRKFNp6Wl4ddff8WUKVNQtmxZmJqaIiIiAs+fPxcly+rSpQv279+v8edgZWUFc3NzhIaG4uzZszAzM4OTkxOKFCmCT58+4fnz54iPjxfNY2ZmhqNHj6pc7oULF1CxYkUhxqSkJPTq1QtDhw6Fk5MTzMzM8O7dO7x8+VJuG+jVq1euDHDz5s0bzJs3D/PmzYOZmRkcHR1hY2MDExMTxMXFISQkRG7fp6+vLxqch348TEpDRERERERERERE9AMKDAxUOXpO7dq15UYiICIiIiIiov/H8yoiIiIiIiIiIiIiItKmVq1a4cCBA3LT//zzT52ut2rVqhg1ahT++ecflfW+fv0qKhcuXBibN2+Gl5eXqLN9WFgYwsLC5OY3MjLCuXPnMGXKFO0ETlnWoUMH+Pn5oU2bNqJEDKmpqQgJCRElLpFVrFgxnDp1Ci4uLliyZAkmTJggvHbnzh3UqFFDq0mHdu7ciWLFimHRokWi6TExMYiJicnSMpcsWQIjIyP89ddfoulv3rzBmzdvFM5jaGiI/2PvvuNrPP8/jr9PhtiSIDaxxd57hKJLKdWqao22Vpe2RqcarRZfqkOXGVpK1WxRVIWaiVSs2ETESASxBUl+f3i4f7kzz0lychJez8fD43Gu+1zXfX3Ofe77uq9z4vqctWvXqmXLlunqMzWNGjXS6NGjNXr0aKviyZ8/v3bt2pXq3yoTc3Jy0vHjx9WyZUtt27bN9NzZs2dNCW9s8euvv6pbt24pJq5JzbJly9SzZ08tWLDA2BYfH6/jx4+nmGwnf/782rlzp8qVK5eueB9GQ4YMUcWKFdW9e3fFxMQY22NiYnT48OE02zs7O6f6fHrPgW3btqlatWq6deuWse3KlSvGD8Mk9sUXXyh37tzpSkojSdu3b1eNGjV05coVxcXF6ciRIzpy5EiydfPly6f9+/crf/78qe6zcuXKCgoKUtOmTU3Ju65cuaJdu3al2K5v376aPXt2ul5HZrp+/Xqa54Crq6v++usvlS1bNouiQnbk5OgAAAAAAAAAAADZS6VKlbR48WI5OfEVMgAAAACkB5+rAAAAAAAAAACArcaNG5dkW9myZbNkIfjXX3+tDRs2qHnz5ipYsKAsFotV7V588UX9888/KlasWIp1nJyc1LRpU504cUItWrTIrJCRQa1bt9bFixf1xhtvKF++fKnWtVgs8vb21qJFi3Tq1Cn5+PhIkoYOHaoJEyaY6gYFBalhw4aZGuvEiRMVEhKiBg0apHluOjs7q379+poyZUqq9b744gsFBgaqatWqqdZzcnLS448/rsjISPn6+toautVGjRql+fPnq1ChQinWcXFx0WOPPaazZ8+qfPnyNvfh5OSkrVu3asmSJSpdunSa9XPnzq0nnnhCgwcPTrFOly5ddPz4cXXt2lVFihRJM4FJYr/++qv++uuvNONxdXVV7969deHChTTfMyTVqVMnRUdHa+DAgcqbN2+a9S0Wi0qWLKnXXntNYWFhqdZN7zlQrlw5nTx5UnXr1k2z3oYNG/T+++9btd+UlC5dWhEREWrVqlWK44jFYlGHDh0UGRlpdeKj2rVr6+LFi+rZs6dcXFxSrVu2bFmtX7/eIQlpXnjhBfXv31+lS5e26h7v6upqjH3t2rXLggiRnVniE6YfRKZp0KCBzp07p+LFi6eYkctuRqc84cj2Rl+2ucnAKm52CATIOX46HJN2pRTE7383EyMBch5LjS+Nx+/6cD/Bw+3LAxm4nwS9nImRADmPpcGsBKUzDosDyB5K/v/D2P2OCwPIDpxrODqCNPn7+6tt27ZGOX/+/Kpataq6deumt956K83s/gAA4J6Ef6Rv06aN/P39HRfMQy40NNT0n6769OkjPz8/xwUE4IHH5yog83l7exu/vlquXDmFhoY6NqCHHHNdAAAAAAAA2NOB/+bprwUvOjqMFD32/C/yqd/L0WHkKDt37tTcuXN1/PhxxcXFqXTp0qpdu7YGDBigXLlyOTo8pOHQoUPG+3fhwgXly5dPJUqUUJMmTdSrV680ky1klbi4OP3666/6999/dfbsWd24cUMFChRQpUqV9Mgjj+jRRx+1eZ/Xrl3TjBkzFBwcrHPnzsnV1VWlSpVS+/bt1b17dzu8itT9888/WrBggcLDw+Xk5KSyZcuqSZMm6tOnT6b2Ex0drZkzZ2rfvn2KiIhQbGysPDw8VLNmTXXu3Fm1a9fO1P7SEhUVpZ9++kkHDhzQ+fPnlTdvXpUsWVKdO3dO1/uKlB07dkw///yzjh8/rsjISMXHx6tgwYKqUKGCWrVqpcceeyxLr/nw8HDNmDFDe/bs0bVr11SiRAlVrFhRAwYMUPHixTO9vxs3bui7777T/v37dfbsWRUuXFj16tXTm2++qdy5c2do36tXr9aff/6pM2fO6MaNGypWrJh8fHw0cOBAeXp6ZtIryDh/f39t3LhRBw8e1KVLlxQbG6uCBQvK29tbjz32mDp06ODoEJGNkJTGTkhKk04kpQFsRlIaIP1ISgP8P5LSAOlHUhogIZLSAIYckJQGAAAAAAAAAAAAAAAAAByFpDQAAABA9ufk6AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANkHSWkAAAAAAAAAAAAAAAAAAAAAAAAAAACQZQp5VnB0CKnK7vEBAAAAWcHF0QEAAAAAAAAAAAAAAAAAAAAAAAAAAADg4VHSu5l6vLZVly8ed3QoSRTyrKCS3s0cHQYAAADgcCSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAQJYq6d2M5C8AAABANubk6AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANkHSWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAaS0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADCSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADCQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCApDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFIaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBpDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANJaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABpLSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABhISgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAUhoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIGkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA0lpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGktIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGEhKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGCwxMfHxzs6iAdRgwYNdO7cORUvXlxBQUGODgcAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAArOLk6AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANkHSWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYXRweAzHegmo+jQ0g3n4MHbG4Tv7ajHSIBcg5Lx7Xpbhs/u3YmRgLkPJZ+e4zHA6u4OTASwPF+OhyT7rbXxpTJxEiAnCf/qFPG4/iglx0YCeB4lgazEpTOOCwOIHso6egAAAAAAAAAAAAAAAAAAAAAAAAA0s3J0QEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAALIPktIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGEhKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEBSGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgaQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADSWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAaS0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADCSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADCQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCApDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFIaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBpDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANJaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABpLSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABhISgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAUhoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIGkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA0lpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGktIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkpQEAAAAAAACAh9zo0aNlsViMf/7+/o4OCVmgb9++pvc9NDTU0SFlS76+vqbjhJTFx8frjz/+UK9evVSpUiUVKFDAdOx8fX0dHSIeQpyDOcu8efOM96tMmTK6deuWo0NKkZ+fn+n88vPzc3RIgEMwzgKZh88eDx9/f3/Tez569GhHhwTYnbe3t3HOe3t7OzqcLDVo0CDjtXfr1s3R4QAAAAAAAAAAAABWcXF0AAAAAAAAAAAAAMjZoqOj1b17d61fv97RoQDIoa5du6YRI0YY5TFjxih37twZ2ufdu3e1d+9eHTlyRGfOnNGNGzfk5OQkd3d3ubu7q1KlSqpZs2aG+wEAAACAtHzyySeaO3eubt68qaVLl+qff/5Ru3btHB0WAAAAAAAAAAAAkCqS0gAAAAAAAABANuPt7a2TJ0+mWc9isSh//vwqVKiQKleurPr16+uZZ55Rs2bNsiBKAPh/PXr0sEtCGmvHw4xo06aN/P397doHgLR98cUXOnPmjCSpcuXK6tOnT7r2c+fOHS1atEjz58/XP//8o5s3b6Za38XFRTVq1NDTTz+tHj16yMfHJ139Asj5+vbtqzlz5pi2OTs7KyQkRFWqVMnQvpYuXaqnn346M8IEMoXFYknz+Vy5cilPnjwqUqSIihcvrqpVq6pWrVpq2bKl6tWrJycnpyyKFgAeDCVLltRrr72myZMnS5LefvttBQcHM54CAAAAAAAAAAAgW+OvWQAAAAAAAACQQ8XHx+vq1asKDw/Xhg0bNHnyZDVv3lz16tXTtm3bHB0e7Mzb21sWi0UWi0Xe3t6ODgcPsVWrVmnt2rVGOX/+/Hrvvfe0ZMkSrVu3zvh3f9EVYC0/Pz9jnLNYLPLz83N0SLCT8+fP6+uvvzbKw4YNk7Ozs837mT9/vipUqKBevXpp5cqVaSakkaS7d+9q9+7dGjNmjKpXr66WLVtqw4YNNvcNZLaE45+vr6+jw3loxcbGatSoUY4OA5nA19fXdF0hdfHx8YqJiVF0dLSOHj2qzZs3a+bMmXr77bfVsGFDlSpVSkOGDNGxY8ccHSpyEOb3gPTOO+8oV65ckqS9e/fqt99+c3BEAAAAAAAAAAAAQOpISgMAAAAAAAAAD5jg4GC1atVKP//8s6NDAfAQmDt3rqm8YsUKjR8/Xl27dlX79u2Nfw0aNHBQhACyu/Hjx+v69euSpGLFiqlPnz42tb969aqeffZZ9erVS+Hh4Umed3JyUtGiReXj46NGjRqpfPnyyps3b7L72rJli9q1a6exY8fa/kIAPJAWLlyoPXv2ODoMIFs5d+6cvvnmG1WpUkUDBw7UpUuXHB0SAOQIpUqV0gsvvGCUx4wZo7i4OAdGBAAAAAAAAAAAAKTOxdEBAAAAAAAAAABSN2nSJNWpUyfJ9tjYWF25ckWHDh3S6tWrtXXrVtNzffv2VbVq1dSoUaNU9z969GiNHj06s8MGHgj+/v6ODiHb27Ztm/G4cuXKatu2babte968ebp582aa9SIiIvTiiy+atq1bt86qPjw8PNIVG3KG+Ph4R4eANERHR2vatGlGuV+/fnJzc7O6/eXLl9WxY0cFBASYtufKlUu9e/dWp06d1LZtWxUsWDBJ25MnT2r16tVauXKlVq1aZVoMGhkZmY5XAzx8HoZxNj4+XiNHjtTy5csdHQpgN7/88ouKFStm2hYTE6NLly7p8uXLOnz4sLZv367g4GDdvn3bqBMXF6dp06Zp3bp1Wr58uWrVqpXVoQPIYUJDQx0dgsMNGjRIfn5+kqSDBw9qxYoVevrppx0aEwAAAAAAAAAAAJASktIAAAAAAAAAQDbXoEED+fr6plrn448/1urVq/Xcc8/p2rVrku4tDhs2bJg2btyYBVECeBjdunVLYWFhRrlGjRqZuv8WLVpYVS+5RW3t27fP1FgA2MfMmTONuYvFYtGrr75qU/u+ffsmSUjTtWtXTZkyReXKlUu1bbly5TRo0CANGjRIhw4d0tixY7VgwQJTchoAkKQVK1Zox44datKkiaNDAeyiRYsW8vb2TrPe5cuXNX36dH3zzTc6deqUsf3EiRNq27atNm3apOrVq9sxUgDI+Zo0aaJatWpp7969kqSvv/6apDQAAAAAAAAAAADItpwcHQAAAAAAAAAAIHM8/vjj+v77703b/v33X509e9ZBEQF40F2+fNlULliwoIMiAZBT/fDDD8bjZs2aqWLFila3nTx5spYtW2ba9u6772rx4sVpJqRJrGrVqpo3b57+/fdflSlTxqa2AB5MzZs3N5U/+ugjB0UCZB+FChXSsGHDdODAAfXs2dP03IULF9StWzddv37dQdEBQM7x0ksvGY/9/f114MABB0YDAAAAAAAAAAAApIykNAAAAAAAAADwAOnZs6cKFSpklOPj47Vv3z4HRgTgQXbr1i1T2WKxOCgSADnRtm3bdOzYMaPcrVs3q9uePXs2SYKI7t27a/LkyRkai5o3b67g4GB16NAh3fsA8GB44403VKJECaO8fv16bdiwwYERAdlHvnz5NH/+fL311lum7YcOHdIXX3zhoKgAIOfo2rWrqTxv3jwHRQIAAAAAAAAAAACkzsXRAQAAAAAAAAAAMo+Li4sqV66snTt3GtvOnz9v935v3LihzZs369SpUzp//rxy584tLy8v1ahRQ3Xq1Mnw/g8fPqzdu3fr7Nmzunr1qlxcXJQvXz6VKlVKFStWVPXq1eXikv2/8o6Li1NAQIB2796tCxcuKF++fCpRooRat26t4sWLOzo8k5s3b2rTpk06ePCgrl27Jg8PD3l7e6tNmzbKly9fpvRx6NAh7d69W+fPn9fly5fl6empkiVLqmXLlvL09MzQvk+ePKldu3YpPDxcV65ckcViMY53hQoVVKNGDeXOnTtTXkdKLl68qP/++09Hjx7V5cuXdffuXeXNm1dFihRR+fLlVaNGDXl4eNg1hsQiIyO1detWnTt3ThcvXlShQoVUrFgxNWnSRGXKlLF5f/Hx8XaIMnvLimsjs8TFxenIkSPav3+/zpw5oytXrsjNzU2enp6qVKmSGjduLDc3t0zvNzIyUv/++69OnDihO3fuqEiRIqpevbqaNm0qZ2fnTO/Pni5cuKCNGzfq9OnTunr1qjw9PVWrVi27vRZ7jouJZYdzef78+aZyly5drG779ddfKyYmxigXKVJEP/zwQ6bE5enpaVMs2d2hQ4e0Z88eRUVF6eLFi8qdO7eKFi0qHx8f1alTR7ly5UrXfsPCwhQQEKCIiAjjfC1evLhatGihokWLZupriI+PV1BQkIKDg3X+/HnlzZtXpUqVUqtWrVSsWLFM7evatWvasmWLzpw5o3Pnzil37txq06aN6tevn2q7rDweGXHjxg1t2bJFp0+fVmRkpJydneXl5aXq1aurfv36Dk0wlx3GpYTy5Mmjjz76SG+88Yax7aOPPtLWrVuzPBZbREdHa9++fTp06JAuXbqk27dvy93dXV5eXmrUqJHKlSuXqf3Za4yJjY3Vzp07dfToUUVGRiomJkZFixZV+fLl1aJFC7vMYWC7SZMmafv27QoICDC2TZkyRcOGDZO7u3uWxHD37l1t2bJFR48eVUREhHLnzq2KFSuqVatWmTp/yuprKzvGZK/rPaNy6ndSkZGR2rdvn44dO6bo6GjdvXvXmD80adLELt8T3b59W//++6/CwsJ07tw55cuXT7Vq1VKrVq3SfA1RUVHavHmzjh8/rpiYGOM8q127dqbHaa07d+5o69at2rdvn6Kjo1WwYEGVKVNGbdq0ybTvW8LCwrRz505FRETo0qVLKlSokDHHy8h7VKlSJVWvXl0hISGS7n02+uyzzzIlZgAAAAAAAAAAACAzZf//oQ8AAAAAAAAAsEnihaR58uRJtf7o0aM1ZswYo7xhwwb5+vpa1dfu3bs1atQorVmzRrdu3Uq2TqlSpfTyyy9rxIgRyp8/v1X7laSYmBh99dVXmj59uo4dO5Zq3Tx58qhZs2Z69tlnNWjQINNzCxYsUM+ePY1y//79NW3aNKvjuO/dd9/VlClTjPL333+vwYMHm+r4+fmpX79+Rnn27Nnq27ev4uLi9MMPP2j8+PEKDw9Psm+LxaKOHTtq0qRJqlmzZoox9O3bV3PmzEmy/eTJk6kuIO7Tp4/8/PxSe3mGy5cva8yYMZo2bZquX7+e5PlcuXLplVde0dixY1WkSBGr9pnQtWvXNHnyZM2ZM0cnTpxIto6zs7NatWqlsWPHqlWrVlbvOy4uTjNmzNB3332nPXv2pFo3V65catCggZ555hm99tprKV4nvr6+2rhxo1G2JgnL+vXrNWHCBK1fv15xcXEp1rNYLKpataq6dOmi119/PV1JYay1bNkyjR8/XgEBASm+hlq1amno0KF66aWX5OTklOK+Eh+ThObMmZPsOdqmTRv5+/unK/aslvg6O3HihLy9ve1+bWSWq1evaunSpVq2bJn8/f116dKlFOu6ubnpqaee0gcffJBmsoWEvL29dfLkSUlSuXLlFBoaKuneYs33339fy5cvT/bcL1y4sD788EO9+eabcnV1TXbfoaGhKl++fLLP9evXzzTGJnb/vUoo4dhoy3l4+PBhDR06VH/99Zfu3r2b5HkvLy8NGzZM77zzjlxcXDJ0L7XXuJgTzuWVK1caj8uUKaNKlSpZ1e7atWv68ccfTdsGDBjg0GsvOVk5B0ksKipK//vf//Trr7/q1KlTKdbLly+fOnTooP79++vxxx9PMyFJXFyc/Pz8NGXKFO3bty/ZOk5OTmrcuLE+/PBDPfXUU6nu777E1/79uUt8fLxmzJihcePGGeNOQs7OzurYsaMmT54sHx8fq/pK6d4eEhKizz77TMuXL9eNGzdMbYYMGZLsOJnZxyOle+zGjRtTfW9GjRql0aNHp7rvLVu2aNy4cfrnn39MCZ0S8vLy0sCBAzV8+HAVKFAg1f3dZ8s4mxPGpcT69++vSZMmGfe6bdu26c8//1SnTp2yNI607Nq1SwsWLNDatWu1e/fuVOesFSpU0JAhQ9S/f/80PyumxF5jjHRvPPj000+1bNkyXbx4Mdk6efPmVbdu3TR27NgU5w1S0s+6CaUWS8Jz2cfHRwcPHpR073o+efKkSpcunebrSCgqKkqlSpXS7du3JUklS5ZUWFhYjkvWlxxXV1dNnDjRNO+5ceOG5s6dq7feesuufcfExOjzzz/X999/r6ioqCTPOzs7q3v37po4caLKli2b4r0mNVl9bVkjp13vmT2/TygnfCeVUHx8vDZv3qzffvtN69at06FDh1Ld7/3P6L169bI66U1Kn0uio6M1duxYzZkzJ9mxtXjx4vr888+TfT+OHz+uDz74QEuWLEn2s1GtWrX0/fffq2XLllbFmNLnSVteT0xMjCZOnKivvvoq2ddz//qfMGFCupIz3b59Wz/88IOmTZtmJI1JzGKxqEGDBho5cqQ6d+5scx+S1LZtW2P/J06cUEhIiKpXr56ufQEAAAAAAAAAAAD2kvL/KAYAAAAAAAAA5DixsbE6cuSIaZu1C7xtER8fr/fee0/169fX8uXLU1z8I0mnT5/Wp59+qsqVK2vz5s1W7T8sLEx169bV+++/n+biH0m6efOm/vnnHw0ePDjJAplu3bqpaNGiRnnBggXJLnZNTUxMjObOnWuU8+bNq169elnV9sqVK+rYsaPeeOONZBPSSPeO55o1a9SkSROtWbPGptgyU0hIiOrUqaMpU6akeIzuL8xp2rRpqguHkvPnn3+qYsWKGj16dIqJF6R757G/v79at26tgQMHJrvoKbFLly4Z9dNKSCPdex3btm3TsGHDdPr0aZteR0ri4+P15ptvqn379lq3bl2qCWnu1z948KAmTJhgt/f98uXLevTRR9W1a1ft2LEj1QWLe/fuVd++fdW0aVOdPXvWLvHkVPa+NjJT+fLl1adPHy1dujTVhDTSvbHt999/V8OGDfX5559nqN/ff/9ddevW1dKlS1M89y9cuKChQ4eqa9euqd43HG3u3LmqXbu2/vzzzxTHn8jISI0YMUKPPPKILl++nO6+7DkuJic7ncuHDx82vebWrVtb3Xbjxo2m426xWPTKK69kanyZwVFzkJ9++knly5fXxIkTU108LknXr1/XsmXL9OSTTyab9CWh06dPq1GjRnrllVdSTMAi3UvUsn37dnXu3FlPPPGErl69mup+U3L79m11795dAwYMSDG22NhYrV69WvXq1dPs2bPT1Y8kzZs3T/Xq1dOvv/6aJCFNSrL6eKTX9evX9dxzz6lly5ZavXp1iglppHtj26effqoqVaooMDAwS+LLTuNScnLlyqVRo0aZto0cOdKqRIVZZerUqapfv74mTpyo4ODgNGM7fvy4hgwZooYNG+ro0aM292evMUaSPvvsM1WtWlWzZs1KMSGNdC/pyS+//KJq1app5syZNr8GWwwYMMB4HBcXp1mzZtm8jzlz5hgJaSTp5ZdffiAS0tzXpk0b1alTx7Rt2bJldu3z/ncGY8eOTTYhjXTvHrFw4ULVrl07xaSaqcnqays7xmTP6z0jctJ3UgkNHz5crVu31tSpU9NMSCP9/2f0tm3bKjIy0qrYk3P48GHVq1dPU6ZMSXFsPXfunF5++WUNGzbMtH3lypWqW7eufvvttxRf2969e9W2bVstXrw43THa4vTp02ratKk++eSTFF/P/eu/YcOG2rVrl03737Fjh6pVq6a33347xYQ00r3zcOfOnerSpYs6d+5s8zxfujd+JvTXX3/ZvA8AAAAAAAAAAADA3khKAwAAAAAAAAAPkEWLFik6OtoolyxZUjVq1MjUPuLj49W3b19NnDgxSeKBIkWKqH79+vLx8VHu3LlNz507d04dO3ZMM/nGzZs31b59e+PX6O9zcnJSuXLl1KBBAzVu3FjVqlWz6leuc+XKZfql56tXr2rBggVptkto6dKlunDhglHu0aOHChYsmGa7O3fuqFOnTlq/fr2xzcvLS/Xr11ft2rWVL18+U/0bN26oe/fudl/AlZzQ0FA98sgjpr7LlSunRo0aycfHJ8miyWPHjqlbt25WJ0aYNm2ann766SQLqfLmzSsfHx81btxYlSpVkpOTU5J23bt3T3WxXXx8vLp06aItW7aYtlssFpUsWVL169dXkyZNVL16dbm7u1sVb3p88sknmjp1apLtnp6eqlOnjpo2bapatWqpRIkSdoshoYsXL8rX11dr165N8lzZsmXVsGFDVahQIckxDwwMVIsWLRxyHmZH9r42MlviBZkWi0VlypRR7dq11bRpU9WoUUN58+Y11YmPj9dHH32ksWPHpqvPlStX6vnnn9fNmzclSa6urqpSpYoaN24sb2/vZOuPGDEiXX3Z24IFC9SvX78kSRvy5cunGjVqqF69eipcuLCxfdOmTerRo0e6khPYc1xMTnY7l//9919TuWHDhulu6+PjowoVKmRKXJnJEXOQt99+W4MGDdK1a9dM252dnVW+fHk1atRItWrVMiXLscaJEyfUvHlz/ffff6btTk5OqlChgho2bKhy5colabd69Wq1bds2zSRZyRk8eLCWLFlilAsWLKjatWurZs2aSeaAMTExevXVVzVv3jyb+1m1apV69+5tJIxwcnJSxYoV1ahRI5UrVy7ZxBGOOB7pERkZqTZt2mjRokVJnitdurQaNGigunXrysPDw/TcuXPn5Ovra/Xi/fTKbuNSSl566SVVq1bNKAcHByd7TB0luWQMBQoUUNWqVdW4cWPVr19fZcqUSVInJCRErVq1sinRgb3GmNjYWPXt21cjR440JW+RpMKFC6t27dpq2LChSpUqZXru9u3bevXVVzVlyhSb+rNFnz59TJ9tZ82alWbyycRmzJhhPM6uidQyqlu3bqbyli1bdOfOHbv0de7cObVt2zbJdwYWi0UVKlRQo0aNVKFCBVksFkn3EnU+9dRTSeqnJSuvrewYk72u94zKad9JJZTc++fh4SEfHx81adJEdevWTfb7gs2bN6tdu3bG5y1bREVFqWPHjkZSN4vFYsxzkvusNnnyZM2fP1/SvUSQ3bp1MxLqubm5qVq1amrYsKG8vLxM7e7evasXX3zRqiQ+GREdHa327dsrODjY2FaqVCk1bNhQNWvWlJubm6l+VFSUOnfurCtXrli1/z/++ENt27ZNkjA0V65cxnVWrVo1ubi4JGnXrl07m5OvNmjQwFTetGmTTe0BAAAAAAAAAACArEBSGgAAAAAAAAB4QKxfv16DBg0ybRs2bFiSxewZNXXqVM2dO9e0rVWrVtqyZYsiIyMVFBSkkJAQnT9/XtOnT5enp6dR7+bNm3rhhRd05syZVPd/5MgRo1y0aFFNmzZNUVFRCg0N1c6dO7Vjxw4dOHBAV65c0dGjR/Xjjz+qY8eOxqKzxAYMGGB6bvr06Ta95sT1+/fvb1W78ePHGwvne/XqpT179igiIkJBQUHavXu3Lly4oFmzZpkWl1+7di3FZA0jRozQunXrtG7dOhUrVszYXqxYMWN7cv+sSf7wyiuv6Ny5c8qTJ48++eQTnT59WqGhoQoICDDez/fee890HHft2mXVsVy/fr0GDx6s2NhYY9tTTz0lf39/Xb58WSEhIdqxY4eOHDmi8+fPa8KECSpQoIBRd/ny5Zo4cWKK+//9999NCQry5cun//3vfzp79qxOnz6toKAgbd++Xfv379elS5cUFhamOXPmqFu3bkkWEqXX6dOnNWHCBNO2gQMHKiQkRBcuXFBwcLC2bdumPXv26MyZM7p48aL+/PNPDRo0SIUKFcqUGBIbMGCAaaGWdO/cPXr0qE6ePKnAwEAdO3ZMp06d0vvvv286FidOnNALL7xges/umzx5snFu/fLLL6bnOnbsmOw5OHnyZLu8xqxgz2vDXqpVq6aRI0dq69atunbtmsLCwrR7925t27ZN+/bt09WrV7Vt2zY9//zzpnZjx45VYGCgTX1dvnxZL730kmJjY1W6dGnNmjVLFy5c0KFDh7Rjxw6dOHFChw8f1pNPPmlq991332n//v1J9le8eHHjvBk+fLjpueHDh6c61hUvXtym2BMLCwtT//79TYtby5Urp99++00XLlzQvn379N9//+n8+fPavHmzWrRoIUlas2aNfv75Z5v6sve4mJzsdi4HBQWZyjVr1rS6bUYS2mS1rJyDfP311/r6669N2ypUqCA/Pz+dP39ex48fV0BAgPbs2aPIyEiFhYVp+vTpateuXaox3L17Vz179lRYWJixzcXFRe+//75OnTqlY8eOKTAwUKGhoTp69KheffVVU/ugoCANHjzY2pcsSfL399esWbMkSRUrVtTy5ct14cIF7d69W3v37tWFCxe0cOFCU4KKuLg4DRgwIMki4rS8/PLLiouLU6FChTR58mRFRETo6NGjCggIUGhoqE6fPq0XXnghS45HwntsQrVr1051/Ovdu3eSfcXFxen55583XWtFixY15kinTp3Szp07tWvXLkVFRRkL3u+7ceOGevbsaUqKlNmy27iUEmdn5ySJ20aNGpXsPMlR3Nzc9Nxzz2nu3Lk6efKkrly5ooMHD2rHjh0KCgpSWFiYoqKi9MMPP6hkyZJGu3Pnzln92cZeY4x0bw4yZ84co+zq6qohQ4Zo//79ioqK0u7duxUYGKjw8HAdPXpUAwcONJ0XI0aM0NatW5Pst3fv3sZ1Urt2bdNzqV1TCeeunp6eevbZZ43yyZMnk036mJJ///3XlNyiQ4cOySaCyOmaNGliKt++fTvZuV5mGDhwoI4fP26U74/B4eHhOnbsmAICAozPOSNGjJCLi4uuXr2q1157zea+suLayo4xZeb1ntnz+5z4nVRCBQoUUL9+/fT7778b3w2EhIRo+/bt2rVrl86cOaPw8HCNHz/elFR3//79ev/999Pcf2LDhw/XyZMnlTt3bo0aNUpnz5415jknTpzQgQMH1KZNmyRtIiIi9Pzzz+v27dsqWbKkZs+erQsXLujAgQMKDAzUuXPntHbtWlMSpFu3btk9+ejQoUN18OBBubi46I033tDRo0cVHh6uwMBAY444adIkubq6Gm3Cw8M1bty4NPe9f/9+9ejRw5T8p1WrVvrzzz91+fJl4zo7cOCALl68qGnTppm+mwsICNA777xj0+spX768KbnRzp07bWoPAAAAAAAAAAAAZAVLfHp+tg9patCggc6dO6fixYsn+Q+V9nagmk+W9peZfA4esLlN/NqOdogEyDksHa3/D2+Jxc+unXYl4AFm6bfHeDywilsqNYEH30+HY9KulIJrY5L+4iLwMMk/6pTxOD7oZQdGAjiepcGsBKWU/2M/8HAomXaVVHh7e+vkyZNGedKkSapTp06SenFxcbpy5YoOHz6sNWvWJPk13WeffVa//vqrnJ2dU+1v9OjRGjNmjFHesGGDfH19k60bHh6uKlWqmBZo9OnTR7Nnz05x8U1oaKhatmyp06dPG9u6dOmiZcuWJVu/cePGRlIENzc3BQcHq1q1aqm+hvsOHjyoqlWrJhvL/WQZ9+3Zs0e1atVKc5/Hjx9XpUqVdP/r9Bo1amjfvn3J1vXz81O/fv1M2ywWi6ZNm5ZkQXJC//77r3x9fY1ECK6urjp9+nSqvzie8DwpV66c8avX1urbt69p4ad0b8HlX3/9pUaNGqXYbty4cfr444+Nct26dbVr164U60dHR8vHx0fnzp2TdO/XxadPn66XX0597hQSEiJfX1+dP39e0r1fpT558mSyi9Kee+45LVq0yCj/888/atu2bar7v+/UqVPy8PBI8RfOfX19tXHjRqOc0p9Vvv/+e73++utG+ZNPPjFdV6m5du2aLl26lOwv2qfX4sWL1b17d9O2GTNm6JVXXkmxzapVq/T000/rzp07xrYpU6bo7bffTrFNaGioypcvb5T79OkjPz+/dMedXonjkFJ+r9KSVdeGvfj7+6c4hidn7ty56tOnj1F+7rnntHDhwlTbJL5PSVL9+vX1119/pThuxcbGqlOnTvrrr7+MbW+//bamTJmSYj+Jx9TZs2erb9++qcaWWML7QZs2beTv759i3WeeeUZLliwxyrVq1ZK/v79pIWtCcXFxevHFF/Xrr78meS61e2lWjItS9j+XW7ZsqS1bthjlsLAwq8fBokWLKioqyihPnjxZ7777bqbHmBpbzs+smIPs27dP9evXN43h3bt31y+//CI3t7S//w4KClKlSpWSTZQ2efJkDRs2zCjnypVLy5cv12OPPZbi/qZPn64BAwaYti1dulRPP/10svWTG8ele2PLhg0bTAn8Ejp//rxatmypw4cPG9ueeOIJrVy5MsXYEt/bpXsL5jds2GDVnDMrjodk2/iVnAkTJpgWrzdp0kQrVqyQl5dXim3i4uI0ZMgQTZ061dj21ltvJUlMkN44s/u4lDi+hO9RfHy86tevb0r4N2vWrCRzf2v2ldn279+vokWLpvreJnTp0iV16NDB9H9Z9u/fr+rVq6fYxp5jzNatW9WqVSvjs1CRIkW0evXqNBOOLVy4UC+88ILRrnbt2tq9e3eK9a2d1ydn69atRjI6SerWrZsWL15sVds+ffqYEmgsWrQoyTw9M2K0VeLPzCdOnMhQspyoqKgk88DFixerW7du6d5ncv744w917tzZKLu6umrp0qVJEiAmbtOtWzfdvXvXtD2tzy5ZcW35+/ubPruOGjVKo0ePdmhM9rzeMzq/z8nfSUn3jk3lypVTnNckdvLkSbVt29ZIuJcnTx6Fh4en+PlESvodn3Qvce+qVavUunXrZNvcvHlTjRo1MiWS8vHx0YEDB+Tj46O///7blOAooYMHD6pu3bqKibn3t+bM/j4rudfj5uamJUuW6Iknnkix3bx58/Tiiy8aZS8vL4WHh5uS1SR09+5d1atXzzTXHjNmjEaOHJlqsqHTp0+rbdu2pkRG//33n+rVq5dim8QaNGig//77zyhHRUWpcOHCVrcHAAAAAAAAAAAA7C1zfx4XAAAAAAAAAJDphg0bpg4dOiT59+ijj+rZZ5/VRx99ZEpIU61aNU2bNk0LFy5MMyGNrb7//nvT4p86depoxowZqS7Q8Pb21qJFi0x1VqxYYVqwkVDCRcVt27a1evGPdO+1pxTLwIEDTeXp06dbtc8ZM2aYFgPa+mvnb731VqoJaaR7v7z87LPPGuU7d+5o/fr1NvWTGWbOnJnqgmBJeu+991S6dGmjHBwcrIiIiBTr//jjj0biBeneouK0Ei9IUvXq1U0LBG/fvm1aIJ1QwnOmWrVqViekkaQyZcqkmJDGFgljkKTXXnvN6rb58+fP1IQ00r0F8wm98cYbqSakke4t4v/0009N27766ivFxsZmamw5kT2uDXuxJSGNJPXu3du0WG/JkiW6fPmyTfsoWLCglixZkurCQ2dn5yQJaFavXm1TP/Z06tQp08LUXLlyafHixaku+HRyctLs2bNVqVIlm/rKinExJdnpXL6/uFa6dyxLlChhddvo6GhTObVzLzvIijnI559/blo83rJlSy1cuNCqxePSvcWwyS0ej42NTZKQ5PPPP081Acv9WAcNGmTalvjelJa8efNq6dKlqS7cLlq0qBYvXiwXFxdj26pVq5Lcl9Pi5+dn1ZzTkcfDFjdu3NDEiRONcokSJbRq1ao0kxg4OTnpq6++UtOmTY1ts2bNSnLNZabsNC6lxmKx6LPPPjNtGzNmjG7fvp2lcSSnRo0aVieokCQPDw/9+uuvcnL6//8ylFZSQXuNMZI0duxYI7GMk5OTli9fnmZCGknq0aOHhg4dapT37Nmjv//+26p4bNW8eXNTMrE//vjDqnMwOjralDzTy8tLXbp0sUuMjubp6Znks/jZs2czvZ9vvvnGVP7oo49STUgjSU899ZQpSZe1suLayo4x2fN6z6ic/J2UdO/YWJuQRrqXsCXhvPHmzZtasGCB1e3vmzRpUooJaaR7yW5Gjhxp2nbgwAHlypVLv/32W4oJaaR7rzlhktM7d+7YbSy+b/z48akmpJGkXr16qUmTJkY5MjIy1R8W/P33300JaQYOHKhPPvkk1fdTkkqVKqXFixebrjFb53iJj6+tSacBAAAAAAAAAAAAeyMpDQAAAAAAAAA8QKpWraoBAwboueeeS3PhhK3i4+M1c+ZM07ZJkyaZFgGnpFmzZurRo4dpXzNmzEi2bsIFRin9gnF6dOnSxbTg/ZdfftGtW7dSbXP37l3Tgi03Nze99NJLVveZ3MKelCQ8PpJMv5KcFRo3bqynn346zXouLi5Jfu0+pYU9sbGx+vbbb41y2bJlTQtH0/LEE0+Yfl168eLFydaz1zlji4QxODIOSQoJCdG2bduMcr58+ZIkm0nJu+++a0qQc/LkSa1duzbTY8xJ7HFtZDcJk9LcvXtXgYGBNrUfNGiQypUrl2a9atWqqXbt2kb5yJEjunbtmk192csvv/xiLIiXpJdfflmVK1dOs52bm5vGjBljdT9ZNS4mJzudy3fv3jUl5ilatKhV8wlJunz5su7evWvaZsvC5/DwcP39999W/cusxfT2noOcPXtWv/32m1F2cnLSrFmzTItj02vNmjU6deqUUS5XrpyGDBliVdtx48Ypb968Rnnz5s06cOCA1X2//vrrKlu2bJr1atasqd69e5u2zZo1y+p+WrZsqUcffdSquo48HraYO3euLl68aJRHjx6dapKthJydnfXBBx8Y5WvXrmnNmjWZHqOUvcYlazz55JNq3ry5UT558qTVSaaym8qVK6tx48ZGeevWrSnWtecYc+DAAdP51aNHD9MxTst7771nun/Ycl+0VcIEY3fu3LEq2ci8efNMnxP69Onj0M8J9uTk5JQk2UZmz/NOnz5tSt7q7u6u9957z6q2H3zwgd0SpSRky7WVVbLL9Z5ROf07qfR65JFHTPNIW8+psmXLWpVc+cknn0zyPj/33HOqWbNmmm0TJ9vatWuXTTHaolSpUnr99detqmvL92xfffWV8Thv3rz64osvrI6pVq1apmOwfPlym5L7Jk7OmXCuCQAAAAAAAAAAAGQHjv+LMQAAAAAAAAAg0xw6dEjvvvuuypYta9NiXGscPHhQkZGRRrls2bJ65JFHrG7/8ssvm8qbNm1Ktl7CXwjetGmTwsLCbIw0eS4uLqYYLl26pN9//z3VNitXrjQtSH/mmWesXtArSe3bt1fhwoWtqlu3bl1TOasXoSRerJMaa2PdvXu3zpw5Y5Sff/55mxd1dezY0Xh88OBBRUVFJamT8JwJCQmx6wKolCT+Zetffvkly2O4b+PGjaZyt27d5O7ublVbV1fXJEkPUrpWHxb2uDaym/Lly5vKtl5D6T1GcXFxOn36tE192cvmzZtN5V69elndtmvXrsqXL59VdbNqXExOdjqXr169akoClD9/fpvaJmbt8ZekZcuWqUOHDlb9y6xEHPaeg6xfv9608PXxxx+3KqmSNRLfU3r37m11AiFPT88kCUdsuackTjSTmj59+pjK/v7+Vrft2bOn1XUdeTxssWrVKuOxi4uLnn/+eZvaP/LII6aF6f/++2+mxZZQdhqXrDVu3Lgk5cTJCXOKhPf/1O799hxjVq9ebSrbkgBUkgoXLqwGDRoYZXudq9K9JH4J7zczZsxQfHx8qm0SJ7149dVX7RJbdpH4fn779u1M3f/WrVtNx/yZZ55R7ty5rWqbN2/eJMmt7MXaaysrZYfrPaNy+ndSGeHt7W08tvWc6tq1q5ydndOslz9/flM/ktS9e3er+qhVq5apbM9j1q1bN6s/P1k7d7hw4YICAgKMcqdOneTh4WFTXAk/o127ds2m9ynx2Hn58mWb+gYAAAAAAAAAAADsjaQ0AAAAAAAAAJDNbdiwQfHx8cn+u3btmo4dO6aFCxfqySefNNpcuXJFr7zyikaNGpVpcezYscNUbtu2rSwWi9XtW7dubVq0u2vXrmQXqXXo0MF4fPnyZbVt21aLFi3SnTt30hG12YABA0wLbKdPn55q/cTPW/Pr0gk1bNjQ6rpeXl6mclYvQrFHrIkXhdrSx31ly5Y1lQ8cOJCkTsJzJjY2Vo899phmzJihGzdu2NxfeiWMQZKGDh2qjz/+WOfOncuyGO5LfK22a9fOpvaJF/Zt3749wzHlZDnpOk4oLi5O//zzj4YOHar27durXLlycnd3l7OzsywWi+lf1apVTW2tTXIi3UtkVKdOHavrZ6djlNDOnTuNxy4uLmrUqJHVbfPkyZNkwWNKsmpcTE52OpcTj8958uSxum2BAgWSbLt+/XqGY7I3e85BEp9XTzzxRDoiTJ6j7imFCxdWzZo1re6nWbNmypUrl1EODg62eu7YuHFjq/vJCffY+Ph4bdmyxShXqVJFBQsWtGkf+fLlMyVWtHacsVV2Gpes5evra5r3nT17Vt9++61DYklORESEvv32W/Xq1Uu1atVSsWLFlCdPniT3fovFol9//dVod+PGjRST69hzjMns++LBgwfTTBSTXoUKFTIleDp69GiqCbB27typ4OBgo9ymTRtVqVLFLrFlF4kTx7m5uWXq/hPO1ySpefPmNrW3tX5C9ri2MiqnXe8Z9SB8J5VQaGioJk6cqGeffVY+Pj4qWrSo3Nzckn3/tm3bZrSz5bOaJFPirrQkTqpcv379dLW7cuWK1X3ayh5zh82bN5vuHVn5GU26lzQroZzw2QYAAAAAAAAAAAAPF+t+sgsAAAAAAAAAkC3ly5dPFSpUUIUKFfTcc89pwYIFevHFF41fth47dqwaNWqkTp06ZbivkydPmsq1a9e2qb2bm5uqVaumffv2SZJiYmIUERGhMmXKmOoNHz5c8+bNMxasHz9+XM8995zc3d316KOPytfXV82bN1fNmjVNi7utUbZsWT3++ONauXKlpHu/en348OFkFweGh4frr7/+MsqVK1eWr6+vTf0lXgCTmnz58pnK9lq4lhJ7xJp4Ec5zzz1ne2CJXLx4Mcm2QYMG6ZtvvlFERIQkKTIyUv3799eQIUPUvn17tW3bVi1atFDdunWt/kVtWzVv3lwdOnTQunXrJEl3797VuHHj9MUXX6h58+Z65JFH1KpVKzVu3DjZhAqZKaPXauIEI9nhl+EdKSddx/ctX75c77zzjk6cOJGu9tHR0VbX9fT0lLOzs9X1s8sxSuju3buKjIw0yhUqVLB5EbWPj48pCURKsmpcTE52PpdtSSBQsGBBOTs7G3MdKfskN0qNPecgx44dM5XTs5A2JY66p9iSkEa6lyCratWq2rt3r6R788wzZ86oXLlyabYtX7681f3khHtsRESEaVwICQmxadF+cqwdZ2yVncel1Hz22WfGnE+SJkyYoEGDBtmc/Ee69/6cOXPGqro1atRQiRIlkn0uKipKI0aM0Ny5c03joy2io6OTTRJmzzEm8X3RlnMiObGxsbpy5YoKFSqUof2kZNCgQZo5c6ZRnj59utq2bZts3YwmOE3JrVu3tHnzZqvq5smTRy1atMiUftMSFxeXJClN/vz5k9T7+++/rd5n+/btTeXTp0+byj4+PjZEaHt9yb7XVnrl1Os9ox6E76Tuv44hQ4ZoxYoV6UqiZctnNUkqWrSo1XUTJ0extm3idva8R2fFd1cjRozQiBEjbA8uAVvmTvZKpgYAAAAAAAAAAABkFpLSZKE//vhDkyZN0rVr1zK8r6CgoEyICAAAAAAAAMCD5vnnn9eePXv0xRdfGNuGDRumJ598MsOLUS9dumQqFylSxOZ9JG5z6dKlJAuAKleurN9//13PP/+86deVo6OjtXDhQi1cuFCS5OHhobZt26p79+56+umnrV7kNXDgQGNBuCTNmDFDEydOTFJv9uzZpgVer776qlX7Tyh37tw2t7kvqxel2CPWCxcupHufKUku8YCnp6f+/PNPde7cWWfPnjW237hxQytWrNCKFSsk3VsU2apVK3Xr1k3du3eXu7t7psY2f/58PfXUU9q+fbuxLS4uTps3bzYWjrq4uKhhw4bq1KmTevXqJW9v70yNQcr4terp6SknJyfFxcUlu7+HTU66jiXpww8/NN0D0iMmJsbquhk5PlL2WICXeGFnehayWzueZNW4mJzsdC4nXrh669Ytq9taLBZ5eHgoKirK2GbLcX3jjTf0xhtvJPucr6+vNm7caPW+bGWvOUjiRa8ZTeqQUMJ7gJOTkzw9PW1qn9zczxqFCxe2qZ/k2kRHR1uVlMaWRCKOOh62cOQ4Y6vsNC7ZonHjxurSpYuWL18u6d41+OWXX2r06NE272vixImaM2eOVXVnz56tvn37Jtl+7Ngx+fr6Kjw83Ob+E0rp/m/PMcZe56u9ktI0bNhQ9evX13///SdJWrJkiS5evJhkLLh+/bp+/fVXo+zh4aFnnnkmU2I4d+6cOnToYFXdcuXKKTQ0NFP6TUtUVFSS67JkyZJJ6lkbu5T0Os/onM3Wz3/2vrbSIydf7xn1IHwnFRAQoI4dO2bovnr79m2b6mfkXpvetva8R+fk765SkjhZTuJkOgAAAAAAAAAAAICjkZQmC02aNElHjx51dBgAAAAAAAAAHnBvvfWWKSHBoUOHtG3bNjVv3jxD+02ccDs9iyQSt0n8S+r3Pf7449q/f78+/fRTzZ8/P9lk35cuXdKSJUu0ZMkSFS1aVCNHjtQbb7yRZvKdJ554QmXKlNGpU6ckSXPmzNG4cePk6upq1ImLi9PMmTONsqura7ILQJE6W3/B2xr3E6Uk1rBhQ+3bt0/jx4/XzJkzk/1V6mvXrmn16tVavXq13n33Xb377rv66KOPTO99RhQpUkSbNm3SjBkz9OWXXyb7N4G7d+9q+/bt2r59uz755BO98MILmjRpkooVK5YpMUgZv1YtFovy5Mmj69evS0r5OkX2M2fOnCQJafLkyaNWrVqpcePGKlu2rIoUKSI3NzflypXLqBMREaEXX3wxq8PNNhIvyk14bKzl5uZmVb2sHBezswIFCpiSX9k6zlSpUsWUlCY4ODgzw7Mbe81BEh+//PnzZ1rMCe8piZMJWcPauV9imdGXtT8WY8s8wFHHwxaMM1njs88+0x9//GEcmylTpujNN99MV0KljLh9+7aeeOKJJAkqKleurDZt2qhq1aoqVaqU8uXLpzx58pg+K/3vf//T2rVr0+zDnmNMTjxfBw0apAEDBki6N4f4+eefNWTIEFOdhQsXmo7bSy+9lOFEftldcj+uVbFixUztI6NzNmvna1LWXFu2yunXe0bl9O+kLly4oCeeeCJJspLatWurVatWqlSpkkqWLKk8efIod+7cpv0MHTpUe/bssfXlwkqOvhclPr/slVgNAAAAAAAAAAAASC+S0mSh+39AdHJyyla/JAMAAAAAAADgwVK8eHFVqFBBx48fN7Zt2bIlw0lpEi9Gup+wwhaJ2xQoUCDFuqVLl9ZPP/2kL7/8Un///bf8/f21adMm7d69W7Gxsaa658+f11tvvaWNGzdq4cKFcnZ2TnG/zs7OevXVVzVq1ChJUmRkpJYvX67u3bsbddatW6eTJ08a5c6dO/O9bjokXqw9fvx4NWjQIEP7rFGjRorPeXp6auLEifrss8/k7++vDRs2aOPGjdq5c6fu3Lljqnv16lWNGTNGf//9t9auXZuuheXJcXV11eDBgzV48GDt3LlT69evl7+/v7Zu3Wr6lXXp3iKlX375xTi/q1atmikxJHet2rJAOj4+3vRL3aldp8g+bt++rffee8+07eWXX9bEiRPTfP8PHTpkz9CyvcSL/qxNZJFQ4us7JVk9LmZXLi4uKlGihE6fPi1JioqK0t27d+XiYt2fr1u1aqWtW7ca5Z07d9olzsxmrzlI4nH62rVrKlKkSKbEnD9/fmPx9I0bN2xub8vcL6HM6MseC+kddTxskXicqV69ur7++usM7TNPnjwZav8gqlmzpp5//nnNnz9f0r37wPjx4/W///0vS+P48ccfdfjwYaNcrFgx+fn56bHHHkuzbcIEWKmx5xiTN29e0z109erVVt8LUlK8ePGMhpWqnj17aujQoUYyi+nTpydJSjN9+nRTuX///naNKTsICAgwld3c3OTj45OpfWR0zmbtfE3KmmvLVjn9es+onP6d1Lhx43ThwgWjXLlyZf3yyy9q3LhxmnFn1vcVSF7i4/v222/rySefzNA+K1SoYHXdM2fOmMply5bNUN8AAAAAAAAAAABAZiMpjQN4eXkl+wtBAAAAAAAAAJBZvLy8TElpTp06leF9enh4mMoJF9NYKyoqKtV9Jidfvnzq0qWLunTpIuneQrLNmzdr5cqV+vXXX3Xp0iWj7uLFizV58mSNGDEi1X2++uqr+vTTT3X37l1J9xYNJlwQPmPGDFP9h2ERoT0kXrxWvnx5tW/f3u795sqVSx07dlTHjh0lSTdv3tT27du1atUqzZ8/37TgZ8uWLRo2bJi+//77TI+jYcOGatiwod577z3FxcVp9+7d+uuvv7Rw4ULt3r3bqHfu3Dl1795du3fvlpOTU4b7Te5atWVR08WLF02/6m3NdQrH8/f3V0REhFHu2LGj1YtPL168aK+wcoT8+fPL1dXVSF6VeFGgNaxt46hxMTvy9vY2ktLExcXpzJkzVo9VrVq10oQJE4xySEiIQkND5e3tbY9QM5U95iCenp6mcmRkZKYdCw8PDyMJS1xcnC5dumTTfSE9c7/k2lkj8dzU3d3d5n2kxVHHwxaJx5n4+PiHdpyxtzFjxui3334zrufvvvtO7777rkqUKGH1Pvz8/OTn55fuGBYsWGAqL126VM2aNbOqrbX3f3uOMUWKFDElCqlfv362TwaaP39+vfjii/rhhx8kSfv379e2bduM475//35t377dqN+0aVPVrFkz0/r39vZWfHx8pu0vsyxevNhUbtWqVbIJhjISe+Jz8cyZM6pbt67V7W2Z42XFtWWrnH69Z1RO/05q4cKFxuPcuXPrr7/+sjpxycP+ec3eEs+dSpQokaVzp8RjU3a55gAAAAAAAAAAAID7Mv4/mwEAAAAAAAAA2c79hfX3xcTEZHif5cqVM5UTJtWwRkxMjA4dOmSU3dzcVKxYMZvjKFiwoJ544gl99913OnXqlPr162d6fsqUKWkudCtZsqQ6depklNetW6fQ0FBJ9xZdLV++3HjO29vbSG4C25QvX95UPnr0qEPiyJMnj9q2bav//e9/Cg0N1UcffWR6fsaMGabFsPbg5OSkevXq6YMPPlBwcLAWL16sPHnyGM/v27dPa9asyZS+MnqtJq6feH/InhIufpak1157zeq2+/fvz+xwcpzq1asbj8+fP28kS7FWcHCwVfWyy7iYHdSuXdtUTjhHSEubNm1UqFAhoxwfH291EiZHs8ccpHLlyqbyzp07MydYOe6esm/fPpv6uXPnTpJ5ZsmSJW3ahzVywj22ePHipjnGyZMnk3w2QOaoVKmS6bPIzZs39dlnn2VZ/3FxcQoMDDTKdevWtTpBhWT9/d+eY0xOvS8OHDjQVJ4+fXqyjyVpwIABWRKTI/n7+2vv3r2mbV27ds30fhLO1yRp165dNrW3dr6WVdeWLR6E6z2jcvJ3UmFhYabEI4899pjVCWlu3rypEydO2BwnrOfIe1F8fLwOHz5slEuVKpUkORQAAAAAAAAAAADgaCSlAQAAAAAAAIAHUFhYmKmcGb8237RpU1PZ39/fpl85//fff00LYuvXr69cuXJlKKZ8+fJp2rRppl8RPnfunFULSAYNGmQ8TriYfc6cOaY4X3nlFVkslgzFaQ9OTv//FX9Gfm3entq2bWsq//PPPw6K5P+5urrqs88+U8uWLY1td+7c0Y4dO7I0jm7dumno0KGmbZs3b86UfSe+Vm097onrJ94fsqeIiAhTuWrVqla3zQ7XZnISjnOSfce6xo0bm8p//vmn1W1DQkJ07Ngxq+pmx3HRURo0aGAqJ17Mnpr8+fMnSQjw008/6cKFC5kSm71l9hykVatWpvKqVasyKVLH3VMuXrxoU2Kabdu26fbt20a5bt26cnV1tbq9tbLyeCR8720Z/1xdXdWiRQujfOPGjSyf5zxMRo4cKTc3N6M8Y8YMI9GUvV24cEF37941yrbc+w8fPmx1AjZ7jjFZeV/MzHlFnTp1TNfvb7/9pitXrigmJkY///yzsb1gwYJ67rnn0t1PTnDnzh299957pm358+fXiy++mOl9ZWS+JkkrVqywql5WXVu2eBCudylj12FO/k4qI5/VEseNzOfIz2jHjx/X9evXjXLDhg2zrG8AAAAAAAAAAADAWiSlAQAAAAAAAIAHzK5du3T+/HnTtipVqmR4v1WrVjX9ivTJkye1YcMGq9vPmjXLVG7Tpk2GY5IkFxcXNWnSxLQtKioqzXYdO3Y0/Rry7NmzdffuXWNhuCQ5Ozsn+dXr7CJfvnzG4xs3bjgwkpQ1btxYHh4eRvmff/5RSEiIAyP6fwkXa0vWnTM5JYbE19bSpUt1+fJlq9reuXPHtJA2uf0he0q8IDNhcobUREREaMmSJfYIKcMSjnOSfce6zp07m8o//vij4uLirGo7depUq/vJzuNiVku86DkoKMim9kOGDDEtJD5//rxef/31TInN3jJ7DvLII4/IxcXFKK9evVpHjhzJlFgT3wN++eUX06L41Fy6dElLly41bWvdurXVfc+dO9fqunPmzDGV7XXvysrjkZG53mOPPWYqf/vttza1h/XKlCmjwYMHG+Xbt29r9OjRWdJ3eu/9kvT9999bXdeeY0zic3XatGl2S76Q2fOKhMnRrl+/rvnz52vJkiW6ePGisf2FF15I0u+DZtiwYQoICEiyrWDBgpneV6NGjUzfSWzfvt3q+UNAQIACAwOtqptV15YtHoTrXcrYdZiTv5PKjucU/l+pUqVUs2ZNo3zs2DGtXr06S/pOPIbZMlcGAAAAAAAAAAAAsgpJaQAAAAAAAADgARIfH6+RI0eatjk5Oenxxx/P8L4tFoteeeUV07bhw4crNjY2zbYBAQFasGCBaV+vvvpqhmO6L/GCn4QL/lNisVg0YMAAo3z69Gm9//77OnTokLHtiSeeUKlSpTItzszk6elpPL5w4YKuXr3qwGiS5+rqqrffftsox8fHa+DAgdniV77Tc87klBh8fHzUvHlzo3zt2jWNGjXKqrZff/21wsLCjLK3t7c6dOiQKXHBvooXL24qb9682ap2b775pmJiYuwRUoYlHOck6cSJE3br6/HHH1fp0qWNcnBwsKZMmZJmuy1btmjatGlW95Odx8WsVqVKFXl7exvlTZs22dS+ZMmS+uyzz0zbFi5cqPfffz/Jwt/sJrPnIMWKFdPzzz9vlOPi4vTKK69YnVgpNR07dlTZsmWN8okTJ6xOxDRy5EjTYvNWrVqpWrVqVvf93Xff6dSpU2nW279/f5IENi+//LLV/dgiK49HwjEwNDTUpjhfffVVubu7G+Xff/9dK1eutGkfsN4HH3yg/PnzG+VffvlFBw8etHu/hQsXNiWP2L59u1VJkoKDg21KcmDPMaZBgwZq27atUT516pQ+/vjjDO83OZk9r+jRo4dp/j59+nRNnz7dVKd///4Z6iM7u379unr16qVvvvnGtL1GjRoaMWKEXfp0dXVNkrBt8ODBunXrVqrtbt68qUGDBlndT1ZdW7Z4EK53KWPXYU7+Tiq9n9VWrVql5cuXZyw4WGX48OGm8ttvv211ct+M2Lhxo6n86KOP2r1PAAAAAAAAAAAAwFYkpQEAAAAAAACAB8T169f16quvJllw2rt3bxUpUiRT+hg8eLDy5MljlP/77z8NGjQo1cXfYWFh6t69u6lOly5dVLFixSR1Dxw4oMGDB+vo0aNWxxQYGCh/f3+j7O7urgoVKljV9uWXX5arq6tRnjx5sun57LyIsEaNGsbj+Ph4LV682IHRpGzIkCGmXzPfvHmzunfvbtPinuvXr+ubb77RzJkzkzx35coV9erVS7t27bJ6f6Ghofr999+NspOTk+rWrWt1++S8/vrr+uOPP6xOhBATE5NkAWeDBg0yFENCQ4cONZW/+eabJAv2E1uzZo0++ugj07a3335bTk78OSknSJiISJLGjx+fZHFkYh9//LEWLVpkz7AyJOE4J0krVqywW/IWZ2dnjR492rRt+PDh+uqrr1K8rv/++2916tRJsbGxslgsVvdl73ExJ3nyySeNx+Hh4Tbd/yVp2LBheuqpp0zbJkyYoB49eliVzCShS5cupXnNZKbMnoN88MEHcnNzM8r//vuvnn/+eauTTu3cuTPZc9DZ2VlDhgwxbXv//fe1fv36VPc3a9asJIvgE9+b0nLjxg117do11cR758+fV7du3UwL8x977DFVrVrVpr6slZXHI+EYGBUVZZrvpqVQoUJ67733jHJcXJx69uypFStWWL0PSQoKClKPHj1savMw8vLyMp0XsbGx2rFjh937dXZ2VpMmTYzy2bNnk4wliR09elRdunSx+X5qrzFGkj799FPTfHPixIkaO3asTQnGwsPDNXz4cAUGBqZYJ/G8IuHngfTIkyePevfubZT/++8/bdiwwSjXr19f9evXz1Af2dHly5c1efJkVa9eXfPnzzc95+XlpSVLlpi+M8hs77zzjimxSWBgoLp06aLIyMhk60dEROipp57Srl27rJ6vZeW1Za0H5XrP6Pw+p34nVbZsWVOywcDAQC1cuDDV/QYEBOjFF1+0Og5kTK9evUzn5+HDh/X444/rzJkzVu/jzp07mjNnjiZMmGB1m4Tnjre3d5JrBAAAAAAAAAAAAMgO+F/EAAAAAAAAAJDNBQUF6e+//07234oVKzR9+nT1799fZcqU0axZs0xtS5UqpS+++CLTYildurQmTpxo2jZjxgy1a9dO27dvN22/fv26Zs6cqQYNGpgWhnt6euq7775Ldv8xMTH68ccfVbVqVbVp00bffvut9u3bl+wvX58/f15ffvmlHnnkEdPzffr0Ua5cuax6PV5eXuratWuyz5UqVUpPPPGEVftxhI4dO5rKgwcP1ptvvqlff/1Va9asMZ0nISEhDory3qLoRYsWmRber1ixQjVq1NDkyZMVFhaWbLtTp07p999/14svvqiSJUtqyJAhySYYiIuL0/z581W/fn01atRIEyZM0H///afbt28nqXvlyhXNnDlTzZo1My2Se+qpp5L8crmttmzZos6dO6t8+fIaNmyY/P39deXKlST17ty5o7/++kstWrQwLZotXry4OnXqlKEYEurWrZueeeYZoxwfH6++fftq0KBBOn78uKnu2bNn9eGHH6pTp06m49a8eXO98cYbmRYT7KtNmzYqV66cUT516pRatGihdevWmRZgxsfHa+vWrerQoYPGjRsnSfLx8cnyeK3h5eWlOnXqGOUjR46oWbNm+vrrr/Xnn38muSfeunUrQ/298sorat++vVGOj4/XO++8ozp16ujzzz/XkiVL9Mcff2jq1Kl69NFH1aFDB0VHRytv3rw2LRi197iYk7zwwgum8rJly2xqb7FYNGfOnCSL/hctWqTKlStr8ODBWrlypa5du5Zs+2vXrmnDhg0aMmSIvL29tX//fpv6z4jMnoNUr15dkyZNMm1btGiRatSooTlz5ujSpUtJ2oSHh2vGjBlq27atGjVqlGwd6V4ipYQL4WNiYvT444/ro48+0tmzZ011jx8/roEDB+rVV181jT09evRQly5drH4998ezoKAg1atXT3/88Ycp8czt27e1aNEi1a9fX4cPHza258mTR1OnTrW6n/TIquOReK7XtWtXvf/++1q0aJHWrl1rGv8S39slacSIEabz6OrVq3r66afVrVs3/fPPP8kmF7h165YCAgL0+eefq0GDBmrYsKF+++231A8IJN1LkuXu7p7l/SZMiiLdSyYxbNiwJAk6oqKiNHnyZDVs2FBhYWGyWCw2JW+y5xjTokULY05y36hRo9SoUSMtWLAg2XaxsbE6cOCApk2bpkcffVTly5fXpEmTdP369RRfQ4cOHUzlTz/9VP369dPPP/+sv/76y3RNBQUFpbifhAYOHJjic9k5wWlytmzZkmRutXLlSs2bN0/fffed3n77bTVt2lReXl4aNmxYkvlKpUqV5O/vrypVqtg1Ti8vL3399dembWvXrlXVqlU1ePBgzZs3T6tXr9Yvv/yiQYMGqWrVqkbisNTer8Sy6tqyxYNwvWd0fp+Tv5NK/P717t1bX3zxRZLvDcLDw/Xxxx+rdevWunTpknLnzi1vb+9k40XmcXZ21uLFi1WoUCFj27Zt21SzZk2NGjXKNN9MKCIiQn/++acGDhyoUqVKqW/fvjpw4IBVfR49etT0fV3Pnj0z9iIAAAAAAAAAAAAAO3FxdAAAAAAAAAAAgNQNGzYsXe1Kly6tdevWZTjZRmKvv/66AgIC9PPPPxvb/P391axZMxUtWlRlypTRrVu3dOLECd28edPUNk+ePJo/f75KliyZah9xcXHatGmTNm3aZLQrVaqUPDw8JEmRkZEKCwtL8mvYlStX1qeffmrT6xk0aFCyi2379esnZ2dnm/aVlTp16qSqVavq0KFDku4tIp46dWqyC7H79OkjPz+/LI7w/7Vq1Upz585Vv379jEVlp0+f1rBhwzRs2DCVKFFCXl5ecnNz0+XLlxUZGZniIrbU7Ny5Uzt37tT777+vXLlyqXTp0vLw8JCzs7MuXLig0NDQJIvJihYtqm+//TZTXqcknTx5UpMnT9bkyZNlsVhUqlQpFS5cWHny5NGVK1d0/PjxJAvrnJ2dNWPGDNMvvmeGadOm6ejRo9q9e7ekewk2fvrpJ/3000/y9vZW0aJFdfHiRZ04cUJxcXGmtuXLl9f8+fOz9TUAM1dXV/3vf//Tc889Z2w7fPiwOnbsKA8PD1WoUEGxsbEKCwvTxYsXjTrFihXTTz/9pNatWzsi7DQNHTrUtIAzKCgoxYXiJ06cyPCCzUWLFql9+/amPvbu3au9e/cmW9/Z2Vl+fn5Jkpm4uKT+Z9isGhezu+bNm6tChQpGQo0lS5bYPO/x8PDQhg0b1Lt3by1fvtzYfn9R8Y8//ihnZ2cVKVJERYoUUYECBXT9+nVdunRJp0+fTjKXuK9z586mJEX2kNlzkDfeeEPHjx/XlClTjG3Hjh1T37595ezsrHLlyqlw4cKKiYnR2bNndf78eav26+zsrPnz56tt27ZGEoI7d+7o888/1/jx41W+fHl5enrq/PnzCg0NTdK+fv36+uGHH2x6Lb6+vnJxcdHMmTN17Ngxde7cWYUKFZK3t7fi4+N14sQJXb161dTGYrHoxx9/VMWKFW3qy1ZZdTx69+6tcePGKSoqSpIUHR2tCRMmJFt31KhRGj16tGmbk5OT5s+fr6efflr+/v6S7s0Fli5dqqVLl8rNzU3lypWTh4eHbt26pejoaIWHhye78B5pc3d31/Dhw/XRRx9lab/9+vXTDz/8oODgYEn33uPJkydrypQpqly5stzd3XXhwgWdOHHC9N5+8MEHOn36tPFZwhr2GmMk6f3331dkZKRp30FBQerZs6ecnJxUtmxZFS5cWNK9a+Hs2bO6ceOG1fuXpHr16qldu3b6559/JN37zOnn55fs56Q2bdoY101qfHx81Lp1a+Nz63158+ZNkngtu7MlwV5Czs7OGjRokD7//HMVLFgwk6NK3osvvqjQ0FCNHDnS2BYdHW3c95Pz9NNPa/jw4abnU5uvZeW1Za0H5XrP6Pw+p34nNWzYMP38888KDw+XdC/B3ocffqhPPvlEVatWVb58+Yy5Q8L9fvPNN5o3b16ycwpkrqpVq2rp0qV65plnjM9ely5d0tixYzV27FgVKVJExYsXV758+XTlyhVFRUXZdO4ntnTpUlM5veMwAAAAAAAAAAAAYG9Ojg4AAAAAAAAAAJC53NzcNHDgQO3bt0/VqlXL9P1bLBbNmTNHw4cPl5OT+Wvm8+fP67///lNISEiSxT/FixfX2rVr9eijj9rc582bN3X06FEFBgYqMDBQJ0+eTLL4p1mzZvr3339VoEABm/bdtm3bJL/mbrFY9Morr9gcZ1ZycXHR4sWL7b7wOrM8//zz2rx5c5JjLUlnz57V7t27FRAQoEOHDiWbeMHZ2TnNhWMJ3b59W8ePH1dQUJACAgJ07NixJIusq1Wrps2bN6tMmTK2vyArxMfHKzw8XLt379b27dsVEhKSJCGNh4eHFi9erCeffDLT+/f09NTGjRvVoUOHJM+FhoYqMDBQx44dS5KQplGjRtqyZYvKlSuX6THBvp599lmNGzdOFovFtP3SpUsKCgpScHCwKSFNmTJl9Pfff9vtGsgML730kkaMGJHkNdmLu7u7/v77b/Xp0yfNukWKFNGyZcv07LPP6tq1a6bnChUqlGb7rB4Xs6vBgwcbj7dv366jR4/avI+CBQtq2bJlmjNnTrLHJDY2VhEREdq/f7+2b9+uvXv3Kjw8PNmENM2bN9e6deu0fPlylS5d2uZYbGGPOciXX36p7777Tnnz5jVtj42N1fHjxxUYGKg9e/bYvIC2QoUK2rJli+rXr2/aHhcXp2PHjikwMDDZxdKPP/64/P39jUXctvj+++/1zDPPGOXLly9r9+7d2rNnT5KENLly5dJPP/1kWuRuT1lxPDw9PbV48WJ5eXmlO85ChQpp3bp1evfdd5MkX4iJidHhw4e1Y8cO7d69WydPnkw2IU12vkdkN0OGDMnQ+5Uerq6uWr58uSpXrmzaHhcXp0OHDmnHjh06evSo6b1999139dlnn6WrP3uNMff3PWfOHLm7uyd5LaGhoUbiimPHjiWbkKZAgQJJ2ib2888/J7luM2rgwIFJtvXo0SPLErQ4SokSJfTOO+/oyJEjmjp1apa/3o8//lizZ89Oczy1WCx6/fXX9dtvvyU5b1Kbr2X1tWWNB+V6z+j8Pqd+J+Xp6akVK1aoWLFipu13797V/v37FRAQoBMnThj7dXJy0pdffqn+/fvbHC/Sr23btgoMDFSjRo2SPBcVFaV9+/Zpx44dOnDgQLLnvsVisXrulDCxUps2bVS9evX0Bw4AAAAAAAAAAADYEUlpAAAAAAAAACAHy5Url4oWLaoaNWqoV69e+v7773X69Gn9+OOPVi2ITy+LxaKJEyfqv//+U+fOneXm5pZi3ZIlS2rkyJE6cuSIWrZsmep+a9eurS1btui9995TgwYNUv3l8vuaN2+uuXPnasuWLUkW91irX79+pnKHDh2S/Bp3dlSjRg3t2bNHs2fPVvfu3VWlShUVKlRIzs7Ojg4tWQ0aNFBISIjmzp2rpk2bphmnm5ub2rVrp0mTJunUqVMaMGBAkjru7u7atWuXRo8erRYtWqR6Lt5Xu3ZtffPNN9qzZ0+yySDS448//tDUqVP15JNPprkYVrp3XQwfPlyHDx9Wly5dMiWG5BQqVEhr167VkiVL1Lhx41QX/tWsWVOzZ8/W9u3bVaJECbvFBPv68MMPtXLlStWpUyfFOgULFtTQoUO1d+9e1axZMwujS58JEyYoODhYw4YNU8uWLeXl5aXcuXPbrT93d3f5+fkpMDBQQ4YMUc2aNeXh4SEXFxd5eXmpXbt2mjJlio4dO6ZOnTpJkinZj2RdUhrJPuNiTvPqq68qX758ku4l85o5c2a699W7d2+dOHFCP//8sx5//HGrzhNXV1fVq1dP77//vg4dOqQtW7aoffv26Y7BVvaYg7z22ms6duyY3nrrrTTnRu7u7urZs6fWr1+fZr+lS5dWYGCgZsyYoRo1aqRYz2KxqEmTJlq+fLlWrVplc8LA+3LlyqVFixZp2rRpKlu2bLJ1nJ2d9eijj2rXrl1Zvmg7K45H69atdfDgQU2dOlVPPfWUypcvrwIFCiRZhJ8aFxcXTZ48WYcOHdKAAQOsSpri7e2tAQMGaO3atckm10Hy8uXLpw8//DDL+y1btqwCAwP1xhtvpDruNW3aVGvWrNHkyZMzlOzNXmOMdG8cDw0N1aeffmrVPN3Dw0Pdu3fX3Llzde7cOdWtWzfV+iVLltT27du1aNEivfDCC6pRo4bc3d2t+tyZku7duydJyPKgJJHIlSuX3N3dVbFiRbVs2VKvvPKKvvrqKwUFBSk8PFxffvmlypcv77D4+vbtq8OHD2vKlClq3bq1SpYsKVdXV+XPn1+1a9fWW2+9pd27d2vq1KlydXW1eb6W1deWNR6U6z2j8/uc+p1UvXr1FBQUpBdffDHFeb/FYlGHDh20fft2vfPOO2n2j8xXsWJFBQQEaMWKFWrXrp1y5cqVan1nZ2c1a9ZMY8eO1dGjR/Xpp5+m2UdAQID27t1rlIcMGZLhuAEAAAAAAAAAAAB7scQn9/NvyLAGDRro3LlzKl68uIKCglLcZg8HqvnYbd/25nPwgM1t4td2tEMkQM5h6bg23W3jZ9fOxEiAnMfSb4/xeGCVtBesAQ+ynw7HpLvttTH8Ui4ebvlHnTIexwe97MBIAMezNJiVoHTGYXEA2UNJRweQ5W7cuKHNmzcrLCxMUVFRcnNzk5eXl2rUqJHmwsDUXL9+Xfv379exY8cUERGh69evy8XFRYUKFVKFChVUr149FS1aNMPx9+7d2/QLxYsWLVL37t0zvF+k7vLly9q+fbvOnDmjqKgo3blzRwUKFJCXl5eqVaumqlWr2px4IiYmRiEhITp69KjOnj2ra9euyWKxqGDBgvL29lbdunVVqlQpO72ie+Lj43X48GEdOXJEYWFhunLlimJjY1WgQAEVL15ctWvXVpUqVWxaUJ5ZIiIitHXrVp07d06XLl1SwYIFVaxYMTVp0iTFBf/IuQ4cOKAdO3YoMjJSd+/eVeHCheXj46OmTZumubAOtmnYsKHx979cuXLp6tWr6TrG9hgXc4Jhw4Zp8uTJkiQvLy+FhYVZlWQsLbdv39bevXt15MgRnT17Vjdu3JCzs7M8PDzk4eGhsmXLqm7dug49pvaeg8THxys4OFgHDhzQ+fPndfXqVeXLl0/FixdX9erVVbNmzXQnswsLC9OOHTsUERGhK1euyMPDQyVKlFDz5s2tSnySUGhoqCmxQZ8+feTn52eU4+LiFBQUpF27dikqKkp58uRRqVKl1Lp1axUvXjxd8We2zDwe9hQfH6/9+/dr//79ioqKUnR0tNzc3FSoUCGVL19e1atXV8mSD9/nmQfFtWvX9O+//+ro0aO6fPmy8uTJozJlyqhp06Z2mevZc4yRpNOnTyswMFCRkZG6cOGCnJycVLBgQZUqVUo+Pj6qWLGiQ+bUCR07dkyVK1fW/f9+VaNGDe3bt8+hMSF5U6dO1ZtvvmmU/fz81KdPH6vaZvW1lR1jsvf1nhE58TupixcvatOmTTp58qRxLMuXL5/t5g24d35t375dp06d0oULF3Tz5k3lz59fRYoUUdWqVeXj42Mk2bTWyy+/rNmzZ0uSqlSpopCQkGybYBoAAAAAAAAAAAAgKY2dkJQmfUhKA9iOpDRA+pGUBvh/JKUB0o+kNMD/IykNkBCLOHOS6OholSxZUjdv3pR0bzF8eHi4XF1dHRwZACAniIqKUsmSJXXnzh1J9xLUBAYGOjiqnCUyMlIVKlTQ9evXJUk//fSTBgwY4OCo7I85yP9LKykNAGRXH374ob744guj/NVXX2nIkCEOjAgp6dq1q5YtW2aU9+3bpxo1ajguIAAPpTNnzqh8+fK6ffu2JGn+/Pnq2bOng6MCAAAAAAAAAAAAUubYnwsCAAAAAAAAAMDBfv75Z2MxuCT169fvoVwMDgBInx9++MFISCNJLVq0cGA0OZOXl5dpAf+kSZMUGxvrwIiyBnMQAMjZ7ty5o1mz/j9Jc548efTSSy85MCKkJDQ0VH/88YdR9vT0lI9Pzv3RLwA515QpU4yENDVr1lSPHj0cHBEAAAAAAAAAAACQOpLSAAAAAAAAAAAeWnfu3NGUKVOMspOTkwYNGuTAiAAAjhQfH29T/d27d+vzzz83bXv55ZczM6SHxgcffKCSJUtKko4cOaI5c+Y4OCL7Yg4CADnfnDlzFBERYZR79uwpT09PB0b08LBlznbnzh316dPHlPCuT58+cnLiv80ByFpnz57Vd999Z5S/+uorxiIAAAAAAAAAAABke/xFCwAAAAAAAADw0Bo3bpxOnDhhlLt16yZvb2/HBQQAcKh58+apd+/e2r17d5p1Fy9eLF9fX926dcvY1q5dO9WuXdueIT6w8ufPr4kTJxrl0aNHm47tg4Y5CADkbBEREfr444+NssVi0dtvv+24gB4y9evX12+//abbt2+nWu/48eNq3769Nm3aZGxzc3PTa6+9Zu8QASCJMWPG6ObNm5Kkrl276pFHHnFwRAAAAAAAAAAAAEDaXBwdAAAAAAAAAAAAWeH48eM6fvy44uPjdebMGS1fvlxLly41nnd2dtaoUaMcGCEAwNHu3r2rn3/+WT///LOqV6+udu3aqU6dOvLy8pKLi4suXryovXv3auXKldq/f7+pbYECBTRjxgwHRf5g6NWrl3r16uXoMDIdcxAAyPn+/vtvSdKNGze0b98+ffvtt4qIiDCef/bZZ1WrVi1HhffQCQ4OVo8ePeTu7q5HH31UjRo1Urly5ZQ/f35dvXpVYWFh8vf31+rVqxUbG2tq+9lnn6lSpUoOihzAw+zHH3/Ujz/+6OgwAAAAAAAAAAAAAJuQlAYAAAAAAAAA8FCYO3euxowZk+Lz77zzjmrWrJmFEQF4mJw9ezZJEpPM0KBBA3l4eGT6fiGFhIQoJCTEqrqFChXS4sWLVb58eTtHhZyIOQgA5HwdOnRI8blChQrpyy+/zMJocF90dLQWLlyohQsXWlX/rbfe0rvvvmvnqAAAAAAAAAAAAAAAeHCQlAYAAAAAAAAA8NDr3LmzPv/8c0eHAeABtmbNGvXr1y/T97thwwb5+vpm+n4fVkWKFJGbm5tiYmKsbtOxY0d99dVX8vHxsWNkeFAxBwGAnC1//vxasmSJSpUq5ehQHiqlSpXS6dOnra5fpkwZjR07Vn379rVfUAAAAAAAAAAAAAAAPIBISgMAAAAAAAAAeOg4OTnJ3d1d9erVU9++fdWrVy9ZLBZHhwUAcLBOnTopMjJSa9as0ebNm7Vnzx6dOHFCFy5c0M2bN5U3b155enrK29tbbdq00VNPPaWGDRs6OmzkIMxBACDnc3NzU7ly5dSxY0cNHTpU3t7ejg7poXPq1Clt27ZNGzZsUEBAgI4dO6YzZ87o2rVrcnJykoeHh7y8vNS0aVM98sgjevrpp5UrVy5Hhw0AAAAAAAAAAAAAQI5DUhoAAAAAAAAAwENh9OjRGj16tKPDAABkcwULFtSzzz6rZ5991tGh4AHBHCRt3t7eio+Pd3QYAJAixqjsxWKxqHnz5mrevLmjQwEAAAAAAAAAAAAA4IHm5OgAAAAAAAAAAAAAgAdd3759FR8fn+n/fH19Hf3SAAAAAAAAAAAAAAAAAAAA8AAiKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEBSGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgaQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADSWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAaS0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADCSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADCQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCApDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFIaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBpDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANJaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABpLSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABhISgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAUhoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIGkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA0lpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGktIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGEhKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEBSGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAwRIfHx/v6CAeRA0aNNC5c+fk5OQkLy8vSVJkZKTi4uJUvHhxBQUFOThCAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAEjKxdEBPKjy588vSYqLi9O5c+ccHA0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAWMfJ0QE8qIYPH65KlSqpePHixj8nJw43AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgOzNEh8fH+/oIB4WDRo00Llz51S8eHEFBQXZrZ8D1Xzstm978zl4wOY28bsG2CESIOew1JuW7rbR75fIxEiAnMd9/Fnj8cAqbg6MBHC8nw7HpLtt/JnvMzESIOexlHzNeBz/zxMOjARwPEu7VQlKZxwWB5A9lHR0AAAAAAAAAAAAAAAAAAAAAAAAAOnm5OgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADZB0lpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGktIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkpQEAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGEhKAwAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkJQGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGAgKQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwEBSGgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAgaQ0AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADSWkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAaS0gAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADCSlAQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYSEoDAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADCQlAYAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAYCApDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADAQFIaAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAICBpDQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANJaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABpLSAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJKUBAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABhISgMAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMJCUBgAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAABgICkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAMBAUhoAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgIGkNAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA0lpAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAGktIAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAwkpQEAAAAAAACAh9zo0aNlsViMf/7+/o4OCVmgb9++pvc9NDTU0SFlS76+vqbjhJTFx8frjz/+UK9evVSpUiUVKFDAdOx8fX0dHSIeYP379zfOtWeffTbVupyXgH0wt0Bm4nzK+TZv3my8f/nz51dERISjQwJS5OfnZxpz/Pz87NYXc1E8iPhuETnRwzrfDA0NNb3uvn37OjqkB96gQYOM492tWzdHhwNkOea/Dx/mhnjYPMzzqwd5Tr1gwQLjdZUpU0Y3btxwdEgAAAAA4FAujg4AAAAAAAAAAAAAOVt0dLS6d++u9evXOzoU2EF0dLT27t2rw4cP69KlS7pz5448PDxUvHhxNWnSRCVKlHBofDt37tSsWbMkSS4uLho3bpxD4wHS68iRIzpw4IBOnTqlq1evKjY2VoUKFZK7u7tKlSqlunXrysPDw9FhAnCgK1eu6ODBgzp58qTOnj2r69evS5Lc3d1VvHhx1a9fX+XKlXNwlFJcXJzefPNNo/zuu++qWLFiDowIAAAAsM25c+cUGBioEydO6OrVq3J1dZWnp6cqVaqkOnXqWP35/JNPPtHcuXN18+ZNLV26VP/884/atWtn5+gBAACQET169NCECRMUHBys8PBwjR8/XmPHjnV0WAAAAADgMCSlAQAAAAAAAIBsxtvbWydPnkyz3v1fnC9UqJAqV66s+vXr65lnnlGzZs2yIEoA+H89evQgIc0DJC4uTv7+/vrjjz/0999/a9++fanWr169ul5//XX17dtXefPmzaIo/9/bb7+tuLg4SVLv3r1VpUqVLI8B9uXr66uNGzemWidXrlxyc3OTh4eHihUrpooVK6p69epq3ry5mjdvrjx58mRRtLbZuHGj/Pz89OeffyoqKirN+uXLl9djjz2mHj16qFWrVnJycsqCKIGHx+nTpxUQEKAdO3YoICBAO3fu1NWrV43ny5Url6W/+Hvjxg35+flp06ZN2rFjh1V9e3t7q1+/fnrjjTfk6elp/yCTMXPmTAUHB0u6lzBn2LBhqdb39/dX27Ztk2wfOXKkzYs9Eu+rS5cuWrZsmU37AICEHuS5KABkldu3bys4ONiYZwcEBOjIkSOKj4836syePVt9+/Z1XJC6953YvHnz9N1332nHjh0p1rNYLKpevbqefPJJffTRRypYsGCKdUuWLKnXXntNkydPlnTve6zg4OAs+zyf3N+bKlSooIMHD8rV1TVD+7p06ZLc3d0zI0wgw1L6XJmQk5OT3NzclC9fPnl5ealUqVKqVq2a6tatqzZt2qhixYpZFC0AwFbW/B8aJycnFSxYUIUKFVKVKlXUoEEDPfXUU2revLnN/VksFn366ad66qmnJEn/+9//1L9/f5UpUyZd8QMAAABATsf/UAMAAAAAAACAHCo+Pl5Xr15VeHi4NmzYoMmTJ6t58+aqV6+etm3b5ujwYGfe3t6yWCyyWCzy9vZ2dDh4iK1atUpr1641yvnz59d7772nJUuWaN26dca/+wsvkL2tWbNGpUuX1iOPPKKvvvoqzYQ0khQSEqLXX39ddevWVWBgYBZE+f9WrVqlLVu2SLr3H0RHjBiRpf1nB9wP7rl9+7auXr2qsLAwBQYGasGCBfrkk0/Uvn17FSlSRD169NCmTZscHaYhICBAzZo1k6+vr/z8/KxKSCNJJ06c0A8//CBfX19VrFhRs2bN0t27d+0cLZLj6+trXHsWi8XR4SADtmzZom7duqlUqVIqXbq0unXrpgkTJmjDhg2mhDSOcObMGb3++utauHCh1clwQkNDNWrUKPn4+GjJkiX2DTAZMTEx+vTTT43y4MGDU12km5qvvvrK6vER2Zefn59pvPTz83N0SMhEzEXvyWlzUQDIKsOGDVPjxo1VoEABNWnSRG+99ZZ++eUXHT582JSQJjs4ePCgGjdurN69e6eakEa697eZ/fv3a+LEiTpz5kya+37nnXeUK1cuSdLevXv122+/ZUrM6XX8+HHNmjXLoTEgcyScZ/v6+jo6nGwvLi5ON2/eVFRUlEJCQrRu3Tp9++23euWVV1SpUiXVrFlTEyZMUHR0tKNDRQ7St29f07WYlcmMAZjFxcUpOjpaJ0+e1Lp16zR+/Hi1aNFCtWrV0ubNm23e35NPPqmaNWtKkm7duqVx48ZldsgA8H/s3XdUFFcbBvBnqSqiiIIFlWLDXlBQbGCNxi6xxx6xJZpYYu/GXmIssXeNscXeFQuKggUVsCCCiiKoKIhI3e8PDvPtbGMXdlnU53cO53CHKXfZnTt37s77XiIiIqIvBpPSEBEREREREREREX1l7ty5g8aNG2P79u2GrgoRfQO2bdsmKh8+fBjz589H586d0aJFC+HHxcXFQDUkbTx+/BivXr1S+jdra2tUqVIF9erVQ9myZZVu27Rp01wNtpw2bZrwe4cOHVCpUqVcOzZ9OT59+oR///0XTZs2RbNmzfDgwQOD1mfu3Llwd3eHn5+f0r8XKVIE5cuXh6urKypVqoTChQsrXS88PByDBg1Cq1at9Fldoq+ev78/Dh48qFEwaV5hZWWFypUrw83NDTVr1oStra3COtHR0fjhhx9yPQHIunXr8Pz5cwCAubk5Ro0ale19xcfHY/78+bqqGhGRQeS1vigRUW7asGED/P39kZycbOiqqHXx4kW4ubnh5s2bouXGxsYoU6YMXFxcULt2bZQpUyZb+7ezs0OvXr2E8syZM5Genp6jOufU7Nmz8fnzZ4PWgSivCQoKwoQJE1C2bFksXboUaWlphq4SERHpwP3799G0aVOsXr1aq+0kEgnGjh0rlDdt2sTEU0RERERE9M0yMXQFiIiIiIiIiIiIiEi9xYsXo2bNmgrL09LSEBcXh4cPH+LEiRO4evWq6G/9+/eHs7Mz6tWrp3b/M2bMwIwZM3RdbaKvgo+Pj6GrkOddu3ZN+L1ChQrw9PQ0YG1IlyQSCVq0aIEePXrAw8MDTk5Oor8/e/YMK1aswPLly4UH9BMTE9G+fXuEhISgVKlSeq3f2bNnRcFCw4YN0+vxKO8YN26cQiKW1NRUvH//Hu/fv0dkZCT8/Pxw48YNxMXFida7cOECXFxcsGnTJnTv3j03qw0A8Pb2xrp16xSWt2nTBt26dUPLli1hZ2en8PfY2FicOXMGJ06cwP79+xEfHy/8LTo6Wq91JvqWFSxYEB8/fjR0NVCtWjW0adMGjRs3hqurK4oXL66wztOnT7F582YsXrwYiYmJADJmB/b29kb9+vXh7Oys93qmpaVhyZIlQrlz585K66qN1atX47ffftN7v4KISFNfcl+UiP5vy5YtuZ68j/7PxMQExsbGSEpKMnRVEBgYiPbt24vusytXroxJkyahbdu2sLa2Fq0fFxcHX19fHDlyBLt379b4OEOHDhU+cw8ePMDhw4fRqVMnXbyEbImMjMSaNWvw66+/GqwORPpUvHhx7NixQ2H5x48f8f79e8TGxuLu3bvw8/PDw4cPIZVKhXXi4+MxZswYnDhxAv/++y+KFCmSm1Unoi+Mg4ODqA2h3KHsGZq0tDTExsbi3r172LdvHx49eiT8LT09HT///DPKlSuH1q1ba3yc7t2749dff0VsbCxSUlKwfPlyLF++XFcvg4iIiIiI6IvBpDREREREREREREREeZyLiws8PDzUrjNlyhScOHEC3bp1E4Im09PTMXbsWFy8eDEXaklE36LPnz/j2bNnQrlq1aoGrA3pipmZGby9vfHbb7/BwcFB5Xply5bF4sWL0aZNG3z//fdCIFFcXBwmTpyIrVu36rWef/75p/C7vb09WrZsqdfjUd5RpUoVtGjRIsv1UlNTsW/fPixbtgw3btwQln/69Am9evWCVCpFjx499FlVkWXLlikkpKlWrRr+/vtvNGzYUO22RYoUQbdu3dCtWzcsWbIES5YswV9//SUKmiOinLG0tISLiwvq1asHV1dX1KtXD0+fPjVowr2SJUvi8ePHKF++fJbrOjo6YtasWejSpQuaNWuG2NhYAEBycjKmT5+OPXv26Lu6+O+//xARESGUf/rppxzvMzExEbNnz8aaNWtyvC8iIl34UvuiRESGIpFIUL58eaGPXa9ePdSpUwffffedwb+7SE5ORq9evUT31r/++isWLFgAU1NTpdsUKlQIbdq0QZs2bbBkyRJIJBKNjuXm5obq1avj3r17ADLGtQyZlAYA5s2bh59++gkFCxY0aD2I9CFfvnwa9dkAICwsDCtWrMDGjRtFiWnPnj2L7777DufOneN5QkSUx6h7hqZHjx6YM2cOlixZgvHjxwtJg9LT0zFmzBi0bNkSRkZGGh0nX7586N27N1auXAkA2Lx5M2bPng1LS0udvA4iIiIiIqIvhWZ3UURERERERERERESU57Vp0warV68WLbt8+TJevXploBoR0dfuw4cPonKhQoUMVBPSFXd3dzx69AgrVqxQm5BGVvPmzbFw4ULRsj179oge4Ne1iIgIHD9+XCj37t1b4wdI6dthYmKCHj164Pr161i4cCFMTP4/Z0t6ejoGDhyI+/fv50pd/Pz88Pvvv4uWeXh44MqVK1kmpJFnbW2NuXPnIiQkBE2aNNFlNYm+Se3bt0dQUBDev3+PCxcuYOHChfDy8oK9vb2hqwYLCwuNEtLIqlWrFubNmydaduzYMXz+/FmXVVNK9n7Uzs4u2wl96tSpA3Nzc6G8ceNGPH36NMf1IyLKTXmpL0pEZCiHDx/Gu3fv8OjRI+zYsQOjRo2Cu7s78uXLZ+iqAchIyhIcHCyUhw8fjqVLl6pMSCMvf/78Wr2WH3/8Ufjdx8cHISEhmldWR9zd3YXfY2JisHz58lyvA1Fe4+TkhOXLlyMwMBC1atUS/e3GjRsYNmyYYSpGRETZJpFIMHbsWIwdO1a0PCgoCFevXtVqX3379hV+j4uLw86dO3VSRyIiIiIioi8Jn8wkIiIiIiIiIiIi+or07NkThQsXFspSqZQBLkSkN/LBzZrODEx5V506dbIVhD9s2DDR9ScpKQk+Pj46rJnYrl27kJ6eLpS7dOmit2PR12HcuHHYs2ePaFliYmKuBZV4e3sjJSVFKDs6OuLIkSOi80ZbdnZ2OH/+PMaPH6+LKhJ9s8qVK4cqVap8VcnNevbsKXo9CQkJePbsmV6PGRkZKbr2d+7cOdt9wzJlymDo0KFCOSUlBTNmzMhhDYmIDMfQfVEiIkNp0qQJrKysDF0NpaKjo0XJHO3t7bFo0SK9HrNz586isiECmufMmSPqpy9evBixsbG5Xg+ivMjJyQnXrl2Dq6uraPmOHTtw9uxZA9WKiIhyYtKkSTAzMxMtO3funFb7qFu3LkqXLi2Ud+zYoZO6ERERERERfUlMsl6FiIiIiIiIiIiIiL4UJiYmqFChAgICAoRlMTExej/up0+fcOXKFTx//hwxMTHIly8fbG1tUbVqVdSsWTPH+3/06BECAwPx6tUrxMfHw8TEBBYWFrCzsxOCSGVnnM6r0tPTcePGDQQGBuLt27ewsLBAyZIl0aRJE5QoUcLQ1RNJTEzEpUuX8ODBA3z8+BFFihSBg4MDmjZtCgsLC50c4+HDhwgMDERMTAw+fPgAa2trlCpVCo0aNYK1tXWO9h0REYHbt2/jxYsXiIuLg0QiEf7fTk5OqFq1qt5n5H337h1u3bqF0NBQfPjwAampqShQoACKFSsGR0dHVK1aFUWKFNFrHeRFR0fj6tWriIqKwrt371C4cGEUL14cbm5uKFOmjNb7k0qleqil9t6/f49r167h1atXePPmDdLT02FlZYVy5cqhZs2asLW1zdZ+9d22yfv48SN8fX3x8uVLREVFIV++fGjatCnq1Kmjdru0tDQEBAQgNDQU0dHRSEpKgo2NDRwdHdGwYUOYm5vrvK7yTE1NUb9+fZw6dUpYps/g9127dgm/29nZwcXFRWf7vn//PgICAhAVFQVTU1PY2dmhfv36cHBw0Mn+k5OT4efnh/DwcMTExCA9PR02NjaoUKEC6tevD2NjY50cJ7s+f/6M4OBghISEICYmBgkJCbC0tETRokVRvXp1VKtW7YtN3NClSxf8+uuvWLZsmbDsypUrOH36NFq1aqW34548eRJ3794VLdu0aRMKFiyY430bGxuLZunU1NOnTxEUFIRnz57hw4cPMDExgbW1Nezt7VG/fn2d1C0v1yk5ORnXr19HREQEYmJi8OnTJ1haWsLe3h7VqlVDuXLldHYsbT179gw3btzA69evhf5RiRIl0LBhQ9jY2ORo34mJiQgMDERwcDBiY2ORmJiI/Pnzo1ChQnBwcICzs3O2+gOU9xQqVAg2NjZ4/fq1sOzNmzeoWLGi3o75zz//iBLGdezYMUf7mzhxIjZs2ICEhAQAGcEeEyZMQOXKlXO0X31KSUnBw4cPERwcjKioKMTHx8PCwgLW1tZwdnZGnTp1dHrfqq8+MJDRFgUEBOD169eIjY1F4cKFhbYor927fm3YF2VfVN/u3r0rjLEBQOnSpVG/fn04OTnlWh0MTV99wcxxv8ePHyM6OhppaWmwtbWFo6Mj3N3dYWpqmqN6R0VF4datWwgPD0dcXBzS09NRoEAB2NrawsnJCdWqVctWn/njx48ICgrCgwcP8PbtWyQmJqJw4cIoVqwY6tSpo7f+g7+/Px4/fozIyEgYGRmhXLly8PT0zDJx5+fPn3HlyhWEhIQgPj4eRYoUgbOzMxo3bmzQ8eHAwEAEBAQgOjoa5ubmKFGiBNzd3XXWfpPubd26FUlJSUJ53LhxKFCggF6PWb58eVSpUgXBwcEAMsa35syZo9djyqtduza6du2Kffv2AQA+fPiARYsW4Y8//sjVemjrxYsXCAoKwtOnT/HhwwcAgLW1Nezs7NCgQQOdjrNnjrU+efIEb968QXx8PAoWLAg7OztUqVIFlStXznYCzk+fPsHX1xeRkZGIjo6GsbExbG1tUaVKFdSpU4dJ3/OAfPny4d9//0Xt2rVFCZumTZuGFi1a5Fo94uPjcfHiRTx79ky4J6tSpYrOx/tz89zKi3XS5/meU7r+Pk9WSkoK7t27h/v37+Pt27dISEiAubm50CeuWLFitvvEhhh3fvv2La5evYrIyEi8e/cOxYoVg6urK2rVqpXltqGhofDz88PLly8hkUhQqlQpeHh4wM7OTuf11NSHDx9w6dIlhIaGIjExEUWLFkX58uXRqFGjXPm+72tjZWWFunXr4urVq8Ky0NBQrfYhkUjQoUMHrF69GgBw9epVREREZGuSDyIiIiIioi9V3n9Cn4iIiIiIiIiIiIi0Iv9wXP78+dWuP2PGDMycOVMoX7hwAR4eHhodKzAwENOnT8epU6fw+fNnpevY2dlh4MCBGD9+vFYPmiUlJWH58uVYv349njx5onbd/Pnzo0GDBvjhhx8wdOhQ0d/++ecf9OzZUyj/9NNPWLduncb1yPTbb7+JAodWr16tMKP1li1bMGDAAKG8efNm9O/fH+np6VizZg3mz5+PFy9eKOxbIpGgVatWWLx4MapVq6ayDv3798fWrVsVlkdERKh9KLJfv37YsmWLupcn+PDhA2bOnIl169YJAaCyzMzMMGjQIMyaNQvFihXTaJ+yPn78iCVLlmDr1q14+vSp0nWMjY3RuHFjzJo1C40bN9Z43+np6diwYQNWrVqlEPwvz8zMDC4uLujatSuGDx+u8jzx8PDAxYsXhbImSVjOnTuHBQsW4Ny5c6LAXHkSiQSVKlVCx44dMWLECL0Ggf/333+YP38+bty4ofI1VK9eHWPGjMGPP/6oNshP/n8ia+vWrUo/o02bNoWPj0+26q5Keno69uzZgxUrVsDf3x9paWlK15NIJKhduzZ69+6NAQMGaPTAsr7aNlWfp+DgYMyZMweHDh3Cp0+fRNuMGjVKZVKa8PBwzJ49G//99x/evXundJ0CBQqgS5cumDVrFhwdHTWqZ3bJ/28zHxjXtYiICNy/f18oe3p66mS/Bw4cwNSpU4XAIFkSiQTu7u5YtGgRGjRokK39379/H7NmzcKJEyfw8eNHpetYWVmhT58+mDp1qtpAcl1fD168eIF//vkHx44dw7Vr10RBWfKKFCmCAQMGYMyYMShVqpTK9fKqmTNnYuPGjYiLixOW/f3333oNBF68eLGo7O7urnFfS1c+f/6MY8eO4cCBAzh//jyioqJUrmtsbIzmzZtj4sSJeq2nIep07do1zJs3D+fOnVNob2U5ODjAy8sLQ4cOFQVgyPebZak797K6Dqanp2PLli1YtmyZqH2TZWRkBFdXV0yaNAnt27dXuS9lQkNDMWvWLBw4cEBp/05WqVKl0Lp1awwZMgT169cX/e27774TJR/z9fWFu7u7VnVJSUlB6dKlER0dDSAj2Ovly5cGCXL62sn3YaysrPR6vKNHjwq/m5mZoWHDhjnaX/HixfHLL79g3rx5ADLOk6lTpwrBs3nFmzdv8O+//+Lo0aO4fPmyyms8AFhYWKBHjx6YMGECypcvn63j6bMPnJycjDVr1mDdunVK+0OZ+3VxccHUqVPRoUMHlfsKDw9X2fccMGCA6L5Z3tOnT+Hg4ID58+dj4sSJwvK5c+di0qRJWb4OeV26dMHBgweF8vHjx9GmTRut96Nv7IuyL6pvO3fuxKxZs/Do0SOlf3d3d8f8+fOFcRgHBwdEREQAAOzt7REeHq6TeqgaO9OU7GdM27GGnPYFVYmOjsasWbPwzz//4O3bt0rXsbS0ROfOnTFr1iytAxf37t2LZcuW4dq1a2rXMzY2Ro0aNdCpUyeMHDlSbcLnR48eYffu3Th58iQCAgKQmpqqct0SJUpg2LBh+PnnnzXus/n4+Iju1adPn44ZM2YgLS0Nq1atwsqVK/H48WOF7QoUKIARI0Zg1qxZCsmk4+PjMWfOHPz999+icyiTjY0N5s2bh0GDBmlUR/m2LPP6o83rAYDdu3dj5syZePjwodJt3dzcsHjxYjRq1EhlXfz8/ETte8uWLXH69GmNXoesFStWYNSoUUJ5/PjxWLBggdb7+VZs3LhR+N3ExATdu3fPleN6enoK1/unT58iODgYVapUyZVjZ5o1axYOHjwo9CUzPzvFixfP1Xqok5qairNnz2Lfvn04e/ascE1SRiKRoH79+hg/fjw6duyY7QQSQUFB+OOPP3Ds2DG145olSpRAx44dMWzYMI0Tl/v6+mLu3Lk4f/68yr6Ora0tvL29MW7cOFhaWqrcl6rx+YsXL6p97Zlt14sXL+Dg4CC8/xUrVlTZhqlz+PBhUTLSbt26Yc+ePVrvJy+yt7fHqFGjhLYeyOhH3L17FzVq1NDrsV+9eoVx48Zh//79Sr+bKFSoEIYPH46pU6eiQIECWvetDHFuZeVLPN/l/++y1H0Po0m/Wpff58mLiorCnDlzsGvXLlHSJWWKFSuGZs2aYeDAgWjdurXK9XJjjFfVd2sPHjzAtGnTcOjQISQnJytsV7t2baxatUrpffSFCxcwadIk+Pn5KfxNIpGgffv2+Ouvv1C2bNks6yc/BpLV9/KqXs+rV68wefJk7Ny5U+nrsbCwwOjRozFx4kS1E8hwDFdR6dKlReU3b95ovQ9PT08hKY1UKsWxY8cwfPhwndSPiIiIiIjoS/BlThtCREREREREREREREqlpaUpPNCf3WA3daRSKX7//XfUqVMHhw4dUpm0AQAiIyMxe/ZsVKhQAVeuXNFo/8+ePUOtWrUwYcKELBPSAEBiYiLOnz+PYcOGKQRRdOnSBTY2NkL5n3/+yTIYV15SUhK2bdsmlAsUKIDevXtrtG1cXBxatWqFkSNHKk1IA2T8P0+dOgU3NzfRQ2K5LTg4GDVr1sSyZctU/o8yAxXr16+vdUDS0aNHUa5cOcyYMUNlQhog43Ps4+ODJk2awNvbW21gTKbY2Fhh/awS0gAZr+PatWsYO3YsIiMjtXodqkilUvz8889o0aIFzpw5ozYhTeb6Dx48wIIFC/T2vn/48AGtW7dG586dcf36dbVJde7du4f+/fujfv36wozpedWDBw9Qq1Yt9OrVC35+fiqDcYGM//OtW7cwZswY/Pnnn2r3q++2TZmdO3eidu3a2L17t9qgOHlz5sxBpUqVsGnTJpUJaYCM2Xd37NgBZ2dnUbCNPsifS0WLFtXLceTPl6ZNm+Zof5nnbteuXVUGYEulUvj6+qJRo0aYPXu2VvtPTU3Fzz//jJo1a2Lv3r1qg9Xfv3+PlStXonz58jh27JhWx8muu3fvomzZshg3bhx8fHzUBgEDGe3t0qVLUaVKFZw4cSJX6qhLlpaWCgED6hJQ5VRcXBwuXLggWvbTTz/p5VjqNGrUCF5eXti1a5fawAAg4zp8+vRpeHp6YuTIkRpdh/N6neLj4+Hl5QV3d3ccOXIky/Y2PDwcixcv1jigNSciIyNRr149DBo0SGVCGiAjEYWfnx86dOiAtm3bIj4+XqP9b9++HdWqVcP27ds16gO/fPkSmzdvxsqVKxX+5u3tLSpv2LBBozrIOnz4sBDMAABeXl5fbDBDXvbw4UNRUFXBggVRsWJFvR3v48ePotmG69Wrl2VyVE2MHz9elEznwIEDuHXrVo73qyuxsbEoWbIkRowYoTbRR6aEhARs3LgR1apV0zhpqCx99YEB4Pr163B2dsbo0aNV9ocy9xsQEICOHTuiQ4cOWt9ba2PgwIEwNTUVyhs3btQoSaisqKgoHDlyRCiXLVtWbTCdIbAvyr6oPvuiQMaYVpcuXdCnTx+VCWmAjJnemzZtiqVLl+qtLoaiz77gv//+i/Lly2PVqlUqE9Jk1mHbtm2oVKkSVq1apVG9M9+7bt26ZZmQBsjoM9++fRvTp09Xe708evQoKlWqhBkzZsDPzy/LvnVUVBSmT5+O6tWrw9/fX6O6K5OQkIA2bdpg1KhRShPSABljGIsWLUKrVq2QmJgoLH/y5AlcXFywcOFCpQlpACAmJgaDBw/Gr7/+mu06aiM5ORl9+vRBr1691CZzuH79Ojw8PNRe++vXry9KcnD27NlsJYJav3698LtEIjHIveeX4smTJ6L3rXr16tlKwJ4d8uNYJ0+ezJXjyqpcuTL69OkjlBMSEvDHH3/kej3U6dGjB9q0aYONGzeqTVABZPSVrl27hs6dO8PLy0vrPmpKSgqGDx+OGjVqYNeuXVkm2o6KisLatWtFCVlUSUhIQLdu3dCoUSOcOHFCbV8nOjoas2fPRsWKFXPU3maldOnS+P7774Xyo0ePcOnSJa33I9vmAMCQIUNyXLe8ZOjQoTAzMxMt+++///R6zDNnzqBy5crYuXOnyv5hXFwc5s+fD1dX12x9t5Wb51ZerJM+z/ec0vf3eWfOnIGzszNWrVqVZUIa4P9JeLO6BzXUuPOhQ4dQp04d7N27V2kCFwC4ffs2mjZtKkpUC2QkC23evLnShDRAxufs8OHDcHNzy1bSruy4fPkyqlWrhs2bN6t8PQkJCZg7dy48PT3Vfi/IMVxF8udTdhJaNWnSRFQ2RB+OiIiIiIjIkJiUhoiIiIiIiIiIiOgrsnfvXrx//14olypVClWrVtXpMaRSKfr374+FCxcqJN0oVqwY6tSpg8qVKyvMZhsVFYVWrVplmXwjMTERLVq0wIMHD0TLjYyMYG9vDxcXF7i6usLZ2RkFCxbMsr5mZmaigJ/4+Hj8888/WW4n6+DBg6LAku7du6NQoUJZbpeSkoJ27drh3LlzwjJbW1vUqVMHNWrUUJjF7NOnT/Dy8sryoUt9CA8PR/PmzUXHtre3R7169VC5cmUYGxuL1n/y5Am6dOmi8QOD69atQ6dOnUQPsAEZCX4qV64MV1dXlC9fXmFGv3Xr1sHLy0vtw5dSqRQdO3aEr6+vaLlEIkGpUqVQp04duLm5oUqVKqKgVl2bNm2a0gBua2tr1KxZE/Xr10f16tVRsmRJvdVB1rt37+Dh4aF0ZuWyZcuibt26cHJyUvif+/v7o2HDhgb5HGriwoULaNCgAe7du6fwNxsbG9SoUUPla1NH322bMsePH0ffvn2FB0yNjIxQrlw51KtXD/b29grnHZDx0G7//v0xdepUhQdTixYtKrx+Ozs70d+Sk5MxePBgLFu2TOt6aiIhIUEh6E1fwe+XL18WlevWrZuj/c2ZM0d07hYoUABVq1ZFrVq1FNqM9PR0TJs2DfPnz9do358+fUK7du2wcuVKhc9ViRIlUKtWLdSpUwe2traiv8XHx6Njx47Yu3dv9l6UFpKTkxXaWDMzM5QrVw61a9eGq6srKlSoABMTE9E6Hz58QLt27RQSrnwJunTpIip/+vQJ169f18uxrl69qvDeywb+5BZlgSylSpVCtWrVhOtT4cKFFdZZtWqV3oKKcqtOL168QIMGDbB//36Fv1laWsLZ2Rmurq6oVKmSQhuvb0+fPoW7u7tC+2lkZAQnJyfUrVsX9vb2CtudOHECnp6eWQaQnDlzBv369VMIeitQoACqVKmC+vXro3bt2nBwcNDomtm+fXuUKlVKKP/7778qg4JVkQ9aY6CsfsyZM0dU7t27t0I7rkvXr18X9Utyem3OZGVlhbFjxwplqVSKKVOm6GTfupCWlqZwP2RsbAxHR0fUqlULbm5ucHZ2hrm5uWidpKQkDBgwQJT4NCv66gMDwJEjR+Dp6amQONTMzAyVKlUS7r/lP0NHjhxBs2bN9JZMw9bWFp07dxbKYWFhOH/+vFb72LJli+g9GjRokNb/H31jX5R9UX32RdPT0+Hl5aUQhAkAJUuWRN26dUXtlFQqxZgxY7Br1y691McQ9NkXXL9+PXr27KmQLLBgwYIqz+OkpCSMHDkSs2bNynL/gwcPVvre2draonbt2qhfvz6qVq0qSsatCWXtdv78+VGxYkW4uLigbt26cHR0VGgvIyMj4eHhgZCQEK2OB2R8tnr06IEzZ84Iy0qVKoW6deuiSpUqCuMfly9fxqhRowBkJGlo1qyZkMhGIpHAyckJ9erVg5OTk8Kxli9fjp07d2pdR23169dPdJwiRYqgRo0aqFOnjsL7npaWhsGDB6tNMjF06FDhd6lUqnViYT8/P1GSSw8PD70kq/9ayL8XNWvWFH6PjIzEH3/8gfr166NkyZLIly8f7OzsUL9+fUyaNCnHSRJdXFxE5ewkA9GF6dOnixIArl27Fs+fPzdIXZRR1lbZ2NigSpUqcHNzQ82aNZUmEjpw4AA6duyYZcL4TB8+fECLFi2wZs0ahW0y20Y3NzdUrlxZo++jZEVHR6Np06ZK+1SlS5eGi4sLatWqpRDkHxUVBQ8PjxwlQs+KbJsDKN6rZyUyMlKUINDJyQnNmjXTSd3yiuLFi6Nhw4aiZfrse168eBEdO3ZUSJKSL18+ODs7w8XFBSVKlBCWBwUFoW3btqIkbprIrXMrL9ZJn+d7Tun7+7ygoCC0b99e4fNlbm4u3Pe7uLigfPnyomuDJgwx7uzr64tu3boJn/98+fKhcuXKqFu3rkLfOCUlBX369BH6kvPnz8eMGTOE+9DChQujRo0aqFWrlsL7HhUVhS5duiAlJSVb9dTUzZs30aZNGyHRTOZ3hZn3w/IJVPz9/dG/f3+V++MYriL5iYOKFy+u9T5sbW1RpkwZoSz/XSUREREREdHXLm99y05ERERERERERERE2Xbu3DmFB0nHjh2r84CrlStXKgTPNW7cGL6+voiOjsbNmzcRHByMmJgYrF+/HtbW1sJ6iYmJ6NWrF16+fKl2/7Kz5drY2GDdunV48+YNwsPDERAQgOvXryMkJARxcXEIDQ3F33//jVatWqmc1WrIkCGiv2n7gG12H7yaP3++8EBS7969cffuXbx+/Ro3b95EYGAg3r59i02bNokS3Hz8+BHjx49Xur/x48fjzJkzOHPmjOhhqeLFiwvLlf2o2p+sQYMGISoqCvnz58e0adMQGRmJ8PBw3LhxQ3g/f//9d9H/8fbt2xr9L8+dO4dhw4YhLS1NWNa+fXv4+Pjgw4cPCA4OxvXr1/H48WPExMRgwYIFsLS0FNY9dOgQFi5cqHL/+/btEz34ZWFhgUWLFuHVq1eIjIzEzZs34efnh6CgIMTGxuLZs2fYunUrunTporPg3MjISCxYsEC0zNvbG8HBwXj79i3u3LmDa9eu4e7du3j58iXevXuHo0ePYujQoUofxtSFIUOG4M6dO6JlP/30E0JDQxEREQF/f388efIEz58/x4QJE0T/i6dPn6JXr16i9yzTkiVLhM/Wjh07RH9r1aqV0s/gkiVLdPKawsPD0bVrV1HyLXNzc4wZMwZBQUGIjo5GYGCg8No+fPiA06dPY8iQIVk+RKzvtk2ZgQMHIj09HYULF8aSJUvw+vVrhIaG4saNGwgPD0dkZCR69eol2mbWrFnYunWrUDY1NcWoUaMQFBSEN2/eCK//xYsXCA0Nhbe3t+i8HT9+PK5evapVPTXx77//imYlLVSokMJD+7py8+ZN4XdjY2NUrlw52/sKDQ0VggGLFy+Obdu24e3bt7h//z5u376NN2/e4OTJkwrHmDx5skYz1Q8bNkyUsKhgwYKYOnUqwsLC8OrVK9y+fRs3b97E69evcefOHXh5eQnrpqWlYdCgQQgNDVXYrz6uB02bNsWyZctw//59JCQkIDQ0FLdu3cL169fx6NEjxMfH47///oOrq6uwTXp6Ovr06YOPHz9m+b/IS1xcXBSCHm/fvq2XY8k/mFy2bFmtg0Z1pWzZshgzZgzOnTuHDx8+IDIyEvfu3ROuT7GxsQgMDMSwYcNE/5/NmzcrDYb9EuqUnJyMrl27IigoSLS8U6dOuHz5MmJjYxESEoLr16/jwYMHiI+Ph7+/PyZPnoyyZcsq7K9v377C+VSjRg3R39Sde8qug6mpqejZsyeePXsmLDMxMcGECRPw/PlzPHnyBP7+/ggPD0doaCgGDx4s2v7mzZsYNmyY2tc/evRoUaC/p6cnfHx8EBcXh6CgIFy7dg23bt3C06dP8fHjR1y9ehVTpkxRGtybWb9BgwYJ5YSEBK2C1iMiIkSByJUqVVKYYZZy5vPnzxg9erSoj2ZjY6NR4HtOyF6bAaBatWo62/fo0aNFCTNOnDihkIzS0OrWrYs//vgDN2/eREJCAsLCwnD79m34+fkhJCQEHz9+xNmzZ9G6dWvRdiNGjNAo8FeffeCgoCB0795dFMjYuHFjHD16FB8+fMCDBw+E++93795h3bp1or7HjRs38Ouvvyrst0SJEkIbOG7cONHfxo0bp7bNlA20lJ/dW5t7efmAfmNjYwwcOFDj7XMD+6L/x76ofvqiy5cvx9GjR0XLOnXqJIxN+Pv7IyQkBDExMVi7dq1wnztixAitgxbzIl33BWXdunULI0aMEAU0ly1bFv/++y/evHmjcB7LJw2fPn26KJhfnr+/v+h6bmJigokTJyI8PByvX7/GrVu3cO3aNdy/fx/R0dGIiorC3r170bdvX+TPnz/L/42xsTG+//57rF27Fo8fP8bHjx/x8OFDBAQEwN/fH2FhYXj//j127NghSjj76dMn9OrVS23yaGW2bdsmfBZ79uyJ4OBgREZGwt/fH0FBQXj9+jWGDx8u2mbDhg24d+8e+vbti2fPniFfvnyYNm0aXr58iSdPnuDGjRt48uQJHjx4oNCnHDt2rF6Dh7dv3y4kPf/uu+9w7do1vH37FoGBgbh58ybevHmDgwcPioKB09LSMHLkSJX77N27t+i6vXnzZqXjcqp8bYHD+iaflMbJyQlSqRR//vknypUrh8mTJ+P69euIiopCUlISXr58ievXr2PevHlwcXHBDz/8gKioqGwd29HRUfReBwQE5Oi1ZJejo6Poc5KUlKT3+wZtFStWDMOHD8exY8cQExOD6OhoBAUFwc/PD3fu3EFMTAweP36MiRMnihKLnTt3Dn/++adGxxgwYIBCYqCmTZvi5MmTiI2NxcOHD+Hn54fg4GB8+PAB9+/fx9y5c7Mcj0xPT0ePHj1E90o2NjbC9xbPnz9HQECAcK24cuWKKKnLp0+f0LNnT9FkCZlkx+dl1ahRQ21frG/fvsK6rVu3hoODg1Det29flolvZW3atEnURg0ePFjl93NfMjc3N1FZX322uLg49OnTR3RfVrRoUaxbtw4xMTEICQlBQEAAXr16hTt37qBDhw4AgLt372LRokVaHy83zq28WCddnu+tW7cWzq1WrVqJ/rZjxw6V56GqxHn6+j4v0/jx40VJq2vVqoWjR48iLi5OuO8PCAjA48ePhQkI5s6di+rVq6vcp6zcHnfu06cPkpOTUapUKWzZsgVv375FcHAw/P398fr1a5w6dUrUn//06RMmTZoEX19fTJ48GQBQr149nD17VujDZbbHq1evFiUXDg4Oxt9//611HbXRrVs3JCQkwNraGkuXLkV0dDRCQ0OF++EXL14oJKE5cuSIyvsJjuGKxcbGKoxdyicJ1JTsOfH+/XulYyZERERERERfK4lU22+HKNtcXFwQFRWFEiVKKNzU6lKIc/YfvDW0yg+yMYvGbf3MzEf0pZDUXpftbd9PyJ0ZmYnyKqv5r4TfvSuaq1mT6Ou39lFS1iupIH25Woc1IfrySEr9/wFJ6fm2BqwJkeFJmh2XKWkXkE309SmV9SpqODg4iGY0W7x4sWimzkzp6emIi4vDo0ePcOrUKYUH6X744Qfs3r1bIdBF3owZMzBz5kyhfOHCBXh4eChd98WLF6hYsaLowch+/fph8+bNKh84DQ8PR6NGjRAZGSks69ixI/777z+l67u6ugoPhZubm+POnTtwdnZW+xoyPXjwAJUqVVJal8xkGZnu3r2r0cN0YWFhKF++vBBsUbVqVdGss7K2bNmCAQMGiJZJJBKsW7dOIZBY1uXLl+Hh4SEEspiamiIyMlJt0Lrs58Te3h7h4eFZvhZZ/fv3FyW2AABra2ucPHkS9erVU7nd3LlzMWXKFKFcq1YttQ/ivn//HpUrVxYe0jcyMsL69euzDAYMDg6Gh4cHYmJiAGTMlB4RESEKUMzUrVs30Wyj58+fh6enp9r9Z3r+/DmKFCmiMljTw8MDFy9eFMqqvlZZvXo1RowYIZSnTZsmOq/U+fjxI2JjY0Wzq+XU/v37RQGFQEYwj+xDiPKOHz+OTp06iYJ2li1bhtGjR6vcJjw8HI6OjkK5X79+2LJlS7brnRV3d3dR8GWpUqVw8uRJjc7ld+/e4dmzZ6hVq5bC33KjbQMUP09ARsDuhQsXNGrnrl69isaNGwttRbFixXDixAnUrVtX7XZ79uxBr169hO1q1KiBwMDALI+nqU+fPqFq1aqidmjYsGFYvVr3YwZJSUkoUKCA8FqcnJzw5MkTrfah7D21t7eHr68v7OzslG7z6dMntGrVShQEX61aNdy9e1flZ2TPnj3o0aOHUC5XrhxOnTqFcuXKqa3fokWLRAG7HTp0wKFDh1Sun9PrQXR0NGJiYhSCJFVJT0+Ht7c3NmzYICxbvXp1lskxckL+3Nm8ebPamUA1Ua1aNVFw6s8//4wVK1bkaJ/K/PDDD9i3b59Qbt++PQ4fPqzz42Tl8uXLcHd3z7JfmOnMmTNo3769EKzg6uqK69evq91Gvm/x9OlTUYCTIeo0ceJEzJ8/XyibmZlhy5Yt6NmzZ5bHS0lJgZ+fHxo3bqz075r2EVRZsmQJxo4dK6rboUOH8N1336ncZv369QozCB88eBCdOnVSWDc4OFh0Xnt6euLs2bMaJaxMT09HaGioKAA50/Pnz+Ho6CgEutSpU0fj5x+mT58uCnJctGiR6H8gS9vPk774+PiI+pXZaWd17fr164iPjxfKnz9/RnR0NAICArB//35ER0cLfytRogSOHz+O2rVr67VOffr0EQVWXbp0SeW5o4z8/1m+T7V8+XJR4pOmTZvCx8cnW/vSpcTERAQGBqJ+/foabzN79mxMmzZNKI8fP14huaU8ffWBU1NTUbt2bdH97cyZMzF16lS1AaWRkZHw9PQUJZO9deuWys+Z/H2yttfxSpUq4dGjRwAy2srIyEgUK1Ysy+0uXLggCuz9/vvvFZJz6KqO2mBfVIx90Qz66ItGRUWhXLly+PTpk7Bs0qRJmDt3rsptwsLC0KhRI7x69Uq0PKv3VpuxxZyeb7KfeXXXA0B/fUGpVIqaNWvi3r17wrKaNWvCx8cHVlZWSveXlJSEtm3b4vz588KykiVL4smTJ0qTyIwfP14UZL5p0yaFMUdV3r59i/T0dJXjik+fPoWRkRHs7e012t/nz5/RtWtXHD/+/++Ajh8/jjZt2qjcRv56nGnx4sUYM2aMyu0GDhyIzZs3C+XKlSsjJCQEBQsWxPHjx1X2Lz59+oS6desiJOT/z5+q6idn0qa/qer1TJ06VW0Sj0ePHqF27dqi8/DOnTtKx/qBjIB02eQyhw8fRvv27VXuP1N8fDxKliwpJAsuWrQoIiMjRQHVmXJ7LE8dfbSvmurYsaPo3nz58uUICgrSKgGevb09Tp48qfF3F7JcXFxw69YtofzmzRsULVpU6/1oQv77ptjYWKGtevXqFcqVKyeMiZqYmCA4OBgVKlTQel+6du3aNdSuXVuUfEKdO3fuoHnz5nj37h0AwM7ODuHh4WqT0q9du1Y00YNEIsHChQtV3qfKkkqlKtsGAFiwYAEmTJgglN3c3HD48GFRwk156enpGDVqFFauXCks++WXX9Qm3NDmuijvjz/+EJIzAMCKFSvw888/Z7mdVCqFk5OT0DcwMTHB8+fPlX53k9M6akMf9+/79u3DDz/8IFr29u1bUcJ6XRgzZgyWLl0qlO3s7HD58mVRey1vwoQJSu8ls2pLc+Pc0qZvmFt10uf5ntMxLH1/n/fhwwcUK1YMqampAABnZ2fcunVLo0SGQMZ37+qudbkxxqvsu7VKlSrh/PnzoiSAsh49eoQaNWoIxzE2NoaTkxMeP34MLy8v7Nq1C6ampkq3lb9nqVmzpkLSIFna9q+UvR4HBwecPXtW7T27fF+xU6dOKpP6fI1juPL9kKzalkzjxo3D4sWLhbKxsTGePXum8rOjjnx7vW/fPnTt2lXr/RAREREREX2JdDs9LhERERERERERERHp3NixY9GyZUuFn9atW+OHH37A5MmTRQlpnJ2dsW7dOuzZs0fjB8A0tXr1alHShpo1a2LDhg1qA9YcHBywd+9e0TqHDx8WBbDJygw2AzKCZ7V5qNvZ2VllXbI7w/qGDRtEQcbazjL7yy+/qE1IA2TMRC/7YGtKSgrOnTun1XF0YePGjWoT0gDA77//jtKlSwvlO3fu4PXr1yrX//vvv0Wzxs6dO1ej2emrVKkiemAvOTlZ9DC2LNnPjLOzs8YJaQCgTJkyKhPSaEO2DgAUZndWp2DBgjpNSANkBLrLGjlypNoHWAGgbdu2mD17tmjZ8uXLtZqVWZ9Onz4tCsY1NzfXOBgXyEi6pCwYF8idtk2VLVu2aNzOzZo1S0jGYmRkhEOHDmWZkAYAunfvLgr6unv3Ls6ePatVPdUZO3as6AF/CwsLUfIqXXr27JloJnrZ9ii7JBIJ9u7dqzIIGAAKFCiAAwcOoHDhwsKy+/fvK8xInEkqlWLGjBmi7TUJAgYyHpCVvSYcOXJEoY3RJVtbW42DgIGMz96qVatEr0U2aPBLIR/MLh94qyuZwRGZ1CWc06fGjRtr1S9s2bIlxo0bJ5Rv3LiB4ODgL6pO7969w19//SVatnLlSo2CkIGMJIHaJNXQRlpamkJQ2R9//KE2IQ2Q0Q+VDaABFK/5meTbDW9vb40S0gAZ57myhDRARt9JNgD51q1bomBKVdLS0rBp0yahbGZmhn79+mlUHxLz9vYW3Re2b98egwYNwpo1a4SENPny5cPQoUNx9+5dvSekATICTmTp4vosa9iwYaJ9Xrx4EadPn9bpMbIjf/78WiWkATKC12XblqyCwPXZB963b58oIY23tzemTZumtg8MZAT87d+/X9SmqGqLdEE2GVdycjK2bdum0Xby9/za3svnFvZF2RcF9NMX3bhxoygRhqenp9qENEBG0s/t27frvC6GoM++4JkzZ0QJaQoUKIDDhw+rTcxgbm6OAwcOiAL2X716hV27dildX/a8K1iwIPr27atRvYGMhCTq7jscHR01TkgDZPQrtm/fLmqDsnPeyY9NKDNnzhzR9SUzyczSpUvV9s0LFCiAqVOnipadOHFC6zpqo2PHjmoT0gBAxYoVFRI8qKtXdsevd+/eLSSkAYAff/xRaUIa+r/379+Lytu2bRP9v+vUqYOFCxfi4MGDOHjwIBYsWKDQr46IiEDbtm0RFxen9fHlg6ANlfiyZMmSokTrqampmD59ukHqIq9BgwYaJ6gAMpLnL1y4UChHRkaqvWdITU3FH3/8IVo2ceJEjRJUABl9OFXfQ3z69ElUl5IlS+L48eNqE9IAGf2c5cuXi+4xNm3apPB51ZWBAweKEjJo2uacOXNG9Jlt166dyoQ0XzplyTB13W/79OkTNm7cKFq2c+dOtQlpAGD+/PnZGrfS97mVHV/y+a4L+v4+7+nTp0JCGiAjiYimCWkAZPn9lSHGnU1NTfHvv/+qTSpSsWJFUWKZtLQ0PH78GOXLl8fWrVtVJqQBMpLKVKpUSSgHBgbq7fsDICNJyt69e7O8Z1+wYIHoXDl9+rTo+zJZHMPNGBtZsmSJwjk2dOjQbCWkAfJOH46IiIiIiMgQmJSGiIiIiIiIiIiI6CtSqVIlDBkyBN26dcsykExbUqlU4cHIxYsXq515LlODBg3QvXt30b5kZ7WWJZsYQt0DYdrq2LEjSpYsKZR37NiBz58/q90mNTVVFCRobm6OH3/8UeNj5s+fXyEgQhXZ/w8AjR4M0yVXV1e1swdnMjExQZcuXUTLVM2slpaWJgoAKlu2bJbBJ7Latm0reth///79StfT12dGG7J1MGQ9ACA4OFgUuGphYaHwcKoqv/32myhBTkRERJ4I+AUyHqiVNX78eI2DcdXJrbZNmUaNGqF169YarRsSEoJTp04J5e7du8Pd3V3jY/3++++i16TqfNLW7t27sWbNGtGyefPmZfuBzqw8f/5cVJZt17PLy8sry4RcQEbArHwbJvtwrqxTp07hwYMHQnnUqFEaBQFnkk3qI5VKVc52aShmZmaiYOXbt28rtIN5nXzA6MePH/VyHPmkNLKBnFlJSEjA2bNnNfrRdcIYAOjTp4+ofPXqVZ0fQ1va1GnDhg2iwMzGjRvnmYQEp06dErVn9vb2GDVqlEbbzp07FwUKFBDKV65cEYJ1ZemzbyKfGEeTwLWTJ0/ixYsXQrljx44GS9L0tTMzM8Mvv/yCsWPH5tr/WB/XZ1nm5uaYNm2aaJm+EuDlht69ewu/R0dHIzQ0VOW6+uoDy++7QIECmDdvnsbbVq9eHR07dhTKhw4d0lsyy/79+4sC6zXpb7979w4HDhwQyiVLlsT333+vl/rlFPui2mNfVDPyCZw0PcebN2+Oli1b6rw+uU2ffUH5dujXX39F2bJls9yucOHCooRRALBu3Tql68p+po2MjDROLqgv1tbWoqBWbe8NJBJJlglcgIwgT/nku/b29holuG7fvr3o/3T79m2t6qgt+eB2VbQZ83VxcRG9/uPHjyMyMjLLY3wpidjyEvkkH5nvi5GREVauXImAgACMGzcOnTp1QqdOnTB+/HjcvHkTS5cuFX3v8vTpU4wePVrr48v3l+X707lpwoQJsLS0FMp79uwRJd76kvTo0UOUGEFdW3XgwAE8e/ZMKDs5OSm00dm1bds20XjQjBkzYG1trdG2xsbGmDhxolD++PGjaExYl0qUKCHq09+7dw/Xr1/Pcrtvqc1RlnBO1/22Q4cO4cOHD0L5u+++Q9OmTTXadv78+TqtiyranFu5Ja+c7zmVG9/n5aXvLzPldNz5hx9+QI0aNbJcr0OHDgrLJkyYIBpbVUYikShsq8++ZdeuXTWagKJIkSJo1aqVUP706ZPo3l/e1z6Ge/PmTYXvSU6dOoU9e/ZgypQpqFy5MsaOHSuafKhBgwZYtGhRto+Zl/pwREREREREuY1JaYiIiIiIiIiIiIi+Ig8fPsRvv/2GsmXLqgxOyq4HDx4gOjpaKJctWxbNmzfXeHv54IFLly4pXU82kcGlS5dEDwrmhImJiagOsbGx2Ldvn9ptjh07Jpr5rGvXrho/PAwALVq0QNGiRTVaV372+tx+iEk+QEIdTesaGBiIly9fCuUePXpo/bCj7MN1Dx48wJs3bxTWkf3MBAcH6z3oRBn5BBw7duzI9TpkunjxoqjcpUsXtbN1yzI1NVVIvKTqXM1NKSkp8PHxEcomJiYYPny4TvadW22bMprO0A4ozqStTYIsIGOmdBcXF6F8+fJlrbZX5saNGwozdrZt2xYjR47M8b5VkX1AH8iYMT6ntJlxvm/fvqLgI/nzLdPx48dFZW3frxo1aohmGNbF+6VrsrP1pqam4v79+wasjfbkPzvJycl6OU58fLyobGFhofG2T58+RcuWLTX6kZ0dWFfkZ2Q2xPVVnjZ1kg/C+OWXX/RSp+yQbzv69u2rUTI0ICMgWD6RoLJrj3zfZOfOndpVUo02bdqIAq937dqFT58+qd1GPnj7aw5aM7Tk5GQsXLgQlSpVwuDBgxXaIX2QvT4bGxtrNcO5pgYMGIDy5csLZX9/f/z33386P05u0LQt02cf+O3bt7hx44ZQbteuHYoUKaLVPmTv1T5+/Ki360TRokXh5eUllENCQnDlyhW122zfvh1JSUlCecCAARq3s7mNfdHsYV9UvZiYGDx69EgolytXDm5ubhpvLx8k+iXSZ19Qvu81YMAAjbft2bMn8ufPL5Rv3ryptB8l25eLi4vDkSNHslFT3ZI97yIjIxETE6PxtjVq1EDFihU1WrdatWqicufOnUVB56oULFgQDg4OQllXY8rKVK9eHVWqVNFo3WrVqomuQVmN+Xp7ewu/p6WlYfPmzWrXDwwMREBAgFB2d3fXuG7fMlVJJRYsWIARI0YoTfgvkUjw66+/KiRY2r59u9afN/nrgPx4V24qWrQofvvtN6Gcnp6ucaL9vMbCwgK2trZCWZsxg2HDhuksUYNsX8zExAQ9evTQavvmzZuLkmzpsy8m2+YAWScsiImJweHDh4VymTJl8N133+mlbnmBsrFnXffb5O9tZJOYZsXd3V3h/lIftDm3ckteOd9zKje+z5MfI/z333+RkpKiXUV1LKfjzrJJStWR71dKJBKFiVdUkU8IrM++pT6+owe+/jHcsWPHKnxP8t1336FHjx6YO3cuHj58KKxrYmKCESNG4Ny5c6L7MW3lpT4cERERERFRbmNSGiIiIiIiIiIiIqI87sKFC5BKpUp/Pn78iCdPnmDPnj2iWb/j4uIwaNAgTJ8+XWf1kJ8h0dPTU+nD2ao0adJE9AD+7du3lT68KTsb84cPH+Dp6Ym9e/fq5AG5IUOGiB7mzeoB25zO+KjJrGaZZB+eBHL/ISZ91FX+YWltjpFJfqbpkJAQhXVkPzNpaWn47rvvsGHDhiwfrNMl+VnEx4wZgylTpiAqKirX6pBJ/lxt1qyZVtvLJ2Tx8/PLcZ1yKiAgQDSbY+3atUVBkjmRW22bMq6urhofR9fn04MHD0SzA2orNDQU7du3F70vzs7O2L59u1b/P23Jn9c5eXgUyHgIWdOZZ4GMGdplg9yioqKUPows+35ZWFjA2dlZ67rJznKqrO3Th0+fPuGff/6Bt7c36tevj1KlSsHS0hJGRkaQSCSiH/mgFWVJw/Iy+SQN5ubmejmO7GzjAJCQkKCX42jjxo0bmDRpEtq2bQsnJydYW1vD1NRU4T2W/5/o8z3WdZ1SU1NF1y8jI6M8FSSVG9dqNzc3FCpUSCgfOHAA3bp108mM90ZGRqJ+cVxcHPbs2aNy/aioKBw9elQoOzo6okWLFjmux7fqzp07onvCuLg4PHr0CDt27ECbNm2E9dLS0rBx40Y0atQIb9++1WudZK/POb02q2JiYoKZM2eKlk2dOhXp6el6OZ62UlJScOTIEfz8889o0qQJSpcujUKFCim9hrZu3Vq0raq2TJ994CtXroj6gvq6V9MVbWf3lv27RCJRSKSYV7AvKsa+qO7IJqgAMgKWtaHt+nmNPvuC4eHheP36tVC2t7dHuXLlNN6+UKFCojY3LS0N/v7+CuvJjzP17t0bS5Yswfv377WvtBrv37/Hxo0bMWDAANSpUwclS5aEhYWFwjknkUgwb9480bbanHeySXKzIp/gu06dOtnaNi4uTuPttKXNddPU1FQUXJ7VmG/Pnj1F/fiNGzeqHb/J6fj1t0pZEsXKlSuLkrOoMmHCBFGyxNTUVK0nCChQoICobOixgt9++010/hw6dEiUwNDQgoKCMHPmTHTs2BEVKlRAsWLFYGZmprStkp1kQF07JT/W2rZtW53UVSqVwtfXVyhXrFhRdE5rwsLCQvR+6LMv1rx5c1SoUEEo//PPP2oTm27dulU09j1w4EDRd25fG2X/i6+p36aPcyunvqTzXRdyY4zQ3t5edJ5fv34d3333Ha5evarVsTSRW+POmvYt5fuVjo6OGifkld82r/QttXmegGO4GWxsbHDlyhWsXLkyx+OWea0PR0RERERElJvy5hQwX7no6GitvmRT5ubNmzqqDREREREREREREX3JLCws4OTkBCcnJ3Tr1g3//PMP+vTpg7S0NADArFmzUK9ePbRr1y7Hx4qIiBCVa9SoodX25ubmcHZ2FmavTkpKwuvXr0WBTgAwbtw47Ny5UwhsDAsLQ7du3WBlZYXWrVvDw8MD7u7uqFatmtYPu5YtWxZt2rTBsWPHAGTMGPfo0SOlM/W+ePECJ0+eFMoVKlSAh4eHVseTfzBMHQsLC1FZNvgwN+ijrvIPS3fr1k37isl59+6dwrKhQ4dixYoVQlBQdHQ0fvrpJ4waNQotWrSAp6cnGjZsiFq1ault9kN3d3e0bNkSZ86cAZARjDB37lzMmzcP7u7uaN68ORo3bgxXV1eFBAW6ltNztWbNmqKyPmf/09STJ09E5ewEzaqSW22bMtrMYip/PmlzziqTlpaGuLg4FC5cWOttX758iVatWiE6OlpYVqZMGZw+fRrW1tY5qpe2cpJYB8i4Lmh7TlarVg1Pnz4Vyk+fPlUblJ2QkJDj4AxlbZ8upaSkYOnSpZg7d67awBN1VAVH+vr6anxNa9SokdLAMH2Qf1hb2czHN2/eRGxsrEb7c3FxUfpAu/w5YciZMy9fvoyRI0fi7t272dpe1wGwgP7qFBUVJXogvFKlSkrfY0PJjWt1vnz58Pvvv2Py5MnCsr1792Lv3r2oXLkyWrVqhaZNm6JBgwbZSnIxaNAgzJw5E6mpqQAygmEHDBigdN3NmzcL62Vuq6sEZmfPntV43a8hiEIZS0tLWFpaokKFCujduzfOnDmD7t27C+3X3bt30a9fP1FQiazg4GC8fPlSo2NVrVoVJUuWVLtOTq/N6vTo0QPz5s0T+l3379/H7t27tZpJPlNsbKzGz94UKVJE5XM+UqkUmzZtwqRJk0R9I22oasv02QeW71uOHz8e48ePz9E+9dlfadSoEapWrYqgoCAAGe3ZihUrlPZlr127JqwHZATpOTk56aQeuugbyGJfNAP7orrvi0ZGRor+XrlyZa3qV65cOZiZmWmccDWv0WdfMKf9OCCjLycbHK2sL/fDDz9g7ty5CA4OBgB8/PgRY8eOxaRJk9C0aVM0a9YMjRo1Qt26dbP1uU1ISMDMmTOxYsUKJCUlab09oN39gY2Njcbrygd6ZndbfY6tajsmY2FhIQRbZ1UvCwsL/Pjjj1i1ahWAjERIZ86cQatWrRTWTUxMxM6dO4VyoUKFdDL+CmSMyYeFhWm0bub3E18SZW3CoEGDNLpmmpiYYODAgZg0aZKw7OLFi1odX9M+c27d7xQqVAi///67qD84efJkYaxbW7q6/t67dw8jR47EpUuXslUPde2U7OfbwsJC62ulKq9fvxb1nYKDg3N8/6vPvphEIsGQIUMwbtw4ABnXh927d2PIkCFK19+wYYPwu5GREQYOHKiTerx69UrUh1enVKlSqFKlik6OmxVl43jy7UdO7y1l+23m5uZafWcAaN/PA/R7bmXXl3i+60JufZ83Y8YM0djJ+fPncf78eTg4OKB169Zo2rQp3N3dYW9vr9XxM+X2uLOm/UNd9SuBvNO31PZ5Ao7hAjExMWjdujX27duX433rc9yTiIiIiIgor2NSmlyUOQiZnp5ukJlRiYiIiIiIiIiI6OvXo0cP3L17VzRz7dixY/H999/n+KEh+UCYYsWKab0P+W1iY2MVEjdUqFAB+/btQ48ePUSzjr1//x579uwRZvAqUqQIPD094eXlhU6dOmk8s5W3t7eQlAbIeIh24cKFCutt3rxZSO4DAIMHD9Zo/7JyEsyU2w816aOub9++zfY+VVH2ELC1tTWOHj2KDh06iGZG/PTpEw4fPozDhw8DyBinb9y4Mbp06QIvLy/RLMW6sGvXLrRv3140E2F6ejquXLmCK1euAMgIWqhbty7atWuH3r17i2a515WcnqvW1tYwMjJCenq60v0ZgvyD9zlNyCIrt9o2ZbSZJVdf55O2SWnevXuHVq1aiQJhbWxscObMGY1ec07JPwj8+fPnHO1PfrbL7Gwj/8B0QkJCtoP6VNFnIpPExES0a9cO58+fz9F+VL3m3r17Kzxcr8rTp0/10i4qI584oFSpUgrrjBkzRuPAsgsXLihNXieflEabc7latWoqr7EzZszAzJkzNd7X2rVrMWzYsBz1L3T9udZnnfR53dAF2WuPkZGR1gm9lF13lJk4cSIiIiKwbt060fKQkBCEhITgzz//BJARqN26dWv06tULbm5uGtWhZMmS6NChAw4cOAAgIwlEcHCwQmCYVCrFxo0bhbKxsbHKwIfsaNmypcbrfitBAy1btsSxY8fQqFEjoT937NgxnDlzRun/a+HChdi6datG+968eTP69++vsLxAgQLCvVtOr83qGBkZYfbs2ejcubOwbMaMGejevTtMTLR7BCowMFDjz0/Tpk3h4+OjsDw9PR39+vXDjh07tDq2PEO0Zbl1r6ZL3t7e+OWXXwD8Pwh/+PDhCuvJBsoCEM0KnlO66BvIYl+UfdFMuu6Lyn8utL3vk0gkKFSokJBE40vzJY4hyDM1NcWRI0fQtm1bPHz4UFienJyMM2fOCIkizM3N0aBBA3Ts2BE9e/ZE8eLFszz+mzdv0KxZM9y7d0/rusvSpq3JybhjbiWL0oa+x3y9vb2FpDRAxrVNWVKaffv2ic733r17K4ybZNe2bds0vuecPn06ZsyYoZPj5hZlSWmaNm2q8fby6wYEBGh1fPkAcvkA80y5eb8zcuRILFu2TBhfP3v2LHx8fLRO1A/o5vp79OhRdO3aNUcJ0lS1U3FxcaKg+2LFiuks6P5L7Gf3798fU6ZMEf5fGzZsUJqU5vLly6JrUuvWrRWSI2bXqVOnNB4r6NevH7Zs2aKT42ZFWeJR+X5bTu8tZdvxQoUKaf1Z1PZ7Ln2eW9n1pZ7vupBb3+f16tULYWFhmDZtmuh6ER4ejrVr12Lt2rUAAHt7e7Rq1Qo9evSAp6enRv8rQ4w7Z7cvlle/t9dnvb7mMVxl4y4fP35EWFgYjh8/jmXLlgnt+IcPH9ChQwdcvHgR9erV07gu8jTtwxEREREREX2NmJQmF40bNw6LFi3Cx48fDV0VIiIiIiIiIiIi+or98ssvoqQ0Dx8+xLVr1+Du7p6j/cqPbWbnIRv5bVTNgN2mTRsEBQVh9uzZ2LVrl9Jx1djYWBw4cAAHDhyAjY0Npk6dipEjR2b5gFzbtm1RpkwZPH/+HACwdetWzJ07F6ampsI66enpogevTE1NlQZfknr6mC0x88FKeXXr1sX9+/cxf/58bNy4UensoR8/fsSJEydw4sQJ/Pbbb/jtt98wefJk0XufE8WKFcOlS5ewYcMGLF26FKGhoQrrpKamws/PD35+fpg2bRp69eqFxYsXaxQ0pKmcnqsSiQT58+cXZhXP7kz1uiRfB13NcA7kbtsmT5vPXm6eT6rEx8fju+++E81aW7hwYZw6dQqVKlXSdfWUkn/IPqefz+wEa8m/3/KfIX28V/p84Hn48OEKQcA2Njbw8PBAzZo1UaZMGRQqVAj58+eHsbGxsM7p06exaNEivdVLnxISEvDo0SPRsnLlyunlWBUrVhSV79y5o5fjqHPhwgWFwAATExM0atQIbm5usLe3h62tLfLlywdzc3PRtto8LJ6X6qTP64YuyLYbumiHVLWFEokEa9euRZcuXTBnzhwhSZ68hw8f4uHDh1ixYgUaNmyI5cuXo27dulnWY+jQoUJAA5Ax0+6yZctE61y4cAFPnjwRyt9//73SwHvSrQYNGqBPnz7Ytm2bsGzLli16O6etrKyEpDRpaWlITEzUOGmotjp16oR69erB398fABAaGorNmzfrNPGIpmbPnq2QkKZQoULw8PCAi4sLypQpAysrK+TLl0/U7wsMDMTYsWOz3L8+27K80LfUVt++fTFhwgR8+vQJQEabI5+UJj4+XkhkC2Tco3Xq1Emv9coJ9kXZF82k676ofFCnmZmZ1vuQ74N9Sb6WMQQnJyfcunULy5cvx+rVqxEZGamwTlJSEnx8fODj44Pff/8d3t7emDt3LiwtLVUe/4cfflBISFOmTBl4enqiSpUqKF26NAoWLIj8+fPDyMhIWGfbtm3Yvn27Ni+Vsql69epwd3fH1atXAQCHDh1CTEwMbGxsROutX79eVDZEf+hLpWwsVv7+XR35cbCPHz9q1QeWb0u0TR6mD/nz58eUKVMwYsQIYdnkyZPh6+ub63V59OgRvLy8RAkqJBIJXF1d4e7uDicnJ5QoUQL58uVTCOTv06cPXr9+rXb/7GeLFStWDF27dsWuXbsAAP7+/ggMDETNmjVF632Lbc7NmzdFZWtra51PdiDbb9N3n03f51Z2fMnnuy7k5vd5U6ZMQcuWLTFr1iycOnVKNDFLpoiICKxfvx7r169HtWrVsHTpUrVjSXlx3JkUfUtjuAULFkSNGjVQo0YNDBw4EK1atUJgYCCAjIQy3bt3x71797KdTCYv9uGIiIiIiIhyC5PS5KJ27dqhXbt2hq4GERERERERERERfeVKlCgBJycnhIWFCct8fX1znJRG/kG9zAfctCG/jboAidKlS2Pt2rVYunSpMCvnpUuXEBgYqPCgXExMDH755RdcvHgRe/bsEQUqyTM2NsbgwYMxffp0ABkzHR46dAheXl7COmfOnBHN5NmhQwedzqr8rZAPsJs/fz5cXFxytM+qVauq/Ju1tTUWLlyIOXPmwMfHBxcuXMDFixcREBCAlJQU0brx8fGYOXMmzp49i9OnT+tsFmFTU1MMGzYMw4YNQ0BAAM6dOwcfHx9cvXpVCNTNlJ6ejh07dgifb10l9VB2rhYtWlTj7aVSqWimN3XnaW6Rr4MuJwDI7bYtuwoUKCD6DJ04cQImJjn7qq9EiRIar5uYmIh27doJAeCZdTp27Bhq166do3poo0yZMqJy5uzN2ZUZ1KwN+fdb/jMk355YW1uLgqPzkjt37mDr1q1C2dTUFAsXLsTw4cOzDIKQfUD5SxMQEKDQl9DX57hx48aickREBN68eaP1rLc5MWbMGFFgwPfff4+///4bpUuXVrudrmc9zs066fO6oQsFCxYUZjrXRTuU1XWndevWaN26NZ4+fYrTp08L/eqXL18qrOvr64uGDRtix44d+OGHH9Tut0WLFihXrpzQHmzfvh3z588XBZl8i0FreUWXLl1ESWkyg5r1oUyZMnj27JlQfvXqFZycnPR2vLlz56JVq1ZCefbs2ejbt2+uJlB4/fo1FixYIFo2ceJETJo0KcsgN2WBX8rosy2T76+MHj0a33//fY72qc/3HMgI9OnevTs2b94MIKMfExAQIEqitWvXLlEb2a9fv2wFduYW9kXZF82k676ofGBcdtoP+TGML8nXNIZQoEABTJo0CRMmTICvry/Onz8PHx8fXL9+XTR2AwDJycn466+/cPr0aVy6dEnpWObhw4fh4+MjOvaaNWvQs2dPUQIaZc6dO6fBqyNd8fb2FvpvycnJ2Lp1qyip3cOHD3H58mWh7OLikqvjM1+6ypUri8oSiUSr8bxChQopLIuNjdU4KY38vVjZsmU1PrY+DR48GIsWLUJ4eDiAjHuIY8eO5bifqK0JEyaI7v9dXV2xdetWODs7Z7ltVpMWALnbz65SpQr+/PPPHO1TXwk/ZXl7ewtJaYCMe/mVK1cK5ffv32Pfvn1CuUSJEmjfvr3e62VoN27cEJX10c4WLlwYb9++BaD/Ppu+z63s+JLPd13I7e/z3NzccOzYMbx69Uo0Rij7TEGm+/fvo3Xr1liyZAl+/fVXpfvLi+POpOhbHcO1tbXFkSNHUKtWLWEym6dPn2LGjBnZTnKbV/twREREREREuYFJaYiIiIiIiIiIiIi+Qra2tqIHyJ4/f57jfRYpUkRUznxIUhtv3rxRu09lLCws0LFjR3Ts2BFAxgOWV65cwbFjx7B7927ExsYK6+7fvx9LlizB+PHj1e5z8ODBmD17NlJTUwFkPGglm5Rmw4YNovW/hgevDEE+4N7R0REtWrTQ+3HNzMzQqlUrIVA1MTERfn5+OH78OHbt2iV6YMzX1xdjx47F6tWrdV6PunXrom7duvj999+Rnp6OwMBAnDx5Env27BFmZQOAqKgoeHl5ITAwMMsAIE0oO1e1eSju3bt3otlXNTlP9c3a2lpUjo6O1tm+DdW2aatYsWKiB8zr1KmTa8mykpOT0bVrV1y6dElYZm5ujv/++w8NGzbMlTpkKlOmDIyMjITP6IsXL3K0P/n3ThPynxH52XGtrKxgYmIiXGMSExNzpe3Ljn///Vf00PjMmTMxevRojbbNfIg3K5mBTHmJbBANkPHwf7169RTWkw3WzC53d3fRZxYAjh8/jr59++Z435p49OgRbt++LZSrVauGAwcOaBSgr+l7nBfrpM/rhi4UKVJESEqTnp6O2NhYra4d2b3uODo6wtvbG97e3gCAsLAwnDt3DgcOHMDp06eFz2lycjL69u0LNzc3tX0IiUSCIUOG4PfffweQ0T4eOHAAPXv2FMoHDx4U1i9dujTatGmj8evUhGwbRmLlypUTlaOiopSut2XLFmzZsiVHx3JwcICvr69QfvHihV4TlLRs2RIeHh5CO/38+XOsWbNG42sYAHh4eOTo83Po0CFR4NeQIUPwxx9/aLRtXmjL5O/VSpYsmWf7K7KGDh0qJKUBMu7lZZPS6PteXhd9A1nsi7IvCuinLyrffihLRKdOfHy83gJ2cxLQrGkip69xDMHIyAiNGzdG48aNMX36dKSkpCAgIAAnT57Erl27EBoaKqz78OFD9O/fH8ePH1fYzz///CMqr127Vui7ZUVf9wekXLdu3TB69Ghh7HvDhg2ipDT6vubNmDEDM2bM0Ok+85IqVaqIylKpFMnJyRonOfz8+bPCMm2Snsu3yw4ODkrXy+37HTMzM8yYMQP9+/cXlk2ZMgVt27bVqv3OyfX348ePOHbsmFAuXrw4Tp48qfF9r+z3RaoUKlRI1F968+YNpFKpTpJuyPezpVJpnu2LyWrSpAkqV66MkJAQAMDOnTuxaNEiISHOzp07Rfc//fv3z3HCdFn9+/cXfe7ygqioKNF9LgB4enoqrJfTe0tra2uhPxEXF4ePHz9mmehUlqb9vNw4t7T1pZ/vumCo7/NKliyJfv36oV+/fgAyPkfnz5/Hf//9h8OHDwsTfUilUowZMwYNGjRA/fr1RfvIi+POpNy3PIZbpkwZLFq0CIMGDRKWrVixAiNGjFDZ/1JH0z4cERERERHR1yjnTzYTERERERERERERUZ6T+bBYJl3MOGZvby8qyybV0ERSUhIePnwolM3NzVG8eHGt61GoUCG0bdsWq1atwvPnzzFgwADR35ctW5blg02lSpVCu3bthPKZM2eEB6Wjo6Nx6NAh4W8ODg5CchPSjqOjo6gsGyCTm/Lnzw9PT09hltfJkyeL/r5hwwa9zwBuZGSE2rVrY+LEibhz5w72798vmuH0/v37OHXqlE6OldNzVX59+f0ZQoUKFUTlgIAAne07r7RtWTHU+ZSWloZevXrhxIkTwjITExP8888/aNmyZa7UQZa5uTkqVaoklJ89e6Y0EEhTz58/1/r8v3fvnqgs/95IJBLR5yoxMVHrINDc4ufnJ/xuZGSEoUOHarxtUFCQPqqkd/Hx8di6datoWZs2bTQOPNNWoUKFFAJW5Gcd1SfZ9xjISMynSWAAoL/3ODfqVKJECVEAz8OHD/PUTMh55Vrt5OSEn376CSdOnEBgYKAoicjnz5+xatWqLPcxYMAA0fsn+/nevn276D5k4MCBMDY2zlZdKedMTU31tu8aNWqIyrJ9I32ZO3euqDxv3jwkJCTo/biZ5Nuy4cOHa7ytpm2ZPvvAeeVeTVuurq6oVauWUN69e7fwvt+5c0f0P2rSpImo35gXsS/Kviign76ofLIF2WBRTQQGBuotaDBfvnyismyAe1ZiYmI0Wk+ffcGc9uOUbZOdvpypqSkaNGiAmTNn4tGjR1i1apUo0fGJEyeExAKyZM+7okWLolu3bhof80s9775U+fLlEwLFgYzPcWay4OTkZFFbYmFhgV69euV6Hb9kderUUVj2+vVrjbeXT3ZlbGyMwoULa7StVCrFo0ePhLKdnZ1CMi1D6tOnD5ydnYXynTt3FBKq6dOtW7eQnJwslHv27KlxkoXQ0FCNvwsrX7688HtCQoLSNjM7SpQoIRr3j4iIUPi+Lq/KTGALAO/fv8fevXuFsuy9vkQi+SYmcvj7778V3rvOnTvr/Diy/TapVKp13+LOnTsarZdb55Y2vvTzXRfyyhhhqVKl0KdPH+zbtw+PHz8WJc2USqVYtmyZwjZ5cdyZVPuWx3D79+8vGrtMTk7G7Nmzs7WvBw8eiMryY6JERERERERfMyalISIiIiIiIiIiIvoKPXv2TFS2tbXN8T7lZ0Dz8fHRKkDl8uXLogc469Spo/HDaapYWFhg3bp1olmooqKiNAqokw12kkql2LhxIwBg69atonoOGjQoz8yYJ0s20MRQs4tlRT4I//z58waqyf+Zmppizpw5aNSokbAsJSUF169fz9V6dOnSBWPGjBEtu3Llik72LX+uavt/l19ffn+GUKdOHdFsw7dv30ZUVJRO9p0X2zZlDHE+SaVSDBw4EPv37xeWGRkZYfPmzejUqZPej6+Ki4uL8HtaWhqCg4OzvS+pVCoEdWkiIiJCNNtziRIllM5cmpvvV06uB7IBVzY2Nho/9J+eno6LFy9qday8Yvr06YiPjxct0yYAOjuUtfeXL1/W6zEzyQfVaROcr6/PbW7UycTEBA0aNBDK6enpOHnypMbH0YTsuQdod/7lxWt1tWrVsG7dOtEyTfomNjY26NKli1D28fER+uIbNmwQlhsZGWHgwIE5ridpLiIiQlTWR9K8TLLXZkAxaYY+uLu7o23btkI5Ojoaf/75p96Pmyk32jJ99oEN1VcBcn7/Knvdjo+Pxz///ANAMenblxAoy74o+6KAfvqi1apVE7Ufly9fxocPHzTe/vDhwzqvU6ZChQqJytokgfD399doPX32BR0cHETX1IiICISFhWm8fXx8vCiBlomJCerWrZujOkkkEgwfPlwhKYmyvpzs/7t8+fIaB5vGxcXh5s2bOaonaU82QQTw/2vdoUOHREmaevToAUtLy1yt25fO0dER1apVEy3T5jMuv27FihU1/g4hLCxMlEwxp22ArhkbG2PWrFmiZdOmTUNaWlquHD+3xjEaN24sKh8/flzjbdUxNTVFw4YNhfKnT5/0+t2D7Ocup/3sfv36iRLqZLY5/v7+osQXzZo1EyW1/RpFREQo3F82btxYIfGfLri6uorKR48e1Xjb2NhYjb9X+lbHCAH9ne+ZvrYxQnt7e+zatUu0LKt+JZA3PlOk2rc8hmtkZKTQt9m+fbvC2KkmZMc8raysREmviIiIiIiIvnZMSkNERERERERERET0lbl9+7bCzMEVK1bM8X4rVaqkEHRx4cIFjbfftGmTqNy0adMc1wnICN5wc3MTLXvz5k2W27Vq1Uo0o/jmzZuRmpoqJKcBMh7AHjBggE7qqWsWFhbC758+fTJgTVRzdXUVBZWdP38+R8kjdEn2wXBAs8/Ml1IH+XPr4MGDGgefpaSkYPv27Wr3ZwimpqZo3ry5UE5NTcXq1at1su+82rbJ++6770TldevW6X2W3ZEjR2Lbtm2iZatXr0afPn30etysyD9EntPAOPnXqM26TZo0Ubqe/Pu1cuVK7SumoZxcD2QfkJedlTYrhw8fxosXL7Q6Vl5w4MABhVldPT090axZM70et02bNgrBbgMGDBAFoemLfBCEpu9zUlKSQvumK7lVJ/nzcMWKFRpvqwnZcw/Q7vyTv1bs2LEDqampGm0bGxuLgwcPipapaou0ld2+iWywrFQqxYYNG3Dt2jXRrMetWrXK9mzNlD1HjhwRlfU5c6+bmxtMTU2Fcm4Frc+dO1cUhLlo0SK8f/8+V46d3bbszp07uHbtmkbr6rMPbGdnJ7o2PXnyBCdOnNDJvuXlpL1UplevXihYsKBQXr9+PRITE0VBc0WKFIGXl1eOjpNb2BfNwL6obpmYmKBNmzZCOSkpSTTepU58fLxWn0ttyfcHbt++rfG2e/bs0XhdffYF5ftyW7Zs0Xjb3bt3IzExUSjXrVtXlEAoJzTpy2X3vNu0aRM+f/6c/cpRtjg7O4s+b/v27UNsbKwocBj4MhKx5UWdO3cWlWWTMmdl7969orKHh4fG28r3lXV1P6dLXl5eqF27tlB+8OCBwpixvmS3ny2VSrFmzRqNjyN/nVizZo3Oxlrl9/3XX3/pZL/K6PJ7IisrK3Tr1k0oX7lyBQ8ePPjm2pzPnz+jW7duCveWs2fP1svx2rdvLypv3bpV4zHD9evXIykpSaN1c+vc0sbXcL4Duh0jzCvf55UvX170vVlW/Uogb4w7k3rf8hhuhw4dULNmTaGckpKCP/74Q6t9vH79WjQWIf9dJRERERER0deOSWmIiIiIiIiIiIiIviJSqRRTp04VLTMyMhIFomSXRCLBoEGDRMvGjRun0QyZN27cEGYvz9zX4MGDc1ynTPIPw2kyu7ZEIsGQIUOEcmRkJCZMmICHDx8Ky9q2bQs7Ozud1VOXrK2thd/fvn2rMNN2XmBqaorRo0cLZalUCm9vb70n0tBEdj4zX0odKleuDHd3d6H88eNHTJ8+XaNt//zzTzx79kwoOzg4oGXLljqpV06NGjVKVF64cKFoRrrsysttmywXFxd4enoK5efPn2PKlCl6ORYATJw4USHoefHixQqzdBtC69atReVLly7laH/79u2Dv79/lutFR0djyZIlomWqZozs1KmTaIbE69ev6+3h/ZxcD0qUKCH8Hhsbq1HisI8fP2LMmDHaVTIPWLx4Mbp37y5aZmFhobPg/qysXbsWJiYmQvnJkyfo1KmT3q/fsu8xoHxWW2WmTp2qMNutruRWnQYOHAhLS0uhfPnyZWGWb12QPfcA4OnTpxpv26pVK5QtW1a0raYJA6ZOnSoKbmncuDGcnZ01PrY62e2beHh4iOqwZcsWhXPraw9ay2tCQkKwefNm0bKOHTvq7XgFCxYUBcIHBASIAu71pVatWqLEI+/fv8eiRYv0flwge21ZWloahg8frtVx9NUHBjL6vLJGjx6tcfCbNnLSXipjaWmJ3r17C+Xr169j6tSpoqDRPn36IF++fDk6Tm5hXzQD+6K6J3/tnTVrFsLCwrLcbty4cXrrBwIZiWFlk7CcOXNGo4Ri/v7+Con51NFnX1D+vn/p0qUaJUqKi4vDjBkzRMt02UfSpC8ne94FBQVp9L+PjIzEzJkzc1w/yp6hQ4cKv3/+/Blz5szBmTNnhGXVq1dXSNhOmhkwYIAoseLevXvx4MGDLLe7e/cu/vvvP9Ey2UQeWbl48aKoLD/OlRdIJBLMmTNHtGzmzJm5Mq6f3TGDNWvW4M6dOxofp2PHjnBwcBDKYWFhCm10dg0ePBhWVlZCed++fTh27JhO9i1Pti8WHh6e4/3JtjkAsGzZMuzevVsoFytWTCGh09ckLCwM7u7uuHHjhmj5wIED9ZaMvnr16qhfv75QfvXqFSZMmJDldo8fP1Y4T9XJrXNLG1/D+Q7k7J43r36fl5ycjLi4OKGcVb8SyBvjzqTetzyGK5FIFL5T3bJlC54/f67xPuS/i8yLfTgiIiIiIiJ9YlIaIiIiIiIiIiIioq9EQkICBg8erPBwa9++fVGsWDGdHGPYsGHInz+/UL516xaGDh2qMBuarGfPnsHLy0u0TseOHVGuXDmFdUNCQjBs2DCEhoZqXCd/f3/4+PgIZSsrKzg5OWm07cCBA0UPnssHeOXlB6+qVq0q/C6VSrWayTU3jRo1SjSb3pUrV+Dl5aVVsGNCQgJWrFihdFbvuLg49O7dW6uZtcPDw7Fv3z6hbGRkhFq1amm8vTIjRozAkSNH1J4LspKSkhRm6XZxcclRHWTJB+mtWLEiy5nOT506hcmTJ4uWjR49GkZGeePrpObNm4tmnUtKSsJ3332ncVDuu3fvVD6orO+2TVdmz54tej8WLlyIWbNmafy5A4AXL15g3LhxagNfFyxYgPnz54uWTZ8+Pc8Ef9rb24vawAsXLuRof1KpFN26dcPLly9VrpOYmIguXbqI2q4qVaqgVatWStc3NjZWmD131KhRWgdBPnr0CEOGDEFkZKTKdXJyPZB94B0Axo8fj/T0dJXrf/r0CV26dNEomDUvSE1NxZ49e+Dm5oZx48YhNTVV+JuxsTG2bt2qs2QeWXF3d8e8efNEy86ePYvGjRvj+vXrWu0rNTVV4+AG+ff477//zrKftXbtWixevFirOmkjt+pkZWWlkMxh5MiRomRi6qSkpODy5csq/y577gEQ9S2yYmxsrFC3CRMm4Ny5QDcoFgABAABJREFUc2q327Rpk0KggKq2+c8//8SqVau0mp1ZPpmHNn0T2aRlr1+/xo4dO4Ry8eLFFWb+/lb1798fEolE+Onfv7/KdePj4zF8+HCNgtxl3b9/H61btxbNUG1vb48ffvghu9XWyPfffy/8npycDF9fX70eL9OsWbNgbGwslK9evZorx5Vvy6ZOnao2EU9aWhoGDhyIa9euaXUcffaBe/fuLWrLHj16hDZt2qjtE8lLSUnB1q1bsWDBApXryLeXhw8fznFAs3yw7Jd0Ly+PfdH/Y19Ut1q1aiVqPz58+IDmzZurbBOSk5MxZswYrF27FkBG0J4+GBsbi4L3EhMTMX78eLXbPHnyBN26ddMoeWwmffYFW7RogRo1agjlhIQEdOzYUe1YV3JyMn744Qe8evVKWFayZEn06tVL6fq9e/dWSFyhTmxsLDZs2CBapqwvJ3veJScnY+LEiWr3GxMTg3bt2mmUvIb0o0uXLrCxsRHKS5cuFY0DfUnXPH0KDw8X9bMlEkmWCTocHR1FSeuTk5PRo0cPvHv3TuU2MTEx6Nmzp6hNr1+/Pjw8PDSuq+x3GQ4ODgp9pbyibdu2osST4eHhWvUTs8vFxQVmZmZC+cCBA1n28Y8ePYrffvtNq+MYGxsrBKfPmzdPoV+pilQqVTkmWbhwYfz+++9COT09HT179sThw4e1quPNmzcVktrJk/38vHnzRvT5yo769eujZs2aQnndunWipIN9+/YVvT9fi6dPn+LXX39FzZo1Fb5rcnd3x19//aXX48v33VeuXInx48eL7utl3bp1Cy1atEB8fLzGfbbcOre08TWc70DOxggB/X+ft2vXLsybNw+xsbEa12nVqlWiMY6s+pVA3hh3pqx9y2O4Xbt2FZ2vycnJCt+FqiN/jW3btq2uqkZERERERPRFyBtPERMRERERERERERGRSjdv3sTZs2eV/hw+fBjr16/HTz/9hDJlymDTpk2ibe3s7BQCoHOidOnSWLhwoWjZhg0b0KxZM/j5+YmWJyQkYOPGjXBxcRHNMmVtbY1Vq1Yp3X9SUhL+/vtvVKpUCU2bNsVff/2F+/fvKw06iYmJwdKlS9G8eXPR3/v166fxQ7G2trYqZ3W0s7PL0w8TyQefDRs2DD///DN2796NU6dOiT4nmsw2ri+FCxfG3r17Rcl/Dh8+jKpVq2LJkiWiWfxkPX/+HPv27UOfPn1QqlQpjBo1SulsZenp6di1axfq1KmDevXqYcGCBbh165bSh3Xj4uKwceNGNGjQQBQo1L59e4UZ/bTl6+uLDh06wNHREWPHjoWPj49oFsFMKSkpOHnyJBo2bChKClKiRAm0a9cuR3WQ1aVLF3Tt2lUoS6VS9O/fH0OHDlUI4Hv16hUmTZqEdu3aif5v7u7uGDlypM7qpAs7duwQzTr58uVLuLq6Yty4cUpnMk5ISMCZM2cwZMgQ2NvbK8xinEnfbZuuNGzYEHPnzhUtmz59OurVq4d//vlH6UPFaWlpCAkJwbp169C6dWs4Ojpi8eLFSEhIUHqMrVu3KszE2qhRIzRq1EjltUjVjz7bHtnAvcjISAQEBGRrP3Z2djAxMUF4eDjq1KmDHTt24PPnz8Lf09PTcerUKdStW1cUXC+RSLBu3Tq1D/336NFD9IBvSkoKhgwZgubNm+Po0aNK34OUlBQEBgZi+fLlaNy4MZydnbF+/Xq1gds5uR706dNH9KD6sWPH0L59e4X1Pn/+jH379qFmzZrCjOyVK1dWWSd9Cw4OVvi8nTp1Cnv27MHatWsxdepUtGrVCkWLFkWPHj0UZjcuWLAg9u7dK2onc8PYsWMxcOBA0bLAwEDUr18f7du3x44dO0RBqrJSUlJw584dzJkzB5UqVcoyMCFT+fLl0aBBA6EcHx+PJk2aYO/evaIgusy6dO/eXUjKpa/3ODfrNG3aNNFs08nJyejZsye6du0KX19fhT5mamoqbt68iSlTpqBcuXKYOnWqyn3Lzzw8e/ZsDBgwANu3b8fJkydFn8+bN28qbD9q1Ci4ubkJ5aSkJLRp0waTJ09W+ByEhYXB29sbgwcPFgWgdu/eHR07dlRav6dPn2LkyJEoVaoU+vXrh4MHD6r8fN25cwc9evQQJcwzMjJS+Lyq069fP+TLl0/p3/r37y/qC+ZFvr6+Sq9l8u/d58+fc+26l5aWhjVr1sDJyQnt27fHtm3b8OTJE6XJ6JKTk3H16lUMGzYMderUEfVRJBIJ/vrrL1ECPn3o0aOH6Lqoqt+la87Ozvjxxx9z5ViyOnfuDEtLS6F869YteHp6KiQaS01NxalTp+Dm5ia03dq2ZfrqAxsbG2P//v0oXLiwsOzatWuoVq0apk+fjkePHind7vXr1zh69Ci8vb1hZ2eH/v37IyQkRGX9bW1tRYGtjx8/RoMGDfDnn3/i6NGjCueSbF9MlVq1asHV1VXp39zc3FC9evUs95EXsC/Kvqg+SSQSbNiwAQUKFBCWhYeHo169evDy8sL69etx7Ngx/Pvvv5g6dSqcnZ2xdOlSAMB3332HsmXL6q1u8kk01q9fj4EDByokO3j37h2WL18OV1dXhIeHa50EVl99QYlEgs2bN4v6N7du3UKtWrWwf/9+0dhKeno6zpw5g7p16+L06dOi/WzatEll/+nYsWPw8PBAlSpVMH36dFy7dk1pssHExETs3bsXbm5uiIiIEJbXrFkT9erVU1i/b9++ovLff/+Nfv36ibYFMvroGzduRI0aNYRERoY8775lZmZmGDBggNK/5cuXD3369MnlGmnn1atXKvvP8uNYytpXbfoH2TFt2jRR0p/AwEC4ubnh8OHDovvTlJQU/Pfff3BzcxNdp/Lly4e///5b4+OFhoaKtu/Zs2cOX4F+yY9D5gYLCwvRNTItLQ1t2rTBunXrFD4Hjx8/xvDhw9GxY0ckJSXB1tYWRYsW1fhYgwYNgpeXl1CWSqUYO3YsmjVrhtOnTyMpKUm0fnp6OoKCgvDHH3+gcuXKKs9NICPZnuz3S/Hx8ejUqRO6dOmC8+fPK+wbyOjz3LhxA3/88QdcXFxQt25d/Pvvv2pfg3xfrHPnzpgwYQL27t2L06dPi84jTRP7yfYh5Q0ePFijfeQFqu7fDx06hK1bt2L58uUYOHAgqlSpgnLlymH58uX4+PGjaB9t2rTBiRMnRP0pfWjRogUGDRokWrZo0SJUrlwZ06ZNw969e3Hs2DGsW7cOXbt2haurK549ewYjIyONk5Pl5rmlqa/lfPfw8BD1C7du3YouXbpg06ZNOHHihOjzpyyBr76/z4uOjsakSZNQunRpeHl5Yffu3Qp9v0yPHj3CyJEjFRLlKPuc5cVxZ8ralz6GmxMSiUQhQdXGjRs1SronlUpFyeUaNGgAR0dHndeRiIiIiIgoLzMxdAWIiIiIiIiIiIiISL2xY8dma7vSpUvjzJkzOU62IW/EiBG4ceMGtm/fLizz8fFBgwYNYGNjgzJlyuDz5894+vSpwkzx+fPnx65du1CqVCm1x0hPT8elS5dw6dIlYTs7OzsUKVIEQMYDdM+ePVMIyKxQoYLCjIJZGTp0qNIHewcMGABjY2Ot9pWb2rVrh0qVKuHhw4cAMh6wXblyJVauXKmwbr9+/bBly5ZcruH/NW7cGNu2bcOAAQOEB0kjIyMxduxYjB07FiVLloStrS3Mzc3x4cMHREdHazVjX6aAgAAEBARgwoQJMDMzQ+nSpVGkSBEYGxvj7du3CA8PVwg4srGx0ekslxEREViyZAmWLFkCiUQCOzs7FC1aFPnz50dcXBzCwsIUHqY1NjbGhg0bdB4ovG7dOoSGhiIwMBBAxgNza9euxdq1a+Hg4AAbGxu8e/cOT58+VZiN3tHREbt27cpz50DZsmVx4MABdOrUSZil+/Pnz1i8eDEWL14MW1tblCxZEmZmZsJ7Lv/aVMmNtk0XJkyYgOjoaCxbtkxYdvPmTfTs2RNGRkYoW7as8DD2+/fv8erVK6WBa6oom/HzypUrCskXNKHPtqd3796YMmWKcB04cOAA6tatq/V+ypcvj6FDh2Lq1Kl4/fo1fvzxRwwdOhSOjo4wNTVFeHi40vZo1qxZotmiVfnrr78QGxsrus6cP38e58+fh4mJCezt7WFtbY3U1FS8f/8ekZGRKmfAVSUn1wNnZ2cMHToUq1evFpYdP34cx48fR5kyZVCyZEl8/PgR4eHhos9RkyZN8OOPPxpsRvZFixZh0aJF2dq2VatWWLlyJSpUqKDjWmlmw4YNKFOmDGbPni1qn44ePYqjR48CyEhyVaxYMRQpUgTJycn48OEDnj17pvAwf6bq1aurfS8WL14MDw8PIaD81atX6NatGwoWLIgKFSrAyMgIL168wOvXr4VtLCwssHPnTtSpU0cXL9tgdTI1NcW+ffvQunVrBAUFCcsPHDiAAwcOwNLSEmXKlIGlpSU+fPiA8PBw0XXayclJ5b5r166NZs2a4fz58wAy+q9btmxR2u41bdpUYSZVY2Nj7Nq1C56enkKSvpSUFPzxxx+YP38+HB0dYW1tjZiYGISHhyvss06dOlizZk2W/4MPHz5g27ZtQjIMGxsb2NrawtLSEp8/f0Z4eLhwTZU1fvx4pbMgq1KkSBF069ZNIWGSRCL5IoLWevfurTIgR9br169VXhP1dd1LSUkRtRGWlpYoUaIErKysIJVKhc+usqQREokE69evz5VZjkuXLg1PT0/hnPjvv//w119/aTxre05Mnz4du3bt0voamhNFixbF1KlTMX78eGHZ9evXUb9+fRQvXhxly5bF58+fERERIUpW6ezsjPnz56tMKKWMPvvAlSpVwsGDB9G1a1ehzxMbG4tZs2Zh1qxZKFasGEqUKAELCwvExcXhzZs3iImJ0bjumcaMGSNKhHDz5k2lCbuAjKRaDg4OWe5z6NChCsk+AOXBcnkV+6Lsi+pbxYoV8d9//6FDhw5CHyc1NRX79+/H/v37lW5Trlw5bNu2TZTQxMREt4+ZtmnTBu3atROubQCwefNmbNmyBRUqVICVlRXevXuHsLAwoT2zsLDAv//+q1X/RJ99wTp16mDVqlUYOnSoUMfw8HB4eXnB0tISDg4OMDY2RkREhNLzeObMmfjuu++yfA0hISFCm2xsbIzSpUvD2toaZmZmeP/+PcLCwhT6AAUKFFBIXJ6pVatWaNu2LY4fPy4sy+wrOjk5wcbGBu/fv8fTp09F7UGvXr1QoUIFzJw5M8s6k+4NGTIEixYtUhgL9/LyEsbL86pTp06pDeSXpa591bR/oC1bW1v8+++/aNOmjXD+h4aGomPHjihcuLBwzKdPnyokIDc2Nsa6detEyfeycvDgQVE5rycVatq0KVq2bCkkhcsts2fPxrFjx4T/eVxcHLy9vTFq1ChUrFgR5ubmePXqFV68eCFsY2xsjC1btmDYsGF4+/atxsfauHEjYmJicPHiRWHZhQsXcOHCBRQoUABlypSBlZUV4uPj8ezZM1HSEnt7e5X7NTIywq5du9CpUydhPEAqleLgwYM4ePAgzM3NYW9vjyJFiuDz5894//49Xrx4oXSCBnX69u2LuXPn4s2bNwAyxoMXLFigdN3p06djxowZWe6zT58+GD9+vEKClkaNGn1RiSTU3b9nxcrKCrNnz8bw4cNFSRT1afXq1Xj58iVOnDghLAsLC1P7neeiRYtgbW2NdevWCcvU9dty89zS1Ndwvtva2uLHH38U9b8yz3V59vb2Ssf5cuP7vE+fPonuA6ysrITxpeTkZDx//lzp/X6PHj3QqVMnpfvMi+POpN6XPoabU926dcOMGTOEMYykpCQsWLAAf/75p9rtAgICRO1QXu/DERERERER6UPujJIRERERERERERERUa4xNzeHt7c37t+/D2dnZ53vXyKRYOvWrRg3bpzCw5gxMTG4desWgoODFZI2lChRAqdPn0br1q21PmZiYiJCQ0Ph7+8Pf39/REREKDyE36BBA1y+fFk0W70mPD09UbFiRdEyiUSiMCthXmNiYoL9+/drPUu0ofTo0QNXrlxR+F8DGQ8pBgYG4saNG3j48KHSYB1jY2OtEn4kJycjLCwMN2/exI0bN/DkyROFB7qdnZ1x5coVlClTRvsXpAGpVIoXL14gMDAQfn5+CA4OVkhIU6RIEezfvx/ff/+9zo9vbW2NixcvKn3wOjw8HP7+/njy5InCA6z16tWDr6+v2odsDalp06bw9fVV+gB8dHQ0AgMD4e/vLwpe04Qh2rbsWrp0KbZu3QorKyvR8vT0dISHhwtBvk+ePFGakMbS0lJh2y+Nvb29aLbhXbt2afV+y5oyZQp++eUXoZyQkID79+/j9u3bCu2RkZERpk+frjCboiqmpqbYs2cPFixYoJB4KjU1FU+ePIG/vz9u376tEPSXqVixYmqTVuX0erBs2TK0a9dOYfnz589x48YNBAcHiz5Hnp6eOHTokM4DU/XJwsICPXv2hK+vL06dOmWwhDRARlszY8YMXLlyRRToK+vdu3d49OgRrl+/jtu3byMsLExpQppy5cph9erVuH37ttrAdHd3d6xfv15hltOPHz/i9u3buHnzpigwoEiRIjh69Chq166dzVeZtdysk52dHXx9fdGhQweFv8XHxyM4OBjXr1/HgwcPFK7TWdm+fXuOAiicnJzg6+ursI/09HShfVAWqNKmTRv4+PhkKwA1JiYGQUFB8PPzw507dxQS0hgbG2PatGmYN2+e1vtWNpu6h4cHypcvr/W+vlbK+oLaio+Px+PHj+Hv74+AgAA8fvxYaUKaChUq4Ny5c7l6XzNs2DDh98jISCFBjb45ODgYJDnFuHHjlH7uX79+DX9/f9y7d08UvFy9enWcOXMGhQoV0vpY+uoDAxnXdn9/f6XXpTdv3uD+/fu4fv06QkJClAaoSSSSLO+pfvzxR4wfP16nSYq6d++u0Ke1tLREjx49dHaM3MC+KPui+tayZUtcuHABVapUyXJdDw8PXLlyBTY2NqIg3MKFC+u8Xlu3blVod6RSKR49eoQbN24gNDRUaM+sra1x/PjxbPW79NkX/Omnn7B7926Fscj4+Hjcu3cPd+7cUTiPzc3NsXLlSkybNk3r15KWloaIiAjcvn0b169fx8OHDxX6AHZ2djh79qza/9XOnTvh6uqqsDwsLEzYr2x70KNHD2zevFnr+pLulCtXDi1atFBY/iUlYtM3+fPXxMQEBQsW1GhbDw8PnDp1Cra2tqLlHz58QGBgIAIDAxUS0hQqVAj//fcffvzxR63qKZsMu2nTphq1zYY2d+7cXD9muXLlsHfvXoX38PPnz7h79y78/f1FgeH58uXDzp070aZNG62PVahQIZw+fRqDBw9W6Kt++vQJDx8+xPXr1xEcHKyQpCUrhQsXxpkzZ/Dbb78p9F2SkpKEsZ/AwEBEREQoTUiTVT/b2toa+/fvV/j85oSlpSV69uypsPxbaHOqV6+ORYsWISIiAiNHjsy1hDQAYGZmhoMHD2Ls2LFZ9nUtLCywYcMG/PbbbwqfS3X9ttw8tzT1tZzvy5cvz3YSJMAw3+e9f/8eDx48gJ+fH27duqX0fv+nn34SXbvk5cVxZ8ratzyGa2RkhMmTJ4uWrV+/HlFRUWq3kz0PLC0tmZSGiIiIiIi+SUxKQ0RERERERERERPQFMzMzg42NDapWrYrevXtj9erViIyMxN9//62XgJFMEokECxcuxK1bt9ChQweYm5urXLdUqVKYOnUqHj9+jEaNGqndb40aNeDr64vff/8dLi4uGgUZubu7Y9u2bfD19UXx4sW1fi0AFGZrbdmypV5mXtW1qlWr4u7du9i8eTO8vLxQsWJFFC5cOMvZ8AzFxcUFwcHB2LZtG+rXr59lPc3NzdGsWTMsXrwYz58/x5AhQxTWsbKywu3btzFjxgw0bNhQ7WcxU40aNbBixQrcvXtXaZKc7Dhy5AhWrlyJ77//XqNkH6VKlcK4cePw6NEjdOzYUSd1UKZw4cI4ffo0Dhw4AFdXV7VBoNWqVcPmzZvh5+eHkiVL6q1OulClShXcu3cPGzduRO3atdW+LmNjY7i7u2PVqlUYM2aM2v3qq23Th759+yI8PByzZ8/W6HNcpEgReHl5Ydu2bYiKikKtWrX0X0k9Gz16tPB7REREjmZt/vPPP7F//361s/26u7vj8uXLGs0oLG/8+PF4+vQpxo4di7Jly2a5fokSJdCnTx8cOHAAL1++zPL6lpPrgZmZGQ4dOoRly5ahRIkSKtdzcHDAypUrcfbs2TyZ1MjU1BQFCxZE2bJlUa9ePfTo0QNz5szBuXPnEBMTg127dsHd3d3Q1RQ0aNAAN27cwPnz59G3b19YW1trtF358uUxcOBA+Pj44PHjxxg2bJhG73O/fv1w6dIlNGnSROU6+fLlw8CBAxEUFAQPDw9NX0q25WadChcujEOHDuHChQto1aoVzMzM1K5fqVIlTJkyRWHGWHmlSpWCn58f9u7di169eqFq1aqwsrLSKlC+dOnS8Pf3x4YNG1C1alWV60kkEri5ueHQoUM4fvx4lokYZ82ahX/++Qd9+vTRKAFfwYIF0adPH9y+fRszZ87UuP6y3N3dUalSJdGybyFoTRvXrl0TfjcxMcHPP/+sct3ChQvj4sWL+P333+Hq6prl5xbIaAubN2+OHTt24N69e/D09NRJvTXVuXNnUSDUhg0bcu3YkydPRoECBXLteJn+/vtvbNu2DU5OTirXsbW1xezZs+Hv74/SpUtn+1j66gMDGYGAN27cwOHDh9GsWbMsP2/GxsZo0KABZs2ahdDQUMyePTvLYyxYsAB37tzB2LFj0ahRI9ja2iJfvnxZbqdKgQIF0L17d9Gynj17wsLCItv7NBT2RdkX1bf69evj9u3b2LlzJzp27AhHR0fky5cP+fLlQ7ly5dC7d28cP34cFy5cQIkSJZCeno4PHz4I2+tjjNHa2hoXLlzAlClTVCaOMDExQZ8+fXDv3j21fcas6KsvCADdunVDaGgoRowYobZPb2lpib59++LBgwcYMWJElvu9ceMGFi5ciObNm2vUrpUvXx6zZ8/Gw4cP0aBBA7XrWllZ4dKlS5g6dara97Zq1arYtWsXdu/erVE/hPRr4MCBonKlSpVydF58ba5evSoq//jjjyhWrJjG2zdp0gQhISEYO3as2uuMtbU1Ro8ejdDQUKVJ1dS5ceMG7t27J5RHjRql1faGUq9ePXTq1CnXj9uqVSv4+/ujffv2KtcxMTGBl5cXAgMDFfqF2jAzM8P69etx69YtdOnSJct2197eHqNGjcLJkyez3LeJiQmWLFmChw8fYsiQIRolj3FwcMCQIUNw+vRppYlq5TVp0gQPHjzAypUr0b59ezg6OsLS0jJHCVXk2xwrKyv88MMP2d5fXmFkZIR8+fKhaNGiqFy5Mlq2bImff/4ZmzdvxpMnT3D37l2MHTs2W4lEdcHc3ByLFi3CvXv3MHHiRNSuXRvFihWDsbExrK2t0bBhQ8yePRtPnjwREtC+e/dOtI+s+m25eW5p6ms43y0tLXHq1CmcPHkSAwcORK1atWBtba2QrEUdfX2f5+3tjUOHDmHw4MEaJR0xNzdH586dcfnyZaxbty7Lcc68OO5M6n3rY7i9evUSnQuJiYlYtGiRyvWTkpKwc+dOoTxgwACDXSeIiIiIiIgMSSKVn0qWvnghzqofUMjrKj8I0Xob6W3FIASib4mk9rpsb/t+Qt4OLCHSN6v5r4TfvStmHbBG9DVb+ygp29tKX67WYU2IvjySUsOF36Xn2xqwJkSGJ2l2XKb00mD1IMobShm6Arnu06dPuHLlCp49e4Y3b97A3Nwctra2qFq1ao4SHyQkJCAoKAhPnjzB69evkZCQABMTExQuXBhOTk6oXbs2bGxsclz/vn37ima42rt3L7y8vHK8X1Lvw4cP8PPzw8uXL/HmzRukpKTA0tIStra2cHZ2RqVKlbQOUkxKSkJwcDBCQ0Px6tUrfPz4ERKJBIUKFYKDgwNq1aoFOzs7Pb2iDJkzez9+/BjPnj1DXFwc0tLSYGlpiRIlSqBGjRqoWLFirs6ymen169e4evUqoqKiEBsbi0KFCqF48eJwc3PTKDgxr3r9+jWuXbuG169f4+3btzAxMUGRIkVQoUIF1KpVK9tBi/pq2/QhMjIS/v7+iI6Oxtu3b2FkZIRChQrBzs4OlStXRrly5QzymdO3unXr4ubNmwCADh064NChQzne57179xAQEICoqCiYmZmhZMmSaNCgARwdHXO870yhoaG4c+cOYmJiEBsbK1zbypYti8qVKxssMVpqair8/f1x9+5dvH37FsbGxihRogRq1aqFmjVrGqRO34rMa0dISAhevHiB+Ph4pKenw8rKClZWVihRogTq1KmDIkWK5PhY4eHh8PX1xatXr5CUlAQrKytUqlQJ7u7uBknoYIg6JSQkwNfXFy9evMCbN2+QlpaGQoUKwdHRETVq1MhR0oicevbsGa5fv47Xr18jLi4ORYoUQcmSJeHu7p6jmc8jIyPx4MEDPH36FLGxsUhKSkKBAgVQtGhRVK1aFdWrV9couZ868fHxKFmyJBISEgAARYsWRWRkZI73+7UIDQ1FhQoVhPKgQYO0StqS2c998uQJXr16hfj4eAAZQUtWVlZwdnZG9erVDR40vnLlSiHZjrm5OcLDw9UmmvhaSKVS3LlzBzdv3sSbN28glUpha2uLatWqoW7dunpJHKqvPjCQ0Q/28/PD8+fP8fbtWyQmJqJgwYIoVqwYKlWqhMqVK+eJ5C9NmjTB5cuXhbK/vz/q1q1rwBrlHPui7IvmBffv30f16tWFcv/+/bF582a9HS85ORmXL1/G48eP8fbtWyFZTtOmTXXS/5Wnr75gWloabty4gcePHyM6Ohrp6emwsbGBk5MT3N3dtQpKlt9vSEgIHj9+jMjISKEPYGlpCTs7O9SqVSvbbcTnz59x7do1hISEIDY2FmZmZihVqhTq1auns0TOpBvTpk0TJYFbtGgRxo4da8Aa5S19+vQRgoVNTU3x8OHDbJ8XKSkpuHHjBoKCgvDmzRuYmprCxsYGzs7OcHV1zfb42sCBA4W2tGLFiggODs6zyeXzmlevXuHy5ct48eIFPn36hEKFCqF8+fJwd3fXS7K6pKQkXLt2DREREYiJiUFycjIsLS1RtmxZVKtWDeXKlcv2vqVSKYKCgoTP1/v372Fubo7ChQvD0dERVapUQalShv+OcdOmTULSEwAYMWIEVq5cacAakSpeXl7Yv3+/UA4PDxcli1Unt8+tvFgnfZ7vOaWv7/NiYmIQHByMsLAwvHv3Dp8+fUKBAgVQpEgRODs7o2bNmtm+38+L486kiGO42tm2bRv69esHIOf9TCIiIiIioi8Zk9J8hZiUhujbwqQ0RNnHpDRE/8ekNETZx6Q0RP/HpDREsgz/wChp7v379yhVqhQSExMBZMxi/+LFi2wHihARUe45fvw4vv/+ewCARCJBSEiIwgyPRESUO9atWwdvb2+hPHr0aCxbtsyANcpbZP8/pqamePz4scaBYl+SpKQklC9fHi9evAAATJw4EX/88YeBa0Vfo4cPH8LZ2Vko16pVC7dv3zZgjYi+Hn/++SdGjx4tlFeuXIkRI0YYrkJE37i0tDQ4ODgI/SszMzO8ePFCJ8navxZ2dnZ4+TLju8mffvoJ69Zl/5lKfXj58iUcHR2RnJwMANi1axd69uxp4FoRqebu7o5r164J5Tt37jBJYB6UkpKC0qVLIzo6GgBgY2Mj/E5EpArHcLVTo0YN3Lt3DwAwZMgQrF271sA1IiIiIiIiMoyvbzpEIiIiIiIiIiIiIiItbN++XUhIAwADBgxgQhoioi9E27Zt0bBhQwAZMw0vXLjQwDUiIvp2yT+QP2QIJ5iRdf78eeH3gQMHfpUJaQDA3Nwc06ZNE8pr1qxBXFycAWtEXyv5YHPZgCoiyr7U1FSsWbNGtCzznouIDOP48eNCQhoA6Ny5MxPSyHjw4IGQkMbMzAxTpkwxcI0ULVu2TEhIU61aNXTv3t3ANSJS7e7du6KENG5ubkxIk0f9888/oiQ07u7uBqwNEX0pOIaruWPHjgkJafLly5cn+5lERERERES5hUlpiIiIiIiIiIiIiOiblZKSIpr5y8jICEOHDjVgjYiISFvLly+HkVHG157btm3Do0ePDFwjIqJvz9mzZ3Hr1i2h3KxZM1SuXNmANcp7Lly4ACAjacvkyZMNXBv9GjRoEGrVqgUAeP/+PRYvXmzYCtFX5927d1i/fr1QLlSoEPr06WPAGhHlXVKpVKv1p06diocPHwrl2rVrC206ERmGfALeESNGGKgmeZNs8sdBgwahbNmyBqyNolevXmHVqlVCWXYciygvYptjGNr22V68eIHffvtNtGzgwIG6rBIRfYU4hqs5qVSKqVOnCuWxY8eiTJkyBqwRERERERGRYXFUm4iIiIiIiIiIiIi+WXPnzsXTp0+FcpcuXeDg4GC4ChERkdbq1q0rPHCfmpr61Qf6ExHlNQkJCRg9erRomXxg1LcuKChImL188ODBX30Ag5GREf766y+hvHTpUrx+/dqANaKvzW+//Yb4+HihPGTIEBQsWNCANSLKu3r16oUlS5bg3bt3atf78OEDRo4cifnz54uW//rrr/qsHhFlYcuWLbhy5YpQrlu3Lho3bmzAGuU9sskfJ02aZODaKJo5cyYSExMBAJ07d0bz5s0NXCMi1S5cuIBdu3YJZTs7O3Tr1s2ANfp2XLp0CR07doSvr2+W6/r4+KBBgwZ48+aNsKxixYr4/vvv9VlFIvrCcQxXO3v27MHt27cBAKVLl8bEiRMNXCMiIiIiIiLDkki1TatMeV6I85ebqbbygxCtt5HeHqKHmhB9OSS112V72/cTSuqwJkRfHqv5r4TfvSuaG7AmRIa39lFStreVvlytw5oQfXkkpYYLv0vPtzVgTYgMT9LsuEzppcHqQZQ3lDJ0BUiJsLAwhIWFQSqV4uXLlzh06BAOHjwo/N3Y2Bh37txBtWrVDFhLIiIiIqK8zdfXF4mJiUhKSsKjR4+watUqPHnyRPi7m5sb/Pz8DFhDIvqaBAcH4+XLl0hPT8fz58+xY8cO+Pj4CH8vWLAgwsLCYGNjY7hKEuVhHh4euHjxIkxNTdGsWTPUr18flSpVgpWVFZKSkhAVFYVr167h0KFD+PDhg2jbdu3a4ciRIwaqOdG359WrVwgKCgIAREdH4+zZs9i2bRvS0tKEdY4fP442bdoYqopE9BWJjY3FzZs3AQDv3r2Dr68v1q1bh8+fPwvrrF69GsOGDTNUFb8pPj4+8PT0BAA4ODigZcuWqFOnDkqUKIF8+fIhNjYWISEhOHXqFG7cuCHa1tjYGL6+vnBzczNE1Ykoj+IYLhEREREREemSiaErQERERERERERERESUG7Zt24aZM2eq/Puvv/7KhDRERERERFno3bs3IiIilP7N1NQUf//9dy7XiIi+ZgsXLsTWrVtV/n3+/PlMSEOkgZSUFJw6dQqnTp3SaP3GjRtj27Zteq4VEck6deoUBgwYoPLvXl5eTEhDRDoTGBiIli1bqvy7m5sbvL29c7FGlCk8PBzr16/XaF0zMzNs3ryZCWmISAHHcImIiIiIiEiXjAxdASIiIiIiIiIiIiIiQ+vQoQP++OMPQ1eDiIiIiOiLZWpqik2bNqFWrVqGrgoRfSOGDRuGESNGGLoaRHmanZ2dVutbWFhgwoQJOHPmDIoUKaKnWhGRttzc3LBx40ZDV4OIvhHly5fH/v37YWTEUJPcUrhwYRQsWFCrbdzc3ODj44NevXrpqVZE9DXiGC4RERERERFlh4mhK0BERERERERERERElNuMjIxgZWWF2rVro3///ujduzckEomhq0VERERE9EUxMzNDqVKl4OnpiV9//RXVq1c3dJWI6CtmbGyMokWLwtXVFUOHDsX3339v6CoR5Xk7d+7E9OnTcerUKfj5+eHhw4d49uwZ4uPjkZKSAisrKxQrVgw1a9aEh4cHfvjhBxQrVszQ1Sb65kkkEhQqVAjVqlVDjx494O3tDVNTU0NXi4i+YgULFkSlSpXQpUsX/PLLL1onSKGcqV27NmJiYnD27FlcunQJt2/fRlhYGGJiYpCYmAgzMzMULVoUpUuXRuPGjdGmTRt4eHgYutpE9IXgGC4RERERERHllEQqlUoNXQnSrRDnyoauQrZVfhCi9TbS20P0UBOiL4ek9rpsb/t+Qkkd1oToy2M1/5Xwu3dFcwPWhMjw1j5Kyva20perdVgToi+PpNRw4Xfp+bYGrAmR4UmaHZcpvTRYPYjyhlKGrgAREREREREREREREREREREREREREREREVG2GRm6AkRERERERERERERERERERERERERERERERERERERERERERESUdzApDREREREREREREREREREREREREREREREREREREREREREREREJmJSGiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiARMSkNEREREREREREREREREREREREREREREREREREREREREREREAialISIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIBk9IQERERERERERERERERERERERERERERERERERERERERERERkYBJaYiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIhIwKQ0RERERERERERERERERERERERERERERERERERERERERERERCRgUhoiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiEjApDREREREREREREREREREREREREREREREREREREREREREREREJmJSGiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiARMSkNEREREREREREREREREREREREREREREREREREREREREREREAialISIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIBk9IQERERERERERERERERERERERERERERERERERERERERERERkYBJaYiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIhIwKQ0RERERERERERERERERERERERERERERERERERERERERERERCRgUhoiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiEjApDREREREREREREREREREREREREREREREREREREREREREREREJmJSGiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiARMSkNEREREREREREREREREREREREREREREREREREREREREREREAialISIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIBk9IQERERERERERERERERERERERERERERERERERERERERERERkYBJaYiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIhIwKQ0RERERERERERERERERERERERERERERERERERERERERERERCRgUhoiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiIiEjApDREREREREREREREREREREREREREREREREREREREREREREREJJFKpVGroShARERERERERERERERERERERERERERERERERERERERERERFR3mBk6AoQEREREdH/2LvzqKjqx//jr/eAqCAg4oJoLFpqalqaWmZqFrmlZS5pi5Zpan1arD7f9iytNLPlY7tm2mJpi0u5lLlrZu6mZmmKa6IiIiAIAvf3Bz9uEOACM1zA5+Mcju+Zec97XvecudwZz3m/AAAAAAAAAAAAAAAAAAAAAAAAAAAAAEoOSmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADZKaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANkppAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA2SmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADZKaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANkppAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA2SmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADZKaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANkppAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA2SmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADZKaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANkppAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA2SmkAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAADZKaQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAANkppAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA2b6cDAAAAAAAAnMnOnTs1f/58+/YNN9yghg0bOpgIAAAAAAAAAAAAAAAAAAAAAAAAAMo2SmkAuFVSUpJ+/fVXxcbGKigoSJdffrmqV6/udCwAAIBSafny5Xrrrbfs248++qjatGnjXCDAIT/88IOGDx8uSTLGaNeuXQ4nApzz1FNP6b777lNkZKTTUQAAAAAAAAAAAAAAAAAAAAAAAFCGUUoDIF+pqak6ePCgfTskJES+vr4Fzj916pQef/xxffTRRzp9+rR9v8vlUvfu3TV+/HjVqlXLo5kBAADKmrVr12rWrFkyxqhcuXKaMmWK05EARyQlJcmyLElSrVq1FB4e7nAiwDmvvvqqXnvtNbVv31733XefevTooXLlyjkdCwAAAAAAAAAAAAAAAAAAAAAAAGUMpTQA8vXuu+/qv//9ryTJ29tbu3fvLrCUJj09XVFRUVq1apW9UTRbRkaGZs2apVWrVmnlypWqW7eux7MDAIrP8uXLHXvttm3bOvbaQHHJyMiQJFmWpbCwMAUEBDicCHBGtWrVJEnGGIWGhjqcBnCeZVlasmSJlixZouDgYPXv31+DBg1SgwYNnI4GAAAAAAAAAAAAAAAAAAAAAACAMoJSGgD5+vbbb+2Cme7du6tWrVoFzn355Zf1888/yxgjY0yuYprs24cPH9bNN9+szZs3y8vLy+P5AU+qU6eOI69rjNGuXbsceW2gIO3bt5cxpthf1xij9PT0Yn9doLjVrFlTUtZ7PigoyOE0gHNyFtGcOHHCwSRAyWJZlmJjY/Xmm2/qzTffVOvWrXXfffepd+/eqlChgtPxAAAAAAAAAAAAAAAAAAAAAAAAUIoZK2d7BABISk1NVWBgoE6fPi1J+uSTT3TnnXfmO/fEiRMKCwtTUlKSXUbTpk0bXXPNNYqNjdU333yjhIQEWZYlY4zefvtt3X///cV2LIAnuFyuPAVMxcEYo4yMjGJ9TeBsOB8Az1qyZImuv/56GWMUHh6u3bt3Ox0JcERcXJxq1qyp06dPq3z58oqNjZWfn5/TsQBHjB8/Xh999JG2bt0qSXZBYPb3bkkKCAjQnXfeqUGDBqlp06aOZQUAAAAAAAAAAAAAAAAAAAAAAEDpRSkNgDw2bNigK6+8UlLW5rZ9+/apVq1a+c6dNGmSBg8ebG98e+aZZzRy5Ej78T179qh169Y6fPiwLMtS06ZNtXHjRs8fBOBB2SUcxSl7gyklHChpOB8Az0pLS1NISIji4+NljNGBAwdUs2ZNp2MBjujevbvmzJkjY4w+/vhjDRgwwOlIgKN+/fVXTZgwQV999ZVOnjyZq5xG+qespnnz5rrvvvvUr18/ypwAAAAAAAAAAAAAAAAAAAAAAABwziilAZDHN998oz59+kiSgoKCdOzYsQLndu3aVfPnz5ck1apVS3v27JGXl1euOdnFNZLYTI0yweVyOfK6lHCgJIqIiCj2Upps0dHRjrwuUNyGDBmiiRMnyhij5557Ti+88ILTkQBH/PLLL2rTpo0kqWbNmtq4caOqVavmcCrAeUlJSfriiy/00Ucfad26dZKUb0GNn5+f+vXrp0GDBqlFixaO5QUAAAAAAAAAAAAAAAAAAAAAAEDpQCkNgDzefvttPfzwwzLGqGHDhtqyZUu+89LT01WlShWdPHlSkvT444/r1VdfzTMvMTFRVapUUUZGhowxmjNnjjp37uzRYwA8admyZY69drt27Rx7bQCAMw4ePKjGjRsrISFBFSpU0MKFC3X11Vc7HQtwxNixY/Xkk0/a31W++uorXXrppU7HAkqM3377TRMmTNAXX3yh+Ph4SVmFNDnLaSTpsssu03333ac77rhDgYGBTsUFAAAAAAAAAAAAAAAAAAAAAABACUYpDYA8xowZo6efflrGGLVq1UqrVq3Kd966devUsmVLSVkb2xYvXlxgYcYll1yiXbt2yRijd955R8OGDfNYfgAAgLJm4cKFuuWWW5ScnCx/f3+9/vrrGjRokNOxAEdMmDBBw4cPV0pKinx8fNS3b1/17dtXLVu2VJUqVZyOB5QIp06d0tdff62PPvpIK1askPRPIU3OgpoKFSqod+/eGjRokNq0aeNYXgAAAAAAAAAAAAAAAAAAAAAAAJQ8lNIAyGP06NF65plnZIxRs2bNtHbt2nzn/e9//9Pw4cMlST4+Pjpx4oTKly+f79yrrrpKa9askTFGr7zyip544gmP5QcAAChL9u3bJ0nasGGDhg0bpsOHD8sYo7CwMN12221q2bKlIiMjFRAQoHLlyp3X2mFhYZ6IDHhEnTp17PGxY8eUmJgo6Z+iDUmqVKnSeZ8Lxhjt2rXLfUGBEmbHjh2aOHGiPv30Ux09elRS1vs+ZzmNJNWvX1+DBw9W//79FRwc7FheAAAAAAAAAAAAAAAAAAAAAAAAlAyU0gDI491339WDDz4oSapVq5b279+f77xbbrlF3333nYwxuvrqq7Vy5coC12zRooXWr18vY4xefvllPfnkkx7JDgAAUNa4XK5cpRuS8hQJFIYxRunp6UXKBhSn7HPBsqxc7/2i/reGMUYZGRlFjQeUeKdPn9asWbP00UcfaeHChfb9Oc8hY4x8fHx066236v7779c111zjRFQAAAAAAAAAAAAAAAAAAAAAAACUAC6nAwAoeWrXrm2PDx06ZP8l9ZxSUlK0cOFCezNou3btzrjm8ePH7XGlSpXclBRAtkWLFsnLy0teXl7y9vZ2Og4AwAP+XRqQXc5RlB+gNPp3GVP2+VCYH+BCUq5cOfXu3VvTp0/X8OHDcxWc5byupKamatq0aWrbtq2uuuoqLViwwOHkAAAAAAAAAAAAAAAAAAAAAAAAcAK71gHkccUVV0iSvSHt888/1/Dhw3PN+eKLL5ScnGzPu+666wpc7/Tp0zpw4IC96bNmzZoeSg5c2CgXAICyj9/1uJCFhYVRJAMUwcqVKzVx4kR98803OnXqVK7z6d/FZ9n3rVmzRp07d1bv3r310UcfUTILAAAAAAAAAAAAAAAAAAAAAABwAaGUBkAeYWFhatKkibZs2SLLsjRixAg1b95cbdu2lSRt3rxZzzzzjF1aU6VKFbVv377A9bZu3aq0tDRJWZvbLr744uI4DAAAgDJhwIABTkcASoQ9e/Y4HQEodY4dO6ZPPvlEH330kf78809J/xTQZH+nN8YoKipKAwcO1O+//67JkyfrwIEDueZ8/fXX2r9/v5YvXy4vLy/HjgcAAAAAAAAAAAAAAAAAAAAAAADFh1IaAPm6//77NXToUBljlJSUpOuuu04NGjRQuXLl9PvvvysjI8PevHbvvffK27vgXyc//fSTPS5fvrwaNmxYHIcAAABQJkyePNnpCACAUmbhwoX66KOPNHv2bKWlpeUqoslWtWpV3XPPPbrvvvtUp04d+/4RI0Zo7ty5Gjt2rH7++We7mGb16tWaMGGChg0bVuzHAwAAAAAAAAAAAAAAAAAAAAAAgOJnrOxdKQCQg2VZuvbaa7Vq1Sp7A1pO2feFhoZq69atqly5coFrtWzZUuvWrZMxRtdcc42WL1/u4fTAhWfRokWKioqSlHV+ZmRkOJwIF4qcG5iLkzFGu3btcuS1AQAASqJDhw5p8uTJmjRpkvbs2SNJucpossft2rXT0KFDdeutt6pcuXJnXHPKlCkaPHiwMjMzJYnv9AAAAAAAAAAAAAAAAAAAAAAAABcQb6cDACiZjDGaM2eOunfvrpUrV+a637IsWZalGjVqaPbs2WcspNm0aZNdSCPJLs0AAJQNe/bsybe8zNOyrysAAAAXMsuyNG/ePE2cOFHz5s1TRkZGriKabJUrV9aAAQM0ZMgQ1a9f/5zXv/vuu7V8+XJNmTJFkrR161a35gcAAAAAAAAAAAAAAAAAAAAAAEDJRSkNgAJVrlxZy5cv19dff61vvvlGO3bsUEpKikJDQxUVFaWhQ4cqKCjojGuMGzdO0j9/nf3mm2/2eG4AQPErzpKY4i7AAQAAKGn27dunSZMmafLkyTp48KCkrM9IxphcZbKtW7fWkCFD1KdPH5UvX75QrxUVFWWX0iQkJLjrEAAAAAAAAAAAAAAAAAAAAAAAAFDCGYtdvQA8KDExUZmZmfbtwMBAB9MAZdeiRYsUFRUlKascJCMjw+FEuFC4XC5HXpf3OQAAuFB16tRJCxcutItnpH8KAi3LUkBAgO666y4NGTJEjRs3LvLr8V0DAAAAAAAAAAAAAAAAAAAAAADgwuTtdAAAZZu/v7/TEQAAHrRkyRKnIwAAAFxQFixYYJfQGGPscporr7xSQ4YMUb9+/eTr6+twSgAAAAAAAAAAAAAAAAAAAAAAAJR2lNIAAACg0Nq1a+d0BADABSwxMVFr167Vxo0bFRsbq/j4eKWmpp7XGsYYTZo0yUMJAc+xLEuVKlVSv379NGTIEDVr1swjr9OoUSNNnjzZI2sDAAAAAAAAAAAAAAAAAAAAAACg5DKWZVlOhwAAAEWzaNEiRUVFScraWJ2RkeFwIgCAp2RmZmrBggVatWpVkYs4du3a5aGUgGetX79e48aN04wZM5Senl7odSzL4rMTSh2Xy6UmTZpo6NChuuOOO+Tv7+90JAAAAAAAAAAAAAAAAAAAAAAAAJRB3k4HAAAAAACcm//97396/fXXdfDgwVz3F6Zr1BjjrlhAsXrppZc0cuRIZWRk2O99Y0yu8+Df7+/8zhHOAZRWv/zyi1q1auV0DAAAAAAAAAAAAAAAAAAAAAAAAJRxlNIAAACgTFm0aJFuvPFGSVmFA+np6Q4nAoouOTlZt9xyixYtWpSrhEPKKts413KN7LmFKbEBSoLRo0fr+eefl5S7VObf58HZSmgsy+I8QKkUExOjw4cP67vvvpMkNWnSRBEREc6GAgAAAAAAAAAAAAAAAAAAAAAAQJlEKQ1wgenQoUOu28YYLVq06Kzz3KWg1wMAwJ0oGkBZ079/fy1cuFCS7FIZl8ul6tWr69ChQ/a86tWryxij48ePKzU11b4/u4yjatWq8vPzK97wgJts3LhRzz77bK5CpjvvvFN33nmnIiIi1KBBA/uxqVOnqnnz5oqLi9O2bdu0ePFizZgxQ6mpqTLGqGHDhvrwww9Vu3ZtJw8JOG8zZszQgw8+aN/esmWLg2kAAAAAAAAAAAAAAAAAAAAAAABQllFKA1xgli5dmmsTZ/b4TPPc5UyvBwAAgPzNnj1bM2bMsD9HBQUFaezYserbt698fX3lcrnsuVOnTrXLBXft2qXFixfr/fff16ZNm2SMkZeXlz744AN17NjRkWMBimL06NF26ZgxRp988onuuuuufOfWqFFD9erVkyRdddVVuvfee3XkyBH95z//0TfffKPt27erT58+WrhwoS699NJiOwagqOLj4+3zoGbNmmrYsKHDiQAAAAAAAAAAAAAAAAAAAAAAAFBWuc4+BQAAAADglLFjx0rKKvirUKGCFi9erIEDB8rX1/eMz6tbt64GDx6sDRs26J133pGPj4+OHDmibt266ZtvvimO6IDbpKWl6fvvv5cxRsYY9e3bt8BCmoJUr15dX331lZ5++mlZlqVDhw6pS5cuSkxM9FBqwP2Cg4MlZRUz1apVy+E0AAAAAAAAAAAAAAAAAAAAAAAAKMsopQEuQJZl2X9Z/VzmuesHAAAA5yc+Pl6rV6+2iziGDx+uJk2anPc6999/v77++msZY5Senq4BAwbor7/+8kBiwDPWrFmj1NRU+3vFww8/XOi1XnrpJV1//fWSpH379umll15yS0agOISGhtrjkydPOpgEAAAAAAAAAAAAAAAAAAAAAAAAZR2lNMAFJjMzM9dPRkbGOc1z109BrwcAAIC8fvnll1wFfwMHDiz0WjfddJPuu+8+SdKpU6f07LPPuiUjUBxylij5+vqqZcuWZ5yflpZ2xsdffPFFSVlFnBMmTFB6enrRQwLFoHnz5nK5XLIsS3v37j3rex0AAAAAAAAAAAAAAAAAAAAAAAAoLEppAAAAAKCEOnjwoD2uWrWq6tSpc8b5p06dOuPjjz76qKSsIo6ZM2cqISGh6CGBYhAXFydJMsYoMjIy3zku1z//xZGamnrG9Vq3bq0qVapIkhISErR69Wo3JQU8KzQ0VO3bt5ckpaSkaO7cuc4GAgAAAAAAAAAAAAAAAAAAAAAAQJlFKQ0AAAAAlFA5izhq1aqV75xy5crZ47OV0lx88cWqXbu2JCk9PV2rVq1yU1LAs3KWzPj7++c7x9/fX5ZlSZJiY2PPumZYWJg93r59exETAsVn+PDh9vipp55ScnKyg2kAAAAAAAAAAAAAAAAAAAAAAABQVlFKAwAAAAClQIUKFfK9PyAgwC7iiImJOes6NWvWtMd//fWXe8IBHhYQEGCPCyrgCAwMtMf79u0765peXl72+NixY0VIBxSvrl276oEHHpBlWdq5c6e6du2qI0eOOB0LAAAAAAAAAAAAAAAAAAAAAAAAZYy30wEAAEDRhYaGasCAAU7HAAC4Wc6SjcTExHznBAUF2YUau3fvPuuaqamp9jghIaGICYHiERoaao+PHz+e75xLLrnELqNZs2bNWdfMeb54e/PfIyhd3n77bfn7+2vs2LFavny5GjZsqP/85z/q27evGjRo4HQ8AAAAAAAAAAAAAAAAAAAAAAAAlAHsugIAoAy49NJLNXnyZKdjAADcLDIyUpJkWZaOHj2a75yGDRvqr7/+kiStWrXqjOulpqZqx44dMsZIknx9fd2YFvCcRo0aSco6Fw4cOKBTp06pQoUKueY0bdpUixYtkmVZWrFihY4fP66goKB811u4cGGucpvq1at7LjzgZh06dLDHwcHBOnr0qOLi4jRq1CiNGjVK/v7+Cg8PV0BAgMqVK3fO6xpjtGjRIk9EBgAAAAAAAAAAAAAAAAAAAAAAQClEKQ0AACVAcnKyPvvsM61cuVKxsbEKCgpS8+bNdfvtt6tmzZpOxwMAOCS7iEOSXTpQpUqVXHOaNWum7777TpZlac2aNdq5c6cuueSSfNf79NNPderUKUlZ5QO1a9f2XHjAjS655BIFBQXp+PHjsixLmzdvVqtWrXLN6dq1q9544w0ZY5SSkqInnnhCEyZMyLNWXFyc/vOf/8gYI8uyJCnPWkBJtnTpUrtcTJI9zn4/JyQkaMuWLbnmnI1lWec1HwAAAAAAAAAAAAAAAAAAAAAAAGWfsbJ3rAC4ICxfvtzpCGrbtq3TEQCP2r59u7788kv79sCBAxUREVHg/J9//lm9evXSkSNH8jzm6+urN954Q4MHD/ZEVKBMWrRokaKioiRlbdLOyMhwOBFQNJGRkdq7d6+MMZo7d646deqU6/Hff/9djRs3tgs2rrrqKs2fP1+BgYG55q1evVqdOnVSYmKiXT7w999/q0aNGsV5OECh9ezZUzNnzpQxRs8995xeeOGFXI9blqWLL75Ye/bssd/jHTt21LBhw1S/fn2lpaVp5cqVGjNmjPbt22cXcDRt2lQbNmxw4IiAwnG5XG4vkMk+Z/jcBAAAAAAAAAAAAAAAAAAAAAAAgGyU0gAXGE9sXjsfxhilp6c79vpAcXjooYf07rvvSpJq1Kihffv2ydvbO9+5u3btUrNmzZSYmChJuc7P7Eu0MUYffvihBg0a5OHkQNlAKQ3KmsGDB2vSpEkyxuiBBx7Q+PHj88xp166dVq5cad+uUaOG+vXrl6uIY8aMGfbnMGOMunXrplmzZhXXYQBF9tlnn2nAgAEyxqhBgwbatm1bnjmzZ89Wjx497JKm/L77ZN9vWZZcLpfmzp2rjh07FschAG7hcrk8si6fmwAAAAAAAAAAAAAAAAAAAAAAAJATpTTABSa7lMapU59NbrgQ1K1bV9HR0TLG6LHHHtPYsWMLnNutWzfNnTvX3jD973Mz+3z19fXV9u3bddFFF3k0O1AWUEqDsubHH39U586dJUnVqlXT/v375ePjk2vOli1b1KJFC50+fVqS8i3jyFnE4e/vr7Vr16pevXrFcxCAGyQlJSkiIkKpqamSpLlz56pt27Z55j399NMaM2bMWT9fSdIrr7yiJ5980sPJAQAAAAAAAAAAAAAAAAAAAAAAAKD08cyfVgZQotFFBXjOkSNH7EIaSerSpUuBc3///Xe7kMayLAUHB+ujjz7S9u3btWLFCt188812gUBKSopeffXV4joMAEAJcsMNN+jWW29V165d1bJlS23cuDHPnMsuu0zffPONfHx8chXSWJZlf/bLvt4EBATom2++oZAGpU6lSpUUGxurxMREJSYm5ltII2UVzUyePFnVq1fP97uPZVkKDw/XV199RSENAAAAAAAAAAAAAAAAAAAAAAAAABTAWLRTABeUF1980ekIGjFihNMRAI9ZunSpOnToIElyuVw6ceKE/Pz88p37zDPPaPTo0ZKyigKWL1+ua665JtecqKgoLVq0SJIUHBysw4cPy+WiUw44k0WLFikqKkpS1rmVkZHhcCKg+OzcuVNPPPGE5s2bp7S0tFyP+fj4qGfPnho1apTq1KnjUEKg+Jw+fVpLly7V6tWrdfjwYVmWpZCQELVu3Vrt2rWTt7e30xEBAAAAAAAAAAAAAAAAAAAAAAAAoMSilAYAADeaMmWKBg4cKEkKCwvTnj17CpzbrFkzbdq0ScYYtWnTRsuWLcszZ+XKlWrbtq2krHKNzZs3q3Hjxh7JDpQVlNIA0smTJ7Vhw4ZcRRzNmzeXr6+v09EAAAAAAAAAAAAAAAAAAAAAAAAAAEApwJ8FBwDAjY4fPy4pqwijSpUqZ5y3efNmGWMkSb169cp33jXXXKNKlSopKSlJkrRlyxZKaQAAZ+Xn56drr73W6RgAAAAAAAAAAAAAAAAAAAAAAAAAAKCUcjkdAACAsiQ5OdkeV6xYscB5q1evlmVZsixLknTjjTfmO88Yo8jISPv20aNH3ZQUAAAAAAAAAAAAAAAAAAAAAAAAAAAAAID8eTsdAACAsqR8+fL2+OTJkwXOW758uT2uWrWq6tevX+BcPz8/e5yUlFTEhAAAAADKGsuytGDBAi1fvlyrV6/Wvn37dPz4cSUmJsrf319BQUEKDw/XVVddpXbt2ikqKsrpyAAAAAAAAAAAAAAAAAAAAAAAACjhKKUBAMCNKleuLClrU+iePXsKnLdw4UJJkjFGbdq0OeOaOcttfHx8ipwRKOtCQ0M1YMAAp2MAAAB4nGVZevvtt/XWW29p7969ue7PFhcXp7i4OO3evVtLlizR6NGjFRERoeHDh+uBBx6QMcaJ6AAAAAAAAAAAAAAAAAAAAAAAACjhKKUBAMCN6tevb48TExO1ceNGXXHFFbnmREdHa/369fbmz/bt259xzdjYWHscGBjovrBAGXXppZdq8uTJTscAAADwqP379+v222/XqlWrcpXQGGMKLJrJnhcdHa2HH35YX331laZOnaqLLrqoWDIDAAAAAAAAAAAAAAAAAAAAAACg9KCUBgAAN7riiivk4+Oj06dPS5LGjBmj6dOn55ozbtw4SVkbQo0xioqKKnC9uLg4HTp0yN5UGh4e7qHkQMmRnJyszz77TCtXrlRsbKyCgoLUvHlz3X777apZs6bT8QC3Wb58udMR1LZtW6cjAPLy8nL09Y0xSk9PdzQDcL5iYmLUvn177dmzx/5eIWV9x8gunnG5XKpYsaJSUlKUmZlpPzfn3JUrV6pDhw5auXKlatSoUfwHAgAAAAAAAAAAAAAAAAAAAAAAgBLLWDn/lDIAFCAzM1MLFizQqlWrtHHjRsXGxio+Pl6pqanntY4xRrt27fJQSqBkuPXWWzVr1ixJWe/5AQMGaNiwYSpXrpw+//xzvfHGG/ZG0Msvv1zr168vcK0ff/xRnTt3ttfavXs3xTQoVbZv364vv/zSvj1w4EBFREQUOP/nn39Wr169dOTIkTyP+fr66o033tDgwYM9ERUodi6Xy74eOIEiDpQULpfL0dc3xigjI8PRDMD5sCxLV111ldauXZurYOaiiy7SXXfdpeuvv15NmzZVlSpV7OfExcVp8+bNWrx4sT777DPt27dPxhi7wKZVq1b65ZdfHDkeAAAAAAAAAAAAAAAAAAAAAAAAlEyU0gA4q//97396/fXXdfDgwVz3F+bXBxs+cSHYsGGDWrZsKcuyZFlWnsKB7HPHGKMvvvhCt912W4Fr3X///frggw8kSSEhIfr77789FxzwgIceekjvvvuuJKlGjRrat2+fvL298527a9cuNWvWTImJiZKU69zJed58+OGHGjRokIeTA56XXUrj1FcyPpehpHDqXMh+Tc4FlDaffvqp7r77bvs9XK5cOY0aNUqPPvpogZ+zckpPT9dbb72lZ599VqdPn7bPgylTpuiuu+4qhiMAAAAAAAAAAAAAAAAAAAAAAABAaUApDYACJScn65ZbbtGiRYtylQFIyrdooyDZc9nwiQvJmDFj9PTTT+c6Z6Tc51D37t01a9asAtfIyMhQrVq1dPToUUlSz5499dVXX3k2OOBmdevWVXR0tIwxeuyxxzR27NgC53br1k1z587Nc95ky76W+Pr6avv27brooos8mh3wNJfL5ejr87kMJUX79u3P+buFpyxZssTR1wfOR9OmTbV161a7kGbWrFnq3Lnzea/z448/qlu3bsrIyJBlWbrsssu0efNmDyQGAAAAAAAAAAAAAAAAAAAAAABAaXT2P58M4ILVv39/LVy4UNI/RQAul0vVq1fXoUOH7HnVq1eXMUbHjx9XamqqfX/2xtKqVavKz8+veMMDDnvyySdVrVo1PfHEE4qLi7PvtyxLXl5eGjhwoMaPH3/GNaZNm6YjR45Iyjqfunbt6tHMgLsdOXLELqSRpC5duhQ49/fff7cLaSzLUtWqVTVmzBhdc801io2N1bhx4zR79mwZY5SSkqJXX31V77zzTnEdCuARI0aMcDoCUCIsXbrU6QhAqXHgwAFt2bJFxhgZY/T4448XqpBGkjp27KjHH39cY8aMkSRt3bpVBw4cUO3atd0ZGQAAAAAAAAAAAAAAAAAAAAAAAKWUsSzLcjoEgJJn9uzZ6tGjh10kEBQUpLFjx6pv377y9fWVy+WyH/vpp5/UoUMHSdKuXbu0ePFivf/++9q0aZOMMapevbqmTJmijh07OnY8gFPS0tK0aNEi7dixQykpKQoNDVWHDh3OaaPn888/ry1btti3P/roIwUHB3syLuBWS5cuta8PLpdLJ06cKLCk7JlnntHo0aMlZZUwLV++XNdcc02uOVFRUVq0aJEkKTg4WIcPH5bL5fLgEQAAAJQs3377rXr37i1J8vLy0v79+xUSElLo9WJiYlS7dm1l//fgV199pZ49e7olKwAAAAAAAAAAAAAAAAAAAAAAAEo3b6cDACiZxo4dK0myLEsVK1bU4sWL1aRJk7M+r27duqpbt64GDx6s9957T4899piOHDmibt266YsvvlCvXr08HR0oUXx8fNS5c2d17tz5vJ87cuRIDyQCis+ePXvsce3atQsspJGk+fPnS8oqpGnTpk2eQhpJGjFihF1KExcXp99//12NGzd2b2gAAIAS7PDhw5KyPjNFREQUqZBGkkJCQhQZGaldu3bJGGOvDwAAAAAAAAAAAAAAAAAAAAAAALicDgCg5ImPj9fq1atljJExRsOHDz+nQpp/u//++/X111/LGKP09HQNGDBAf/31lwcSAwBKouPHj0vK2jRdpUqVM87bvHmzjDGSVGCB2TXXXKNKlSrZt7ds2eLGtAAAACXfiRMn7PGZPl+dj5zrJCQkuGVNAAAAAAAAAAAAAAAAAAAAAAAAlH6U0gDI45dffpFlWbIsS5I0cODAQq9100036b777pMknTp1Ss8++6xbMgIASr7k5GR7XLFixQLnrV69Otd158Ybb8x3njFGkZGR9u2jR4+6KSkAAEDpkF0gY1mWjhw54pY1c36mCgoKcsuaAAAAAAAAAAAAAAAAAAAAAAAAKP0opQGQx8GDB+1x1apVVadOnTPOP3Xq1Bkff/TRRyVlbZqbOXMmf3kdAC4Q5cuXt8cnT54scN7y5cvtcdWqVVW/fv0C5/r5+dnjpKSkIiYEAAAoXUJCQuzxvn37FB0dXaT1oqOjtWfPHhlj8qwPAAAAAAAAAAAAAAAAAAAAAACACxulNADyiIuLkyQZY1SrVq1855QrV84en62U5uKLL1bt2rUlSenp6Vq1apWbkgIASrLKlStLyiol27NnT4HzFi5cKCnrutOmTZszrpmz3MbHx6fIGQEAAEqT1q1byxhjl8iMHTu2SOtlP9+yLBlj1Lp16yJnBAAAAAAAAAAAAAAAAAAAAAAAQNng7XQAACVbhQoV8r0/ICBAx44dkzFGMTExZ12nZs2aOnDggCTpr7/+cmtGoLjs27cvz31hYWHnNM9d8ns9oKSqX7++PU5MTNTGjRt1xRVX5JoTHR2t9evX2xur27dvf8Y1Y2Nj7XFgYKD7wgIASryBAwd6ZF1jjCZNmuSRtQF3q1atmlq2bKk1a9bIsixNmDBBrVq10t13333ea3322WeaMGGC/TmsRYsWqlatmpsTAwAAAAAAAAAAAAAAAAAAAAAAoLSilAZAHjk3+ScmJuY7JygoSMeOHZMk7d69+6xrpqam2uOEhIQiJgScERERYW/YlLI2MKenp591nrsU9HpASXXFFVfIx8dHp0+fliSNGTNG06dPzzVn3LhxkiTLsmSMUVRUVIHrxcXF6dChQ/b5FR4e7qHkQMlSp04dj6xrjNGuXbs8sjbgCVOmTHH7Z6zs6w+lNChNnnjiCd16660yxsiyLN17773atGmTRo4cqYCAgLM+PzExUSNGjND48eMl/XMePPnkk56ODgAAAAAAAAAAAAAAAAAAAAAAgFKEUhoAeURGRkrK2ph29OjRfOc0bNhQf/31lyRp1apVZ1wvNTVVO3bssDeQ+vr6ujEtUPwsy3LrPKCsqlSpkrp06aJZs2ZJkr755hsNHDhQw4YNU7ly5fT555/r/ffft68Pl19+uRo0aFDgemvXrpX0z8bp+vXre/wYgJJgz549dvGAO3miQA0oyf59DnEOoLS65ZZbFBUVpZ9++sm+Prz99tv6+OOPdeutt6pDhw5q0qSJqlatKj8/P508eVLHjh3T5s2btXjxYs2cOVNJSUn2Z6rsYsCbb77Z6UMDAAAAAAAAAAAAAAAAAAAAAABACWIsdswD+JeDBw/qoosukpS1UfPo0aOqUqVKrjkjR47UCy+8IElyuVzavn27LrnkknzXmzhxooYMGWKvN336dPXq1ctzBwB4iMvlylUKYIxRRkbGWee5S0GvB5RkGzZsUMuWLWVZlr3xOaec59MXX3yh2267rcC17r//fn3wwQeSpJCQEP3999+eCw6UINnXFXfIPg+z/+W6gtLE5XIVeY3scynn5zTOBZRGCQkJateunTZv3pznO8rZ5JxrWZYuv/xyLVu2TP7+/h7NDAAAAAAAAAAAAAAAAAAAAAAAgNLF2+kAAEqeWrVqKTw8XHv37pUkrVmzRp06dco1p1evXnrhhRdkjFFmZqYGDBig+fPnKzAwMNe81atX67///W+uTXLXXntt8RwI4GYDBgxw6zzgQtCsWTO99NJLevrpp/MUARhj7OtDt27dzlhIk5GRoRkzZthrtGnTxvPhgRIiLCzsvEtpkpOTdfz4caWnp0v6p6QgKChIAQEBbs8IFIfo6Ojzfk5ycrKOHj2qtWvXavr06Vq3bp2MMbr44os1adIkhYWFeSAp4HkBAQFavHixBg8enOszkqQzlmPm/PxlWZZ69uypCRMmUEgDAAAAAAAAAAAAAAAAAAAAAACAPIx1pp0qAC5YgwcP1qRJk2SM0QMPPKDx48fnmdOuXTutXLnSvl2jRg3169dP9evXV1pamlauXKkZM2bk2gzdrVs3zZo1q7gOAwBQQkyaNElPPPGE4uLict3v5eWlgQMHavz48SpfvnyBz586daruuusuSVnXk48//pgCKOAsMjIytGnTJk2bNk0TJkxQYmKigoOD9dlnn+UpHAQuFDNmzNDgwYMVHx+vmjVrauHChWrQoIHTsYAi+fbbb/XGG2/ol19+yXX/mYpqWrdurccee0w9evQolowAAAAAAAAAAAAAAAAAAAAAAAAofSilAZCvH3/8UZ07d5YkVatWTfv375ePj0+uOVu2bFGLFi10+vRpSVmb3HJuest5n2VZ8vf319q1a1WvXr3iOQgAQImSlpamRYsWaceOHUpJSVFoaKg6dOig2rVrn/W5zz//vLZs2WLf/uijjxQcHOzJuECZsnfvXt16663auHGjvL29NW3aNN16661OxwIcsXbtWrVv314pKSmKiIjQpk2bFBAQ4HQsoMi2b9+uZcuW6ddff9XevXt1/PhxJSUlqVKlSgoKClJ4eLiuuuoqtWvXjjImAAAAAAAAAAAAAAAAAAAAAAAAnBWlNADylZGRodtuu02pqamSpGeffVatWrXKM2/OnDnq3bu3UlNT7UKa7F8rOW8HBATo66+/VlRUVDEdAQAAAHI6evSomjdvrgMHDqhixYrauHEjZYG4YI0YMUKjRo2SMUYPPfSQ3nzzTacjAQAAAAAAAAAAAAAAAAAAAAAAAECJQikNgCLbuXOnnnjiCc2bN09paWm5HvPx8VHPnj01atQo1alTx6GEAAAAkKTJkyfr3nvvlTFG3bp106xZs5yOBDji0KFDqlWrliTJ19dXR48eVcWKFR1OBQAAAAAAAAAAAAAAAAAAAAAAAAAlB6U0ANzm5MmT2rBhgw4fPizLshQSEqLmzZvL19fX6WgAAACQlJSUpODgYJ0+fVoul0sHDx5UjRo1nI4FOKJu3bqKjo6WMUbz5s1Tx44dnY4EAAAAAAAAAAAAAAAAAAAAAAAAACWGy+kAAMoOPz8/XXvtterVq5d69+6ta6+9lkIaAACAEqRSpUqqU6eOJMmyLK1YscLhRIBzqlWrZo//+OMPB5MA56dOnTqqU6eO6tatq19++aVIa61atSrXegAAAAAAAAAAAAAAAAAAAAAAAEA2b6cDAAAAAACKT2BgoD3eu3evg0kAZyUlJdnj5ORkB5MA52fPnj2SJGOMUlJSirRWSkpKrvUAAAAAAAAAAAAAAAAAAAAAAACAbJTSAABwjgYOHOjo6xtjNGnSJEczANn27duX576wsLBzmucu+b0egLM7evSoPU5PT3cwCeCc+Ph47dixwy7hqFy5srOBgPNEgQwAAAAAAAAAAAAAAAAAAAAAAAA8jVIaAG53+vRpxcXFKSgoSD4+Pk7HAdxmypQpjm3+tCyLUhqUKBEREbnOB2NMvuUW/57nLgW9HoAz27Vrl6Kjo+3zsmrVqg4nApwxduxY+zpijFFkZKTDiQAAAAAAAAAAAAAAAAAAAAAAAACgZHE5HQBA2fHNN9/o6quvlq+vr0JDQ1WxYkVddtllevvtt2VZltPxAAAeYFmW/XOu89z1A+D8WJal4cOH22NJatSokZORgGKXmZmpMWPG6NVXX7XLmcqXL6927do5nAxwxunTp+1xuXLlHEwCAAAAAAAAAAAAAAAAAAAAAACAksbb6QAASqavvvpKjz/+uCTJGKMFCxaofv36Bc5/7LHH9NZbb0lSrqKAbdu26ZFHHtG3336refPmydfX16O5AU+jCAMAUBrt3LlTjzzyiObPny9jjCzLUtWqVdWqVSunowHnbPny5YV6XkpKig4fPqyNGzdq5syZ2r9/v/2Zzhije++9VxUrVnRnVKDUOHTokD329/d3MAkAAAAAAAAAAAAAAAAAAAAAAABKGkppAOTr888/14EDByRJ11xzzRkLaaZNm6Y333xTUtamzuyNzsYYSVklHitWrNCdd96pGTNmeD484CHR0dFORwBKjAEDBrh1HoCCjRw5slDPy1nE8dtvv9n3Z39Oe+qpp+zPa0Bp0L59+yK/Z3OW0ViWpbp16xb6HAPKgjlz5kjKOifCwsIcTgMAAAAAAAAAAAAAAAAAAAAAAICSxFjZO7IA4P+zLEtBQUFKTEyUJL3xxht6+OGH852bnp6uevXqac+ePfbGTj8/PzVu3FixsbHatWtXrpKaOXPmqHPnzsV5OAAAAKWay+UqUhFHzhKO7Ns33XSTZs2aJZfL5ZaMQHHIPhcK+98YOc8BSbriiis0Y8YMhYeHuy0j4C779u0r8LGIiAj7/fz555/rmmuuOed1LctScnKyoqOj9c0332jKlCmSss6PgQMHauLEiUXKDQAAAAAAAAAAAAAAAAAAAAAAgLKDUhoAefzxxx9q2LChpKyNadu2bVODBg3ynTtnzhx1797d3hDXpUsXTZ06VQEBAZKkmTNnqm/fvkpPT5ck3XjjjZo/f34xHAUAAEDZUNRSmmyWZcnlcun+++/Xa6+9pvLly7shHVB83FWidMUVV2jw4MEaPHiwvLy83LIm4G5n+t2f87/yinp9yFlc9sMPPygqKqpI6wEAAAAAAAAAAAAAAAAAAAAAAKDs8HY6AICS56+//rLHFSpUUP369QucO336dElZG9n8/Pz0ySef2IU0ktSjRw898cQTeumllyRJixYtUkJCQq45AAAAOLOidIkaY1SvXj116dJF9957r10+CJQ2I0aMOO/nGGNUoUIFBQQEKDw8XM2aNVONGjU8kA7wjLP9/i/q9cEYI8uyFBUVRSENAAAAAAAAAAAAAAAAAAAAAAAAcqGUBkAe+/fvl5S1QS0sLOyMf3l90aJF9uO9evVSlSpV8sy577777FKajIwMbdq0SW3btvVAcgAAgLJnyZIl5/2cnEUcF110kfz8/DyQDChehSmlAVAwy7JUrlw53XXXXXrrrbecjgMAAAAAAAAAAAAAAAAAAAAAAIAShlIaAHkkJSXZ48DAwALn7dixQzExMXYpTffu3fOdV7t2bYWGhurvv/+WJO3cuZNSGgAAgHPUrl07pyMAAIrZgAEDCnzsk08+sb+HR0VFqWbNmue8rsvlkp+fn6pUqaJGjRqpffv2qlatWpHzAgAAAAAAAAAAAAAAAAAAAAAAoOyhlAZAHmlpaec075dffpGU9dfVXS7XGTdM5yyliY+PL3JGAAAAAADKqsmTJxf42CeffGKP/+///k8dOnQojkgAAAAAAAAAAAAAAAAAAAAAAAC4wFBKAyAPf39/exwXF1fgvKVLl0qSjDG69NJLVaVKlQLnulwue5yamlr0kEApdezYMcXGxio+Pr5Q50Lbtm09kAoAAABAaWJZlowxTscAAAAAAAAAAAAAAAAAAAAAAABAGUYpDYA8qlWrJilrk9vevXuVkpKiihUr5ppjWZZ++OEHexPc2Yoy4uPj7bGvr697AwMl3I8//qjJkydr1apVOnjwYKHXMcYoPT3djcmAwhs4cKCjr2+M0aRJkxzNAAAA4ITJkyfb40aNGjmYBAAAAAAAAAAAAAAAAAAAAAAAAGUZpTQA8mjSpImkfwow5syZo969e+ea89NPP+nw4cP2vPbt259xzUOHDtnj7NIboKz7888/dfvtt2vTpk2SssqcgLJiypQpdjFZcbMsi1IaAABwwRowYIDTEQAAAAAAAAAAAAAAAAAAAAAAAHABMBY75AH8i2VZql27tmJiYmRZlurWravVq1crODhYkpSYmKj27dtr48aNkqTy5csrJiZGgYGB+a63c+dO1a9fX1JWgc3PP/+sq666qngOBnDIunXrdN111yk5Odku0Mi+5OYs8vj3ZfjfJR85HzfGKCMjw4OpgXPncrkcL6XhfACAsmH58uVOR1Dbtm2djgAAAAAAAAAAAAAAAAAAAAAAAAAAJYq30wEAlDzGGA0YMEBjxoyRMUa7d+9Wo0aN1LNnT5UrV05z5sxRdHS0PffWW28tsJBGklasWJFr7UaNGnn8GAAnxcfHq0ePHjp58qRd2hEQEKDOnTsrMjJSo0ePlpR1Ptx1112qVauW4uLitG3bNq1fv16nTp2yn9ewYUP17t3bsWMBzoRuQ6BoRo4c6XQEPf/8805HANS+fXvHis6krM9k6enpjr0+4G4HDx5UXFycTpw4oczMTDVt2vSM39kBAAAAAAAAAAAAAAAAAAAAAACA/BiL3cQA8pGYmKgGDRooJiZGUlbxQPZG0eyxZVkqX768Nm/erHr16hW4VteuXTV//nwZY9S4cWNt3ry5WI4BcMqoUaM0YsQI+5zp3LmzPvvsMwUFBUmSXC6X/dhPP/2kDh062M9NTEzUpEmTNHLkSMXHx8sYozvuuEMff/yxvL3pkkPJsXfvXqcjKDw83OkIQJHkvB44JSMjw9HXB6R/zgWn/nvCGMO5gFItLS1Nn3/+ub799lutWrVKCQkJuR7/93eObHPnztWxY8ckSdWrV1enTp2KJS8AAAAAAAAAAAAAAAAAAAAAAABKB3a3A8iXv7+/5s2bp+uvv15xcXG5Nkxnbxh1uVz64IMPzlhIc+jQIS1cuNB+fn4b4YCy5v3337fPk/r162vGjBny8fE5p+f6+/vrkUceUe/evXXTTTdp8+bNmjp1qjIzM/X55597ODlw7iiEAZxRUGlHfuU2Z5qbs3AQKAkKU0hzru/78zk/gNLmm2++0YMPPqgjR45IyvvePtPv+k2bNun555+XJJUvX16HDh1SYGCg58ICAAAAAAAAAAAAAAAAAAAAAACgVKGUBkCBmjZtqt9//10vvviivv32W3uTm4+Pj9q1a6fnnntObdq0OeMa48eP1+nTpyVlbYbr1q2bx3MDTtq5c6diYmJkjJExRs8///w5F9LkVKtWLS1YsEBNmzZVTEyMvvzyS3Xv3l19+vTxQGoAgFPcUcRhWdY5l9WcaS7glBEjRpzXfMuy9Omnn2rv3r32bW9vbzVu3FiXXXaZgoOD5efnp5MnT+rYsWP67bfftG3bNqWnp9vnRGRkpPr37+/2YwGK0/DhwzV+/Hj793p+ZbJncv/99+uVV15RSkqKUlNTNX36dN13330ezQwAAAAAAAAAAAAAAAAAAAAAAIDSw1jsSARwjk6cOKGUlBRVrVpV3t7n1mm1ZMkSJSQk2LdvuukmeXl5eSoi4Ljp06erX79+kiQvLy/Fx8fLz88v1xyXy2VvGP3xxx91ww03FLjepEmTNHjwYBlj1KhRI/3222+eCw8AKFbLli07r/mHDx/Wk08+qb1799pFA5dffrnat29fYBHHsmXLtGnTJklZBQUREREaM2aMqlevLklq166dW48J8LSEhAT17t1bCxculGVZqlatmp555hndcccdCg4OLvB5x44d0+eff65XXnlFR48elTFGN954o7766iv5+/sX4xEA7jFq1Ci70Cm7gKZ69eq67rrrFBYWptdee81+7KefflKHDh3yXadnz56aOXOmjDHq2rWrvvvuu2I7BgAAAAAAAAAAAAAAAAAAAAAAAJRslNIAAOBG7777rh588EEZYxQZGam//vorz5ycpTSzZs1St27dClwvKSlJVatWVVpamowx+vPPP3XxxRd7LD8AoGTaunWrOnfurL///luWZSkqKkqvvfaamjRpctbnbt68Wf/973+1cOFCGWNUu3Zt/fDDD7r00kuLITngPhkZGerQoYNWrlwpSbruuuv07bffKjAw8JzXOHHihHr27KnFixfLGKNrr71Wixcvlsvl8lRswO22bNmiK664wi4oq1ChgsaNG6fBgwfbBbI5v3OcqZTm888/V//+/SVJ/v7+iouLo0gWAAAAAAAAAAAAAAAAAAAAAAAAkiR2XQEA4EYnTpywx8HBwfnO8fX1tTeQJiUlnXG9SpUqKTIy0r69du1aN6QEAJQmx44dU6dOnXTw4EFJ0rPPPqsff/zxnAppJKlp06ZasGCBnnnmGVmWpf3796tjx46Ki4vzZGzA7V5//XWtWLFCknTZZZdp/vz551VII0mBgYGaN2+emjRpIsuytGLFCr3++uueiAt4zHPPPafMzExZlqVy5crphx9+0LBhw+xCmvPRunVre5yUlKSdO3e6MyoAAAAAAAAAAAAAAAAAAAAAAABKMUppAABwo4oVK9rj7OKZfwsICLDHBw4cOOualStXtsd///134cMBAEqlp59+Wn///beMMbr11ls1cuTIQq0zatQo3XrrrZKkgwcP6plnnnFnTMCjMjIycpXHvPHGGypXrlyh1vLx8bHXsixL48aNU2ZmpltyAp528uRJ/fDDDzLGyBijxx9/XNdee22h16tTp478/f3t29u3b3dHTAAAAAAAAAAAAAAAAAAAAAAAAJQB5/8nlAEAQIGCg4PtcWJiYr5zatasqZiYGEnS77//ftY1jx49ao/T0tKKmBAoOY4dO6bY2FjFx8crNTX1vJ/ftm1bD6QCSpakpCR9+umn9u3CFtLkfP6MGTNkWZY++eQTjRs3Tn5+fkWNCXjc6tWr7c9EQUFB6tChQ5HW69Chg6pUqaK4uDjFxsbql19+0TXXXOOOqIBHrVixwv5OYIzRf/7znyKvGRISYn93yf6eAgAAAAAAAAAAAAAAAAAAAAAAAFBKAwCAGzVo0ECSZFmW9u/fn++cpk2bauPGjbIsS0uWLDnjevv27dPu3btljJGUtQkbKM1+/PFHTZ48WatWrdLBgwcLvY4xRunp6W5MBpRMK1asUGpqqowxCg0N1aWXXlqk9Ro2bKjatWvrwIEDSk1N1fLly9W5c2c3pQU8Z+vWrZKyfv+Hh4cXeb3sdeLi4uz1KaVBaXDgwAFJWe/hiIgIhYSEFHnNypUr2+OEhIQirwcAAAAAAAAAAAAAAAAAAAAAAICygVIaAOcsMTFRa9eu1caNGxUbG6v4+Hilpqae1xrGGE2aNMlDCQHnNWrUSN7e3kpPT1dKSop2796tOnXq5JrTpk0bTZkyRZK0f/9+TZs2TX379s13veeff15SVsmNMabIZQSAU/7880/dfvvt2rRpk6Ss9zSAs9u5c6c9Dg0NdcuaNWvWtEsNdu7cSSkNSoXs8hhJOn36tFvWzLlOzvWBkiw2NtYeV69e3S1r5iz6c7lcblkTAAAAAAAAAAAAAAAAAAAAAAAApR+lNADOav369Ro3bpxmzJiRa7Pa+cou1aCUBmWZn5+frrzySq1evVqStHjx4jylND179tQDDzygtLQ0WZaloUOHyuVyqXfv3jLGSJISEhL01FNP6dNPP5UxRpZlKTAwUFdffXWxHxNQVOvWrdN1112n5ORk+1qQLef430U1OR/L73HgQnDy5El7HB8f75Y1c66Tc32gJAsICJCUdS2Ijo5WamqqypcvX+j1UlNTtXv3bvta4+/v75acgKf5+fnZY3f9Ds9ZdBMcHOyWNQEAAAAAAAAAAAAAAAAAAAAAAFD6UUoD4IxeeukljRw5UhkZGXYZQHZBRrZzKQ349xygLOvUqZNdSvP9999r0KBBuR4PDAzUgw8+qHHjxskYo4SEBPXr10//+c9/dPHFFystLU3btm1TWlqapH8KnR566CH5+PgU+/EARREfH68ePXro5MmT9rUgICBAnTt3VmRkpEaPHi0p6zpx1113qVatWoqLi9O2bdu0fv16nTp1yn5ew4YN1bt3b8eOBXBC1apVJf1TxBEbG2vfVxixsbGKjo62z6uirAUUp9q1a9vj5ORkffPNN7rjjjsKvd7XX3+t5ORkSVnXoIsuuqjIGYHiUL16dUlZ14X9+/cXeb0jR45o//799nUhJCSkyGsCAAAAAAAAAAAAAAAAAAAAAACgbHA5HQBAyTV69Gg9//zzSk9Pz3V/dkFG9o9lWbl+JOV6PPs5wIWiX79+krLe9/Pnz893s+gLL7ygxo0b2+eTZVmKjY3Vr7/+qg0bNig1NdV+TJJatmypZ555pliPA3CHt99+WwcPHrTfy126dNHu3bv1xRdf6OWXX5b0T3HZgAED9Morr+iDDz7QihUrdPjwYb3xxhsKDAyUZVnavn27du3apWeeeUYjRoxw7JiA4lSnTh1JWedJRkaGPvjggyKt99577yk9Pd3+bJa9PlDStW/fXj4+Pvbnpscff1wxMTGFWuvQoUP673//a19/fHx81L59ezemBTzn0ksvtccJCQnasGFDkdb77rvvJP3zPb9Vq1ZFWg8AAAAAAAAAAAAAAAAAAAAAAABlB6U0APK1ceNGPfvss7mKZ+6880798MMP+uOPP3KVzEydOlV//PGHVq1apYkTJ6pfv37y8fGx5zRs2FArVqxQdHS0du/e7dQhAcXmkksu0ffff6+vv/5aX375pTIzM/PM8fX11ZIlS9SuXbtcZU7Z/+YsdOrSpYt++OEHlStXrvgOAnCT999/376O1KtXTzNmzFBQUNA5Pdff31+PPPKItmzZoqZNm8qyLE2dOlV33323Z0MDJUi7du1UpUoVSVnXhJdfflnLli0r1FpLly7VK6+8Yl9jgoKC1K5dO7dlBTwpMDBQt9xyi12ccfjwYV1zzTXnXcixfv16tWnTRocPH7bXuvnmmxUYGOih5IB7NWnSRDVr1rR/l7/zzjuFXuv06dMaN26c/f3jsssuU3BwsLuiAgAAAAAAAAAAAAAAAAAAAAAAoJSjlAZAvkaPHi3LsuyyjE8++USffvqpbrzxRtWrVy/X3Bo1aqhevXq66qqrdO+992rq1Knau3evevXqJcuytH37dvXp00fJyckKDw934nCAYte1a1f17NlTPXv2LPB9HxwcrCVLlmj27Nnq16+fIiIiVLFiRVWoUEERERG644479OOPP2rOnDlslEaptHPnTsXExEjKKlt6/vnn5ePjc97r1KpVSwsWLFBISIgsy9KXX36pr776yt1xgRLJ29tbgwcPtsszUlNT1blzZ7311lvKyMg4pzUyMjL05ptvqkuXLjp9+rS91n333Sdvb28PHwHgPq+//roCAgIkZV1XoqOj1bJlS/Xr10/z5s3TyZMn831eUlKS5s2bp759+6pVq1aKjo62Cz38/f31+uuvF9sxAO5w22232d/XP/30Uy1YsKBQ6zz44IPasWOH/b1/0KBB7owJAAAAAAAAAAAAAAAAAAAAAACAUs5Y2TtPAOD/S0tLU2BgoNLS0iRJffv21dSpU3PNcblc9kbOn376SR06dMh3rWeffVavvPKKJCk8PFy//fab/P39PZgeAFBSTJ8+Xf369ZMkeXl5KT4+Xn5+frnm5Lye/Pjjj7rhhhsKXG/SpEkaPHiwjDFq1KiRfvvtN8+FB0qQU6dOqUmTJtq1a5ck2aUytWrVUt++fdW+fXs1btxYwcHB8vX1VXJysmJjY7V161YtW7ZM06ZN08GDB+3nWZaliy++WL/99psqVKjg8NEB52fevHnq1auXUlNTJf1zPkhZRTVhYWF5zoX9+/fbpRs5z4Py5cvr22+/VZcuXRw7HqAwYmNjVbduXSUlJcmyLPn6+mry5Mnq3bu3PedM39mPHz+uBx54QNOnT5eUdV6EhoZq9+7dhSoQBAAAAAAAAAAAAAAAAAAAAAAAQNlEKQ2APFauXKm2bdtKytrY+csvv6hly5a55pxrKY0kRUVFadGiRTLG6PHHH9err77qufAAgBLj3Xff1YMPPihjjCIjI/XXX3/lmZPzejJr1ix169atwPWSkpJUtWpVpaWlyRijP//8UxdffLHH8gMlSXR0tDp06KC9e/fahRqS7PPnTHLOtSxL4eHhWrRokerUqePRzICnLF68WHfeeadiYmLsc+Df/7WR8zzJeV/23OrVq2vq1Km6/vrriyc04GY5y/qyy5auvvpq9e/fX82bN1eLFi0kZb3vJ0+erIYNG+qvv/7S4sWLNW3aNJ08edI+R7y8vDRv3jxFRUU5eUgAAAAAAAAAAAAAAAAAAAAAAAAoYVxOBwBQ8uQsDfD19c1TSPNvaWlpZ3z8xRdflJS1+XPChAlKT08vekgAQIl34sQJexwcHJzvHF9fX3tDdFJS0hnXq1SpkiIjI+3ba9eudUNKoHSIjIzUqlWr1LFjR7t8IGfBRkE/Uu4ijhtuuEErV66kkAalWocOHbR9+3YNHTpUFSpUyPVez3lu/Pu2ZVkqX768hgwZou3bt1NIg1Lt3nvv1TPPPGNfEyzL0i+//KJhw4bl+g5vWZbuuecetWrVSnfccYcmTZpkf+bKPjdee+01CmkAAAAAAAAAAAAAAAAAAAAAAACQB6U0APKIi4uTlLVBLefm/5xcrn9+faSmpp5xvdatW6tKlSqSpISEBK1evdpNSQEAJVnFihXtcXZhwL8FBATY4wMHDpx1zcqVK9vjv//+u/DhgFKoZs2amj9/vqZNm6ZWrVrlKp6R8hZwSP8U1rRq1UpffvmlFixYoFq1ajkRH3CrwMBAvffee/r777/13nvvqU+fPgoPD5ekPMVM4eHh6t27t9599139/fffev/99xUUFORkfMAtRo0apUmTJuX5zJWzvCy7sObfZWWWZalixYr68ssv9cgjjzh0BAAAAAAAAAAAAAAAAAAAAAAAACjJvJ0OAKDkyVky4+/vn+8cf39/nThxQsYYxcbGnnXNsLAwu+xm+/btatOmjXvCAgBKrODgYHucmJiY75yaNWsqJiZGkvT777+fdc2jR4/a47S0tCImBEqnPn36qE+fPvrjjz+0dOlSrVmzRtHR0YqPj1dSUpL8/PxUuXJl1alTRy1atFD79u116aWXOh0b8IjAwEANHTpUQ4cOlZRVyJGQkGCfC4GBgbmKmoCy5p577lH79u316quv6rPPPlNKSook5SqgyS6hyb7f29tbd911l5599tkCi2gBAAAAAAAAAAAAAAAAAAAAAAAASmkA5BEQEGCPk5OT850TGBioEydOSJL27dt31jW9vLzs8bFjx4qYEABQGjRo0EBS1ubn/fv35zunadOm2rhxoyzL0pIlS8643r59+7R79267XCAoKMi9gYFSpkGDBmrQoIFdxgEgq4AjMDBQgYGBTkcBik1kZKQ++OADjR49WkuXLtWKFSu0fft2HTt2TPHx8fL19VXVqlUVGRmp6667TjfccIOqV6/udGwAAAAAAAAAAAAAAAAAAAAAAACUcJTSAMgjNDTUHh8/fjzfOZdccoldRrNmzZqzrrl792577O3Nrx5cODIzMzV37lzNnz9fGzZs0P79+3XixAmlpKSc91rGGKWnp3sgJeAZjRo1kre3t9LT05WSkqLdu3erTp06uea0adNGU6ZMkSTt379f06ZNU9++ffNd7/nnn5eUVXJjjNGll17q0fwAAAClSVBQkHr06KEePXo4HQUAAAAAAAAAAAAAAAAAAAAAAABlgMvpAABKnkaNGknK2vR/4MABnTp1Ks+cpk2b2nNWrFhRYHmNJC1cuDDX4/xFdlwoZs2apYiICN1yyy368MMPtWbNGh06dEjJycmyLKtQP0Bp4ufnpyuvvNK+vXjx4jxzevbsqfLly8sYI8uyNHToUH311Ve53u8JCQl64IEH9Omnn8oYI0kKDAzU1Vdf7fmDAAAAKEGWL1+uW2+91f5ZuXKl05EAAAAAAAAAAAAAAAAAAAAAAABQRlFKAyCPSy65REFBQZKySmc2b96cZ07Xrl0lScYYpaSk6Iknnsh3rbi4OP3nP/+xSwQkqVWrVh5IDZQsL730knr27KkDBw7Y5RrGmFznwrkqzHOAkqJTp072+Pvvv8/zeGBgoB588EFZliVjjBISEtSvXz/VqFFDrVu31pVXXqkaNWrogw8+kCR73kMPPSQfH59iOw4AAICSYO3atZo1a5Zmz56tefPmqUmTJk5HAgAAAAAAAAAAAAAAAAAAAAAAQBllrOyd8gCQQ8+ePTVz5kwZY/Tcc8/phRdeyPW4ZVm6+OKLtWfPHrsgoGPHjho2bJjq16+vtLQ0rVy5UmPGjNG+ffvsUo2mTZtqw4YNDhwRUHzmzJmj7t27S/qnUCb7chsWFqbq1avLz8+vUGsvWbLEPSGBYrJz507Vr19fkuTt7a1du3bpoosuyjUnOTlZV111lbZu3SpjTK4ip5wfVbNvt2rVSsuXL1e5cuWK70AAACXa6dOndeLECaWkpKgw/80RFhbmgVSA+40dO1ZPPvmkJOniiy/Wjh07HE4EAAAAAAAAAAAAAAAAAAAAAACAsopSGgD5+uyzzzRgwAAZY9SgQQNt27Ytz5zZs2erR48edklAdvlGTtn3W5Yll8uluXPnqmPHjsVxCIBjGjVqpO3bt9vv/dDQUD333HPq06ePgoKCnI4HFLu5c+fq1KlTkqQrr7xS4eHheeYcO3ZMvXr10rJlyyQpzzUl+yNrly5dNHXqVAUGBno4NVDy/fHHH9q/f3+Rijj69+/vgWSA5x0/flyff/655s+frw0bNujo0aOFXssYo/T0dDemAzwn53f1K6+8Ur/++qvTkQAAAAAAAAAAAAAAAAAAAAAAAFBGUUoDIF9JSUmKiIhQamqqpKxCgbZt2+aZ9/TTT2vMmDF2ecC/f6Vkl3JI0iuvvGL/RXegrPrjjz/UsGFD+71/6aWXatmyZapatarT0YBS4fvvv9e0adO0evVqHT58WJZlKSQkRK1bt1b//v0VFRXldETAUT/++KMmTJigBQsWKDk5ucjrZWRkuCEVULzeeustPffcc/Y5UNT/1jDGcC6g1FiyZImuv/56GWMUHh6u3bt3Ox0JAAAAAAAAAAAAAAAAAAAAAAAAZRSlNACK7JNPPtGTTz6pw4cP5/t4eHi4XnvtNfXq1auYkwHF79NPP9Xdd98tKWuD85IlS/ItdAIA4HzEx8fr7rvv1vfffy+paCUc2cVpFHGgNBo6dKgmTpxonwMFlWOeD84FlCZpaWkKCQlRfHy8jDE6cOCAatas6XQsAAAAAAAAAAAAAAAAAAAAAAAAlEHeTgcAUPoNGDBAt99+u5YuXarVq1fr8OHDsixLISEhat26tdq1aydvb37d4MJw5MgRe1ytWjUKaQAARZacnKyOHTtq3bp1dplMdrFMYdBLitLqk08+0YQJEyT9U65kWZaCgoJ02WWXqXr16vLz83M4JeBZPj4+6t27tyZOnChJ+vDDD/XCCy84GwoAAAAAAAAAAAAAAAAAAAAAAABlEi0RANyiXLlyioqKUlRUlNNRAEdlZGRIytooHR4e7nAaAEBZMHr0aK1duzZXGU2FChV044036vLLL6eIAxeM559/XtI/hTRNmzbVmDFjFBUVJZfL5XA6oPg8//zz+uqrr5SQkKDXXntNHTt21NVXX+10LAAAAAAAAAAAAAAAAAAAAAAAAJQxlNIAAOBGoaGh9jglJcXBJACAsiAtLU1vvfWWXcIhSYMHD9arr76qypUrOxsOKEYbN27U/v37ZYyRJLVu3Vo//fSTKlas6HAyoPjVqlVLX3/9tW655RYlJyerU6dOev311zVo0CCnowEAAAAAAAAAAAAAAAAAAAAAAKAMMVb2zkYAAFBkO3bsUIMGDSRJAQEBiouLk8vlcjgVAKC0Wrx4sW644Qa7iOPuu+/WpEmTHE4FFL/Jkyfr3nvvlSQZY7R+/XpdfvnlzoYCHLJv3z5J0oYNGzRs2DAdPnxYxhiFhYXptttuU8uWLRUZGamAgACVK1fuvNYOCwvzRGQAAAAAAAAAAAAAAAAAAAAAAACUQt5OBwAAoCypV6+emjdvrvXr1ysxMVFLly5Vhw4dnI4FACil/vrrL0mSZVlyuVx6+eWXHU4EOOPo0aP2ODQ0lEIaXNAiIiLssjIpq6jJsizt3btXr732WqHXNcYoPT3dHREBAAAAAAAAAAAAAAAAAAAAAABQBlBKA+C87dixQ2vWrFF0dLTi4+OVlJSkSpUqqXLlyoqMjFTLli1Vr149p2MCjnn55ZfVqVMnSdJzzz2n6667LtemUeBClZmZqblz52r+/PnasGGD9u/frxMnTiglJeW812LTNC4Ux44dk5T1nr/44osVEhLicCLAGdmfpYwxqlWrlsNpgJLBsqxc50b2fQAAAAAAAAAAAAAAAAAAAAAAAIA7UEoD4JwcOHBAH3zwgSZPnqyYmJizzg8JCdE999yjIUOG6KKLLiqGhEDJceONN+rxxx/XuHHjtHr1ag0ePFgffvihvLy8nI4GOGbWrFl66KGHdPDgQUlsmAbOVcWKFe1xUFCQg0kAZ4WFhdnjpKQkB5MAJQufqQAAAAAAAAAAAAAAAAAAAAAAAOApxmL3CoAzyMzM1Ouvv64XX3xRKSkpeTa8Zf81dinvZjhjjCpUqKARI0boscceo5ADF5ynnnpKr776qowxatGihUaOHKkbb7zR6VhAsXvppZc0YsQI+zpxpmvH2RhjZFmWjDHKyMhwa06gJJozZ466d+8uY4zq1q2rHTt2OB0JcMT+/fsVHh4uSfL19VVcXJx8fHwcTgU445577vHY2pMnT/bY2gAAAAAAAAAAAAAAAAAAAAAAAChdKKUBUKC0tDT16tVLc+fOzVMkcKZfHf+eY4xRp06dNGPGDJUvX97DqQHnDRw40B4vW7ZM0dHR9nkRGBioZs2aqXr16qpQocJ5rWuM0aRJk9yaFfC07EINKe/1ISwsTNWrV5efn1+h1l6yZIl7QgIlWEJCgqpXr660tDR5eXnpyJEjCgoKcjoW4Ijrr79eS5YskTFGM2bM0M033+x0JAAAAAAAAAAAAAAAAAAAAAAAAAAosyilAVCgm2++Wd9//72krCKB7F8XderUUbt27dS4cWMFBwfLz89PJ0+e1LFjx7RlyxYtX75cu3fvzvU8Y4y6du2q7777zrHjAYqLy+Wyyzey5bzc/vuxc5F9HmVkZBQ5H1CcGjVqpO3bt9vXg9DQUD333HPq06cPxRrAORo4cKCmTJkiY4zefPNNPfTQQ05HAhyxdu1atW7dWpmZmbr00ku1bt268y75AwAAAAAAAAAAAAAAAAAAAAAAAACcG0ppAOTrk08+0T333GOXZ1iWpauvvlqvvvqq2rRpc9bnr1ixQk888YRWr16dq5hm8uTJ6t+/v6fjA47Kr5SmqCilQWn0xx9/qGHDhvZ14NJLL9WyZctUtWpVp6MBpUpMTIwaN26s48ePq0qVKlq7dq0iIiKcjgU4Yvz48XrkkUdkjFGnTp00bdo0+fv7Ox0LAAAAAAAAAAAAAAAAAAAAAAAAAMocl9MBAJRML730kl0iIEmPPvqofv7553MqpJGka6+9VqtWrdIjjzxil2lYlqVRo0Z5MjZQYliW5dYfoDRas2aNpH9Kld5//30KaYBCCAkJ0cyZM1WxYkUdO3ZM119/vTZu3Oh0LMARDz30kN5//32VK1dOP/zwg5o0aaIJEybo+PHjTkcDAAAAAAAAAAAAAAAAAAAAAAAAgDLFWOx0B/AvGzZs0JVXXiljjCTppptu0uzZswu9Xvfu3TVnzhxJkjFGa9euVbNmzdySFSiJ9u7d67G1w8PDPbY24G7jxo3T//3f/0mSqlevrpiYGIcTAaXbhg0b1KdPH+3evVve3t7q1auX+vTpoyuvvFLVq1eXj4+P0xEBj+rQoYM93rlzpw4ePCgp6zuGMUYRERGqXr26KlSocF7rGmO0aNEit2YFAAAAAAAAAAAAAAAAAAAAAAAAgNLO2+kAAEqerVu3SpIsy5IxRi+//HKR1nv55Zc1Z84cu+Rm69atlNKgTKM4BsiSkZEhKWuzP+cFUHheXl65bhtjlJ6erunTp2v69OmFXjd7HaC0WLp0qf2dQpI9tixLlmVp9+7dio6OPq81s7/zAAAAAAAAAAAAAAAAAAAAAAAAAAByczkdAEDJc+jQIXscGhqqxo0bF2m9yy67TLVq1ZJlWXnWBwCUXaGhofY4JSXFwSRA6ZZduJH9WUrKKuPIeX9hf4CywBhj/wAXgpUrV8rLy8v+WbJkSaHWWbx4sb2Gt7e31q9f7+akAAAAAAAAAAAAAAAAAAAAAAAAKM0opQGQh7e3t6SszZ05CwWKombNmnnWBwCUba1atZKUVaixb98+ZWZmOpwIKL2yyzbcUShDcQdKM3eUMVHMhNLuww8/tN/DLVq00HXXXVeodTp06KArrrhClmUpMzNTEydOdHNSAAAAAAAAAAAAAAAAAAAAAAAAlGY0QwDIIzw83B7Hx8e7Zc0TJ07Y47CwMLesCQAo2erVq6fmzZtr/fr1SkxM1NKlS9WhQwenYwGlTtu2bSmSASTKzQBlnQfz5s2zrwt33HFHkdbr37+/NmzYIGOMvvvuO33wwQfuiAkAAAAAAAAAAAAAAAAAAAAAAIAywFj8aXAA//L333/roosukmVZ8vb21qFDhxQcHFzo9WJjYxUaGqr09HQZY7R//36Fhoa6MTEAoKRasGCBOnXqJGOMrrrqKq1cuZJyDQAAgELavHmzrrjiCkmSMUY7d+5UnTp1Cr3erl27dMkll9jr/f7776pfv75bsgIAAAAAAAAAAAAAAAAAAAAAAKB0czkdAEDJExoaqhtuuEGSlJGRoffff79I67333nt2IU1UVBSFNLhg/fnnn5o4caLuvfdeRUVF6corr1T9+vV15ZVXKioqSoMGDdJHH32kHTt2OB0VcJsbb7xRjz/+uCzL0urVqzV48GBlZGQ4HQsAAKBU2r59uz2uXLlykQppJKlu3bqqXLmyfXvbtm1FWg8AAAAAAAAAAAAAAAAAAAAAAABlh7fTAQCUTGPHjlXLli2Vnp6ul19+Wddee63atWt33ussXbpUr7zyiowx8vHx0WuvveaBtEDJ9t133+n111/XypUrc91vWZY9NsZIkiZPnixJatu2rR577DHddNNNxRcU8JCxY8fKy8tLr776qiZPnqytW7dq5MiRuvHGG52OBgAAUKrExMRIyvr+UKtWLbesWbt2bcXHx0uSDh486JY1AQAAAAAAAAAAAAAAAAAAAAAAUPpRSgMgX02bNtWUKVM0YMAApaamqnPnznrllVf04IMPysvL66zPz8jI0Pjx4/XMM88oLS1N3t7emjJlii677LJiSA+UDCdOnNCwYcM0ffp0Sf+U0GQX0GT/m1P2nGXLlmn58uXq27ev3nvvPQUGBhZTasC9Bg4caI8jIyMVHR2ttWvXqnPnzgoMDFSzZs1UvXp1VahQ4bzWNcZo0qRJ7o4LAABQoiUnJ9tjPz8/t6yZc52kpCS3rAkAAAAAAAAAAAAAAAAAAAAAAIDSz1jZu98BIB9Lly7VPffco71799p/ib1v375q3769GjdurODgYPn6+io5OVmxsbHaunWrli1bpmnTpungwYOyLEsRERGaPHmy2rVr5/ThAMUmKSlJHTp00Pr162VZll1Acy6X3ZxzjTFq3ry5lixZ4rZNp0BxcrlceQqYcp4H+ZUznU32uZGRkVHkfAAAAKXJu+++qwcffFDGGNWtW1c7duwo8pr169fXzp07ZYzRG2+8oYcfftgNSQEAAAAAAAAAAAAAAAAAAAAAAFDaUUoDXGC8vLwK/dzsXxfnUiBQ0FxjjNLT0wudASgtunbtqvnz50vKet9bliVfX1/dcsstuv7669W0aVNVrVpVfn5+OnnypGJjY7V582YtXrxYM2fOVHJysv08Y4y6dOmi77//3uGjAs5ffqU0RUUpDQAAuFB99dVX6tu3rySpfPnyiouLU8WKFQu9XnJysqpUqaLTp09Lkj777DPdfvvtbskKAAAAAAAAAAAAAAAAAAAAAACA0o1SGuAC43K5zvs5/y4TOJdfGwU9hxIBXAjmzJmj7t272+eBZVkaOnSoXn75ZQUFBZ31+fHx8Xr22Wf1/vvv2883xui7775T165dPZodcLfCXHfOBdcTAEC23bt3a+PGjYqNjVV8fLxSU1PPe43nn3/eA8kA9/vtt990+eWXS8r6PDRjxgzdfPPNhV5v5syZ6tmzp73eqlWr1KpVK3dEBQAAAAAAAAAAAAAAAAAAAAAAQClHKQ1wgXG5XDLGnFOxjCdQIoALQevWrbV69WpJWe/5Dz/8UIMGDTrvdT7++GMNHjxYUlYxzdVXX62ff/7ZrVkBT9u7d6/H1g4PD/fY2kBJ98cff2jVqlVFKuIwxmjRokUeSgh41sGDB/XOO+9oypQpOnLkSJHX4zsKSpOQkBAdPXpUlmWpWbNmWrduXaHXat68uTZt2iTLshQUFKTY2Ng8JbMAAAAAAAAAAAAAAAAAAAAAAAC4MHk7HQBA8Wrbti0bzAAPio2N1Zo1a+zzbNCgQYUqpJGkgQMH6tdff9XEiRMlSb/++qtiY2NVtWpVt+UFPI3iGMC9Zs+erTFjxmjNmjVFWseyLD4TotT6/PPP9cADDygpKemMZZvZ7/GC5mSXdXIuoLS55ZZbNGHCBEnSxo0b9eijj+qNN94473UeffRRbdy4UcYYGWN0yy23cD4AAAAAAAAAAAAAAAAAAAAAAADAZqwz7eACAADnZfbs2erRo4ekrI3Of/31lyIjIwu9XnR0tOrWrWtvDp05c6a6d+/ulqwAgNIjIyNDQ4YM0eTJkyXlLpXJ+ZUuvzKB/B7Pfn5GRoYnYwNu98knn2jgwIH2e7ig939+/9VR0OOcCyht9u/fr0suuUSnT5+2z4U77rhD77zzjgICAs76/ISEBD3wwAP64osvJGWdDz4+Ptq+fXuRvrsAAAAAAAAAAAAAAAAAAAAAAACgbPF2OgAAAGVJTEyMpKzNzeHh4UXe1BkZGamIiAjt2bNHxhgdOnTIHTEBAKXMI488oo8//liS8pTRFLaIAyhtdu3apaFDh+YqpLn22mt1xx13KCIiQp06dbLf7+PGjVPjxo0VFxenbdu2afHixfrll18kZZ0TDRs21JgxY+Tv7+/kIQGFctFFF+mpp57Siy++aJ8LU6dO1Xfffaf+/furS5cuuvLKK1W1alX7ObGxsVq3bp3mzZunzz77TAkJCbmuI0888QSFNAAAAAAAAAAAAAAAAAAAAAAAAMiFUhoAANwoLi7OHlerVs0ta1arVk179uyRJB0/ftwtawIASo9ly5bp3Xfftcs2fHx89N///ld33nmnIiIiVKFCBfuxhQsXqk2bNrmKOD7++GMdPnzYLuKYOnWqmjZt6uQhAYUyZswYpaam2rdHjhypZ599Nt+5l19+uTp06GDfHjVqlDZt2qT7779fq1ev1vbt2/Xkk09qwYIFCg0N9Xh2wN1GjBihrVu36ttvv7WLaRISEvTuu+/q3XfflZRVNuPr66vk5ORcpWQ5y2gsy1KfPn304osvOnIcAAAAAAAAAAAAAAAAAAAAAAAAKLlcTgcAgGyrVq1SnTp1VKdOHdWtW9fpOEChBAYG2uOcBTVFkXOdgIAAt6wJACg9Ro8eLSmrRMDLy0vff/+9Ro4cqXr16snHxyfPfB8fH4WEhOj666/Xyy+/rL179+rxxx+XJG3fvl1t27bVihUrivUYgKLKyMjQtGnTZIyRMUYdO3YssJCmIJdffrlWrlypu+66S5Zlafv27eratatOnz7todSAZ3355Zd6+OGHZVmWfW5IWdcLy7KUmZmppKQkZWZm2vdJsudJ0mOPPabPP//ckfwAAAAAAAAAAAAAAAAAAAAAAAAo2bydDgAA2VJSUrRnzx5JuTfJAaVJjRo1JGVtBN2zZ49iYmIUEhJS6PViYmIUHR1tnxPZ6wOl3Z9//qnly5dr9erV2rdvn44fP67ExET5+/srKChI4eHhuuqqq9S2bVvVq1fP6biAY06ePKlFixbZ14H77rtPN9xww3mt4ePjo7Fjx6pu3boaNmyYEhMT1aNHD23bto3rCkqNDRs26OTJk5Kyviv897//LdQ6LpdLkydP1u+//67169frt99+07hx4/TUU0+5My5QLLy9vfXmm2+qU6dOevHFF7V69Wr7sfy+U+cspmnTpo1GjBih66+/vtjyAgAAAAAAAAAAAAAAAAAAAAAAoHShlAYAADdq2bKlpKxNoJmZmXrnnXf00ksvFXq9d955R5mZmfaarVq1cktOwCnfffedXn/9da1cuTLX/dkbpKV/NlFPnjxZktS2bVs99thjuummm4ovKFBCrF69WhkZGZKyzo2hQ4cWeq0hQ4Zo8eLF+vrrr3X8+HE999xzmjBhgruiAh71xx9/2OPy5curXbt2Z5x/+vTpAh9zuVx66aWX1LlzZ1mWpfHjx+vJJ5+kGBOlVseOHdWxY0etXbtWCxYs0IoVK7Rr1y7FxcXZpX9VqlTRJZdcomuvvVadOnXSFVdc4XRsAAAAAAAAAAAAAAAAAAAAAAAAlHCU0gAA4EYXXXSRGjdurG3btsmyLL322mtq166doqKiznutxYsX67XXXpMxRpZlqVGjRqpdu7YHUgOed+LECQ0bNkzTp0+X9E8JTXYBQH5FANlzli1bpuXLl6tv37567733FBgYWEypAeft3bvXHgcGBqpx48ZnnJ+amnrGx5955hl9/fXXsixLU6dO1fjx41WhQgW3ZAU8KS4uTlLW9SIyMlIulyvPnJzXklOnTp1xvRtuuEEBAQFKSEjQkSNHtG7dOrVo0cK9oYFi1qJFC97HAAAAAAAAAAAAAAAAAAAAAAAAcJu8u7gAAECRPPLII7IsS8YYnT59Wt27d9dbb72lzMzMc3p+Zmamxo8fr5tuuknp6en2WsOHD/dwcsAzkpKSFBUVpenTp+cpo7Esq8Cf7HnZxUzTpk1TVFSUTp486dixAMUtZxFHWFhYvnO8vf/pGj1bEUeTJk1Uo0YNe+6qVavclBTwrJy/+wsqJ6tUqZJ9/YiPjz/jel5eXgoPD7dvb926teghAQAAAAAAAAAAAAAAAAAAAAAAAKAMoZQGAAA3u+eee3TFFVdIyioRSE1N1WOPPaaLL75YL7zwgpYvX55no/SJEye0fPlyvfDCC6pXr56GDx9uFwsYY9SsWTPdc889xX0ogFvcdtttWrdunV2wZFmWKlasqNtvv12TJk3SunXrtGfPHh09elR79uzRunXrNGnSJN1xxx2qWLFiruetX79effv2dfqQgGKTnp5uj/38/PKd4+/vbxdxHDly5Kxr1q5d2x7/+eefRUwIFA9/f397nJqamu+cgIAAe7x///6zrlmhQgV7fC7nDgAAAAAAAAAAAAAAAAAAAAAAAABcSLydDgAAQFljjNF3332n1q1ba//+/XaZxp49ezRq1CiNGjVKkuRyueTr66vk5GRlZmbaz88uFsh+Xnh4uGbPnu3IsQBFNWfOHM2fP1/GGElZ7++hQ4fq5ZdfVlBQUJ75wcHBCgsLs4uY4uPj9eyzz+r999+3z4l58+Zp7ty56tq1a3EfDlDsAgMD7XFSUlK+cypXrqzjx49Lkvbu3XvWNXNec/5dkgaUVCEhIfb4xIkT+c6pU6eODh48KElav379Wdfct2+fPc6+TgEAAAAAAAAAAAAAAAAAAAAAAAAAsricDgAAQFlUq1YtLVu2TC1btpRlWTLG2IUa2T8ZGRlKTExURkZGrvtzzm3VqpUWL16s0NBQpw8JKJRXXnlF0j9lSxMmTNB7772XbyFNfipXrqx33nlHEydOlPRPWVP2ukBZFxYWJinrHIqLi8t3ToMGDezxr7/+esb1MjIytHPnTruAo3z58m5KCnhWw4YNJWWdC/v27dPp06fzzGnatKk9Z+nSpUpOTi5wvTVr1ujw4cP2uVC1alUPpAaKl2VZ2rBhg6ZOnaq3335bL730kkaOHKk9e/Y4HQ0AAAAAAAAAAAAAAAAAAAAAAAClEKU0AAB4SEREhH7++We99tprql27tl3KIckunsn5k82yLF100UV6/fXX9fPPPysyMtKJ+ECRxcbGas2aNfZ7fNCgQRo0aFCh1ho4cKAGDRpkn0e//vqrYmNj3RkXKJEaNWpkj//++28lJSXlmXP55ZdLyrp+rFy5Un///XeB682cOVNJSUn2uVSzZk33BgY8pH79+qpUqZKkrHKlbdu25Zlz4403Ssr6nJWQkKDRo0fnu1ZqaqoefvhhSf+UpjVv3twTsYFisXnzZvXv31+VK1dWixYt1L9/fz3yyCMaMWKEXnzxRe3evTvf540dO1YDBw7UwIED9cILLxRvaAAAAAAAAAAAAAAAAAAAAAAAAJR4lNIAAOBBXl5eeuyxxxQdHa25c+fq//7v/9SuXTuFh4crICBALpdLAQEBCg8PV7t27fTEE09o3rx52r17t4YPHy6Xi0s1Sq+ff/5ZmZmZ9ob/J598skjrZT/fGCPLsrRq1aoiZwRKuoiICIWEhNi3161bl2fOLbfcIinr3EhPT9ewYcOUnp6eZ97u3bv18MMP5ypCu+aaa9wfGvAAb29vXXvttfbtH3/8Mc+cTp06qXr16pKyymZeeeUV3X///dqyZYvS0tKUlJSkH374QW3atNGvv/5qnwt16tRR06ZNi+dAADdKS0vTAw88oGbNmmnq1KlKTEyUZVm5fs4kJCREU6ZM0SeffKKXXnpJe/bsKZ7gAAAAAAAAAAAAAAAAAAAAAAAAKBXY6Q4AQDFwuVzq3LmzxowZoyVLlmj37t06fvy4Tp8+rePHj2v37t1asmSJRo8erU6dOlFGgzIhJiZGUlZRRnh4uCIjI4u0XmRkpCIiIuwN1ocOHSpyRqA06NChgz2eN29ensdbtGihyy67zL49Z84cNW/eXG+//bYWLFigOXPm6Mknn1SzZs0UExMjy7JkjFHbtm0VFhZWLMcAuMNNN91kj2fOnJnncW9vb7388sv2e9yyLH344Ye6/PLLVbFiRQUGBqpr165av369/bgxRi+88EIxHgXgHsnJyWrXrp0++OCDfMtnchaQFeT2229XtWrV7AKbqVOneiIqAAAAAAAAAAAAAAAAAAAAAAAASil2vAMAAMAj4uLi7HG1atXcsmbOdY4fP+6WNYGS7tZbb7XHX3/9db7lA++88449tixLW7Zs0SOPPKLOnTvr5ptv1muvvaaEhAR7Trly5fTqq696NjjgZn369JGXl5csy9KaNWu0Zs2aPHPuvfde3XHHHXbhjCS7cCP7J2dZx5AhQ3THHXcU2zEA7tKvXz/9+uuv9m1jjHr06KH3339fc+bMyfda8W/e3t7q0aOHfXv+/PkeyQoAAAAAAAAAAAAAAAAAAAAAAIDSiVIaAAAAeERgYKA9zllQUxQ51wkICHDLmkBJ17VrVzVr1kwNGzZUpUqVtGrVqjxzrr32Wr333nuSsooJjDG5Sjiy77csS+XKldOECRPUsmXLYj0OoKiCg4N19OhR+6dJkyb5zvv00081YsQIVahQId9iDsuyVKlSJY0dO9Y+b4DS5Pvvv9f3339vFyxdcskl2rRpk7799lsNGTJEXbp0kaRcBUwF6datmyTZZU8pKSmeCw4AAAAAAAAAAAAAAAAAAAAAAIBSxdvpAAAAACibatSoISlrk/OePXsUExOjkJCQQq8XExOj6Ohoe4N19vpAWVe+fHmtW7furPOGDBmievXq6bHHHtOmTZtyPZZdzHH11Vdr3Lhxuvrqqz0RFfC4ypUrn3WOMUYjRozQsGHDNHv2bK1evVqHDx+WZVkKCQlR69at1aNHD1WpUsXzgQEPGDVqlCTZ7+mlS5eqZs2ahVqrRYsW9jgjI0Pbt29Xs2bN3JLz/7F339FRVesbx58zSSippBAgQEKQjkpELqJIMQqogHTByhVEsQKKvSFybSgKFrh4RcVCESleikoHG03KjyYxBIJgeiENQpLz+yMr5ybSUmYyk+T7WWsW58zs8+53r5WTmWGt/QQAAAAAAAAAAAAAAAAAAAAAAABVG6E0AAAAcIjOnTtLKgwHKCgo0Pvvv68pU6aUu97777+vgoICq+ZVV11llz6B6uS6667Tb7/9pqioqHMGcVxyySXObhGoNMHBwRozZozGjBnj7FYAu4mPj9eOHTuskL5XXnml3IE0UuF9Ur9+fSUmJkqSfv/9d0JpAAAAAAAAAAAAAAAAAAAAAAAAIIlQGgAAADhI06ZNdemll2rfvn0yTVNTp05Vjx491KtXrzLXWrdunaZOnSrDMGSaptq3b68mTZo4oGugemjZsqVatmzp7DYAAHb2008/yTRNSZKHh4dGjBhR4ZpBQUFWKE1SUlKF6wEAAAAAAAAAAAAAAAAAAAAAAKB6IJQGAIBSat68eYlzwzAUHR190XH2cr75AFc2fvx43XvvvTIMQ2fOnNEtt9yi1157TY8++qhsNttFry8oKND777+vp59+Wnl5eTJNU4ZhaMKECZXQPQAAgGuJi4uTVPjdoEWLFvLy8qpwTV9fX+s4MzOzwvUAAAAAAAAAAAAAAAAAAAAAAABQPRBKAwBAKR05ckSGYcg0TUmFG0FLM85ezjcf4MruueceffDBB9q1a5cMw9Dp06f1+OOPa8aMGbr77rsVGRmpyy+/XPXq1bOuSU9P1+7du7Vu3Tp98cUXiomJscJoDMNQx44ddc899zhvUQAAAE6Snp5uHRcPk6mIrKws67hu3bp2qQkAAAAAAAAAAAAAAAAAAAAAAICqj1AaAADKqLSBM/YMkbF3wA1QWQzD0LfffqtrrrlGx44ds+6fI0eO6JVXXtErr7wiSbLZbPL09FR2drYKCgqs64uHQJmmqbCwMC1btswpawEAAHA2f39/67h4QE1FxMXFWceBgYF2qQkAAAAAAAAAAAAAAAAAAAAAAICqj1AaAABKKTQ0tFRBM6UdB9QUjRs31saNGzVixAht3brVuj+Khy3l5+crIyPjrGuLj73qqqv01VdfKSQkpHIaBwAAcDENGjSQVPjZKCYmRrm5uapVq1a560VFRSkpKck6b9q0aYV7BAAAAAAAAAAAAAAAAAAAAAAAQPVAKA0AAKV05MgRu44DapJmzZrpp59+0rvvvqsZM2bo2LFj1mvnCnEqCqwxTVOhoaEaN26cxo0bJ5vNVmk9A44WGxvr7BYUGhrq7BYANW/e3KnzG4ah6Ohop/YAlFanTp2s49zcXK1bt0433nhjuet9+eWX1nGtWrXUpUuXCvUHAAAAAAAAAAAAAAAAAAAAAACA6sMwi3b8AoCT/fbbb3r88cet8/Xr1zuxGwCAoxQUFOj777/Xxo0btWXLFh09elSpqanKzMyUt7e3/P39FRYWpi5duqhHjx7q3bs3YTSolmw22zlDmSqLYRjKy8tz2vxAkaJ7wVn/PWEYhvLz850yN1Ael156qQ4cOCBJ6t69+zm/Oxd/j1m9erUiIyPPGvPXX3/p0ksvVVpamiQpMjJSq1evdlzjAAAAAAAAAAAAAAAAAAAAAAAAqFIIpQEAAAAAJyCIAyjkzIAm0zS5F1DlTJ8+XRMmTLDum8mTJ+u5554rMeZioTQZGRm68cYb9csvv0gqfE/49ttv1bdv30pYAQAAAAAAAAAAAAAAAAAAAAAAAKoCQmkAAAAAwAkIpQEKNWvWzGmhNEViYmKcOj9QFmfOnFHr1q119OhRK1hp7NixevXVV+Xn5yfpwqE033//vcaPH69Dhw5Zz/3jH//Qr7/+WrkLAQAAAAAAAAAAAAAAAAAAAAAAgEsjlAbAOZ0+fVoHDhywHvHx8crMzFRmZqZycnLk7e2tevXqqV69emrRooU6deqkiIgI1apVy9mtA063adMm67hDhw7WxtDySE9P1+7du63z7t27V6g3AIDruOeee5zdgj755BNntwAAKIctW7YoMjJSp06dsoJpPD091b9/f1155ZV64oknJBUGkI0fP16NGjXSH3/8oXXr1ik6Otq6xjRNBQQEaNu2bQoPD3fyqgAAAAAAAAAAAAAAAAAAAAAAAOBKCKUBYMnMzNQ333yjxYsXa82aNTp16lSZrnd3d1fPnj01ZswYDRgwQB4eHg7qFHBtNptNhmFIklavXq3IyMhy11q7dq169+4tqXBDaV5enl16BAAAAFC1/fe//9WIESOs7+5FQTNFx0WKniv+fFEgjZ+fn5YsWaKePXtWXuMAAAAAAAAAAAAAAAAAAAAAAACoEmzObgCA8+Xm5mratGlq3ry5Ro0apeXLlysnJ0emaV7wIanE+ZkzZ7RmzRoNHz5cTZo00Zw5c5y8MsB57Jn59vf7DgAAAAD69++vrVu3ql27diUCaaTC0JmiR/HvEsWfa9++vbZs2UIgDQAAAAAAAAAAAAAAAAAAAAAAAM6JUBqghjt48KAiIiL0xBNPKCkp6ayNaud7SGf/5fXizycmJmrMmDHq2bOnYmJiKn9hgJMV3xAKAAAAAI7Qvn177dq1S1999ZU6d+4sSecMlC3+fPv27fXZZ59p9+7datWqlbNaBwAAAAAAAAAAAAAAAAAAAAAAgItzd3YDAJxn7969uu6665SSkmL9VfW/b1r7u6Ix/v7+GjlypHJycpScnKz9+/crKipKZ86cKTFu06ZN6tKli77//ntFRERU0sqA6omgG7iS5s2blzg3DEPR0dEXHWcv55sPAACgpnFzc9OIESM0YsQIpaSk6Mcff9SBAweUnJystLQ0eXp6KigoSOHh4bruuusUEhLi7JYBAAAAAAAAAAAAAAAAAAAAAABQBRjmhdInAFRbOTk5uuyyy3T48GErQKZWrVoaOnSo+vfvrw4dOigwMFBnzpxRQkKCtm/frnnz5mn9+vVWMEbv3r21aNEieXl5SZJyc3P1ww8/aN68eVq4cKEKCgokFf419uDgYG3dulWhoaFOWzNQWWw2m3WfrF69WpGRkeWu9e2332rgwIGSJC8vL2VkZNijRaDCin7Oiz5KGoah/Pz8i46zl/PNBwAAAAAAAAAAAAAAAAAAAAAAAAAAAKDiCKUBaqjJkydr0qRJVlBAhw4dNH/+fLVu3fqC1/3www+66667lJSUJEm67bbb9MUXX5w1bu/evRozZoy2bNlizdGtWzdt3LjRIesBXIk9Q2nefPNNPf3005Kkxo0b69ixY3bpEaio4j/npmleNJTGni40HwCg6svMzFRubq51XqtWLXl7e593/M8//6w777yzzPO89NJLGjlyZLl6BAAAAAAAAAAAAAAAAAAAAAAAAIDqjlAaoAYyTVMhISFKSEiQaZpq0aKFtmzZIn9//1Jdv3v3bl199dU6deqUDMPQsmXL1K9fv7PGnTp1SgMGDNDq1aslSYZhaOHChRoyZIhd1wO4GnuF0qSnp+uqq67SoUOHZBiGIiMjrfsJcLZmzZqdFTYTExNTqnH2cq75gJokNzdXcXFxZb6uUaNG8vDwcEBHQMWlp6erefPmSktLs55btGiRBg0adN5r1q5dq169ellhmKVVv359RUVFydfXtyItAwAAAAAAAAAAAAAAAAAAAAAAAEC15O7sBgBUvh07dig+Pl5SYVDM22+/XepAGknq0KGDHn74Yb311luSpA8//PCcoTR16tTRggUL1KZNGyUmJkqSpk+fTigNqoXJkyeXatzcuXP1448/lrquaZrKzs5WTEyM1q5dW2JD9rXXXlvWNgGHOXLkiF3HAZA2bNiglJQU67xZs2bq2LHjecdv3rxZvXv3LvM8EydO1BtvvFGuHgFHe/vtt5WammqdP/DAAxcMpPm70gahmaappKQkvfrqq3r99dfL3CdQVZ05c0YpKSny9/dXrVq1nN0OAAAAAAAAAAAAAAAAAAAAAAAAXJhhluXPiAOoFubMmaN7771XkuTr66vk5GS5ubmVqcauXbusTdJubm5KT0+Xp6fnOcfOmDFD48ePlyTZbDbFxcUpKCio/AsAXIDNZjvvpufib62l3Rh9vjqGYcg0TdWqVUsHDhxQeHh4uesBAFxXdHS02rZtq/z8fElS3bp1tXXrVrVr1+6816xdu1a9evUq81y1atXSvn37dMkll5S7X8ARTp06pQYNGigzM1OmaapZs2b6/fff5eHhccHriu6Fos9dpflvjqLPWAEBAfrrr78uOgdQ1S1atEhvv/22tm/froKCAklSu3btdN999+nhhx+u0PcWAAAAAAAAAAAAAAAAAAAAAAAAVE82ZzcAoPIlJiZKKtyIGR4eXuZAGklq1aqVdVxQUKDY2Njzjh02bFiJDaJbtmwp83xATVS0WdowDL399tsE0gBANTZp0iTl5eVZYRrvvPPOBQNpijMMo9QPSTpz5oxeeOEFh60FKK/ly5crIyPD+vzz3HPPlSkspuj+Wb9+/QUfs2bNssampqZq6dKljlgO4DALFy5UaGioQkNDFRYWpt9///2C4x9//HENHz5cW7duVX5+vkzTlGma2rdvn8aPH6/rrrtO2dnZldQ9AAAAAAAAAAAAAAAAAAAAAAAAqgp3ZzcAoPIVD6Gx2cqXTfX3v6KelJR03rGNGjVSo0aNdOLECUlSTExMueYEXE3RZuaKjjkfb29vXX/99ZowYYK6d+9e7joAANeWkJCg+fPnW5+vrrnmGo0ZM6ZMNUr7flMUeLZkyRKlpKQoICCgzP0CjrJgwQLrOCwsTP/85z/LVadHjx4Xff3zzz/XTz/9JEmaP3++hg0bVq65AGf44osv9Oeff0qSunbtqtatW5937Pz58/XOO+9I+l+IWVHwk1T4/rF582bdeeedWrx4seObBwAAAAAAAAAAAAAAAAAAAAAAQJVBKA1QA9WvX19S4eazI0eOlNiQVlrR0dElzv39/S84vmHDhlYozcmTJ8s0F+CK1q9ff87nTdNUZGSkdU9NnTpVV155Zanr2mw2eXl5KSAgQGFhYWW+NwFXs2nTJuu4Q4cO8vPzK3et9PR07d692zonrAnVxTfffKP8/HxJhYEBkyZNKtP1RZ/limqcz6FDh9S+fXsVFBQoNzdXX331lR5++OHytg3Y3aZNm6zPPkOGDCkRpmlvd955p3766SeZpqlffvnFYfMA9maaZol7ZejQoecdm5eXp2effVbS/0LJvLy8dOmllyopKUnR0dHW88uWLdOqVat00003Vco6AAAAAAAAAAAAAAAAAAAAAAAA4PoIpQFqoBYtWljHaWlpWrNmjXr16lWmGl9//XWJ85CQkFJf6+7Orx5UfT169CjVuIiIiFKPBaqjnj17WpumV69ercjIyHLX2r59u3r37i2pcGN1Xl6eXXoEnK3oc5VhGOrUqZOuv/56h8zTqlUrDRkyRAsXLpRhGFq8eDGhNHAZx48fV2JioqTCe8HRwRh9+vSxjuPj43Xs2DE1bdrUoXMC9vD7779bQa+GYZT4Wf677777TkeOHLE+i/Xt21dffvmlfH19JUlLlizRiBEjrM9UM2bMIJQGAAAAAAAAAAAAAAAAAAAAAAAAFpuzGwBQ+bp06SIfHx/rL6JPnDhR2dnZpb4+KipK06ZNsza2tW/fXv7+/he8JiEhwToOCAgoX+NAFWKapkzTdHYbgEuw571QdG9xf6G6ME1T27Ztsz5XDRw40KHzDRs2zJp3x44dDp0LKIvdu3dbxzabTd27d3fofM2aNZOXl5d1/n//938OnQ+wlz/++MM6rlOnjlq3bn3esQsWLJBU+Dvf09NTn332mRVII0mDBg3SU089ZX22Wrt2rRV4AwAAAAAAAAAAAAAAAAAAAAAAABBKA9RAbm5uuvXWW2WapgzD0N69e3XjjTfqxIkTF71227ZtuuGGG5SVlWVdP2jQoAtek5qaqj///NM6Dw8Pr/AaAFdWUFBgPSIjI53dDuB0RWEbAM526NAh63OVJN18880Ona9Hjx7WcWZmpvbv3+/Q+YDSSkxMtI4DAwPl7u7u8DmbNGliHScnJzt8PsAejh07Jqnw81VoaOgFP2etXbtWhmHIMAwNHTr0nAGx9913n3Wcn5+vXbt22b1nAAAAAAAAAAAAAAAAAAAAAAAAVE2E0gA11IsvvqjatWtLKvyr6T/++KPatm2rRx99VKtXr1ZCQoLy8vJ0+vRpHTt2TIsXL9att96qrl276tixY9bGN19fX02YMOGCc23YsKHEeUREhCOWBACoQQi6QXWxb98+69jDw0MdOnRw6HxBQUEKDAy0zg8dOuTQ+YDSSktLk1T4+71BgwZlurZWrVoKDAxUYGDgOUM3zsfHx8c6Tk1NLdOcgLNkZmZax35+fucdd+jQIcXFxVnnt9xyyznHNWnSRCEhIdZ5VFSUHboEAAAAAAAAAAAAAAAAAAAAAABAdeD4Pz0OwCU1bdpUH3zwge69915rY39GRoY++OADffDBB+e9zjRNGYZh/fvBBx+oXr16F5zr66+/llS4wbR169YlNkIDAFBaWVlZ1nHdunWd2AlgP8WDMIKDgytlzgYNGig5Ofms+QFnKh60URSeWVrdunVTYmJimecsHnCWkZFR5usBZ8jNzS3VuF9++UVS4Xd4m82mHj16nHdsSEiITpw4Iel/AVEAAAAAAAAAAAAAAAAAAAAAAAAAoTRADTZq1CjFx8fr+eeflyQrbOZ8DMMoEUjz2muv6fbbb7/gHHFxcVq8eLGkws1wffv2td8CABeVl5enn3/+2Tpv0aKFQkJCylzn+PHjio6Ots67detWYvM0UNMcPHjQOr5YIBpQVRRt/jcMQw0aNCjTtQ0aNNCQIUPKPKe3t7d1TCgNXEVREI1pmkpKSqqUOYvPU6tWrUqZE6goHx8f6zglJeW84zZs2CCp8P2lbdu2CggIOO9Ym81mHZ8+fbriTQIAAAAAAAAAAAAAAAAAAAAAAKBaIJQGqOGeeeYZRURE6OGHH1ZMTIwknTf0oiiwJjw8XDNnzlTv3r0vWt/Ly0s7duywzps0aWKHrgHXtnjxYt12222SCjd4HjhwoFx1srKyFBkZad17y5YtU79+/ezWJ1CVpKena86cOZIK36fatGnj5I4A+zh16pR1nJeXV6ZrL730Un399ddlnjM3N/ec8wPOVDxsrLJCaRITE61jf3//SpkTqKj69etLKvx+fvToUeXk5Khu3bolxpimqe+++876bt+9e/cL1iwKSJMkT09P+zYMAAAAAAAAAAAAAAAAAAAAAACAKotQGgC66aabFBUVpYULF2rZsmXauHGj4uLiSoxp2rSpunXrpoEDB2rw4MEl/pL6hfj4+Kh9+/aOaBtwWR9//LEVJNOvXz+1aNGiXHVatWqlm2++WcuXL7fqEkoDVzR58uRSjZs7d65+/PHHUtc1TVPZ2dmKiYnR2rVrS2yYvvbaa8vaJuCSioIETNOstCCO4vPUqVOnUuYELiYsLMw6zsrK0r59+xz6PeLgwYPKzMw85/yAK7v88sslFYb05eXlafny5Ro2bFiJMatXr1Z8fLw1rmfPnhes+ddff1nHRaE3AAAAAAAAAAAAAAAAAAAAAAAAAKE0ACRJNptNI0aM0IgRIyRJeXl5Sk5Ols1mU0BAgNzc3JzcIVA1ZGdna9OmTTIMQ5Kse6q8br/9diuUZu3atTpz5ow8PDwq3CdgT5MmTbJ+5s+lKKTp888/L/ccpmlac3h4eOjuu+8udy3AlQQHB1vHSUlJys/Pd+jnrvz8fCUmJlr3E+EDcBUdOnSQJOtnc+3atQ4NpVmzZk2J86KgD8DVtWvXTo0aNVJcXJxM09Szzz6ryMhIBQYGSpIyMjL0zDPPWONr1aql3r17n7deVFSUMjIyrPNLLrnEcc0DAAAAAAAAAAAAAAAAAAAAAACgSrE5uwEArsnd3V0NGjRQ/fr1CaQBymDXrl06ffq0FcJx/fXXV6he8euzsrK0Z8+eCtUDqirDMKxgmrffflvh4eHObgmwi+I/y7m5ufrll18cOt/WrVtLvE9xL8FVBAcHq0WLFpIKg8j+85//OHS+jz/+2Dpu3ry5GjRo4ND5AHsxDEMjR460PhcdPnxY7du310MPPaTx48friiuu0K5du6yxgwcPlp+f33nrbd68uURtR4ZBAQAAAAAAAAAAAAAAAAAAAAAAoGohlAYAADv6/fffreOQkBAFBQVVqF79+vUVEhJinR88eLBC9QBHMU3znI/SjCnNw8vLSwMGDND69ev10EMPOWmVgP1FRETIZrPJMAxJ0urVqx063w8//GAd22w2RUREOHQ+oCwGDhxovXfs27dPX331lUPmWbBggXbv3i3DMGQYhgYNGuSQeQBHeeaZZ9SoUSPrPCEhQbNmzdJ7772nw4cPW8/XqlVLL7300gVrffPNN5L+F0jj4+PjmKYBAAAAAAAAAAAAAAAAAAAAAABQ5bg7uwEAAKqTlJQUSYWbOoODg+1Ss0GDBjpx4oQkKTEx0S41AXtav379OZ83TVORkZFW2MbUqVN15ZVXlrquzWaTl5eXAgICFBYWZtUBqhMvLy/94x//0JYtWyRJ//73v/XUU0/J09PT7nNlZ2dr1qxZ1r105ZVXytvb2+7zAOV13333adq0aVYg2SOPPKKOHTuqTZs2dpvj999/10MPPSTDMGSapmw2m8aMGWO3+kBl8PHx0cqVK3X99dcrJSWlxGek4j/bs2bNUqtWrc5b56+//tKaNWus6yMjIx3eOwAAAAAAAAAAAAAAAAAAAAAAAKoOQmmAGio1NVX+/v6VOmdKSorGjRunzz//vFLnBSpTbm6udezm5maXmsXrZGdn26UmYE89evQo1biIiIhSjwVqkqFDh2rLli0yDEOJiYl68803NWnSJLvP89Zbbyk+Pl5SYWjBsGHD7D4HUBEtWrTQ8OHDNW/ePBmGodTUVPXp00f//e9/dfnll1e4/t69e3XLLbeUCBG89dZb1bJlywrXBipbhw4dtH//fr388sv65ptvlJCQIEmqVauWevTooRdeeEHXXnvtBWvMmDFDZ86ckVR4P/Tv39/hfQMAAAAAAAAAAAAAAAAAAAAAAKDqMEzTNJ3dBIDKFxISog8++ECDBg2qlPnmz5+vcePGKSkpSfn5+ZUyJ+AMs2fP1tixYyVJoaGhOnLkSIVrhoeH6+jRozIMQ++9954efPDBCtcEKovNZrOO16xZo8jISCd2A7im1NRUNW3aVDk5OTJNUzabTQsWLNCQIUPsNsfSpUs1dOhQmaYp0zTl6empY8eOVXpIIXAxJ06c0OWXX67U1FRJkmmaqlWrlp577jmNHz9ePj4+Za6ZmZmp6dOna8qUKVaAoGma8vf31549e9S4cWO7rgFwhvT0dOXk5CgoKEju7qXLoF6/fr1Onjxpnffr189uwZoAAAAAAAAAAAAAAAAAAAAAAACo+gilAWoom80mwzA0dOhQvf/++6pfv75D5jl+/LgeeOABrVixQqZpyjAMQmlQrS1ZssQKEbDZbDpx4oSCg4PLXS8hIUGNGjWyzufPn69hw4ZVuE8AgGuZNGmSJk+eLMMwZJqm6tSpo2nTpllBZxUxe/ZsTZgwQadOnbI+jz333HOaPHmyHToH7G/FihUaOHCgCgoKJMn6ufX09NSwYcPUt29fderUSWFhYeetERsbq+3bt2vlypVauHChsrKyrDqmacrNzU2LFy9W//79K2tZAAAAAAAAAAAAAAAAAAAAAAAAAFClEEoD1FBFoTSSFBAQoOnTp+v222+36xyzZs3S008/rYyMDBX9qiGUBtXdsWPHFBYWZt1f7733nh588MFy1/vggw/0yCOPSCq8f/bv36/WrVvbpVcAgOs4ffq0/vGPf2jfvn2S/hfCcd111+nZZ59VZGRkmWuuX79er732mtauXWvVk6S2bdtq+/btqlOnjl3XANjT559/rnvvvVd5eXmSVOL7RBFfX18FBQXJz89PXl5eysrKUnp6upKTk5Wenm6NK36taZry8PDQ7NmzNXLkyEpcEVC1/fbbb5o4caKkwntp7dq1Tu4IAAAAAAAAAAAAAAAAAAAAAAAAjkYoDVBDFQ+lKdqk3LdvX82aNUshISEVqh0VFaUxY8Zo8+bNJTZAm6apHj16aP369RXuH3BlrVu31h9//CHTNNWwYUMdPHhQvr6+Za6Tnp6utm3bKj4+XqZpqlmzZjp8+LADOgYAuIJDhw6pW7duSkpKkqQSn6PCw8N10003qVOnTurQoYPq169/ziCOXbt2afv27fruu+8UHR1doo5pmgoKCtLmzZsJOEOVsHXrVt16662KjY0t8Z3iXIp+xs/3WtG1TZs21YIFC9SlSxfHNA1UU2vXrlWvXr0kETYLAAAAAAAAAAAAAAAAAAAAAABQUxBKA9RQ77//vp555hllZWVZGzgNw5Cvr6/eeustjR49usw1CwoK9Oabb2ry5Mk6ffp0iQ3QPj4+euONNzR27FgHrAZwLa+++qqef/55awN09+7dtXLlStWtW7fUNXJycnTTTTdp06ZNkgo3fr7wwguaNGmSI1oGHCYvL08///yzdd6iRYtyhZ8dP37cCtiQpG7duln3GFCd7NmzRwMGDNDRo0fPCuEoy8/8368xTVOhoaFaunSpIiIi7Ns04ECpqal66aWXNGfOHGVnZ0sq373g6empUaNGadKkSQoICHBIr0B1RigNAAAAAAAAAAAAAAAAAAAAAABAzUMoDVCDHTlyRGPGjNHatWtLbFg2DEPXX3+9PvroI4WFhZWq1q5duzR69Gjt2rXLqlFU7+abb9asWbPUpEkTh60FcCVZWVkKDw9XcnKy9VyrVq00a9Ys9ejR46LXb9iwQWPHjlVUVJSkwvsoMDBQhw8flo+Pj8P6Bhxh4cKFuu222yRJNptNBw4cUIsWLcpc59ChQ2rXrp0VLrBs2TL169fPrr0CriItLU0jR47Uf//73xLhG2X56vb3QJu+fftq7ty58vf3t2+zQCVJSUnRhx9+qG+//VY7d+4sVSCGm5ubrrjiCg0YMEAPPPAAYTRABRBKAwAAAAAAAAAAAAAAAAAAAAAAUPMQSgNAH330kZ544gmdPHlShmFYm5e9vLz02muv6eGHHz7vtadPn9akSZP09ttvKz8/3wqkMU1TQUFBevfdd3X77bdX1lIAl7Fs2TINGTJEpmla95RhGLr00kt10003qVOnTgoODpa3t7cyMzOVkJCg7du3a9WqVdq7d2+Je8lms2nRokUaOHCgcxcFlEOfPn20evVqSdKAAQO0ZMmScte65ZZbtHz5crvUAqqCxYsXa+rUqdqyZYv1XPGQmvMp/hWvc+fOevLJJzV48GCH9Ag4Q1ZWln755RcdOnRIKSkpSklJUUZGhnx8fBQQEKCAgAC1atVKV199tby8vJzdLlAtEEoDAAAAAAAAAAAAAAAAAAAAAABQ8xBKA0CSdPz4cd13331atWqVtdm5KBSja9eu+vjjj9WyZcsS12zatEljxozRH3/8YY0tum7EiBGaMWOGgoKCKn0tgKuYNm2aJk6cWOLekC4cKFB8TNF9NW3aNI0bN87xDQN2lp2drcDAQOXm5kqSvvrqKw0fPrzc9ebPn28FnXl7eys5OVkeHh526RVwZT/99JOWLVumzZs367ffftOZM2fOO9bDw0MdO3ZUt27dNGDAAHXt2rUSOwUAVFeE0gAAAAAAAAAAAAAAAAAAAAAAANQ8hNIAKGHu3Ll67LHHlJKSYoViSFLdunU1adIkPf7448rKytKTTz6pjz76SAUFBZL+F6DRuHFjzZw5U/369XPmMgCXsXTpUo0aNUppaWklwmjO9fb799cDAgL06aefcj+hyvr555917bXXSir8+Y6Pj69QWFliYqIaNGhg1du6dauuvPJKu/QKVBU5OTmKiYlRSkqKUlJSlJGRIR8fHwUEBCggIEDh4eGqW7eus9sEAFQzhNIAAAAAAAAAAAAAAAAAAAAAAADUPITSADhLfHy8HnjgAS1dutQKyTBNU4ZhKCIiQgkJCTpx4oT1XNG/Y8aM0dSpU+Xj4+PkFQCuJSEhQe+++65mzZqltLS0i44PCAjQgw8+qEcffbRCAR6As33yyScaPXq0JKlx48Y6duxYhWs2adJEJ06ckGEYmjt3ru64444K1wQAAMCFEUoDAAAAAAAAAAAAAAAAAAAAAABQ87g7uwEArqdBgwZavHixFixYoEcffVSJiYlW+MzOnTutcUXPtWjRQh999JF69OjhxK4B1xUcHKxXX31VL7/8srZu3arNmzcrOjpaKSkpysjIkI+PjwICAtSyZUt169ZN//jHP+Tuzls0qr6UlBRJhe8XwcHBdqnZoEEDnThxQpKUmJhol5oAAAAAAAAAAAAAAAAAAAAAAAAAAAAASmLHO4DzGj58uG644QbdcMMN2r17txVCU/Svm5ubHnvsMU2aNEl16tRxdruAy/Pw8FDXrl3VtWtXZ7cCVIrc3Fzr2M3NzS41i9fJzs62S00AAAAAAAAAAAAAAAAAAAAAAAAAAAAAJdmc3QAA15WSkqJx48Zpz549MgxDkqx/Jck0TeXm5so0TWe1CABwYYGBgZIK3y8SEhLsUjMxMdE6rlevnl1qAgAAAAAAAAAAAAAAAAAAAAAAAAAAACiJUBoA5/T111+rXbt2mjdvnhU6Y5qmdWwYhgoKCjR9+nRddtllWr9+vTPbBQC4oPr161vHf/75Z4WDaRISEhQbG2sFpBWvD9Q0f/zxh6ZOnapbbrlFLVu2VGBgoNzd3eXu7q5169ad85q//vpLsbGxio2NLRHwBAAAAAAAAAAAAAAAAAAAAAAAAAAA8HeE0gAoIT4+XkOGDNGIESOUkJAg0zRlGIZM01Tnzp21YMECtWnTpkRQzeHDh3XDDTfo/vvvV0ZGhpNXAABwFZ06dZIk631k0aJFFar39ddflwhIu/zyyyvcI1DVHD58WIMGDVKbNm309NNPa8WKFYqOjlZqaqoKCgqs++NcJk2apPDwcIWHh+uyyy5Tfn5+JXYOAAAAAAAAAAAAAAAAAAAAAAAAAACqEkJpAFg+++wztWvXTkuXLrXCaCSpTp06euutt/Tzzz9r2LBh2rVrl5599lm5ubnJMAwrbOA///mP2rVrpxUrVjh5JQAAV9C0aVO1bNlSUmGI2ZQpU3Ty5Mly1UpPT9e//vUv670pLCxMrVu3tluvQFXwzTffqGPHjvr2229VUFBQ4rWie+NCHn/8cUmF92NiYqKWL1/ukD4BAAAAAAAAAAAAAAAAAAAAAAAAAEDV5+7sBgA4359//qn77rtP33//vRVGUxQ00717d3388ce65JJLrPEeHh6aMmWKhg4dqlGjRmnXrl3W+OPHj+uWW27RbbfdpunTpyswMNCJKwNcR2Jionbs2KFjx44pPT1dOTk5Mk2zzHVefPFFB3QHOM7IkSP1/PPPyzAMxcfHa8CAAVq5cqXq1q1b6ho5OTkaMGCA4uLiJBWGb9x9992OahlwSStXrtSIESOUn59vBdCYpqmGDRuqadOm2rZt20VrtGrVSl27dtWPP/4oSVq8eLEGDBjg0L4BAAAAAAAAAAAAAAAAAAAAAAAAAEDVZJjl2REPoNqYNWuWnn76aWVkZFiBNKZpysfHR6+//roeeOCBC16fn5+v119/XVOmTFFubq4kWXWCgoI0Y8YMDR8+vDKWAricgoICffTRR5o9e7Z27dpll5r5+fl2qQNUlqysLIWHhys5Odl6rlWrVpo1a5Z69Ohx0es3bNigsWPHKioqSlLhe0xgYKAOHz4sHx8fh/UNuJLExES1aNFCGRkZ1me1YcOG6fnnn9dll10mSbLZbFZYzerVqxUZGXnOWtOnT9eECRMkSY0aNdLx48crZxEAgCpt7dq16tWrl6TCgEC+lwAAAAAAAAAAAAAAAAAAAAAAAFR/hNIANdThw4c1evRobdq0yQqRkQo3+/fp00ezZ89W06ZNS11v//79GjVqlLZu3VqilmEY6t+/v2bOnKlGjRo5ZC2AK4qNjdXgwYO1c+dOSYX3Q5Gie6Qsiu4nNn+iKlq2bJmGDBki0zSte8EwDF166aW66aab1KlTJwUHB8vb21uZmZlKSEjQ9u3btWrVKu3du7dEaJrNZtOiRYs0cOBA5y4KqESPPvqo3n//fUmF984bb7yhiRMnlhhT2lCaPXv2KCIiwqoVExOj0NBQxzUPAKgWCKUBAAAAAAAAAAAAAAAAAAAAAACoeQilAWooLy8vnTp1qsRGf39/f02bNk0jR44sV03TNPXOO+/oxRdfVHZ2tlXXMAz5+vrq7bff1qhRo+y8EsD1JCcnq0uXLjp8+PBZoU8VweZPVGXTpk3TxIkTz7ofLhTSVHxM0b00bdo0jRs3zvENAy6ioKBAQUFBSk9PlyQNHTpUCxYsOGtcaUNp8vLy5O3trdzcXBmGoZUrV6pPnz6OWwAAoFoglAYAAAAAAAAAAAAAAAAAAAAAAKDmcXd2AwCcIycnR4ZhWBv9Bw4cqJkzZ6pBgwblrmkYhh577DENGDBAo0eP1qZNm6zN0enp6RozZozmzZun1atX22sZgEt68cUXFR0dXeIea9KkiQYPHqyIiAgFBwfLy8vL2W0Cleqxxx5T8+bNNWrUKKWlpZUIozlXYFPR/VP0ekBAgD799FP169ev0noGXMGvv/6qtLQ0SYX3xfPPP1+heu7u7mrcuLFiYmIkSbGxsRVtEQAAAAAAAAAAAAAAAAAAAAAAAAAAVEOE0gA1mGmaCg4O1nvvvadhw4bZre4ll1yiDRs26MMPP9QzzzyjjIwMK5hj3bp1dpsHcEXZ2dn6+OOPrZ95m82mV155RU899ZTc3Nyc3R7gVAMHDtQ111yjd999V7NmzbKCNs6lKKgmICBADz74oB599FEFBQVVUqeA64iKirKOg4ODddlll1W4Zr169azj9PT0CtcDAAAAAAAAAAAAAAAAAAAAAAAAAADVD6E0QA12xx136N1331VgYKBD6j/44IPq16+f7rvvPv3www8OmQNwNRs3blRubq4Mw5BhGHryySf17LPPOrstwGUEBwfr1Vdf1csvv6ytW7dq8+bNio6OVkpKijIyMuTj46OAgAC1bNlS3bp10z/+8Q+5u/ORFTVXYmKiJMkwDDVp0sQuNYvfU3l5eXapCQAAAAAAAAAAAAAAAAAAAAAAAAAAqhd2+AI11H//+1/17dvX4fOEhobqu+++0yeffKLHHntMJ0+edPicgDMdOXJEkmSaptzd3fXUU085tyHARXl4eKhr167q2rWrs1sBXJrNZrOOCwoK7FIzJSXFOvb397dLTQBA5YiKitKqVaus8xtuuEHt2rVz+Lx169ZVWFiYw+cBAAAAAAAAAAAAAAAAAAAAAACA6yCUBqihKiOQprh77rlHN954ox588MFKnReobKmpqZIkwzDUqlUr+fn5ObkjAEBVVr9+fUmFYWdxcXEVrpedna2jR4/KMIwS9QEAVcN3332nCRMmSCr8zhEdHV0p815zzTWKiYmplLkAAAAAAAAAAAAAAAAAAAAAAADgGmzObgBAzdGoUSMtWbLE2W0ADuXt7X3OYwAAyqNZs2bWcVxcnI4ePVqheuvXr1deXp5M05QkRUREVKgeAKByZWZmyjRNmaapkJAQhYWFObslAAAAAAAAAAAAAAAAAAAAAAAAVFPuzm4AgOs6cOCAfvrpJ/300086cOCAUlJSlJKSooyMDPn4+CggIEABAQFq27atunbtqq5du6pt27bObhtwqjZt2ljHCQkJTuwEAFAddOnSRd7e3srKypIkffrpp3rppZfKXe+dd96xjkNDQ9W8efMK9wgAqDz169eXJBmGoZCQECd3AwAAAAAAAAAAAAAAAAAAAAAAgOqMUBoAJZw6dUpz587V9OnTdfDgQet50zRLjCsKqJGkbdu2ae7cuZKk1q1ba/z48br77rtVp06dymsccBHdu3eXl5eXsrKydPToUcXFxalhw4bObgsAUEV5eHjoxhtv1KJFiyRJ06ZN08iRI9WsWbMy1/rPf/6jdevWyTAMSdKtt95qz1YBAJWgeBBNenq6EzsBAAAAAAAAAAAAAAAAAAAAAABAdWeYf0+aAFBjLV68WA888ICSkpLOCqEp2rx8LucaGxgYqA8//FBDhw51SK+AK3vsscf07rvvyjAMvfTSS3rxxRed3RLgUhITE7Vjxw4dO3ZM6enpysnJOeu9pDS4t1BT7Nu3Tx06dJBpmjJNUy1bttT3339fIpjGZrNZn9dWr16tyMjIEjX+/e9/a9y4cTpz5oxM05Snp6diYmJUv379ylwKAKCCUlJS1KhRI505c0a1a9dWUlKSvLy8nN0WAAAAAAAAAAAAAAAAAAAAAAAAqiFCaQAoJydHDz/8sD799FMrFKB4CE1pfk2ca7xhGLr77rv1wQcfyNPT085dA67r5MmTuvzyyxUbGytPT09t2rRJHTt2dHZbgFMVFBToo48+0uzZs7Vr1y671MzPz7dLHaAqeOSRR/TBBx/IMAyZpikvLy+NHz9ed911l1q1alUilGbNmjW67rrrFBcXp3Xr1un999/Xli1bSnxGe+ONNzRx4kRnLgkAUE633HKLli9fLsMwNGfOHI0cOdLZLQEAAAAAAAAAAAAAAAAAAAAAAKAaIpQGqOEyMzN1880366effpJpmtZm5qJfDXXr1lWHDh10xRVXqGHDhvLz85OXl5eysrKUnp6uuLg47dy5U7t371ZOTo4klahhGIauvvpqfffdd/L29nbOIgEn2L9/v66//nrFx8crMDBQc+bMUf/+/Z3dFuAUsbGxGjx4sHbu3CmpZNhZ8VCz0ip6fyGUBjVJfn6++vbtqx9++MEKpim6f7y8vJSZmSmp8J4KCAhQTk6O9dlM+t99Y5qmhg8frnnz5jllHQCAivvll1907bXXSpIaNWqknTt3qn79+k7uCgAAAAAAAAAAAAAAAAAAAAAAANUNoTRADWaapm655RatWLFCkqyNym5ubhowYID++c9/6qabbpKbm9tFa+Xn52vVqlX65JNP9O2336qgoMCawzAM3XTTTVq+fLlD1wO4itjYWElSTEyMRo8ercOHD8swDHXu3FnDhw9Xp06dFBwcrDp16pS5dmhoqL3bBRwqOTlZXbp00eHDh88ZflZehNKgJsrJydHYsWP1+eefn3UvFX2OO5fir91///1677335O7uXjlNAwAc4s0339TTTz8twzDUrl07LVy4UG3btnV2WwAAAAAAAAAAAAAAAAAAAAAAAKhGCKUBarD33ntP48aNK7GpuXPnzpo5c6auuOKKctfduXOnxo4dq23btlmboA3D0LRp0zRu3Dh7tQ+4LJvNZt1XRYqHcZSXYRjKy8urUA2gsj300EOaOXNmifeaJk2aaPDgwYqIiFBwcLC8vLzKVbtHjx72bBWoMhYsWKCXX35ZBw8elKQLvr8Ufd1r0aKFXnnlFQ0fPrxSegQAON7s2bM1YcIE5eTkqFatWhoxYoRGjBihzp07KyAgwNntAQAAAAAAAAAAAAAAAAAAAAAAoIojlAaoodLS0tSsWTNlZGRYm5VHjhypjz/+WDabrcL1CwoKNGrUKM2dO9cKpvH19dWRI0dUr169CtcHXFlRKM25gmgq8rZrGIby8/Mr2h5QabKzsxUQEKAzZ87INE3ZbDZNnjxZTz31lNzc3JzdHlDlff/991q5cqU2b96sAwcO6PTp09Zr7u7uCgsL03XXXacbb7xRAwcOtMtnPACA8zVv3tw6Tk5OVkZGhqSSIWXe3t7y9fWVh4dHqesahqHo6Gj7NQoAAAAAAAAAAAAAAAAAAAAAAIAqzd3ZDQBwjlmzZunkyZMyDEOGYWjEiBH65JNP7FbfZrPp008/VW5urubPny9JysjI0KxZs/T000/bbR7A1ZH9hpps48aNys3Ntd5rnnzyST377LPObguoNvr06aM+ffpY59nZ2UpLS5OnpychgABQjR05cqRECGZRGE3x7x4ZGRlWWE1p/T1QEwAAAAAAAAAAAAAAAAAAAAAAADWbYbJbHqiRLr30Uh04cECmaaphw4bav3+/QzYvp6WlqV27doqPj5dpmmrbtq327dtn93kAV3LPPfc4rLY9w6MAR5s5c6YeeughSZK7u7sSExPl5+fn5K6AqiUqKkqrVq2yzm+44Qa1a9fOiR0BAJzNZrPZPUCmKOAmPz/frnUBAAAAAAAAAAAAAAAAAAAAAABQdbk7uwEAlS8uLk779++3/qL6+PHjHRJII0n16tXTuHHj9Mwzz0iSDh48qL/++kuNGjVyyHyAKyA4BiiUmpoqSTIMQ61atSKQBiiH7777ThMmTJBUeC9FR0c7uSMAgLOFhobaPZQGAAAAAAAAAAAAAAAAAAAAAAAA+DtCaYAaaOfOnZL+95fQhw8f7tD5RowYYYXSFM1PKA0AVH/e3t7nPAZQepmZmTJNU5LUuHFjhYWFObkjAICzHTlyxNktAAAAAAAAAAAAAAAAAAAAAAAAoAawObsBAJWv+Aa2gIAAh29uDgsLU1BQ0DnnBwBUX23atLGOExISnNgJUHXVr19fkmQYhkJCQpzcDQAAAAAAAAAAAAAAAAAAAAAAAAAAqCkIpQFqoPT0dEmVu7m5+DwnT56slDkBAM7VvXt3eXl5yTRNHT16VHFxcc5uCahyin+GKvoMBwAAAAAAAAAAAAAAAAAAAAAAAAAA4GiE0gA1kGma1rG7u3ulzGmz/e/XTfH5AQDVV506dTRmzBjrfPbs2U7sBqiaunTpIg8PD5mmqSNHjigrK8vZLQEAAAAAAAAAAAAAAAAAAAAAAAAAgBqAUBqgBvLx8ZFUGA5z/PjxSpnzr7/+Omt+AED1N2nSJIWGhso0Tb355pv67bffnN0SUKUEBASoT58+kqTc3FwtWrTIyR0BAAAAAAAAAAAAAAAAAAAAAAAAAICagFAaoAYKCwuzjpOSkhQXF+fQ+eLi4pSQkCDDMCRJoaGhDp0PAOA6fH19tXLlSjVo0EDZ2dnq06eP/vvf/zq7LaBKeeaZZ6zPUc8995wSExOd3BEAAAAAAAAAAAAAAAAAAAAAAAAAAKjuDNM0TWc3AaByxcbGqlmzZtbm5nfffVePPPKIw+abMWOGxo8fL0kyDEOHDx8uEYwDVDfNmzd3SF3DMBQdHe2Q2oCjxMbGSpJiYmI0evRoHT58WIZhqHPnzho+fLg6deqk4OBg1alTp8y1CTlDTfLmm2/q6aeflmEYateunRYuXKi2bds6uy0AAAAAAAAAAAAAAAAAAAAAAAAAAFBNEUoD1FAtWrRQTEyMTNNUeHi49u7dq7p169p9nuzsbF166aU6evSoNRehGqjubDabDMOQvd9iDcNQfn6+XWsCjlZ0PxRnmuZZz5WVYRjKy8urUA2gqpk9e7YmTJignJwc1apVSyNGjNCIESPUuXNnBQQEOLs9AIATZWRkaNu2bdq5c6eSkpKUlpam06dPl6mGYRj6+OOPHdQhAAAAAAAAAAAAAAAAAAAAAAAAqhpCaYAa6vnnn9err75qhQI8/PDDmj59ut3nefTRR/X+++9LKtzg9swzz2jKlCl2nwdwJecK4SivovCOon8JpUFVUzyk6VzhNOXF/YCapHnz5tZxcnKyMjIyJKnEPeXt7S1fX195eHiUuq5hGIQFAkAVt2PHDr311ltavHhxhQL7+L4BAAAAAAAAAAAAAAAAAAAAAACAvyOUBqih4uLi1Lx5c50+fdrafGbvwJjiwTemaapOnTqKjo5Wo0aN7DYH4IqaNWtW5lCa7OxspaamWhtJi66vV6+efH19rXExMTH2axSoBMVDaeyJTdOoSc4X7lTR+4r7CACqtilTpmjy5MnKz8+33hP+/rmrNKGAhGACAAAAAAAAAAAAAAAAAAAAAADgXNyd3QAA52jYsKGeeOIJvfLKK9YGtNdee00HDx7U9OnT1bhx43LXPnHihB555BEtXbpU0v/+4vrEiRMJpEGNcOTIkXJdl5+fr127dmn+/PmaPXu2MjIyZLPZNHPmTN144432bRKoJCNHjnR2C0C18fdggbIGoBVHNikAVG2vvfaaXnzxRUk6K7DsYgFmf3+d9wQAAAAAAAAAAAAAAAAAAAAAAACci2Gy8wSosc6cOaNu3bpp69atJf4yure3t+677z7985//VPv27Utdb9++ffrkk0/00UcfKTMzs8RmuE6dOunHH3+Uh4eHo5YDVCtHjx7V4MGDtXPnTrm7u2v+/PkaPHiws9sCADhBs2bNKhRAcyExMTEOqQsAcJydO3eqU6dO1rlpmrrzzjt15513qlmzZmrTpo31vvHFF1/oyiuvVEpKivbt26d169Zp8eLFOn36tAzDUNu2bfXvf/9bTZo0kSSFhYU5ZU0AAAAAAAAAAAAAAAAAAAAAAABwPYTSADVcfHy8unfvrqioKCuYRvrfX05v2bKlrrzySl1xxRVq1KiR/Pz85OXlpaysLKWnpysuLk47d+7U9u3bFRUVJUklapimqZYtW2rjxo1q2LChcxYJVFGJiYm68sor9eeff6pu3brauXOnWrVq5ey2AAAAADjRrbfeqkWLFkkq/N796aef6q677rJet9ls1nf61atXKzIyssT1CQkJevjhh7Vo0SIZhqGGDRtqzZo1atu2beUtAgAAAAAAAAAAAAAAAAAAAAAAAC6PUBoASkhI0IgRI7RhwwZr41rxXw1Fz13Iucabpqnu3btr/vz5BNIA5fTJJ59o9OjRMgxD/fv319KlS53dEgAAAAAnyc3NlZ+fn3JzcyVJI0aM0JdffllizMVCaYo8//zzevXVVyVJYWFh2rNnj3x8fBzYPQAAAAAAAAAAAAAAAAAAAAAAAKoSm7MbAOB8wcHBWrt2rV577TV5eHjINE0ZhmE9TNO86OPv493d3fWvf/1L69evJ5AGqIBhw4ZZ9+Xy5csVHx/v7JYAAAAAOMnWrVt1+vRpKxh23Lhx5a41ZcoUXX/99ZKk2NhYTZkyxS49AgAAAAAAAAAAAAAAAAAAAAAAoHoglAaAJMkwDD311FP6448/9MQTT6hevXpW4EzR6+d7SLLG+vn5aeLEifrjjz/0zDPPWK8DKB9vb281b95cUuF9tnnzZid3BAAAAMBZ/vjjD+vY09NTnTt3vuD43NzcC77+8ssvSyr8rjF79mzl5eVVvEkAAAAAAAAAAAAAAAAAAAAAAABUC+7ObgCAa2nSpIneeOMNTZo0SevWrdOPP/6on376SQcPHlRaWlqJDWpubm7y9/dX69atde2116pr166KjIyUp6enE1cAVD9+fn7W8dGjR53YCQDAlWVkZCgzM1Pe3t7y8fFxdjsAAAdISUmRVBgcGx4efs4xNpvNCpg9ffr0Betdc801CggIUEpKik6ePKlff/1V1157rX2bBgAAAAAAAAAAAAAAAAAAAAAAQJVEKA2Ac6pbt6769u2rvn37lnj+5MmTysjIkI+Pj3x9fZ3UHVCzJCYmWsfFg6EAADVXRkaG5s2bpw0bNmjr1q06evSoCgoKrNdtNpvCwsLUuXNn9ejRQ7fddhuf3QCgGigeMnO+ADIfHx+lp6fLMAwlJSVdtGZoaKgVdnPgwAFCaQAAAAAAAAAAAAAAAAAAAAAAACCJUBoAZeTr68uGZqASRUdHKyYmRoZhSJKCgoKc3BFQNs2bN3dIXcMwFB0d7ZDagCs7efKknnvuOX3yySfKycmRJJmmeda4/Px8HT58WDExMVqwYIEef/xx/fOf/9S//vUv+fn5VXbbAAA7Kf59PDs7+5xj/Pz8lJ6eLkmKjY29aE03NzfrODk5uYIdAgAAAAAAAAAAAAAAAAAAAAAAoLoglAYAABdlmqYmTJhgHRuGofbt2zu5K6Bsjhw5IsMwzhmaURFFQU1ATbJx40bdfvvtiouLs+4pwzAuej+Ypqns7GzNnDlTS5Ys0RdffKHrrruuMloGANhZSEiIdZyamnrOMS1btrTCaLZu3XrRmocPH7aO3d35r0IAAAAAAAAAAAAAAAAAAAAAAAAUsjm7AQDVR0ZGhuLj45WXl+fsVoAqLyoqSv369dPy5cutsIGgoCBdddVVTu4MKJ+i4IyKPIrqADXRd999p5tvvll//fWXFVQmFQbOmKYpHx8fhYWFqV27dgoLC5OPj4/1miQrHOqvv/5S3759tWrVKmcuBwBQTkUhlaZp6s8//9SpU6fOGtOhQwdrzObNm88bXiNJa9asKfF6cHCwnTsGAAAAAAAAAAAAAAAAAAAAAABAVWWYRbsUAaCMTNPU/Pnz9cUXX+jHH39UZmam9VrDhg3Vp08f3X333erZs6fzmgScYPLkyeW6LicnR/Hx8dq5c6f27NkjSSXCBN5++22NHz/eXm0ClaJZs2ZlDpLJzs5WamqqFXJWdH29evXk6+trjYuJibFfo4ALO3HihNq1a6eTJ0+WCKOJjIzUyJEj1bNnTzVt2vSs62JjY7Vx40Z99tlnWrduXYlrfX19tX//foWEhFTqWgAAFWOapoKCgpSamirDMPTzzz+fFVy5fv16XX/99dbv/dGjR2v27Nln1UpJSdE111yjqKgoK/Bs//79at26daWsBQAAAAAAAAAAAAAAAAAAAAAAAK6NUBqghjJNU8uXL7cCL9zc3NS3b99SXx8TE6MhQ4Zo9+7dVr2/K9oAN2TIEM2ZM0fe3t526BxwfTabrcwhHMUVD6IpOu/Xr5+WLl0qm81mlx4BV5efn69du3Zp/vz5mj17tjIyMhQYGKjPP/9cN954o7PbAyrV7bffrvnz58swDJmmqeDgYM2dO1e9e/cudY3vv/9eI0eOVGJiohU8MHz4cH311VcO7BwA4AhDhgzRkiVLZBiGXnjhBU2aNKnE66ZpqkWLFjpy5Ij1O79Pnz564IEH1Lp1a+Xm5urHH3/U66+/rtjYWOt7R4cOHfTbb785YUUAAAAAAAAAAAAAAAAAAAAAAABwRYTSADXU1q1b1aVLF2vz2YABA7R48eJSXRsbG6urrrpKCQkJZ4VnFFf8tS5duuiHH36Ql5eXnVYAuK6KhtIUMU1TNptNDz74oKZOnaratWvboTug6jl69KgGDx6snTt3yt3dXfPnz9fgwYOd3RZQKU6ePKkGDRooNzdXpmnK399fW7du1SWXXFLmWn/88YeuuuoqpaWlyTRN1a5dW/Hx8fL19XVA5wAAR/n88881cuRIGYahNm3aaN++fWeNWbZsmQYNGmQFmp3vO3vR6zabTStWrFCfPn0qYwkAAAAAAAAAAAAAAAAAAAAAAACoAmzObgCAc6xatUrS/4JjJkyYUKrrTNPUkCFDFB8fL6kwcKZoE9vfH8Vf+/XXX/XUU085ZjGACzrXPVHahyS1atVKEyZM0J49ezRjxgwCaVCjhYWF6bvvvlOTJk2Ul5enu+66S4cOHXJ2W0Cl2LBhg06fPm19tnrjjTfKFUgjSS1atNDrr79uvdfk5uZq/fr19mwXAFAJBg0apICAAHl6eio2NlabNm06a8yAAQP09NNPlwikOdd39qL3hClTphBIAwAAAAAAAAAAAAAAAAAAAAAAgBIMs2j3CYAaJTIyUhs2bJBUuEG5tJv7P/nkE40ePbrEX1m32WwaOXKkbr31VjVr1kxZWVnatm2b3nvvPe3bt8/a6Obm5qZdu3apffv2jlgS4DI2btxY5msMw1CdOnXk6+urpk2bysvLywGdAVVb8feg/v37a+nSpc5uCXC4Dz74QI888ogkqW7dukpMTJSnp2e562VnZ6t+/fo6deqUJGnGjBl66KGH7NIrAMD1fPbZZ3r66aetYNm/CwsL09SpUzV06NBK7gwAAAAAAAAAAAAAAAAAAAAAAACuzt3ZDQCofKZpaseOHVawTFk2n02bNq1EHXd3dy1ZskR9+/YtMe6KK67QPffcozvuuEOLFi2SJBUUFGjOnDl6++237bAKwHX16NHD2S0A1dKwYcM0duxYnTlzRsuXL1d8fLwaNGjg7LYAhzp58qSkwvCy8PDwCgXSSJKnp6fCw8O1f/9+GYZh1QcAVE8jR47U7bffrg0bNujXX39VfHy8TNNUw4YNdc0116hHjx5yd+e/BwEAAAAAAAAAAAAAAAAAAAAAAHA2dp0ANdDhw4eVkZEhqXCD80033VSq63bs2KF9+/bJMAyZpinDMPTYY4+dFUhTxMPDQ59//rm2b9+uo0ePyjRNLViwgFAaAEC5eHt7q3nz5vr9999lmqY2b95cpmA1oCoKCAiwjuvUqWOXmsXr+Pv726UmAMB1eXh4qFevXurVq5ezWwEAAAAAAAAAAAAAAAAAAAAAAEAVYnN2AwAqX3R0tHVss9l05ZVXluq6VatWlTivVauWnnrqqQteU7t2bU2cOFGmaUqS/vrrL504caKMHQOuwc3NTW5ubnJ3d9e6deuc3Q5QI/n5+VnHR48edWInQOW49NJLJUmmaSo2NtYuNYvfO5dddpldagIAAAAAAAAAAAAAAAAAAAAAAAAAgOqFUBqgBiq+oblx48by9PQs1XUbNmywjg3DUJ8+feTv73/R6wYNGmRdI0m7d+8uQ7eA6zBN03pcCOE1gOMkJiZax3l5eU7sBKgcXbp0Uf369SVJycnJ2rx5c4Xqbdq0ScnJyTIMQ0FBQbr66qvt0SYAAAAAAAAAAAAAAAAAAAAAAAAAAKhmCKUBaqCTJ09KKgyJCQgIKNU1pmlq69atMgzDCuS44YYbSnVto0aNFBwcbF134sSJcnQNuIaicKULKW14DYCyiY6OVkxMjHUfBgUFObkjwPHc3Nz02GOPWeePP/648vPzy1UrLy9Pjz/+eIlaNhtfCQEAAAAAAAAAAAAAAAAAAAAAAAAAwNnYgQjUQDk5OdZx7dq1S3XNgQMHlJmZWeK5rl27lnrORo0aWccZGRmlvg5wJcU37l8scKY04TUASs80TU2YMME6lqT27ds7syWg0jz++OO65pprZJqmduzYoUGDBikrK6tMNbKysjRo0CDt2LFDUuHnuOIBNQAAAAAAAAAAAAAAAAAAAAAAAAAAAMURSgPUQF5eXtZxenp6qa7ZunVrifNatWrpsssuK/WcderUsY6zs7NLfR3gSvz8/KwwjKSkJCd3A9QcUVFR6tevn5YvX24FPgUFBemqq65ycmdA5XB3d9eKFSt0ww03yDRNrVixQq1bt9a///1vpaWlXfDatLQ0zZo1S61bt9bKlSslSb1799aKFSvk5uZWCd0DAAAAAAAAAAAAAAAAAAAAAAAAAICqyN3ZDQCofP7+/pIk0zQVExOjgoIC2WwXzqj65ZdfSpxffvnlcncv/a+Q4hum69atW/pmARfSpEkTpaamSpK+//57DR8+3MkdAa5v8uTJ5bouJydH8fHx2rlzp/bs2WM9b5qmDMPQM888YwXUANVd0X109dVXKyoqSkePHtWJEyf04IMPaty4cbr88st16aWXKjAwUJ6ensrOzlZSUpL27t2r//u//9OZM2esULVmzZqpS5cueuedd0o9/4svvuiQdQEASnJ2WJhhGMrLy3NqDwAAAAAAAAAAAAAAAAAAAAAAAHAdhNIANVDLli2t49zcXP3666+65pprLnjNDz/8IMMwrDCAbt26lWnOpKQk69jPz69sDQMuolu3bvq///s/maapuXPnqlmzZrrvvvvUsGFDZ7cGuKxJkyZVKDymKEjDMAzrfahv37569NFH7dUi4PL+fh8VHZumqdzcXG3fvl07duw467qi+6f4NUePHtUrr7xSpvkJpQGAylH89zYAAAAAAAAAAAAAAAAAAAAAAADgbIbJjhegxjl16pR8fX2Vn58vSbrrrrv06aefnnf8zz//rGuvvbZEKM23336rvn37lmq+uLg4hYSESCrcEP3999/rhhtuqPA6gMq2Z88eRURElLgXJKlevXry9fW1zo8cOWIdN2jQQHXq1LHL/IZhKDo62i61gMpis9kqFEpTxDRN2Ww2Pfjgg5o6dapq165th+6AqsFe91FZFb3XFX1mBAA4VtHv+8r+r7ri32/4nQ8AAAAAAAAAAAAAAAAAAAAAAIAihNIANdTNN9+s7777TlLhxrcVK1aoT58+5xx7/fXXa/369dZGtXr16ik+Pl4eHh6lmmvRokW69dZbJRVudouJiVFoaKh9FgJUsokTJ2ratGlWOEBlvo2ySRRVkc1mq9D1hmGoVatWuvnmmzV69Gi1a9fOTp0BVUdF76OK4L0HACpPz549nRJCVtz69eudOj8AAAAAAAAAAAAAAAAAAAAAAABch7uzGwDgHPfff7++++47GYahgoICDR48WJMmTdK9994rf39/SdLhw4f11FNPlQikMQxDd9xxR6kDaSTphx9+sI79/f0JpEGVNnXqVAUEBGjKlCk6deqU9XzxzaPFg2rstamUDDlUVeXZ2GwYhurUqSNfX181bdpUXl5eDugMqDoICACAmmHDhg3ObgEAAAAAAAAAAAAAAAAAAAAAAACwGCa73IEa6/rrrz8rcMZmsykoKEj5+flKTk6WJOs10zRVt25dRUVFKSQkpFRznDp1Sg0bNlRGRoYkqX///lq6dKmjlgRUmtTUVC1cuFC//PKLDh06pLS0NJ06dUqmaero0aNWGE1wcLDq1Kljt3ljYmLsVgsAAAAAAAAAAAAAAAAAAAAAAAAAAAAAgHMhlAaowWJjY3XNNdfor7/+klQYPnMuReEakjR9+nQ9/PDDpZ7jyy+/1F133WXVmTZtmsaNG1eBrgHXZ7PZrPtm9erVioyMdHJHAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACUns3ZDQBwntDQUG3atElt2rSRaZoyDOOcD9M0ZZqmXnjhhTIF0pimqddee61EqM2AAQMcsRQAQCVzc3OTm5ub3N3dtW7dOme3AwAAAAAAAAAAAAAAAAAAAAAAAAAAAMCOCKUBarjmzZtr9+7dmj59ujp27GgF0BQ9ateurb59+2rz5s2aNGlSmWrPnz9f+/fvl2makqQOHTqoWbNm9l8E4IKKfu6B6qr4e8WFEF4DAAAAAAAAAAAAAAAAAAAAAAAAAAAAVD3uzm4AgPO5u7vrkUce0SOPPKLU1FQdP35cGRkZqlevnpo3b67atWuXq27v3r0VExNjnXt7e9urZcClvfTSS9Zx8+bNndgJ4FiGYVx0DAFNAAAAAAAAAAAAAAAAAAAAAAAAAAAAQNVDKA2AEvz9/eXv72+XWoGBgQoMDLRLLaAqKR5KU1l+/vln3XnnnZIKg0Kio6MrvQfULDabzQqcuVjwTGnCawAAAAAAAAAAAAAAAAAAAAAAAAAAAAC4DkJpAACoBnJycnTkyBFJBICgcvj5+Sk1NVWGYSgpKcnZ7QAAANQYo0aNckhdwzD08ccfO6Q2AAAAAAAAAAAAAAAAAAAAAAAAqh5CaQAAAFBmTZo0UWpqqiTp+++/1/Dhw53cEQAAQM3w6aef2j2I0jRNQmkAAAAAAAAAAAAAAAAAAAAAAABQgs3ZDQAAAKDq6datm6TCDcxz587V5MmTFRcX5+SuAAAAcDGmaZZ4AAAAAAAAAAAAAAAAAAAAAAAAAOdimOw+AQCgylu7dq169eolSTIMQ/n5+U7uCNXdnj17FBERIcMwZJqmDMOQJNWrV0++vr7W+ZEjR6zjBg0aqE6dOnaZ3zAMRUdH26UWAABAVWKzVTxjuujzWfH/FuR7BAAAAAAAAAAAAAAAAAAAAAAAAIpzd3YDAAAAqHouv/xyPfbYY5o2bVqJTc2pqalKTU0tMbZos3NcXJzd5i+aEwAAoKaJiYkp8zXZ2dlKTEzUtm3btGDBAm3fvl2GYahFixb6+OOPFRoa6oBOAQAAAAAAAAAAAAAAAAAAAAAAUJUZZvE/iQwAAKqktWvXqlevXpIKwzry8/Od3BFqAtM09dprr2nKlCk6deqU9XzxwJjiHzXtFSRjmiY/5wAAABWwePFijRkzRmlpaWrUqJHWrFmjNm3aOLstAAAAAAAAAAAAAAAAAAAAAAAAuBBCaQAAqAYIpYEzpaamauHChfrll1906NAhpaWl6dSpUzJNU0ePHrXCaIKDg1WnTh27zRsTE2O3WgAAADXNtm3b1LNnT+Xk5KhZs2batWuXfH19nd0WAAAAAAAAAAAAAAAAAAAAAAAAXAShNAAAVAOE0sBV2Ww2K5Rm9erVioyMdHJHAAAAKPLSSy/plVdekWEYevTRR/XOO+84uyUAAAAAAAAAAAAAAAAAAAAAAAC4CJuzGwAAAAAAAABQ+caOHStJMk1TH330kXJycpzcEQAAAAAAAAAAAAAAAAAAAAAAAFwFoTQAAABwKNM0nd0CAAAAzqFRo0YKDw+XJOXk5GjTpk1O7ggAAAAAAAAAAAAAAAAAAAAAAACuwt3ZDQAAAKD6eumll6zj5s2bO7ETAAAAnEv9+vUVExMjSTp48KD69Onj5I4AAAAAAAAAAAAAAAAAAAAAAADgCgilAQAAgMMUD6WpLD///LPuvPNOSZJhGIqOjq70HgAAAKqKzMxM6zg7O9uJnQAAAAAAAAAAAAAAAAAAAAAAAMCVEEoDAACAaiUnJ0dHjhyRVBhKAwAAgHNLS0vToUOHrM9M9erVc25DAAAAAAAAAAAAAAAAAAAAAAAAcBk2ZzcAAAAAAAAAoPK9+eabysvLk2makqTw8HAndwQAAAAAAAAAAAAAAAAAAAAAAABX4e7sBgAAAAAAAABUnoKCAr355pt64403ZBiGTNNU7dq11aNHD2e3BgAAAAAAAAAAAAAAAAAAAAAAABdBKA0AAAAAAABQRWzatKlc1+Xk5Cg+Pl47d+7UkiVLdOzYMZmmKUkyDEOjR49W3bp17dkqAAAAAAAAAAAAAAAAAAAAAAAAqjBCaQAAAAAAAIAqomfPnjIMo0I1iofRmKapSy65RJMnT7ZHewAAAAAAAAAAAAAAAAAAAAAAAKgmbM5uAAAAAAAAAEDZmKZZrockK9TGNE1dccUVWr16tfz9/Z25HAAAAAAAAAAAAAAAAAAAAAAAALgYQmkAAAAAAACAKqQoXKa81xaF0Xz44YfaunWrwsLC7NgdAAAAAAAAAAAAAAAAAAAAAAAAqgN3ZzcAAAAAAAAAoHReeumlMl9jGIbq1KkjX19fhYWFqWPHjmrQoIEDugMAAAAAAAAAAAAAAAAAAAAAAEB1QSgNAADVgL+/v3r06OHsNgAAAAA4WHlCaQAAAAAAAAAAAAAAAAAAAAAAAICyIpQGAIBqoGPHjlq/fr2z2wAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAVAM2ZzcAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAHAd7s5uAACA6ub06dM6cOCA9YiPj1dmZqYyMzOVk5Mjb29v1atXT/Xq1VOLFi3UqVMnRUREqFatWs5uHQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAQmkAALCHzMxMffPNN1q8eLHWrFmjU6dOlel6d3d39ezZU2PGjNGAAQPk4eHhoE4BAAAAAAAAAAAAAAAAAAAAAAAAAAAAALgwm7MbAACgKsvNzdW0adPUvHlzjRo1SsuXL1dOTo5M07zgQ1KJ8zNnzmjNmjUaPny4mjRpojlz5jh5ZQAAAAAAAAAAAAAAAAAAAAAAAAAAAACAmsowi3bGAwCAMjl48KAGDx6s33//3QqaMQzjotf9/a23+DXF63Tr1k2ffPKJwsPD7dg1UP2tXbtWvXr1klR4L+Xn5zu5IwAAgIvbtGmTs1tQ9+7dnd0CAAAAAAAAAAAAAAAAAAAAAAAAXAShNAAAlMPevXt13XXXKSUlRaZpyjCMs8Jm/q5ojL+/v0aOHKmcnBwlJydr//79ioqK0pkzZ0qMk6T69evr+++/V0REhKOXBFQbhNIAAICqyGazlSrk0lEMw1BeXp7T5gcAAAAAAAAAAAAAAAAAAAAAAIBrcXd2AwAAVDU5OTkaOHCgkpOTrU2jHh4eGjp0qPr3768OHTooMDBQZ86cUUJCgrZv36558+Zp/fr1MgxDaWlpOnDggBYtWiQvLy9JUm5urn744QfNmzdPCxcuVEFBgSQpMTFRN954o7Zu3arQ0FCnrRkAAABA5SA/GgAAAAAAAAAAAAAAAAAAAAAAAK7AMNnpAgBAmUyePFmTJk2SYRgyTVMdOnTQ/Pnz1bp16wte98MPP+iuu+5SUlKSJOm2227TF198cda4vXv3asyYMdqyZYs1R7du3bRx40aHrAeobtauXatevXpJkgzDUH5+vpM7AgAAuDibzVau64qCMos713/3XWwcn5sAAAAAAAAAAAAAAAAAAAAAAABQHKE0AACUgWmaCgkJUUJCgkzTVIsWLbRlyxb5+/uX6vrdu3fr6quv1qlTp2QYhpYtW6Z+/fqdNe7UqVMaMGCAVq9eLalwg+jChQs1ZMgQu64HqI4IpQEAAFXRyy+/XKbxpmlq7ty5Onr0qHXu7u6uSy+9VJdddpkCAwPl5eWlrKwsJScna8+ePdq3b5/y8vKsgJpmzZrp7rvvtmq+9NJL9lsQAAAAAAAAAAAAAAAAAAAAAAAAqjRCaQAAKIPt27erc+fOkgrDLpYuXar+/fuXqcaTTz6pt956S4ZhqE+fPlq5cuU5x6WlpalNmzZKTEyUJHXt2lWbNm2q2AKAGoBQGgAAUN2dPHlSw4YN05o1a2SapurXr6/nnntOd9xxhwIDA897XXJysr744gu9+uqrSkxMlGEY6t27txYuXCgfH59KXAEAAAAAAAAAAAAAAAAAAAAAAABcHaE0AACUwZw5c3TvvfdKknx9fZWcnCw3N7cy1di1a5c6duwoSXJzc1N6ero8PT3POXbGjBkaP368JMlmsykuLk5BQUHlXwBQA/z22296/PHHrfP169c7sRsAAAD7ys/PV2RkpH788UdJ0nXXXadvvvlGfn5+pa6Rnp6uIUOGaN26dTIMQ926ddO6detks9kc1TYAAAAAAAAAAAAAAAAAAAAAAACqGHaaAABQBomJiZIkwzAUHh5e5kAaSWrVqpV1XFBQoNjY2POOHTZsmAzDkCSZpqktW7aUeT6gpunYsaPWr19vPQAAAKqTt99+W5s3b5YkXXbZZVq1alWZAmkkyc/PTytXrtTll18u0zS1efNmvf32245oFwAAAAAAAAAAAAAAAAAAAAAAAFUUoTQAAJRB8RAam618b6NFITNFkpKSzju2UaNGatSokXUeExNTrjkBAAAAVH35+fklwmOmTZsmDw+PctWqVauWVcs0Tb311lsqKCiwS58AAAAAAAAAAAAAAAAAAAAAAACo+tyd3QAAAFVJ/fr1JRVu2jxy5IhM0zwrZOZioqOjS5z7+/tfcHzDhg114sQJSdLJkyfLNBfgbKdPn9aBAwesR3x8vDIzM5WZmamcnBx5e3urXr16qlevnlq0aKFOnTopIiJCtWrVcnbrAAAALufXX39VYmKipMLvEZGRkRWqFxkZqYCAAKWkpCgpKUm//PKLunbtao9WAQAAAAAAAAAAAAAAAAAAAAAAUMURSgMAQBm0aNHCOk5LS9OaNWvUq1evMtX4+uuvS5yHhISU+lp3d9664foyMzP1zTffaPHixVqzZo1OnTpVpuvd3d3Vs2dPjRkzRgMGDJCHh4eDOgUAAKha9u7dK0kyDENhYWEVrldUJyUlxapPKA0AAAAAAAAAAAAAAAAAAAAAAAAkyebsBgAAqEq6dOkiHx8fGYYh0zQ1ceJEZWdnl/r6qKgoTZs2TYZhSJLat28vf3//C16TkJBgHQcEBJSvcaAS5Obmatq0aWrevLlGjRql5cuXKycnR6ZpXvAhqcT5mTNntGbNGg0fPlxNmjTRnDlznLwyAAAA11AUHiNJZ86csUvN4nWK1wcAAAAAAAAAAAAAAAAAAAAAAEDNRigNAABl4ObmpltvvVWmacowDO3du1c33nijTpw4cdFrt23bphtuuEFZWVnW9YMGDbrgNampqfrzzz+t8/Dw8AqvAXCEgwcPKiIiQk888YSSkpKssBnDMC74kGSNLT6+6PnExESNGTNGPXv2VExMTOUvDAAAwIX4+vpKKvycFBMTo9OnT1eo3unTp3X48GHr85ePj0+FewQAAAAAAAAAAAAAAAAAAAAAAED1QCgNAABl9OKLL6p27dqSCjeD/vjjj2rbtq0effRRrV69WgkJCcrLy9Pp06d17NgxLV68WLfeequ6du2qY8eOWRs+fX19NWHChAvOtWHDhhLnERERjlgSUCF79+5Vt27d9Pvvv1uBS1Lh/XG+R9Hr/v7+Gj9+vO6//34NHTpUbdu2lbu7e4lQG9M0tWnTJnXp0kW7du1y1jIBAACcrkmTJtZxdna2Fi1aVKF6X3/9tbKzs63PXk2bNq1QPQAAAAAAAAAAAAAAAAAAAAAAAFQfhlm06wQAAJTanDlzdO+995YI3yg6Pp+iMUX/fv7557r99tsveM3tt9+u+fPnyzAMtW7dWvv377fbGgB7yMnJ0WWXXabDhw9bP9+1atXS0KFD1b9/f3Xo0EGBgYE6c+aMEhIStH37ds2bN0/r16+37pnevXtr0aJF8vLykiTl5ubqhx9+0Lx587Rw4UIVFBRIKryHgoODtXXrVoWGhjptzQAAAM6Snp6uBg0a6MyZMzJNUw0aNNDOnTvVsGHDMtf666+/1LFjRyUkJMg0TdWuXVtxcXHy8/NzQOcAAAAAAAAAAAAAAAAAAAAAAACoamzObgAAgKpo1KhR+te//mWdF4VxnO9RfIxhGHrttdcuGkgTFxenxYsXSyoM4+jbt6/jFgSU09SpU0sE0nTo0EG7d+/WF198oeHDh6tNmzaqX7++QkJCFBERoXvvvVdr167Vd999p6CgIEnSDz/8oPvvv9+qWatWLfXr109ffvmldu7cqc6dO1v3TkJCgu666y5nLRcAAMCp/Pz8NHDgQOuzUXx8vLp27arffvutTHV27Niha6+9VvHx8VatAQMGEEgDAAAAAAAAAAAAAAAAAAAAAAAAi2EW7ZQHAABltmrVKj388MOKiYmRVBg8cy5Fb7fh4eGaOXOmevfufdHaGRkZio2Ntc6bNGnCJlG4FNM0FRISooSEBJmmqRYtWmjLli3y9/cv1fW7d+/W1VdfrVOnTskwDC1btkz9+vU7a9ypU6c0YMAArV69WlLhfbZw4UINGTLErusBAACoCo4fP6727dsrIyNDUuFnMpvNpmHDhumuu+5Sjx495OXlddZ1mTcEXYQAADapSURBVJmZ2rRpk+bOnatFixapoKDAChb09fXVvn371Lhx48peDgAAAAAAAAAAAAAAAAAAAAAAAFwUoTQAAFRQQUGBFi5cqGXLlmnjxo2Ki4sr8XrTpk3VrVs3DRw4UIMHD5bNZnNSp4B9bd++XZ07d5ZUGBSzdOlS9e/fv0w1nnzySb311lsyDEN9+vTRypUrzzkuLS1Nbdq0UWJioiSpa9eu2rRpU8UWAAAAUEWtXLlSQ4cO1enTpyUVBtMUBWQahqHQ0FAFBgbK09NT2dnZSkpK0rFjx6ywzKLxpmmqdu3a+uabb3TzzTc7bT0AAAAAAAAAAAAAAAAAAAAAAABwPYTSAABgZ3l5eUpOTpbNZlNAQIDc3Nyc3RLgEHPmzNG9994rSfL19VVycnKZf9537dqljh07SpLc3NyUnp4uT0/Pc46dMWOGxo8fL0my2WyKi4tTUFBQ+RcAAABQha1bt0533nmn4uLirECav/83X1HwzN+fKxobHBysL7/8Utdff33lNA0AAAAAAAAAAAAAAAAAAAAAAIAqw+bsBgAAqG7c3d3VoEED1a9fn0AaVGuJiYmSCjc2h4eHl+vnvVWrVtZxQUGBYmNjzzt22LBhJTZRb9mypczzAQAAVBeRkZE6cOCAxo4dqzp16ljhM4ZhWI9znZumqdq1a+v+++/XgQMHCKQBAAAAAAAAAAAAAAAAAAAAAADAORFKAwAAgHIpHkJjs5XvY2XR5ugiSUlJ5x3bqFEjNWrUyDqPiYkp15wAAADVhZ+fnz788EOdOHFCH374oW699VaFhYVJKgyfKXpIUlhYmIYNG6YPPvhAJ06c0MyZM+Xv7+/M9gEAAAAAAAAAAAAAAAAAAAAAAODC3J3dAAAAAKqm+vXrSyrc8HzkyBGZpnlWyMzFREdHlzi/2Mbohg0b6sSJE5KkkydPlmkuAACA6srPz09jx47V2LFjJRV+Pjt58qQyMzPl5eUlPz+/Mn9OAwAAAAAAAAAAAAAAAAAAAAAAQM1mc3YDAABUNampqZU+Z0pKiu66665Knxe4kBYtWljHaWlpWrNmTZlrfP311yXOQ0JCSn2tuzv5igAAAOdiGIb8/PzUuHFj1atXj0AaAAAAAAAAAAAAAAAAAAAAAAAAlBmhNAAAlFH79u21ZMmSSptv/vz5atu2rb766qtKmxMojS5dusjHx0eGYcg0TU2cOFHZ2dmlvj4qKkrTpk2zNkm3b99e/v7+F7wmISHBOg4ICChf4wAAAAAAAAAAAAAAAAAAAAAAAAAAAAAuiFAaAADKKC4uTkOHDtXw4cOVmJjosHmOHz+uW265RXfccYdD5wHKy83NTbfeeqtM05RhGNq7d69uvPFGnThx4qLXbtu2TTfccIOysrKs6wcNGnTBa1JTU/Xnn39a5+Hh4RVeAwAAAAAAAAAAAAAAAAAAAAAAAAAAAICzGaZpms5uAgCAqsRms8kwDElSQECApk+frttvv92uc8yaNUtPP/20MjIyVPRWbRiG8vPz7ToPUFHHjh1Tq1atlJuba/2s+vj4aOTIkerfv786dOiggIAA5efnKyEhQdu2bdP8+fO1dOlS5eXlyTAMmaYpPz8/xcTEqF69eueda8mSJRoyZIikwvshISFBgYGBlbFMAAAAAAAAAAAAAAAAAAAAAAAAAAAAoEYhlAYAgDIqHkpjmqYMw1Dfvn01a9YshYSEVKh2VFSUxowZo82bN1u1i+bp0aOH1q9fX+H+AXubM2eO7r333rPuiwspGlP07+eff37RcKfbb79d8+fPl2EYat26tfbv32+3NQAAAFQHZ86cUXp6unJyclSe//ILDQ11QFcAAAAAAAAAAAAAAAAAAAAAAACoityd3QAAAFXNjBkz9MwzzygrK8sK1VixYoXat2+vt956S6NHjy5zzYKCAr355puaPHmyTp8+XSKww8fHR2+88YbGjh3rgNUAFTdq1CjFx8fr+eeflyTrZ/d8DMMoEUjz2muvXTSQJi4uTosXL5ZUGGjTt29f+y0AAACgikpNTdUXX3yhVatW6bffflNiYmK5axmGoby8PDt2BwAAAAAAAAAAAAAAAAAAAAAAgKrMMMvzZ5MBAKjhjhw5ojFjxmjt2rUyDEOSrICN66+/Xh999JHCwsJKVWvXrl0aPXq0du3aZdUoqnfzzTdr1qxZatKkicPWAtjLqlWr9PDDDysmJkaSrJ/lvyv6+BkeHq6ZM2eqd+/eF62dkZGh2NhY67xJkyby8/OzQ9cAAABV07vvvqsXXnhB2dnZknTBUMDSMAxD+fn59mgNAAAAAAAAAAAAAAAAAAAAAAAA1QChNAAAVMBHH32kJ554QidPnpRhGNZGUC8vL7322mt6+OGHz3vt6dOnNWnSJL399tvKz8+3AmlM01RQUJDeffdd3X777ZW1FMAuCgoKtHDhQi1btkwbN25UXFxcidebNm2qbt26aeDAgRo8eLBsNpuTOgUAAKi6xo4dq48++sj6/lE82LK8CKUBAAAAAAAAAAAAAAAAAAAAAABAcYTSAABQQcePH9d9992nVatWldgMahiGunbtqo8//lgtW7Yscc2mTZs0ZswY/fHHH9bYoutGjBihGTNmKCgoqNLXAthbXl6ekpOTZbPZFBAQIDc3N2e3BAAAUKV99tlnuueeeySpRDCmv7+/LrvsMgUHB8vLy6tctT/55BO79QkAAAAAAAAAAAAAAAAAAAAAAICqjVAaAADsZO7cuXrssceUkpJSYnNo3bp1NWnSJD3++OPKysrSk08+qY8++kgFBQWS/reRtHHjxpo5c6b69evnzGUAAAAAcGFhYWE6duyY9T2iQ4cOev3119WrVy/ZbDZntwcAAAAAAAAAAAAAAAAAAAAAAIBqglAaAADsKD4+Xg888ICWLl0qwzAkSaZpyjAMRUREKCEhQSdOnLCeK/p3zJgxmjp1qnx8fJy8AgAAAACuaufOnbryyiut7xpXX321Vq9erbp16zq5MwAAAAAAAAAAAAAAAAAAAAAAAFQ3/PlkAADsqEGDBlq8eLHmzZunoKCgEuEzO3fu1PHjx0s816JFC61bt06zZs0ikAYAAADABe3atUtSYfClJL3//vsE0gAAAAAAAAAAAAAAAAAAAAAAAMAhCKUBAMABhg8frv3796tDhw5WCI0k6183Nzc9+eST2rNnj3r06OHMVoEKSU1NrfQ5U1JSdNddd1X6vAAAAM6WmJhoHYeEhCgiIsJ5zQAAAAAAAAAAAAAAAAAAAAAAAKBaI5QGAAAHSElJ0bhx47Rnz56zAmkkyTRN5ebmyjRNZ7UI2EX79u21ZMmSSptv/vz5atu2rb766qtKmxMAAMBVFP9u0bhxYyd3AwAAAAAAAAAAAAAAAAAAAAAAgOqMUBoAAOzs66+/Vrt27TRv3jwrdMY0TevYMAwVFBRo+vTpuuyyy7R+/XpntgtUSFxcnIYOHarhw4crMTHRYfMcP35ct9xyi+644w6HzgMAAODKQkNDrePMzEwndgIAAAAAAAAAAAAAAAAAAAAAAIDqjlAaAADsJD4+XkOGDNGIESOUkJAg0zRlGIZM01Tnzp21YMECtWnTpkRQzeHDh3XDDTfo/vvvV0ZGhpNXAJTfokWL1K5dO3311Vd2rz1r1iy1b99eK1assO4fAACAmuiaa66RVPhd4siRI8rNzXVyRwAAAAAAAAAAAAAAAAAAAAAAAKiuCKUBAMAOPvvsM7Vr105Lly61wmgkqU6dOnrrrbf0888/a9iwYdq1a5eeffZZubm5yTAMK7TmP//5j9q1a6cVK1Y4eSVA+SUnJ+uuu+7SLbfcohMnTlS4XlRUlHr27KmHHnpIJ0+elCTr3urevXuF6wMAAFQ1TZs21XXXXSdJysnJ0apVq5zcEQAAAAAAAAAAAAAAAAAAAAAAAKorQmkAAKiAP//8UzfffLNGjRql1NRUSbKCZrp37649e/bosccek81W+Jbr4eGhKVOmaOvWrerQoYMVYGOapo4fP65bbrlFd955p5KTk525LKDUZsyYIU9PzxI/yytWrFD79u318ccfl6tmQUGBXn/9dXXo0EGbN28uUdvb21sffvih1q9fb+eVAAAAVA2vv/663NzcJEnPPfecTp065eSOAAAAAAAAAAAAAAAAgP9v786DrKzu/PG/n25W2cFCwAhIRMclQdzGqJjRWBBtjRLEZUxcMBIXHP0aTbSiRmOMkxiNRKPEDbdJ1CTGcXRcMkZicBnUIDEIiXFDUTbZmsUNnt8f/LhDu3ZDN03r61V1i3Pvfc75fE7VvcVzqTpvAAAAgE8ioTQAsJbGjRuX7bbbLvfff//7QjN+/vOfZ8KECfnsZz/7gXO33377PPHEE7ngggvSpk2bFEVRmf+rX/0q22yzTW677bb1vCNouDFjxuSZZ57Jl770pcr3IEkWLVqU0aNHZ+jQoXn55Zfrvd7TTz+dnXfeuc4B69Xfjf322y9Tp07N8ccf3yR7AQBoCXbeeedccsklKcsy06ZNy4gRI1JbW9vcbQEAAAAAAAAAAAAAAPAJU5RlWTZ3EwDQkrzwwgs59thj8/DDD9cJ4SjLMsOGDcvVV1+dzTbbrN7rPfvssxk1alQmTZpUZ62iKHLAAQfkqquuSu/evZtkL9CYrrnmmpxxxhlZvHhxJUgmSTp06JCLLrooY8aM+dC5b731Vs4777xccsklWbFiRZ2gp4033jiXXXZZ/vVf/3V9bQUAYIP3i1/8Iqecckreeeed9O3bN2eddVZGjhyZbt26NXdrAAAAAAAAAAAAAAAAfAIIpQGABurQoUPefPPNOqEZ3bp1y6WXXpqjjjpqrdYsyzI//elPc+6552bZsmWVdYuiSOfOnXPJJZdk1KhRjbwTaHwzZ87M6NGjc++9974vZGn33XfPddddl4EDB9aZ8/DDD+e4447LP/7xj/cFPR122GH52c9+lo033ni97wUAYEO09957V8bPPfdcZs6cmSQpiiJFUaR///7p2bNn2rVr16B1i6LIgw8+2Ki9AgAAAAAAAAAAAAAA0HIJpQGABqqqqqoTmnHQQQflqquuyiabbLLOaz///PM59thj8/DDD78v0GPvvffO73//+3WuAevDTTfdlNNOOy3z58+vhCwlSfv27XPeeeflW9/6VpYuXZpvf/vbueaaa7Jy5cokqVy76aab5qqrrsr+++/fnNsAANjgrPl7ZE1r/hPfB73/UVb/5lixYsU69wcAAAAAAAAAAAAAAMAng1AaAGigqqqqJEnPnj1z+eWXZ+TIkY1e48orr8xZZ52V2traSkiHQ6K0NLNnz84JJ5yQO++8830hS9tvv33mzJmT1157rfLa6j+PO+64XHzxxenUqVMz7wAAYMPzYaE068LvDQAAAAAAAAAAAAAAAN5LKA0ANFBVVVWOOOKIXHbZZenRo0eT1ZkxY0ZGjx6dBx54IEkcEqXFuu222/Jv//ZvmTt3biV8Zk2rX9tiiy1yzTXX5Itf/GIzdQoAsOFbHZLZ2PzeAAAAAAAAAAAAAAAAYE1CaQCgge65557U1NSst3rjx4/PaaedlsWLFzskSov1xhtvZJ999smUKVMqITSr/2zVqlVOO+20nHfeeWnXrl1ztwoAAAAAAAAAAAAAAAAAAJ96TfNfKwPAJ9j6DKRJkmOOOSbPPvtsvvKVr6zXutBY5s+fn1NOOSV/+ctfUhRFklT+TJKyLPP2229HViIAAAAAAAAAAAAAAAAAAGwYitLpXwAAmsivf/3rnHzyyZk7d27KskxRFJXwmTWDaZJk8803zzXXXJO99tqrOVoFAAAAAAAAAAAAAAAAAAD+f0JpAKAJTJs2LY888kgeeeSRTJs2LfPnz8/8+fNTW1ubTp06pXv37unevXu23nrr7L777tl9992z9dZbN3fb0Ghmz56dE088MXfeeWedEJqyLLPLLrvkW9/6Vs4777xMmzbtfUE13/jGN/KTn/wknTp1as4tAAAAAAAAAAAAAAAAAADAp5ZQGgBoJG+++WZuuummjB07NtOnT6+8/lF/1RZFURlvtdVWOfXUU3PkkUemXbt2TdorNKUbb7wxp512WhYuXJiyLCuhM+3bt88FF1yQU089NVVVVXnnnXdy/vnn58c//nFWrFiRJJXr+/Tpk3HjxqWmpqaZdwMAAAAAAAAAAAAAAAAAAJ8+QmkAoBHccccdOeGEEzJv3rz3hdCsGTzzXh90bY8ePXLllVfm4IMPbpJeoam8+uqrGT16dO6///5KuEyy6nO+55575rrrrstnP/vZ9817+umnM2rUqDz99NOVAJtk1ffh8MMPz9ixY9OjR4/1uhcAAAAAAAAAAAAAAAAAAPg0q2ruBgCgJVu+fHmOPfbYjBw5MnPnzq0Ecax+JKsCOT7skaTO9WVZZt68eTn00ENzzDHHZNmyZc25Pai3cePGZbvttqsTSFOWZTp27Jif//znmTBhwgcG0iTJ9ttvnyeeeCIXXHBB2rRpU+f78Ktf/SrbbLNNbrvttvW8IwAAAAAAAAAAAAAAAAAA+PQqytUn4gGABlmyZEn222+/PPLII5UQjiSVsJn27dtn0KBBGTx4cHr16pUuXbqkQ4cOWbp0aRYtWpRZs2Zl8uTJmTJlSpYvX54kddYoiiJf+MIXct9996Vjx47Ns0n4GC+88EKOPfbYPPzww+/7HgwbNixXX311Nttss3qv9+yzz2bUqFGZNGnS+74PBxxwQK666qr07t27SfYCANCSvfDCC5k8eXLmzZuXhQsX5q233mrwGueee24TdAYAAAAAAAAAAAAAAEBLJJQGANZCWZb5yle+knvuuSfJqjCZsixTXV2dAw88MEcffXT23XffVFdXf+xaK1asyL333pvx48fnrrvuysqVKys1iqLIvvvum7vvvrtJ9wNrq0OHDnnzzTcrn9eyLNOtW7dceumlOeqoo9ZqzbIs89Of/jTnnntuli1bVlm3KIp07tw5l1xySUaNGtXIOwEAaHlmzpyZK664IjfccEPmzJmzzuutWLGiEboCAAAAAAAAAAAAAADgk0AoDQCshcsvvzynnHJKiqJIsipEY5dddslVV12VwYMHr/W6kydPzvHHH58nnniiThDHpZdemlNOOaWx2odGU1VVVed7cNBBB+Wqq67KJptsss5rP//88zn22GPz8MMP16lRFEX23nvv/P73v1/nGgAALdUtt9ySk046KUuWLMlH/fPemvdRH/b+6nssoTQAAAAAAAAAAAAAAACsJpQGABpo4cKF6d+/f2praysHO4866qhcd911qaqqWuf1V65cmVGjRuWmm26qHBDt3LlzXnrppXTt2nWd14fGtPoz37Nnz1x++eUZOXJko9e48sorc9ZZZ6W2ttahaQCAJDfeeGNGjRpVuS9a85/3VofQJB8cRPNh77u/AgAAAAAAAAAAAAAAYE3rfnIeAD5lxo0bl8WLFydZdXDz8MMPz/jx4xslkCZZFfJxww035LDDDqscEq2trc24ceMaZX1obEcccUSmTp3aJIE0SXLiiSfmmWeeydChQz/wYDUAwKfJ888/n+OPP75OIM2QIUMybty43HfffXXul37yk5/kvvvuyy9/+ct897vfzRe+8IWUZVm5Zptttsldd92Vhx56KH/4wx+aa0sAAAAAAAAAAAAAAABsgIrSyV4AaJDtttsu06ZNS1mW6dWrV5599tl07dq10essXLgw22yzTWbPnp2yLLP11ltn6tSpjV4H1sU999yTmpqa9VZv/PjxOe2007J48eKsWLFivdUFANhQHHfccbnuuuuSrArJPP/883P22WdX3q+qqkpRFEmS3//+99l7773rzH/66adz4okn5vHHH09RFNl6663zwAMPpE+fPutvEwAAAAAAAAAAAAAAAGzwqpq7AQBoSWbNmpVnn302yaoDoKeeemqTBNIkSdeuXXPKKadkdX7c9OnT8/rrrzdJLVhb6zOQJkmOOeaYPPvss/nKV76yXusCAGwIVqxYkVtvvTVFUaQoigwbNqxOIE19bL/99pk4cWK+/vWvpyzLTJs2LTU1NXnnnXeaqGsAAAAAAAAAAAAAAABaIqE0ANAAkydPTpJKUMyhhx7apPUOO+ywD6wPn2a9e/fO7373u+ZuAwBgvfvzn/+cpUuXVn6PnHHGGWu1TlVVVcaPH58dd9wxZVnmL3/5S37yk580ZqsAAAAAAAAAAAAAAAC0cEJpAKABXnrppcq4e/fu6devX5PW69evXzbeeOMPrA8txbRp03LttdfmmGOOya677pott9wyG2+8cdq2bZuNN944W265ZXbdddccc8wxufbaazNt2rTmbhkAYIM0ffr0yrht27b54he/+JHXv/POOx/6XlVVVX7wgx8kWRW6+bOf/awSdgMAAAAAAAAAAAAAAACtmrsBAGhJFi1alCQpiiJ9+vRZLzX79OmTefPmJUkWL168XmrCunrzzTdz0003ZezYsXUOT7/3oPP8+fMzf/78JMkTTzyRm266KUmy1VZb5dRTT82RRx6Zdu3arb/GAQA2YKvvm4qiyOabb56qqvfnTRdFURm/+eabH7nePvvsk86dO2fx4sWZM2dOnnzyyey8886N2zQAAAAAAAAAAAAAAAAt0vtPrgAAH2rNQI1WrdZPttuaB03fG+gBG6I77rgj/fr1ywknnJBp06alLMvKoyiKD32sed306dNzwgknpG/fvvnNb37T3FsCANggLF26tDLu0qXLB17TsWPHyu+GhQsXfuR61dXV6devX+X5X//613VvEgAAAAAAAAAAAAAAgE8EoTQA0ACdOnVKsiocZubMmeul5uuvv/6++rAhWr58eY499tiMHDkyc+fOfV8ITZI6wTPvfSR5X0jNvHnzcuihh+aYY47JsmXLmnN7AADNbs3fA2+99dYHXtO5c+fK+JVXXvnYNdu1a1cZz5kzZx26AwAAAAAAAAAAAAAA4JOkVXM3AAAtSb9+/SrjefPmZdasWenVq1eT1Zs1a1bmzJlTCfTo27dvk9WCdbFkyZLst99+eeSRRyphNEkqYTPt27fPoEGDMnjw4PTq1StdunRJhw4dsnTp0ixatCizZs3K5MmTM2XKlCxfvjxJ6qxx00035bnnnst9992Xjh07Ns8mAQCa2Zq/PRYtWvSB1wwYMKASoPnUU0997JozZsyojFfffwEAAAAAAAAAAAAAAIBQGgBogEGDBiVZdVizLMv8+te/zsknn9xk9W6//fZKqEdRFJX6sCEpyzKHH354Jk6cmOT/vh/V1dU58MADc/TRR2ffffdNdXX1x661YsWK3HvvvRk/fnzuuuuurFy5srLeY489lsMOOyx33313U28JAGCDtM022yRZdf81Y8aMvPPOO2ndunWdawYNGpQ//elPKcsyEyZMyLJly7LRRht94HqTJk3K7NmzK2E0G2+8cdNuAAAAAAAAAAAAAAAAgBajqrkbAICWpG/fvhkwYECSVQdBL7vssixfvrxJai1btiyXXXZZ5YBo//79069fvyapBeviiiuuyD333JOiKCoBMrvssksmTZqU3/zmN9l///3rFUiTJNXV1dl///3z29/+NpMmTcpOO+2Usiwr6957770ZO3ZsE+8IAGDDtNVWW6Vjx45JVoX5TZ069X3XDB06NMmqoMDFixfnoosu+sC13nrrrZxyyilJUgnC3HHHHZuibQAAAAAAAAAAAAAAAFogoTQA0ECHHXZYJSTjpZdeyplnntkkdc4888y89NJLlVqHH354k9SBdbFw4cKcc845ldCYsixz1FFH5dFHH83gwYPXae3Bgwfnsccey5FHHlknmOZ73/teFi5c2DgbAABoQVq1apUhQ4ZUnt9///3vu+bLX/5yevbsmWRV2MwPf/jDnHjiiXnmmWfy9ttvZ8mSJbnvvvuyxx575H//938rIZgDBgzIoEGD1s9GAAAAAAAAAAAAAAAA2OAJpQGABhozZkzatWuXZNUhzyuuuCJnn312o9Y4++yzc8UVV1QOiLZt2zYnnXRSo9aAxjBu3LgsXrw4SSrhSePHj09VVePcZlZVVeWGG26ohEElSW1tbcaNG9co6wMAtDT7779/Zfy73/3ufe+3atUqF154YZ1Qv1/84hfZfvvt0759+3Tp0iU1NTV56qmnKu8XRZHzzjtvPe4CAAAAAAAAAAAAAACADZ1QGgBooF69euWMM86oc8jzoosuysEHH5yZM2eu09qvvfZaRowYkYsuuihJKjVOP/309O7duzHah0Z1yy23VL4Hm2yySX7+8583SZ0rr7wyvXr1qtS6+eabm6QOAMCG7pBDDkl1dXXKssykSZMyadKk911z7LHH5ogjjqj8nkhW/bZY87H69ST55je/mSOOOGK97QEAAAAAAAAAAAAAAIANX1GWZdncTQBAS/POO+9kyJAhmTRpUiUkoyiKdOzYMaNHj87RRx+dbbfdtt7rTZ06NePHj88111yTJUuW1DkkutNOO2XixIlp3bp1U20H1sqsWbPSp0+fymf1oosuyre//e0mq/ejH/0oZ511VpKkKIq8+uqrwpoAgE+lhQsXZsWKFUmSDh06pF27du+7pizLfP/738+Pf/zjLF++/APX6dSpU84555ycfvrpTdovAAAAAAAAAAAAAAAALY9QGgBYS7Nnz86ee+6Z5557rhJMk6QS0DFw4MDsuOOOGTx4cHr37p0uXbqkQ4cOWbp0aRYtWpRZs2Zl8uTJefLJJ/Pcc88lSZ01yrLMwIED88c//jG9evVqnk3CR7j33ntTU1OTZNVn9oUXXki/fv2arN7LL7+czTffvFLvv/7rv7Lffvs1WT0AgE+COXPm5D//8z/z+OOPZ/bs2SnLMr169cpuu+2W4cOHp3v37s3dIgAAAAAAAAAAAAAAABsgoTQAsA7mzJmTww47LBMmTKiE0az5V+vq1z7KB11flmX23HPP3HrrrQJp2GBdddVVOemkk5IkPXr0yNy5c5u8Zs+ePTNv3rwURZHLL788J554YpPXBAAAAAAAAAAAAAAAAACAT5uq5m4AAFqynj175sEHH8xFF12U1q1bpyzLFEVReZRl+bGP917fqlWrXHjhhXnooYcE0rBBW7RoUZJVYUp9+vRZLzXXrLN48eL1UhMAAAAAAAAAAAAAAAAAAD5thNIAwDoqiiLf+c538o9//CNnnHFGunbtWgmcWf3+hz2SVK7t0qVLTj/99PzjH//IWWedVXkfNlSrP+NJ0qpVq/VSs6rq/25f16wPAAAAAAAAAAAAAAAAAAA0nvVzehgAPgU+85nP5Ec/+lHOO++8/OEPf8jEiRPzyCOPZPr06Vm4cGHefffdyrXV1dXp1q1bttpqq+yxxx7Zfffds/fee2ejjTZqxh1Aw3Tq1CnJqnCYmTNnrpear7/++vvqAwAAAAAAAAAAAAAAAAAAjUsoDQA0svbt26empiY1NTV1Xl+8eHFqa2vTqVOndO7cuZm6g8bTr1+/ynjevHmZNWtWevXq1WT1Zs2alTlz5qQoiiRJ3759m6wWAAAAAAAAAAAAAAAAAAB8mlU1dwMA8GnRuXPnbLrppgJp+MQYNGhQkqQoipRlmV//+tdNWu/2229PWZYpy7JOfQAAAAAAAAAAAAAAAAAAoHEV5epTvQAA0EBbbLFFXnzxxZRlmc033zx//etf0759+0avs2zZsmy33XZ5+eWXK7Wef/75Rq8DANBcBgwY0Kz1i6JwfwUAAAAAAAAAAAAAAEBFq+ZuAABIamtrs2zZsvTo0SOtWvnrmZbjsMMOyw9/+MMURZGXXnopZ555ZsaOHdvodc4888y89NJLSVYdmD788MMbvQYAQHN66aWXUhRFmis/uiiKZqkLAAAAAAAAAAAAAADAhqmquRsAgE+jsizzq1/9KjU1NenSpUu6du2aPn36pG3bttl0000zatSoTJgwobnbhI81ZsyYtGvXLsmqz/UVV1yRs88+u1FrnH322bniiisqB6Xbtm2bk046qVFrAABsKIqiWO8PAAAAAAAAAAAAAAAAeK+ibK7/fhkAWqiyLHP33Xdn9V+h1dXVqampqff8F198MSNGjMiUKVMq673X6oOhI0aMyPXXX5+OHTs2QufQNL73ve/lggsuSFEUKcsyRVFk+PDhGTt2bDbddNO1Xve1117LySefnDvvvLPyPSmKIt/97nfz/e9/v7HaBwDYIPTv37/ZA2JefPHFZq0PAAAAAAAAAAAAAADAhkMoDQA00KRJk7LrrrtWDoweeOCBueOOO+o1d8aMGfnnf/7nzJkzp07Ixnut+d6uu+6aBx54IB06dGikHUDjeueddzJkyJBMmjSpTjBNx44dM3r06Bx99NHZdttt673e1KlTM378+FxzzTVZsmRJZb0k2WmnnTJx4sS0bt26qbYDAAAAAAAAAAAAAAAAAACfekJpAKCBzj///Jx//vlJVoXGTJgwIUOGDPnYeWVZZpdddslTTz1VJ4jmg/4qXv3+6jCOE044IVdccUUj7QAa3+zZs7PnnnvmueeeqwTTJP/3WR44cGB23HHHDB48OL17906XLl3SoUOHLF26NIsWLcqsWbMyefLkPPnkk3nuueeS1A1nKssyAwcOzB//+Mf06tWreTYJAAAAAAAAAAAAAAAAAACfEkJpAKCB9t5770yYMCFJssUWW+Tvf/97veaNHz8+xx57bJ1Amqqqqhx11FE55JBD0r9//yxdujRPPPFELr/88kydOrUSxlFdXZ2nn3462267bVNsCRrFnDlzcthhh2XChAl1gpVWW/Oz/2E+6PqyLLPnnnvm1ltvFUgDAAAAAAAAAAAAAAAAAADrQVVzNwAALUlZlnnqqadSFEWKosjBBx9c77mXXnppnXWqq6tz55135tprr83QoUOz5ZZbZvDgwRk9enT+/Oc/5+CDD64EdKxcuTLXX399o+8HGlPPnj3z4IMP5qKLLkrr1q1TlmXlu7I6YOnjHu+9vlWrVrnwwgvz0EMPCaQBAAAAAAAAAAAAAAAAAID1pChXn3YHAD7W888/n4EDByZJiqLIhAkTMmTIkI+d99RTT2XnnXeuBG0URZEzzjgj//7v//6hc956661svfXWefnll1OWZfr06ZNXX3210fYCTenVV1/N5ZdfnmuvvTYLFiyovF4UxYfOWfO2tGvXrvnGN76Rk08+OZtttlmT9goAAAAAAAAAAAAAAAAAANQllAYAGuCBBx7Il7/85SRJdXV1Fi1alI022uhj5/3gBz/IueeeWwmladu2bV577bV069btI+ddeeWVGTNmTJJVYR6vvPJK+vTps+4bgfVk+fLl+cMf/pCJEyfmkUceyfTp07Nw4cK8++67lWuqq6vTrVu3bLXVVtljjz2y++67Z++9967XdwsAAAAAAAAAAAAAAAAAAGh8rZq7AQBoSWbMmFEZb7rppvUOzZgwYUJlXBRFhg0b9rGBNEkyfPjwjBkzJkVRJEmmTJkilIYWpX379qmpqUlNTU2d1xcvXpza2tp06tQpnTt3bqbuAAA2XEuWLMnbb79ded6mTZt07NjxQ69/9NFH87Wvfa3Bdb73ve/lqKOOWqseAQAAAAAAAAAAAAAA+OQSSgMADbB48eIkq4JlunfvXq85ZVlm0qRJKYoiZVmmKIrss88+9Zrbu3fv9OzZM3PmzElRFHnttdfWunfYkHTu3FkYDQDAh1i0aFEGDBiQhQsXVl77zW9+k+HDh3/onOXLl+ell16q/O6or29/+9sZPny4ezMAAAAAAAAAAAAAAADqqGruBgCgJVm+fHll3LZt23rNmTZtWpYsWVLntd13373eNXv37l0Z19bW1nseAADQMl1yySVZsGBByrJMWZY5/vjjPzKQ5r2KoqjXI0nmzZuXH/7wh021FQAAAAAAAAAAAAAAAFoooTQA0AAdOnSojBctWlSvOZMmTarzvE2bNvnc5z5X75rt2rWrjJctW1bvedBS1dbWZvbs2Xn33XebuxUAgPXuzTffzNixYyuhMf37989ll13W4HVWB9p81KMoipRlmWuvvTbvvPNOI+8EAAAAAAAAAAAAAACAlqxVczcAAC1Jt27dkqw64Pniiy9m5cqVqar66Iy3xx57rM7zz3/+82nVqv5/BS9cuLAybt++ff2bhRaiLMvceuutueWWWzJx4sQsWbKk8l6vXr0ybNiwHHnkkfmXf/mX5msSAGA9ufvuu1NbW5skKYoi3/3ud9O6det6z18dNvPQQw995HV/+9vfcvzxxydJFixYkDvvvDMjR45c+8YBAAAAAAAAAAAAAAD4RBFKAwANMHDgwMr47bffzuOPP57ddtvtI+c88MADKYqicjh0yJAhDao5b968yrhLly4NaxiaWFmWufvuu1OWZZKkuro6NTU19Z7/4osvZsSIEZkyZUplvTW9/vrrufHGG3PjjTdmxIgRuf7669OxY8fG2wAAwAbmtttuq4z79euXo48+eq3W+eIXv/ix799888155JFHkiS33nqrUBoAAAAAAAAAAAAAAAAqqpq7AQBoSXbYYYe0atUqRVEkSa6++uqPvP7RRx/Nyy+/XOe1vfbaq971Zs2alTfeeKPyvG/fvg3oFpreE088kQMPPDDDhw/P8OHDc91119V77owZM7LbbrtlypQpKcuyEtz03sfq9377299m2LBhWbp0aRPuCACgeT388MOV+6ARI0akurq6yWp97WtfS7IqGPCxxx5rsjoAAAAAAAAAAAAAAAC0PEJpAKAB2rVrl3322acSknHLLbfk/vvv/9DrzznnnDrPu3TpkqFDh9a73sSJE+s833LLLRvWMDSxe++9N8mqg8xJ8v/+3/+r17yyLDNixIjMnj07Sd4XQLPmY833Hn/88XznO99pms0AADSzmTNnZu7cuZV7q3333bdJ6w0bNqwynj17dl555ZUmrQcAAAAAAAAAAAAAAEDLIZQGABrom9/8ZpJVIRorV67MV7/61Vx88cVZsGBB5ZoXXnghI0eOzEMPPVQJ0yiKIkcccURat25d71oPPPBAZdytW7f07du38TYCjeCPf/xjZfzZz342Q4YMqde8G264IU899VSKoqi8VlVVlVGjRuW+++7L9OnT89RTT2XcuHHZZpttKt+hsizzi1/8IlOnTm30vQAANLcpU6ZUxlVVVdlzzz2btF7//v3ToUOHyvNnnnmmSesBAAAAAAAAAAAAAADQcgilAYAGOvDAA7PXXntVQjKWL1+eM888Mz179kzv3r3Ts2fPDBw4MHfccUedee3atctZZ51V7zpvvvlmbr/99hRFkaIossceezT2VmCdlGVZCZYpiiIHH3xwvedeeumlddaprq7OnXfemWuvvTZDhw7NlltumcGDB2f06NH585//nIMPPjhlWSZJVq5cmeuvv77R9wMA0Nzmzp1bGffo0SOtWrVq8pqf+cxnKuM33nijyesBAAAAAAAAAAAAAADQMgilAYC1MH78+PTp0ydJUhRFyrLMihUrMnv27MybNy9lWVZCa1Zf86Mf/agypz5++9vfZvHixZUgjr322qvxNwLr4IUXXkhtbW3lM7rvvvvWa95TTz2VqVOnVr47RVHktNNOS01NzQde37p169x8883p379/Zc5tt93WaPsAANhQLFy4MMmq3w+bbLJJg+a2adMmPXr0SI8ePdK9e/d6z+vUqVNlvGDBggbVBAAAAAAAAAAAAAAA4JNLKA0ArIW+ffvm4Ycfzj/90z9VQjU+6LE6nOacc87JmDFj6r1+WZa56KKLKqE2SXLggQc2xVZgrT3//POVcVVVVXbcccd6zbv33nvrPG/Tpk2+853vfOSctm3b5vTTT68E4Lz++ut57bXXGtgxAMCGbcmSJZVx27ZtGzR3yJAhmTt3buVRX2v+5qitrW1QTQAAAAAAAAAAAAAAAD65hNIAwFoaMGBApkyZkrFjx2aHHXaoBNCsfrRt2zY1NTX505/+lPPOO69Ba99666159tlnKwEcgwYNSv/+/Rt/E7AOZsyYURlvuumm2Wijjeo1b8KECZVxURQZNmxYunXr9rHzhg8fXpmTJFOmTGlAtwAAG77VQTRlWWbevHnrpeaaddq0abNeagIAAAAAAAAAAAAAALDha9XcDQBAS9aqVaucfPLJOfnkk7NgwYLMnDkztbW16dq1awYMGFA5VNpQQ4cOzYsvvlh53rFjx8ZqGRrN4sWLk6wKienevXu95pRlmUmTJqUoipRlmaIoss8++9Rrbu/evdOzZ8/MmTMnRVHktddeW+veAQA2RF27dq2M11cozdy5cyvj+gQFAgAAAAAAAAAAAAAA8OkglAYAGkm3bt0a7RBnjx490qNHj0ZZC5rK8uXLK+P6BjBNmzYtS5YsSVEUldd23333etfs3bt35syZkySpra2t9zwAgJagX79+lfHSpUszderUbLvttk1Wb/r06VmyZMkH1gcAAAAAAAAAAAAAAODTraq5GwAAoGXq0KFDZbxo0aJ6zZk0aVKd523atMnnPve5etds165dZbxs2bJ6zwMAaAkGDRqUJJUAvwcffLBJ6/3P//xPneef//znm7QeAAAAAAAAAAAAAAAALYdQGgAA1kq3bt2SJGVZ5sUXX8zKlSs/ds5jjz1W5/nnP//5tGrVqt41Fy5cWBm3b9++3vMAAFqCnj17Zosttkiy6h7r2muvbdJ61113XWU8YMCAbLLJJk1aDwAAAAAAAAAAAAAAgJZDKA0AAGtl4MCBlfHbb7+dxx9//GPnPPDAAymKImVZpiiKDBkypEE1582bVxl36dKlQXMBAFqCgw46KGVZJkmmTp2aX/7yl01S57bbbsuUKVNSFEWKosjw4cObpA4AAAAAAAAAAAAAAAAtk1AaAADWyg477JBWrVqlKIokydVXX/2R1z/66KN5+eWX67y211571bverFmz8sYbb1Se9+3btwHdAgC0DKNHj05VVVUlyO/kk0/O9OnTG7XG3/72t5x00kl1wgKPO+64Rq0BAAAAAAAAAAAAAABAyyaUBgCAtdKuXbvss88+KcsyZVnmlltuyf333/+h159zzjl1nnfp0iVDhw6td72JEyfWeb7llls2rGEAgBZgiy22yKGHHloJi1mwYEGGDRuWv/zlL42y/l//+tfsu+++mT9/fqXGIYcckoEDBzbK+gAAAAAAAAAAAAAAAHwyCKUBAGCtffOb30ySFEWRlStX5qtf/WouvvjiLFiwoHLNCy+8kJEjR+ahhx5KURSVw89HHHFEWrduXe9aDzzwQGXcrVu39O3bt/E2AgCwAbn44ovTvXv3JKvus1555ZXssssuueCCC1JbW7tWay5ZsiQXXnhhdt5557z88sspiiJJ0rVr11x88cWN1jsAAAAAAAAAAAAAAACfDEVZlmVzNwEAQMv1pS996X2BM1VVVdl4442zYsWKvPHGG0lSea8sy7Rv3z7PPfdc+vTpU68ab775Znr16lU5hH3AAQfkzjvvbKotAQA0u3vuuScHHXRQVq5cmeT/7qU22mijjBw5MjU1Ndlpp53Sr1+/D11jxowZefLJJ/Pf//3fuf3227N06dI692TV1dW54447csABB6yvbQEAAAAAAAAAAAAAANBCCKUBAGCdzJgxI7vttltef/31JKsOTH+Qoigq47Fjx2bMmDH1rvEf//Ef+frXv15Z59JLL80pp5yyDl0DAGz4br755nzjG9/Iu+++m+T/7rPWvK/q3LlzNt5443Tp0iUdOnTI0qVLs2jRorzxxhtZtGhR5bo155ZlmdatW+fqq6/OUUcdtR53BAAAAAAAAAAAAAAAQEshlAYAgHX2wgsv5IADDsi0adPqHJJe0+rbznPPPTfnnXdevdcuyzKf+9znMm3atJRlmaIo8vzzz6d///6N0DkAwIZt0qRJOeSQQzJjxozKfdZHhQB+XEBgWZbZbLPNctttt2XXXXdtmqYBAAAAAAAAAAAAAABo8aqauwEAAFq+AQMGZMqUKRk7dmx22GGHlGVZ59G2bdvU1NTkT3/6U4MCaZLk1ltvzbPPPls5YD1o0CCBNADAp8Yuu+ySyZMnZ8yYMWnfvn3lnqgoivc9Pur1sizTvn37jBkzJpMnTxZIAwAAAAAAAAAAAAAAwEcqyg/775MBAGAtLViwIDNnzkxtbW26du2aAQMGpG3btmu11htvvJElS5ZUnnfs2DE9evRorFYBAFqM+fPn58orr8xdd92VyZMnZ8WKFR87p7q6OoMHD86BBx6YE044Id27d18PnQIAAAAAAAAAAAAAANDSCaUBAAAAgBZm6dKleeyxx/L3v/898+fPz/z581NbW5tOnTqle/fu6d69e7bccst84QtfSIcOHZq7XQAAAAAAAAAAAAAAAFoYoTQAAAAAAAAAAAAAAAAAAAAAAFRUNXcDAAAAAAAAAAAAAAAAAAAAAABsOITSAAAAAAAAAAAAAAAAAAAAAABQIZQGAAAAAAAAAAAAAAAAAAAAAIAKoTQAAAAAAAAAAAAAAAAAAAAAAFQIpQEAAAAAAAAAAAAAAAAAAAAAoEIoDQAAAAAAAAAAAAAAAAAAAAAAFUJpAAAAAAAAAAAAAAAAAAAAAACoEEoDAAAAAAAAAAAAAAAAAAAAAECFUBoAAAAAAAAAAAAAAAAAAAAAACr+P1+4wYuiwnlcAAAAAElFTkSuQmCC",
"text/plain": [
""
]
},
"metadata": {},
"output_type": "display_data"
}
],
"source": [
"vlimit = np.max(np.abs(data))\n",
"linewidth = 2.\n",
"\n",
"h = ma.Heatmap(data, \n",
" linewidth=linewidth,\n",
" vmin=0, vmax=1,\n",
" height=1*len(gam_tasks), # height depends on the number of tasks\n",
" width=2*len(trajectory), # width depends on the number of ordered cell types\n",
" cmap='YlOrBr',\n",
" cbar_kws={'title' : 'Min-max scaled\\nmetabolic score', 'orientation' : 'horizontal'}, \n",
" )\n",
"\n",
"h.group_cols(cell_cat, order=['Basal', 'Proliferative / Folicular', 'Secretory / Luteal'], spacing=0.01)\n",
"h.add_top(mp.Chunk(['Basal', 'Prolif. / Folicular', 'Secretory / Luteal'], cell_palette, padding=30, fontsize=30, fontweight='bold', c='white'), pad=0.1)\n",
"h.add_left(mp.Colors(task_cat, palette=task_colors, label='', legend_kws={'title' : 'Metabolic task group'}, linewidth=linewidth, linecolor='white'), size=.5, pad=.1)\n",
"h.add_bottom(mp.Labels(cell_labels, fontsize=30, rotation=90, rotation_mode='anchor', ha='right', va='center'), pad=.1)\n",
"h.add_right(mp.Labels(task_labels, fontsize=30, rotation=0, rotation_mode='anchor', ha='left', va='center'), pad=.1)\n",
"h.add_dendrogram(\"left\", method='ward', metric='euclidean', size=1, linewidth=2., add_base=True, pad=0.05)\n",
"h.add_legends(stack_size=3, stack_by='col', align_legends='left')\n",
"h.render()\n",
"\n",
"# To save the figure, uncomment the following line\n",
"# h.save(f'{sc.settings.figdir}/Heatmap.pdf', dpi=300)"
]
}
],
"metadata": {
"kernelspec": {
"display_name": "Python (single_cell)",
"language": "python",
"name": "single_cell"
},
"language_info": {
"codemirror_mode": {
"name": "ipython",
"version": 3
},
"file_extension": ".py",
"mimetype": "text/x-python",
"name": "python",
"nbconvert_exporter": "python",
"pygments_lexer": "ipython3",
"version": "3.10.14"
},
"widgets": {
"application/vnd.jupyter.widget-state+json": {
"state": {},
"version_major": 2,
"version_minor": 0
}
}
},
"nbformat": 4,
"nbformat_minor": 5
}